

Here, Meng et al. investigated how interferon regulatory factor 3 (IRF3) activation, a key signal mediator/transcriptional factor of the antiviral-sensing pathway, is regulated. They demonstrate that Mst1, a stress response kinase, represses cytosolic antiviral sensing and defense through the repression of RNA virus-induced activation of TBK1 and interference with the IRF3 homodimerization and chromatin binding via direct phosphorylation of IRF3 Thr253 and Thr75 residues.
Keywords: Mst1, IRF3, TBK1, phosphorylation, host antiviral defense
Abstract
Cytosolic RNA/DNA sensing elicits primary defense against viral pathogens. Interferon regulatory factor 3 (IRF3), a key signal mediator/transcriptional factor of the antiviral-sensing pathway, is indispensible for interferon production and antiviral defense. However, how the status of IRF3 activation is controlled remains elusive. Through a functional screen of the human kinome, we found that mammalian sterile 20-like kinase 1 (Mst1), but not Mst2, profoundly inhibited cytosolic nucleic acid sensing. Mst1 associated with IRF3 and directly phosphorylated IRF3 at Thr75 and Thr253. This Mst1-mediated phosphorylation abolished activated IRF3 homodimerization, its occupancy on chromatin, and subsequent IRF3-mediated transcriptional responses. In addition, Mst1 also impeded virus-induced activation of TANK-binding kinase 1 (TBK1), further attenuating IRF3 activation. As a result, Mst1 depletion or ablation enabled an enhanced antiviral response and defense in cells and mice. Therefore, the identification of Mst1 as a novel physiological negative regulator of IRF3 activation provides mechanistic insights into innate antiviral defense and potential antiviral prevention strategies.
Metazoans have developed innate defense mechanisms to resist pathogen infection, which are initiated by recognizing pathogen-associated molecular patterns (PAMPs) and followed by cellular host defense countermeasures. In the case of viral infection, viral RNA is sensed by cytosolic RIG-I-like receptors (RLRs) (Akira et al. 2006; Wu and Chen 2014; Chan and Gack 2015; Yoneyama et al. 2015) or endosomal Toll-like receptors (TLRs) (Gilliet et al. 2008), whereas viral DNA is recognized by various cytosolic sensors such as cGAS (Civril et al. 2013; Gao et al. 2013; Li et al. 2013; Sun et al. 2013), IFI16 (Unterholzner et al. 2010), and DDX41 (Zhang et al. 2011). Facilitated by either mitochondria-associated MAVS (also known as VISA, IPS-1, or Cardif) or endoplasmic reticulum-associated STING (also known as ERIS, MITA, MPYS, or TMEM173), binding of viral dsRNA or DNA to cytosolic sensors leads to activation of the Tank-binding kinase 1 (TBK1) and/or IκB kinase-related kinase ε (IKKε), which phosphorylate and thus activate the signaling mediator IRF3 (Fitzgerald et al. 2003; Sharma et al. 2003). Cytosolic RNA/DNA sensing also induces the NF-κB pathway, which often leads to production of proinflammatory cytokines (Akira et al. 2006; Chan and Gack 2015). IRF3 and NF-κB cooperate to activate interferon-β expression, which initiates an antiviral response for production of hundreds of interferon-stimulated genes (ISGs) to establish an antiviral state for both survival in acute viral infection and modulation of adaptive immune responses (Akira et al. 2006; Wu and Chen 2014; Chan and Gack 2015).
Cytosolic RNA/DNA-sensing pathways induce a potent and danger response to host cells. Thus, sensing of cytosolic nucleic acid is tightly regulated by multiple consecutive processes, such as (de)phosphorylation, ubiquitylation, and oligomerization of CARD domains of RLR and MAVS (Gack et al. 2007, 2010; Nistal-Villan et al. 2010; Zeng et al. 2010; Jiang et al. 2012; Wies et al. 2013). IRF3 serves the key signal mediator and transcription factor for cytosolic RNA/DNA sensing and antiviral defense. Upon C-terminal phosphorylation by TBK1 and/or IKKε, IRF3 homodimerizes and translocates into the nucleus, where they form a functional transcriptional complex by recruiting the coactivators of CBP/p300 and GRIP1 to elicit potent transcription (Yoneyama et al. 1998; Reily et al. 2006). Even though a few negative regulation loops of cytosolic RNA/DNA sensing have been proposed (Chen et al. 2013), still little is known regarding the termination of antiviral response that is important for cell survival after the acute viral infection. It is also unclear whether there is a layer of regulation on IRF3 itself and how the activation status of IRF3 is maintained or terminated during antiviral response.
Mammalian sterile 20-like kinase 1 (Mst1; also known as STK4 or KRS2) is a ubiquitously expressed and highly conserved serine/threonine kinase in the Mst family. Mst1 is a critical regulator in various cellular processes, including morphogenesis, proliferation, stress response, apoptosis, and immune cell development (Ling et al. 2008; Thompson and Sahai 2015). Mst1 along with Mst2 also constitute core components of the mammalian Hippo/YAP pathway, which includes Mst1/2 and Lats1/2 kinases and their downstream effectors, YAP and TAZ transcription coactivators (Zhou et al. 2009; Song et al. 2010; Zhao et al. 2010). Mst1/2 are activated by cell and tissue architecture even though the underlying mechanism is still less known in mammalian cells. Cellular oxidative stress also leads to activation of Mst1/2 through a few distinct pathways involving thioredoxin-1 (Trx1) (Chae et al. 2012) and peroxiredoxin-1 (Prdx1) (Morinaka et al. 2011). The major substrates of Mst1/2 are Lats1/2 and Mob1a/b, but other substrates have also been reported. Under stress conditions, Mst1/2 phosphorylate the FOXO transcription factors to protect cells from oxidative stress (Lehtinen et al. 2006). LC3, one of the main players in autophagy, was also recently identified as a substrate of Mst1/2 (Wilkinson and Hansen 2015). Most intriguingly, loss of Mst1 renders mice predisposed to autoimmune disorders (Du et al. 2014; Fukuhara et al. 2015; Halacli et al. 2015; Thompson and Sahai 2015), and human studies suggest that single-nucleotide polymorphisms (SNPs) in Mst1 are associated with both Crohn's disease and colitis (Waterman et al. 2011; Nimmo et al. 2012). These observations indicate a potential negative function of Mst1 in autoimmunity and host defense.
In this study, we discovered that Mst1 inhibited the functions of the IRF3 transcriptional factor and TBK1 kinase, two key components in antiviral defense. The Mst1-mediated phosphorylations reduced the ability of IRF3 homodimerization and chromatin binding. Activation of endogenous Mst1 or ectopic Mst1 expression retarded the signaling of cytosolic RNA/DNA sensing and weakened antiviral defense in cultured cells and zebrafish. Furthermore, knockout or silencing of Mst1 expression significantly enhanced antiviral sensing and resistance to virus infections. This study provides molecular insights for Mst1-driven regulation of cytosolic RNA/DNA sensing, and therefore reveals a physiological function of Mst1 in innate antiviral immunity.
Results
Mst1 attenuates cytosolic RNA/DNA sensing
In an attempt to systemically analyze phosphorylation events in cytosolic nucleic acid sensing, we screened a cDNA library comprised of the human kinome (92% coverage) by measuring a MAVS- or STING-stimulated pathway. A reporter assay with the IRF3-responsive IFNβ promoter revealed that the Mst family of STE stress kinases, particularly Mst1 but not Mst2, strongly inhibited the IFNβ reporter activity stimulated by RIG-I-N (caRIG-I) (Fig. 1A). Mst1, the kinase well defined in Hippo/YAP signaling, exhibited a striking inhibition at low levels (5–10 ng of plasmids per 106 cells) on either caRIG-I- or STING-stimulated IRF3 activation on both IFNβ and the artificial ISRE promoter (5xISRE) in a dose-dependent manner (Fig. 1B,C). Mst1 also blocked IRF3 activity that is directly stimulated by TBK1 or IKKε, implying that Mst1 might function at the level of TBK1/IKKε or downstream (Fig. 1D).
Figure 1.

Mst1 attenuates cytosolic RNA/DNA sensing. (A) Transfection of Mst family stress kinases, particularly Mst1, elicited a strong suppression of the IRF3/7-responsive IFNβ promoter in HEK293 cells, which was stimulated by activated RIG-I (caRIG-I). The expression of tagged Mst1–4 and YSK1 were revealed by immunoblotting. n = 3 experiments. (*) P < 0.001, compared with control, by Student's t-test. (B,C) Dose-dependent inhibition of both IFNβ (B) and 5xISRE (C) reporters by low-level Mst1 expression in response to RIG-I or STING-stimulated activation in HEK293 cells. (D) Transfection of TBK1 or IKKε exerted a robust activation of the IRF3-responsive IFNβ reporter, which was suppressed by Mst1 expression, also in a dose-dependent manner. (E,F) Two kinase-dead forms of Mst1, K59R and T183A, did not inhibit the IRF3-responsive IFNβ promoter, which was stimulated by either activated RIG-I (E) or STING (F). n = 3 experiments. (*) P < 0.001, compared with caRIG-I without Mst1; (**) P < 0.001, compared with wild-type Mst1 expression, by Student's t-tests. (G) Mst1 did not inhibit Wnt or Hedgehog signaling, which was, respectively, activated by LiCl treatment or Gli1 cotransfection and assessed by reporter assays from the TCF or Gli1 promoter. (H) siRNA-mediated knockdown of Mst1 in HEK293 cells, as evidenced by anti-Mst1 immunoblotting, boosted IRF3 responsiveness by MAVS, or TBK1 stimulation. (I) Similarly, siRNA-mediated Mst1 depletion effectively enhanced the STING pathway. n = 3 experiments. (*) P < 0.01, compared with control siRNA, by Student's t-test. (J) Treatment of mouse embryonic fibroblasts (MEFs) with H2O2 decreased IRF3 phosphorylation at Ser396 and the formation of IRF3 homodimer in Native-PAGE, which was stimulated by Sendai virus (SeV) infection. (K) SeV infection induced the expression of antiviral genes IFIT1 and ISG15 in control or Mst1−/− MEFs. The effects of H2O2 in ISG inductions were significantly stronger in control MEFs than in Mst1−/− MEFs. (*) P < 0.01, compared with control MEFs; (**) P < 0.01, compared with SeV infection.
Point mutation of Lys59 to Arg (K59R) or of Thr183 to Ala (T183A) was known to catalytically inactivate Mst1 (Glantschnig et al. 2002). To verify the involvement of Mst1 kinase activity in antiviral sensing, we generated these two kinase-dead mutants and examined their effects on IRF3 responsiveness. The fact that kinase-dead Mst1 lost its most inhibitory effects indicates the necessity of Mst1 catalytic activity for suppression of IRF3 responsiveness when stimulated by activated RIG-I, STING (Fig. 1E,F), or kinases TBK1 or IKKε (Supplemental Fig. S1A). In contrast, Mst1 did not suppress IFNβ or ISRE promoter activities in resting cells (Supplemental Fig. S1B) or inhibit the TCF or Gli1 promoter upon activation of Wnt or Hedgehog, respectively (Fig. 1G). These data suggest that the inhibitory effects of Mst1 on cytosolic nucleic acid sensing are specific. Complementary to the overexpression experiments, siRNA-mediated knockdown of Mst1 in HEK293 cells could effectively promote IRF3 responsiveness by MAVS or STING (Fig. 1H,I), demonstrating an enhancement of intracellular RNA/DNA sensing in the absence of Mst1. Taken together, these observations suggest a negative role of Mst1 in cytosolic nucleic acid sensing in a manner dependent on its catalytic ability.
Mst1 can be physiologically activated by exposure to reactive oxygen species (ROS) (Graves et al. 1998). To verify the role of endogenous Mst1 in cytosolic nucleic acid sensing, we treated cells with H2O2 to activate Mst1. As shown in Figure 1J, H2O2 treatment reduced IRF3 Ser396 phosphorylation and dimerization in response to Sendai virus (SeV) infection, thereby supporting an inhibitory function of Mst1 in virus-induced IRF3 regulation. Consistently, we observed a lower expression of antiviral products such as well-defined ISGs, including IFIT1 and ISG15 (Akira et al. 2006; Xu et al. 2014), in response to combined treatment of SeV infection and H2O2 (Fig. 1K). However, only a marginal decrease of antiviral genes by H2O2 treatment was observed in Mst1−/− mouse embryonic fibroblasts (MEFs), while a similar level of IRF3 and TBK1 expression was detected (Supplemental Fig. S1C), illustrating that ROS-induced reduction of antiviral response was mainly mediated by Mst1. Together, these data indicate that Mst1 is a negative regulator for antiviral response and may be important for antiviral regulation in the ROS-rich microenvironment.
Mst1−/− cells and mice exhibit an enhanced antiviral response and antiviral defense
To assess Mst1 function in an innate antiviral response, we isolated MEFs from wild-type or sibling Mst1−/− mice and measured the mRNA production of antiviral proteins upon SeV infection. As shown in Figure 2A, a drastic induction of mRNA of antiviral products IFIT1, ISG15, IRF7, and IFNβ was detected by quantitative RT–PCR (qRT–PCR) at 12-h post-infection (hpi), and a significantly higher mRNA induction was seen in Mst1−/− MEFs, illustrating that the absence of Mst1 empowered antiviral response against SeV infection (Fig. 2A). To verify these effects, we also generated Mst1 knockout cell lines by CRISPR/Cas9 technology based on NMuMG, the mouse mammary gland epithelial cell (Fig. 2B, bottom panel). Mst1−/− NMuMG cells displayed a higher IRF3 transactivation in response to MAVS stimulation, and the enhanced IRF3 activation was reverted upon re-expression of Mst1 (Fig. 2B). SeV infection elicited a robust antiviral response in NMuMG cells, as increased antiviral products measured at 6 hpi (Fig. 2C, left panel). As expected, Mst1−/− NMuMG cells displayed substantially stronger antiviral responses (Fig. 2C).
Figure 2.

Mst1 negatively regulates host antiviral defense in primary/cultured cells and mice. (A) Antiviral response of control or Mst1−/− MEFs against SeV infection was measured by mRNA induction at 12 hpi of various ISGs, including IFIT1, ISG15, IRF7, and IFNβ. Mst1−/− MEFs exhibited stronger antiviral responses than control MEFs. (*) P < 0.05, compared with control MEFs, by Student's t-test. (B, bottom panel) Mst1−/− NMuMG epithelial cells were generated by the CRISPR/Cas9 method, and the Mst1 knockout (KO) and rescue were verified by immunoblotting. Cytosolic RNA-sensing signaling in Mst1−/− and control NMuMG was evaluated by IFNβ reporter assay. (Top panel) Stronger IRF3 transactivation was observed by Mst1 ablation but was reversed by the reintroduction of wild-type (WT) Mst1. (C) Antiviral response in Mst1−/− and control NMuMG cells to SeV infection at 6 hpi was measured by quantification of ISGs and IFNβ, and stronger antiviral responses were observed by Mst1 deletion in NMuMG cells. n = 4 Mst1−/− clones. (*) P < 0.01, compared with control NMuMG cells, by Student's t-test. (D) Mst1−/− and control NMuMG cells were infected with GFP-tagged vesicular stomatitis virus (gVSV). Reduced virus replication, as evidenced by a reduced level of GFP+ cells, was observed in Mst1−/− NMuMG cells by microscopy (top panels) or FACS assay (bottom panels). (E) Survival of ∼8-wk-old Mst1+/+ and Mst1−/− mice given intravenous tail injection of gVSV (2 × 107 plaque-forming units [pfu] per gram). n = 12 mice for each group. P < 0.01, by paired Student t-test. (F) Determination of gVSV loads in mouse organs at 12 hpi in Mst1+/+ and Mst1−/− mice, which were intravenously tail-injected, with gVSV. n = 3 mice for each group. (*) P < 0.01, compared with control Mst1+/+ group, by Student t-test.
To examine the effect of Mst1 deletion to cellular viral resistance, we infected NMuMG cells with GFP-tagged vesicular stomatitis virus (gVSV). By either microscopy or FACS analysis for GFP strength, a drastic reduction of GFP+ cells, which should have active VSV replication, was observed in Mst1−/− NMuMG cells (Fig. 2D), indicating a boosted virus resistance in cells without Mst1. Similarly, in the Mst1−/− MEFs, we observed a lower infection of Herpes simplex virus 1 (HSV-1), which was a DNA virus with double-integrated GFP and Luciferase ORFs (Supplemental Fig. S2A), suggesting that Mst1 knockout also promoted defense against the DNA virus. Expression of MAVS is known to activate antiviral response and endow cells with viral resistance (Seth et al. 2005). As expected, prior expression of MAVS drastically reduced gVSV replication in 293T cells (Supplemental Fig. S2B). However, coexpression of wild-type Mst1, but not its kinase-dead mutant, resulted in an impairment of MAVS-driven viral resistance and a restoration gVSV replication (Supplemental Fig. S2B). Taken together, Mst1 ablation enhances innate antiviral response and virus defense, proposing Mst1 as a negative regulator of antiviral defense at the cellular level.
To evaluate the physiological function of Mst1 for antiviral defense in vivo, we challenged wild-type and Mst1−/− mice by intravenous tail injection of gVSV. As shown in Figure 2E, VSV infection resulted in rapid death of Mst1+/+ mice at the interval between 10 and 12 hpi, and no mice survived beyond 36 hpi. In contrast, Mst1−/− mice exhibited a significantly higher resistance to gVSV infection, as fewer mice died at 12 hpi, and some survived longer than 72 hpi. At the same time, a significantly milder gVSV replication and a stronger IFNβ and ISG production were recorded in Mst1−/− mice based on quantification of virus load in the livers, spleens, and lungs or mRNA in peripheral blood mononuclear cells (PBMCs) of sacrificed animals by qRT–PCR (Fig. 2F; Supplemental Fig. S2C), illustrating an enhancement of antiviral defense in mice without Mst1.
Forced Mst1 expression sensitizes zebrafish to VSV infection
Knowing that Mst1 ablation has a drastic effect on antiviral response and defense, we next attempted to verify Mst1's physiological function by ectopic expression in an additional animal model. We developed a strategy in zebrafish to have a fast and real-time assessment of antiviral gene function by combining genetic manipulation and microinjection of GFP-tagged VSV at the early stage of zebrafish embryogenesis. We thus ectopically expressed mammal Mst1 or MAVS in zebrafish embryos by mRNA microinjection at the one-cell stage followed by gVSV infection 48-h post-fertilization (hpf). As shown in Supplemental Figure S2D, zebrafish embryos underwent a visible VSV infection mainly in brain tissue but also in muscle and gut tissues. These infections started to cause embryo death from 48 hpi. Consistent with our findings in cultured cell, MAVS expression promoted resistance of embryos to VSV infection, as shown by ∼20% fewer embryo deaths, while Mst1 expression markedly decreased the resistance of embryos to VSV infection (Supplemental Fig. S2E), supporting a potential role of Mst1 in the suppression of antiviral defense in zebrafish and suggesting a cross-species function of Mst1 on antiviral defense.
Mst1 suppresses RNA virus-induced TBK1/IKKε activation
To unveil the molecular basis for Mst1-mediated inhibition of cytosolic RNA/DNA sensing, we first analyzed the effect of Mst1 on virus-induced activation of TBK1 and IKKε. Upon infection of SeV or HSV-1, we detected a stronger activation of TBK1 in Mst1−/− MEFs, as indicated by the enhanced level of phospho-Ser172 on TBK1 (Fig. 3A; Kishore et al. 2002). Consisting with this, we observed an increase of VSV-induced activation of TBK1 in Mst1−/− NMuMG epithelial cells, which was generated by the CRISPR/Cas9 technique (Fig. 3B). siRNA-mediated knockdown of Mst1 in HEK293 cells also confirmed an enhanced activation of TBK1, as evidenced by the IRF3-responsive reporter and TBK1 Ser172 phosphorylation (Supplemental Fig. S3A). All of the above observations reveal an enhanced level of virus-induced TBK1 activation in the absence of endogenous Mst1.
Figure 3.

Mst1 suppresses RNA virus-induced activation of TBK1. (A) Control and Mst1−/− MEFs were subjected to infection by SeV or HSV-1, and virus-induced activation of endogenous TBK1 was visualized by immunoblotting of Ser172 phosphorylation. Stronger TBK1 activation was seen with Mst1 knockout (KO). (B) Similarly, VSV infection-induced activation of endogenous TBK1, also visualized by Ser172 phosphorylation, was stronger in Mst1−/− NMuMG cells. (C) TBK1 activation detected by immunoblotting of phospho-Ser172 (first panel) and IRF3 activation detected by immunoblotting of phospho-Ser396 (third panel) or phospho-Ser386 (fourth panel) were eliminated by cotransfection of wild-type Mst1 (WT) but not by kinase-dead Mst1 mutants (K59R or T183A). (Fifth panel) Also, an obvious mobility shift of IRF3 was seen after coexpression of wild-type Mst1. (D) Similar Mst1-mediated loss of IRF3 phosphorylation and mobility shift were observed when IRF3 was stimulated by activated RIG-I (caRIG-I). (E,F) IKKε activation, which can be recognized by the anti-TBK1 phospho-S172 antibody, was dose-dependently inhibited by Mst1 (E) but not by its kinase-dead mutants (F). (G) Cotransfection of Mst1 with increasing levels of TBK1 showed that Mst1 still suppressed IRF3 transactivation even though Mst1 could not abolish TBK1 activation when it was expressed overwhelmingly. n = 3 experiments. (*) P < 0.001, compared with TBK1 alone control, by Student's t-test.
Intriguingly, we observed that cotransfection of wild-type Mst1 completely suppressed TBK1 autophosphorylation and activation, while kinase-dead Mst1 had only a minor effect (Fig. 3C; Supplemental Fig. S3B), suggesting that kinase activity of Mst1 is required to inhibit TBK1 activation. Furthermore, we observed an obvious mobility shift of IRF3 in the presence of wild-type but not kinase-dead Mst1, implying an Mst1-mediated modification of IRF3 (Fig. 3C, fifth panel). As expected, TBK1-induced IRF3 activation, revealed by IRF3 phospho-S396 and phospho-S386 residues, was lost in the presence of wild-type Mst1 (Fig. 3C; Supplemental Fig. S3B). IRF3 could be robustly activated by coexpression of activated RIG-I (caRIG-I) or MAVS. We found that coexpression of Mst1 strongly blocked IRF3 activation by RIG-I or MAVS (Fig. 3D; Supplemental Fig. S3C). A similar effect was also seen on IKKε activation, whereas the kinase-dead Mst1 showed only marginal effects on IKKε activation (Fig. 3E,F). Together, all of these observations propose that Mst1 expression blocks TBK1/IKKε activation and also leads to IRF3 modification even though how Mst1 interferes with TBK1 activation is currently unknown. More intriguingly, as shown in Figure 3G, we detected a striking suppression of IRF3 transactivation along with the loss of TBK1 activation in the presence of Mst1. However, the Mst1-mediated blockade of IRF3 responsiveness was still robust even though Mst1 cannot effectively disrupt TBK1 activation when TBK1 was overwhelmingly expressed (Fig. 3G, third and fourth columns). These data indicate the presence of additional mechanisms for Mst1-driven suppression.
Mst1 associates with and directly modifies IRF3
As shown in Figure 3, C and D, we observed a striking mobility shift of IRF3 in the presence of Mst1, suggesting a possible connection between Mst1-guided IRF3 modification and functional inhibition. To confirm this possibility, we examined the effect of Mst1 on constitutively active IRF3. Phosphomimetic mutation on the C terminus of IRF3 (IRF3 5SD) endowed IRF3 activity in transcription (Lin et al. 1998). As expected, IRF3 5SD drove a robust transcription on the IRF3-responsive promoter (Fig. 4A). Intriguingly, we found that Mst1, but not the kinase-dead mutant, still drastically inhibited the transcriptional activity of IRF3 5SD (Fig. 4A). We also observed a strong mobility change of IRF3 forms on Phos-Tag SDS-PAGE by the presence of Mst1 expression (Fig. 4B), while λ phosphatase treatment eliminated this IRF3 mobility shift (Supplemental Fig. S4A). Likewise, an obvious mobility shift was still seen on IRF3 5SD in the presence of wild-type Mst1 (Fig. 4C, third lane). These results thus suggest an Mst1-driven modification on activated IRF3, which may abolish IRF3 transcriptional activity. Furthermore, we identified PPM1B as an IRF3 phosphatase for eliminating multiple phosphorylation modifications (Fig. 4C; W Xiang, R Zhou, and P Xu, unpubl.). PPM1B cotransfection could eliminate Mst1-driven IRF3 phosphorylation, as indicated by the IRF3 mobility shift (Fig. 4C, second and fourth lanes). Corresponding with its dephosphorylation, PPM1B completely rescued Mst1-mediated suppression of IRF3 5SD (Fig. 4D), thus strongly suggesting the critical role of dynamic phosphorylation in the regulation of IRF3 function by Mst1.
Figure 4.

Mst1 associates with and phosphorylates IRF3 to negate its transcriptional activity. (A) Expression of Mst1 wild-type (WT), but not its kinase-dead form, blocked transcriptional activity of constitutively active IRF3 (IRF3 5SD). n = 3 experiments. (*) P < 0.001, compared with IRF3 5SD alone; (**) P < 0.001, compared with wild-type Mst1, by Student's t-test. (B) Phos-Tag SDS-PAGE of transfected IRF3 showed the loss of a faster migrated band in the presence of wild-type Mst1 as another indication for Mst1-induced IRF3 modification. (C) Coexpression of wild-type Mst1 resulted in a clear mobility shift of IRF3 5SD (third lane), which can be negated by cotransfection of phosphatase PPM1B (fourth lane) that we identified as a phosphatase of IRF3 (second lane). (D) Coexpression of PPM1B completely restored Mst1-driven suppression of IRF3 5SD. n = 3 experiments. (*) P < 0.001, compared with wild-type Mst1, by Student's t-test. (E) Lats1/2 double-knockout (dKO) cells were generated by the CRISPR/Cas9 method and verified by immunoblotting of Lats1 expression and TAZ mobility shift and degradation. Cotransfection of Mst1 resulted in the loss of the IRF3 faster migrated band, as in the wild-type or Lats1/2 double-knockout cells. (F) Coimmunoprecipitation assay by differential tags showed the interaction between IRF3 and Mst1, which was enhanced in the presence of Mst1 adaptors such as RASSF proteins and SAV1. (G) Endogenous Mst1 and IRF3 interaction in NMuMG cells was detected by coimmunoprecipitation assay using an anti-Mst1 antibody and was visualized by IRF3 immunoblotting. Note the smaller molecular weight of IRF3 in mouse cells. (H) In vitro kinase assay with separately purified IRF3 and Mst1 displayed the signal of IRF3 phosphorylation in the presence of Mst1 adaptor protein SAV1. Mst1 autophosphorylation was also seen and was boosted by SAV1. (I) Amino acid sequences around the Thr75 and Thr253 residues of IRF3 were similar to a reported consensus motif for Mst1 (Miller et al. 2008). (J) Nano-liquid chromatography-tandem mass spectrometry (nano-LC-MS/MS) assay, which analyzed the modifications on purified IRF3 that was coexpressing with kinase-dead or wild-type Mst1, showed Mst1-induced phosphorylation of IRF3 at the Thr75 and Thr 253 residues as well as reduced phosphorylation at the Ser396 residue. (K) Transfection of phosphomimetic IRF3 mutants revealed that the transcriptional activity of IRF3 5SD was nearly lost when either Thr75 or Thr253 was phosphorylated to an extent comparable with Mst1 cotransfection. n = 3 experiments. (* and **) P < 0.001, compared with control of IRF3 5SD expression, by Student's t-tests. (L) Phosphatase PPM1B, which restored Mst1-driven suppression on IRF3 5SD, failed to restore the transcriptional activity of IRF3 5SD with mimetic phosphorylation on Thr75 or Thr253. (*) P < 0.001, compared with control of IRF3 5SD expression, by Student's t-tests.
To unveil whether Mst1 indirectly promotes IRF3 phosphorylation through its well-defined Lats1 and Lats2 kinases, we analyzed the Lats1/2 double-knockout HEK293 cells generated by the CRISPR/Cas9 strategy. While ablation of both Lats1/2 kinases diminished TAZ phosphorylation and blocked Mst1-mediated TAZ degradation, deletion of Lats1/2 had little effect on Mst1-induced IRF3 mobility shift (Fig. 4E). Indeed, by using coimmunoprecipitation, we detected an interaction between Mst1 and IRF3 5SD (Fig. 4F) even though Mst1 was known for weak interaction with its substrates (Chan et al. 2005). As expected, the interaction between Mst1 and IRF3 was increased in the presence of Mst1 adaptor proteins, including members of the RASSF family and SAV1, which are known to enhance Mst1 interaction with the substrate (Khokhlatchev et al. 2002; Callus et al. 2006; Polesello et al. 2006). The endogenous complex of Mst1 and IRF3 was also detected by coimmunoprecipitation in NMuMG cells (Fig. 4G). Moreover, we detected a phosphorylation signal, albeit weak, on purified IRF3 in an in vitro kinase assay in the presence of purified Mst1 and its adaptor, SAV1, suggesting IRF3 as a direct substrate of Mst1 (Fig. 4H). These data suggest a model in which Mst1 may physically associate with and directly phosphorylate IRF3, leading to functional inhibition of IRF3.
Mst1 phosphorylates IRF3 on Thr75 and Thr253 to abolish its function
Mst1 has been known to preferably modify a consensus sequence featured by a Thr residue along with basic residues at +2/+3 sites (Miller et al. 2008). Interestingly, proximal sequences of the Thr75 and Thr253 residues of IRF3 match the Mst1 consensus sequence (Fig. 4I). Subsequently, we performed a mass spectrometry analysis of purified IRF3 from cells in the presence of wild-type or kinase-dead Mst1 and revealed that both the Thr75 and Thr253 residues, among other sites, were phosphorylated by Mst1 (Fig. 4J). These data, coupled with observations from the in vitro kinase assay, proposed a direct Mst1-induced modification on IRF3 at the Thr75 and Thr253 residues. Furthermore, the Mst1-induced mobility shift, which was apparent on IRF3 5SD by Phos-Tag SDS-PAGE, was compromised when Thr253 was mutated into Asp (Supplemental Fig. S4B), illustrating that Thr253 modification caused Mst1-driven IRF3 mobility shift.
Strikingly, phosphomimetic mutations of singular (T75D or T253D) or double (T75D/T253D) Thr75 and Thr253 resulted in a complete loss of transcriptional activity of the constitutively active IRF3 (Fig. 4K) to a level comparable with Mst1 coexpression. Our data show the critical role of Thr75 and Thr253 phosphorylation in regulating of IRF3 function. The functional inactivation of all three phosphomimetic IRF3 mutants could not be rescued by PPM1B (Fig. 4L), further supporting an inhibitory role of Thr75 and Thr253 phosphorylation in IRF3 regulation. Intriguingly, IRF3 (T75D/T253D) had a stronger interaction with Mst1, implying phosphorylation-induced conformational changes (Supplemental Fig. S4C). Taken together, our data indicate that Mst1 directly modifies IRF3 on Thr75 and Thr253 residues, thereby inhibiting its transcription function.
Thr75/253 phosphorylation disrupts IRF3 homodimerization and DNA binding
Given the profound effect of Mst1-mediated IRF3 phosphorylation, we attempted to understand the underlying mechanism for phosphorylation-mediated IRF3 inhibition. We first focused on Thr253 phosphorylation. We found that the phosphomimetic IRF3 (T253D) could still be efficiently phosphorylated by TBK1 at its C terminus, as confirmed by in vitro kinase assay with purified TBK1 or cotransfection in cells (Fig. 5A,B), suggesting that Mst1-mediated IRF3 Thr253 phosphorylation did not interfere with IRF3 phosphorylation by TBK1. Intriguingly, we observed that IRF3 bearing the T253D mutation, despite being phosphorylated in the C-terminal region (Fig. 5C, first panel), could not be dimerized, as determined by the IRF3 homodimer band on a Native-PAGE gel (Fig. 5C, second panel). We observed an obvious IRF3 dimerization induced by virus infection, while a stronger effect of IRF3 homodimerization was observed in the Mst1−/− cells in response to SeV or VSV infection (Fig. 5D,E). Earlier studies showed that the R211A/R213A and K360A/R361A mutants of IRF3 were still phosphorylated by TBK1 at Ser385 and Ser386 positions, but no longer dimerized after phosphorylation (Qin et al. 2003; Takahasi et al. 2003). Notably, through structural modeling, we observed that Thr253 was located at the same interface as Arg211, Arg213, Lys360, and Arg361, and the Mst1-mediated Thr253 phosphorylation would likely generate both steric hindrance and electrostatic repulsion, thus blocking IRF3 dimer formation (Fig. 5F). The above observations strongly support a model in which Mst1-mediated Thr253 phosphorylation impairs IRF3 homodimerization that is critical for IRF3 function.
Figure 5.

Mst1-mediated IRF3 phosphorylation disrupts IRF3 homodimerization and DNA binding. (A,B) Phosphomimics of Thr253 did not prevent TBK1-mediated IRF3 phosphorylation on its C terminus, as measured by phospho-S396 immunoblotting of samples from the in vitro kinase assay (A) or from cell lysates (B). (C) Electrophoresis on a Native-PAGE gel exhibited a severely impaired formation of IRF3 dimerization when Thr253 was phosphomimicked (fourth lane). Note that there was a comparable level of IRF3 phospho-Ser396 modification (first panel) but with a drastically reduced IRF3 homodimer level on phosphomimetic T253D (second panel). (D,E) A SeV-induced (D) or VSV-induced (E) homodimer of endogenous IRF3 was observed and enhanced in Mst1−/− NMuMG cells, revealing a robust IRF3 homodimerization with Mst1 ablation. Note that E and Figure 3B were from the same experiment. (F) Ribbon representation of the C-terminal IAD of IRF-3 showing that Thr253, Arg211, Arg213, Lys360, and Arg361 are located at the same dimer interface. Thr253 phosphorylation by Mst1 would likely impair IRF3 dimer formation. (G) An immunofluorescence assay showed that IRF3 nuclear import, which was stimulated by MAVS cotransfection, was largely similar between the wild type and the T75D mutation. (H, second lane) In a biotin-labeled DNA pull-down assay, activated IRF3 (IRF3 5SD) was shown to bind the ISRE sequence. Mst1 coexpression weakens DNA binding of activated IRF3, while a phosphomimetic of the Thr75 residue (T75D) also displayed severely impaired binding to the ISRE sequence. As a control, Mst1 did not block binding of Smad3 to its SBE DNA sequence. (I) Ribbon representation of the IRF3:DNA complex (left panel) or the modeled Thr75 phosphorylated IRF-3:DNA complex (right panel). Side chain conformational changes of Thr75 were potentially disruptive for the IRF3 DNA binding by disturbing hydrogen bonds between Arg78 and nucleotides.
The Thr75 residue is proximal to the reported nuclear localization sequence (NLS) of IRF3, including Lys77/Arg78 (Kumar et al. 2000). We thus performed an immunofluorescence assay and found that the Thr75 phosphomimetic did not hinder MAVS-driven IRF3 nuclear translocation (Fig. 5G). Since the Thr75 residue is also located in the DNA-binding domain (DBD) (Lin et al. 1998; Panne et al. 2007), we examined whether Thr75 phosphorylation affects IRF3's DNA binding. By using an in vitro pull-down assay with the ISRE sequence, we indeed observed that the presence of Mst1 severely compromised IRF3 5SD to bind the ISRE element but did not affect the binding of Smad3 to the SBE element, which was included as a control (Fig. 5H). Intriguingly, the DNA-binding capability of activated IRF3 was severely diminished when the IRF3 Thr75 residue was mutated into Asp (T75D) to mimic its phosphorylation (Fig. 5H, fourth lane). Therefore, both observations support the negative regulation of Mst1 on IRF3 promoter binding through IRF3 Thr75 phosphorylation. Furthermore, we modeled the IRF3 DNA-binding surface close to the Thr75 residue according to the reported structure of the IRF3:DNA complex (Panne et al. 2007). As shown in Figure 5I, Thr75 phosphorylation would likely interfere with the Arg78 residue to disrupt its hydrogen bonds with nucleotides, thus forfeiting IRF3's DNA binding. All of these observations are consistent with the hypothesis that IRF3 is phosphorylated by Mst1 at both the Thr75 and Thr253 residues, which prevents homodimerization and promoter binding of activated IRF3, thus terminating its transcription function.
Here we propose a working model for Mst1 regulation on cytosolic RNA/DNA-sensing signaling by which Mst1 impedes TBK1/IKKε activation and directly phosphorylates IRF3 on the Thr75 and Thr253 residues to block IRF3 dimerization and promoter binding, thus serving as a physiological negative regulator in the antiviral response (Fig. 6).
Figure 6.
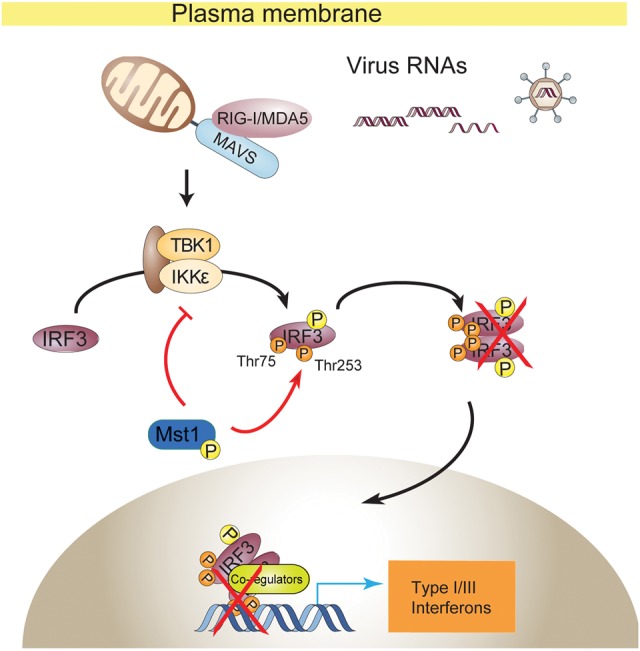
Model for Mst1-driven suppression of cytosolic RNA/DNA sensing and antiviral defense. Mst1 impedes cytosolic RNA/DNA sensing by a dual mechanism. Mst1 directly modified IRF3 at the Thr75 and Thr253 residues, which severely disrupted the activated IRF3 for homodimerization and binding to ISRE elements. Meanwhile, Mst1 also prevented the RNA virus-induced activation of TBK1 kinase by an unexplored manner, thus further keeping IRF3 at rest. In accordance with this, expression of Mst1 dampened antiviral host defense at both the cellular and whole-animal levels, while knockout or knockdown of Mst1 boosted the antiviral sensing and response.
Discussion
Extensive studies have proposed that host antiviral sensing is strictly regulated by a variety of intercellular molecules such as ubiqutin E3 ligases, kinases, phosphatases, and membrane-associated adaptor proteins. In the present study, we show that the stress kinase Mst1 (a key component of the Hippo/YAP pathway and a regulator of cell and tissue homeostasis) is an inherent repressor for signaling of cytosolic antiviral sensing. Through direct phosphorylation, Mst1 abolishes both DNA binding and homodimerization of IRF3, thereby blocking the cytosolic RNA/DNA-sensing signaling. Moreover, by two distinct animal models, we identified Mst1 as a negative physiological regulator of antiviral host defense. Forced Mst1 expression weakens zebrafish resistance against RNA virus infection, while mice with Mst1 ablation are more resistant to an RNA virus attack. Thus, our study reveals that the inherent level and activity of a stress kinase can integrate and coordinate the innate host defense and influence outcome of antiviral immunity.
Pathogenic nucleic acid is recognized in the cytoplasm by RIG-I-like receptors or sensor molecules such as cGAS, DDX41, and IFI16, which activate the response of a danger signal through adaptors MAVS or STING, respectively (Wu and Chen 2014). Our data show that the level and activity of Mst1 are important in the regulation of cytosolic RNA/DNA-sensing signaling through a dual mechanism for direct phosphorylation and inhibition of IRF3 as well as the impairment of TBK1 activation upon virus infection (Fig. 6). Mechanistically, Mst1-mediated phosphorylation at the IRF3 Thr75 residue disrupts IRF3's ability to bind to DNA elements, while phosphorylation on the Thr253 residue eliminates the capacity of IRF3 for homodimerization, a step necessary for its transcriptional function. It thus reveals a new mode of regulation of the cytosolic nucleic acid-sensing pathway.
The mechanism of Mst1 regulation on cytosolic nucleic acid sensing through IRF3
IRF3 is central for signaling of cytosolic nucleic acid sensing and thus the innate antiviral defense. Acting downstream from RIG-I and MDA5 that sense intracellular dsRNA, MAVS coordinates and links the activation of cytosolic RNA sensors to the activation of NF-κB and TBK1/IKKε-IRF3 for induction of antiviral cytokines such as type I and type III IFNs and a variety of ISGs (Wu and Chen 2014; Chan and Gack 2015; Yoneyama et al. 2015). However, very little has been explored regarding how activated IRF3 is regulated to maintain its appropriate level of activation, which is critical to the survival of host cells. Our hypothesis of Mst1-mediated inactivation of IRF3 thus presents a new mode of antiviral defense regulation to ensure the appropriate level of antiviral response by the direct effect of a stress kinase and new modification sites in IRF3. It may also link cellular environments with the host antiviral potential.
Inhibition of IRF3 function by Mst1 involves a dual mechanism; i.e., inhibition of IRF3 homodimerization and elimination of its promoter binding as a consequence of direct phosphorylation at distinct residues. Even though a mass spectrometry scan has suggested that Thr75 and Thr253 of IRF3 might be phosphorylated in cells (Shu et al. 2013), the function implications of the phosphorylation and the kinases responsible for Thr75 or Thr253 modification have not been explored. The Thr75 residue is located in the DBD of IRF3; thus, it is not surprising that its phosphorylation disrupts DNA binding. Indeed, through modeling based on the known IRF3:DNA structure (Panne et al. 2007), we revealed that Thr75 phosphorylation likely disrupts its proximal Arg78 to form hydrogen bonds to nucleotides, thus impairing IRF3's binding to DNA. In addition, by similar molecular modeling based on the IRF3 IAD structures (Qin et al. 2003; Takahasi et al. 2003), we found that Thr253 is located at the same dimer interface as basic residues Arg211, Arg213, Lys360, and Arg361. Mst1-mediated Thr253 phosphorylation likely generates both steric hindrance and electrostatic repulsion to block IRF3 dimer formation. These structure-based insights are consistent with our observations from biochemistry assays. As a result, Mst1 drives a robust inhibition of the signaling of cytosolic RNA sensing. Likewise, the phenotypes characteristic of viral resistance can be detected at both cellular and animal levels when Mst1 was genetically ablated.
Currently, still very little is known about how Mst1 activity and levels are regulated by intracellular conditions or extracellular cues. It is thought that, during tissue growth, Mst1/2 activity gradually increases as a result of changes in tissue structure and mechanics (Thompson and Sahai 2015). Rho signaling and actin stress fibers can also regulate Mst1/2 (Densham et al. 2009), and metabolic control of Mst1/2 signaling may also be achieved by phosphorylation of their adaptor protein, Sav, by Sik2 (Wehr et al. 2013). It will be interesting to examine cytosolic RNA/DNA sensing under these conditions.
Function of Mst1 in host antiviral defense and beyond
Our current data illustrate that Mst1 directly phosphorylates IRF3, rendering its loss on transcriptional function. Thus, Mst1 is identified as the first kinase that negatively regulates IRF3. Our data obtained from both mice and zebrafish consistently support the negative role of Mst1 in antiviral defense, which is also conserved from fish to mammals. Since MAVS, TBK1 and/or IKKε, and IRF3 are widely expressed and since IRF3 can also be activated by DNA damage, membrane fusion, and ER stress (Goubau et al. 2013; Collins and Mossman 2014), the repression of cytosolic RNA/DNA sensing and IRF3 responsiveness by Mst1 may substantially affect many cellular processes. In accordance with this, the level, activity, and subcellular distribution of Mst1 could be important factors in influencing the pathophysiology of MAVS and TBK1/IKKε as well as hundreds ISGs either directly or indirectly. The apoptosis-associated gene reprogramming, the metabolism alteration, and the release of proinflammatory cytokines that are features of virus infection may be particularly relevant to Mst1 status. For example, an increased apoptosis and inflammation by depletion of Mst1 has been observed in some systems (Thompson and Sahai 2015). Loss of Mst1 also renders mice predisposed to autoimmune disorders (Du et al. 2014; Fukuhara et al. 2015; Halacli et al. 2015; Thompson and Sahai 2015). Furthermore, SNPs in Mst1 are associated with both Crohn's disease and colitis (Waterman et al. 2011; Nimmo et al. 2012). Since host defense imbalance accounts for one of the main causes of autoimmune diseases, it is worthwhile to examine whether Mst1's regulation of host defense is integrated into these situations.
The function of Mst1 in the Hippo/YAP pathway has long been established. However, Mst1 also serves as an important regulator in multiple cellular processes. For example, Mst1 was found to directly phosphorylate the Ser14 residue of Bcl-xL, which facilitates Mst1-induced apoptosis (Del Re et al. 2014). Mst1 is also a major player in T-cell development even though controversial observations were made in ex vivo and in vivo studies (Zhou et al. 2008; Abdollahpour et al. 2012; Nehme et al. 2012). Mst1 also is involved in autophagy regulation by LC3 phosphorylation (Wilkinson and Hansen 2015). Thus, Mst1 serves as a key regulator of Hippo/Yap signaling, oxidative stress, T-cell development, autophagy, and apoptosis regulation. Our present study adds new dimensions to the biology of Mst1 in the negative control of cell host antiviral defense.
In conclusion, our study provides novel function and signal integration of a stress kinase in cytosolic RNA/DNA-sensing signaling through an unexplored mechanism and adds a new complexity to this regulation at the layer of IRF3. Our model indicates that the level and activity of Mst1 can be factors for host defense against RNA viruses.
Materials and methods
Expression plasmids, reagents, antibodies, and mice
Expression plasmids encoding Flag-, Myc-, HA-, or eGFP-tagged human TBK1, IRF3, IRF3 5SD, caRIG-I, MAVS, STING, IKKε, Smad3, and the IRF3/7-responsive reporters IFNβ_Luc and 5xISRE_Luc have been described (Xu et al. 2014). ORFs of Mst1-4 and YSK1 were obtained from the Invitrogen ORF Lite Clone Collection cDNA library by PCR, and Flag-, Myc-, or HA-tagged mouse Mst1 was constructed on pRK5 mammal expression vector. Site-directed mutagenesis to generate expression plasmids encoding Mst1 K59R, Mst1 T183A, and IRF3 5SD with T75, T253, or their combination replaced by Asp or Ala was performed using a kit from Stratagene. Detailed information will be provided on request. All coding sequences were verified by DNA sequencing. GFP and Luciferase double-tagged HSV-1 was a gift from Dr. Jiahuai Han (Xiamen University, Xiamen), and GFP-tagged VSV was a gift from Dr. Zhijian J. Chen (University of Texas Southwestern Medical Center, Dallas). SeV (Cantell strain) was from Charles River Laboratories. The monoclonal anti-Mst1, anti-IRF3, anti-pIRF3(S386), anti-pIRF3(S396), anti-TBK1, anti-pTBK1(S172), anti-HA, and anti-Myc antibodies were from Cell Signaling, and anti-α-tubulin and anti-Flag antibodies were from Sigma. Wild-type and Mst1−/− C57BL/6 mice have been described (Song et al. 2010).
Cell culture, transfections, and infections
MEFs and HEK293 and NMuMG cells were cultured in DMEM with 10% FBS. Primary wild-type or Mst1−/− MEFs were isolated, expanded, and cultured by conventional methods. Xtremegene HP (Roche) or Polythylenimine (PEI, Polyscience) transfection reagents was used for transfection. Infection of SeV, VSV, and HSV-1 was as described (Xu et al. 2014).
Luciferase reporter
Cells were transfected with IRF3/7-responsive IFNβ_Luc reporter, 5xISRE_Luc reporter, or other reporters (as indicated in the figures) bearing an ORF coding firefly luciferase, along with the pRL-Luc with Renilla luciferase coding as the internal control for transfection and other expression vectors as specified. After 24 h of transfection with indicated treatments, cells were lysed by passive lysis buffer (Promega). Luciferase assays were performed using a dual-luciferase assay kit (Promega), quantified with POLARstar Omega (BMG Labtech), and normalized to the internal Renilla luciferase control.
qRT–PCR assay
Cells were lysed, and total RNA was extracted using the RNAeasy extraction kit (Invitrogen). cDNA was generated using the one-step iScript cDNA synthesis kit (Bio-Rad), and quantitative real-time PCR was performed using the EvaGreen qPCR MasterMix (Abm) and CFX96 real-time PCR system (Bio-Rad). Relative quantification was expressed as 2−Ct, where −Ct is the difference between the main Ct value of triplicates of the sample and that of an endogenous L19 or GAPDH mRNA control. The mouse or VSV primer sequences used are listed in the Supplemental Material.
Coimmunoprecipitations and immunoblotting
HEK293T cells transfected with plasmids encoding C-terminal HA-tagged Mst1, Myc-tagged IRF3 5SD, and Mst1 adaptors or NMuMG cells were lysed using MLB buffer (Xu and Derynck 2010). Lysates were subjected to immunoprecipitation using anti-Mst1 (Cell Signaling), anti-Myc (Cell Signaling), anti-Flag (Sigma), or anti-HA (Sigma) antibodies for transfected proteins. Preclear procedure was used for coimmunoprecipitation of endogenous proteins. After three or four washes with MLB buffer, adsorbed proteins were analyzed by 10% SDS-PAGE and immunoblotting with the indicated antibodies. Cell lysates were also analyzed by SDS-PAGE and immunoblotting to control protein abundance.
RNAi
Double-stranded siRNA (RiboBio) to silence endogenous Mst1 expression in 293T cells targeted the human Mst1 mRNA sequences si-Mst1-a (5′-CCTCCAGGAGATAATCAAA-3′) and si-Mst1-b (5′-CCGCATCAGCACCGATTTA-3′). Cells were transfected with siRNA using Lipofectamine RNAiMAX (Invitrogen) for 48 h before further assay. The reverse transfection method was used to reach optimal efficiency.
In vitro kinase assay
293T cells were transfected with the Myc-TBK1, Flag-eGFP-IRF3, HA-SAV1, wild-type, or kinase-dead HA-Mst1 expression plasmids. Thirty-six hours after transfection, cells were lysed in MLB buffer, and immunoprecipitation was performed with anti-Myc, anti-Flag, or anti-HA antibody. Beads were washed three times with MLB buffer and once with kinase assay buffer (20 μM ATP or ATPγS, 20 mM Tris-HCl, 1 mM EGTA, 5 mM MgCl2, 0.02% 2-mercapto-ethanol, 0.03% Brij-35, 0.2 mg/mL BSA). Immunoprecipitated Myc-TBK1 or Flag-eGFP-IRF3 with or without wild-type or kinase-dead HA-Mst1 (with HA-SAV1) was incubated in kinase assay buffer for 60 min at 30°C on a Thermo-Shaker. EDTA and PNBM (Abcam) were then added, and the reaction was incubated for 30 min at 25°C with mild shaking. The reaction was stopped by addition of 2× SDS loading buffer and subjected to SDS-PAGE and immunoblotting with the indicated antibodies. The integration of γ-S was detected by immunoblotting with anti-thiophosphate ester antibody (Abcam).
DNA pull-down assay
The DNA probes labeled with biotin were synthesized by Sangon Biotech. The sequence of triple repeats of IRF3-recognized and bound ISRE contained biotin-3xISRE-F (biotin-TAGTTTCACTTTCCCTAGTTTCACTTTCCCTAGTTTCACTTTCCC) and 3xISRE-R (GGGAAAGTGAAACTAGGGAAAGTGAAACTAGGGAAAGTGAAACTA). Single strains of DNA were thermally annealed to form dsDNA prior to pull-down experiments. 293T cells were transfected with plasmids for expressing IRF3 5SD or IRF3 5SD-T75D in the absence or presence of Flag-Mst1 expression plasmid. Forty-eight hours after transfection, lysates were mixed with biotinylated DNA probe and incubated for 30 min at 25°C. Streptavidin sepharose beads were then added and incubated for another 15 min. The beads were washed three times with MLB and resolved by 2× SDS loading buffer for SDS-PAGE and immunoblotting.
Nano-liquid chromatography-tandem mass spectrometry (nano-LC-MS/MS) analysis
Nano-LC-MS/MS analysis for protein identification and label-free quantification was performed by Phoenix National Proteomics Core service as described (Ding et al. 2013). Tryptic peptides were separated on a C18 column and analyzed by LTQ-Orbitrap Velos (Thermo). Proteins were identified by using the search engine of the National Center for Biotechnology Information against the human or mouse National Center for Biotechnology Information RefSeq protein databases.
CRISPR/Cas9-mediated generation of knockout cells
CRISPR/Cas9 genomic editing for gene deletion was described (Ran et al. 2013). Guide RNA sequences targeting the Mst1 exon (set1, 5′-TGCAATCAAGCAAGTGCCCGTGG-3′; set2, 5′-AATGTGGGAGGAGGGTTTGTAGG-3′) were cloned into the plasmid pX330. Constructs, together with puromycin vector, were transfected into NMuMG or HEK293 by PEI. Twenty-four hours after transfection, cells were selected with 1.5 μg/mL puromycin for 72 h, and single colonies were obtained by serial dilution and amplification. Clones were identified by immunoblotting with anti-Mst1 or anti-Lats1 antibody, and four clones of Mst1−/− NMuMG cells and one clone of HEK293 Lats1/2−/− cells were used for the indicated analyses.
Ectopic expression of Mst1 and VSV challenge in zebrafish
We developed a system of GFP-tagged VSV challenge in zebrafish embryos to rapidly assess the gene function on antiviral defense. Forced expression of exogenous genes was obtained by microinjection of mRNA in the one-cell stage of embryogenesis; i.e., in the first 20 min after fertilization. At this stage, exogenous mRNAs distribute most evenly into the majority of cells through cell division and last for 72–96 h in zebrafish embryos (Beis and Stainier 2006; Xiong et al. 2014). Zebrafish embryos were incubated at 28.5°C in E3 egg water. mRNA of Mst1 and MAVS were in vitro transcribed with mMessage mMachine SP6 transcription kit (Life technology), and 25 pg of Mst1 or MAVS mRNA was injected into AB wild-type embryos. Injected embryos with normal development were selected and used for the gVSV virus injection (1 × 103 plaque-forming units [pfu] per embryo) in the embryo yolk at 48 hpf. The infection and death rate of injected embryos were recorded at the desired stages. To detect expression of Mst1 or MAVS by immunoblotting, tissue samples were homogenized and lysed in MLB buffer and subjected to SDS-PAGE and immunoblotting. Care of experimental animals was in accordance with Zhejiang University guidelines.
Mouse VSV challenge and measurement
Eight-week-old Mst1−/− and wild-type C57BL/6 mice (12 of each) were intravenously injected through their tails with GFP-tagged VSV at a dose of 2 × 107 pfu per gram of animal weight. The survival rate of injected mice was monitored at the indicated stages, as shown in the figures. In a parallel experiment, a total of six Mst1−/− and Mst1+/+ mice was injected with a half-dose of gVSV; all mice were sacrificed at 12 hpi, and organs (including the liver, lungs, and spleen) were collected. All organs were lavaged with 1 mL of cold PBS and snap-frozen in liquid nitrogen for RNA and protein extraction. Virus load was measured by qRT–PCR with primer targeting VSV. To assess the antiviral response to gVSV infection, Mst1–/– and wild-type mice (five of each) were intravenously tail-injected at a dose of 1 × 107 pfu per gram of gVSV and sacrificed at 6 hpi. The PBMCs were isolated from animal blood using Percoll (Sigma), and mRNA expression of ISGs was analyzed by qRT–PCR. Care of experimental animals was in accordance with Zhejiang University guidelines.
Immunofluorescence and microscopy
To visualize the subcellular localization of IRF3 and MAVS, HEK293A cells were treated as indicated in the figures, fixed in paraformaldehyde, permeablized, and blocked by horse serum. Cell slides were then incubated sequentially with primary antibodies (anti-IRF3 and anti-MAVS) and Alexa-labeled secondary antibodies followed by extensive washing. Slides were then mounted with VectorShield and stained with DAPI (Vector Laboratories). Immunofluorescence images were obtained and analyzed using a Nikon Eclipse Ti inverted microscope.
Structural modeling
The crystal structures of the DBD of IRF3 bound to DNA and the C-terminal regulation domain of IRF-3 were downloaded from Protein Data Bank (ID: 2O6G and 3A77), analyzed, and modeled using Coot (Emsley and Cowtan 2004). Based on these structures, Thr75 and Thr253 were changed to phosphothreonine (TPO), and the side chain of Arg78 was manually modeled to form an electrostatic interaction with TPO75. The geometries of the modeled structures were refined using RefMac (Collaborative Computational Project, Number 4 1994). All structural illustrations were prepared with Pymol (http://www.pymol.org).
Statistics
Quantitative data are presented as mean ± standard error of mean from at least three independent experiments. Data are presented as fold change, or percentages were log-transformed before statistical analysis. When appropriate, statistical differences between groups were analyzed using an unpaired or paired Student's t-test by Sigmaplot 10.0. Differences were considered significant at P < 0.05.
Supplementary Material
Acknowledgments
We are grateful to Dr. Yingzi Yang for Mst1−/− mice, Dr. Jiahuai Han for the glHSV-1 virus, and Dr. Zhijian Chen for the gVSV virus. This research was supported by the Chinese Ministry of Science and Technology (MoST) 973 Program (2015CB553800 to P.X., and 2012CB966600 to X.-H.F.), the National Natural Science Foundation of China Project (81472665 to P.X. and 91540205 to X.-H.F.), and the Fundamental Research Funds for the Central Universities (to the Life Sciences Institute at Zhejiang University). P.X. is a scholar in the National 1000 Young Talents Program.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.277533.116.
References
- Abdollahpour H, Appaswamy G, Kotlarz D, Diestelhorst J, Beier R, Schaffer AA, Gertz EM, Schambach A, Kreipe HH, Pfeifer D, et al. 2012. The phenotype of human STK4 deficiency. Blood 119: 3450–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124: 783–801. [DOI] [PubMed] [Google Scholar]
- Beis D, Stainier DY. 2006. In vivo cell biology: following the zebrafish trend. Trends Cell Biol 16: 105–112. [DOI] [PubMed] [Google Scholar]
- Callus BA, Verhagen AM, Vaux DL. 2006. Association of mammalian sterile twenty kinases, Mst1 and Mst2, with hSalvador via C-terminal coiled-coil domains, leads to its stabilization and phosphorylation. FEBS J 273: 4264–4276. [DOI] [PubMed] [Google Scholar]
- Chae JS, Gil Hwang S, Lim DS, Choi EJ. 2012. Thioredoxin-1 functions as a molecular switch regulating the oxidative stress-induced activation of MST1. Free Radic Biol Med 53: 2335–2343. [DOI] [PubMed] [Google Scholar]
- Chan YK, Gack MU. 2015. RIG-I-like receptor regulation in virus infection and immunity. Curr Opin Virol 12: 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EH, Nousiainen M, Chalamalasetty RB, Schafer A, Nigg EA, Sillje HH. 2005. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene 24: 2076–2086. [DOI] [PubMed] [Google Scholar]
- Chen W, Han C, Xie B, Hu X, Yu Q, Shi L, Wang Q, Li D, Wang J, Zheng P, et al. 2013. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 152: 467–478. [DOI] [PubMed] [Google Scholar]
- Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, Hopfner KP. 2013. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498: 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Collins SE, Mossman KL. 2014. Danger, diversity and priming in innate antiviral immunity. Cytokine Growth Factor Rev 25: 525–531. [DOI] [PubMed] [Google Scholar]
- Del Re DP, Matsuda T, Zhai P, Maejima Y, Jain MR, Liu T, Li H, Hsu CP, Sadoshima J. 2014. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of Bcl-xL. Mol Cell 54: 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Densham RM, O'Neill E, Munro J, Konig I, Anderson K, Kolch W, Olson MF. 2009. MST kinases monitor actin cytoskeletal integrity and signal via c-Jun N-terminal kinase stress-activated kinase to regulate p21Waf1/Cip1 stability. Mol Cell Biol 29: 6380–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding C, Chan DW, Liu W, Liu M, Li D, Song L, Li C, Jin J, Malovannaya A, Jung SY, et al. 2013. Proteome-wide profiling of activated transcription factors with a concatenated tandem array of transcription factor response elements. Proc Natl Acad Sci 110: 6771–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Shi H, Li J, Dong Y, Liang J, Ye J, Kong S, Zhang S, Zhong T, Yuan Z, et al. 2014. Mst1/Mst2 regulate development and function of regulatory T cells through modulation of Foxo1/Foxo3 stability in autoimmune disease. J Immunol 192: 1525–1535. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. 2003. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4: 491–496. [DOI] [PubMed] [Google Scholar]
- Fukuhara T, Tomiyama T, Yasuda K, Ueda Y, Ozaki Y, Son Y, Nomura S, Uchida K, Okazaki K, Kinashi T. 2015. Hypermethylation of MST1 in IgG4-related autoimmune pancreatitis and rheumatoid arthritis. Biochem Biophys Res Commun 463: 968–974. [DOI] [PubMed] [Google Scholar]
- Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446: 916–920. [DOI] [PubMed] [Google Scholar]
- Gack MU, Nistal-Villan E, Inn KS, Garcia-Sastre A, Jung JU. 2010. Phosphorylation-mediated negative regulation of RIG-I antiviral activity. J Virol 84: 3220–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, et al. 2013. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153: 1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliet M, Cao W, Liu YJ. 2008. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol 8: 594–606. [DOI] [PubMed] [Google Scholar]
- Glantschnig H, Rodan GA, Reszka AA. 2002. Mapping of MST1 kinase sites of phosphorylation. Activation and autophosphorylation. J Biol Chem 277: 42987–42996. [DOI] [PubMed] [Google Scholar]
- Goubau D, Deddouche S, Reis e Sousa C. 2013. Cytosolic sensing of viruses. Immunity 38: 855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves JD, Gotoh Y, Draves KE, Ambrose D, Han DK, Wright M, Chernoff J, Clark EA, Krebs EG. 1998. Caspase-mediated activation and induction of apoptosis by the mammalian Ste20-like kinase Mst1. EMBO J 17: 2224–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halacli SO, Ayvaz DC, Sun-Tan C, Erman B, Uz E, Yilmaz DY, Ozgul K, Tezcan I, Sanal O. 2015. STK4 (MST1) deficiency in two siblings with autoimmune cytopenias: a novel mutation. Clin Immunol 161: 316–323. [DOI] [PubMed] [Google Scholar]
- Jiang X, Kinch LN, Brautigam CA, Chen X, Du F, Grishin NV, Chen ZJ. 2012. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 36: 959–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokhlatchev A, Rabizadeh S, Xavier R, Nedwidek M, Chen T, Zhang XF, Seed B, Avruch J. 2002. Identification of a novel Ras-regulated proapoptotic pathway. Curr Biol 12: 253–265. [DOI] [PubMed] [Google Scholar]
- Kishore N, Huynh QK, Mathialagan S, Hall T, Rouw S, Creely D, Lange G, Caroll J, Reitz B, Donnelly A, et al. 2002. IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2: comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. J Biol Chem 277: 13840–13847. [DOI] [PubMed] [Google Scholar]
- Kumar KP, McBride KM, Weaver BK, Dingwall C, Reich NC. 2000. Regulated nuclear-cytoplasmic localization of interferon regulatory factor 3, a subunit of double-stranded RNA-activated factor 1. Mol Cell Biol 20: 4159–4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB, DiBacco S, de la Iglesia N, Gygi S, Blackwell TK, Bonni A. 2006. A conserved MST–FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 125: 987–1001. [DOI] [PubMed] [Google Scholar]
- Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. 2013. Pivotal roles of cGAS–cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341: 1390–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Heylbroeck C, Pitha PM, Hiscott J. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol 18: 2986–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling P, Lu TJ, Yuan CJ, Lai MD. 2008. Biosignaling of mammalian Ste20-related kinases. Cell Signal 20: 1237–1247. [DOI] [PubMed] [Google Scholar]
- Miller ML, Jensen LJ, Diella F, Jorgensen C, Tinti M, Li L, Hsiung M, Parker SA, Bordeaux J, Sicheritz-Ponten T, et al. 2008. Linear motif atlas for phosphorylation-dependent signaling. Sci Signal 1: ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinaka A, Funato Y, Uesugi K, Miki H. 2011. Oligomeric peroxiredoxin-I is an essential intermediate for p53 to activate MST1 kinase and apoptosis. Oncogene 30: 4208–4218. [DOI] [PubMed] [Google Scholar]
- Nehme NT, Pachlopnik Schmid J, Debeurme F, Andre-Schmutz I, Lim A, Nitschke P, Rieux-Laucat F, Lutz P, Picard C, Mahlaoui N, et al. 2012. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood 119: 3458–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmo ER, Prendergast JG, Aldhous MC, Kennedy NA, Henderson P, Drummond HE, Ramsahoye BH, Wilson DC, Semple CA, Satsangi J. 2012. Genome-wide methylation profiling in Crohn's disease identifies altered epigenetic regulation of key host defense mechanisms including the Th17 pathway. Inflamm Bowel Dis 18: 889–899. [DOI] [PubMed] [Google Scholar]
- Nistal-Villan E, Gack MU, Martinez-Delgado G, Maharaj NP, Inn KS, Yang H, Wang R, Aggarwal AK, Jung JU, Garcia-Sastre A. 2010. Negative role of RIG-I serine 8 phosphorylation in the regulation of interferon-β production. J Biol Chem 285: 20252–20261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panne D, Maniatis T, Harrison SC. 2007. An atomic model of the interferon-β enhanceosome. Cell 129: 1111–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polesello C, Huelsmann S, Brown NH, Tapon N. 2006. The Drosophila RASSF homolog antagonizes the hippo pathway. Curr Biol 16: 2459–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, Derynck R, Lin K. 2003. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol 10: 913–921. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR–Cas9 system. Nat Protoc 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reily MM, Pantoja C, Hu X, Chinenov Y, Rogatsky I. 2006. The GRIP1:IRF3 interaction as a target for glucocorticoid receptor-mediated immunosuppression. EMBO J 25: 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 122: 669–682. [DOI] [PubMed] [Google Scholar]
- Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300: 1148–1151. [DOI] [PubMed] [Google Scholar]
- Shu C, Sankaran B, Chaton CT, Herr AB, Mishra A, Peng J, Li P. 2013. Structural insights into the functions of TBK1 in innate antimicrobial immunity. Structure 21: 1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Mak KK, Topol L, Yun K, Hu J, Garrett L, Chen Y, Park O, Chang J, Simpson RM, et al. 2010. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc Natl Acad Sci 107: 1431–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP–AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahasi K, Suzuki NN, Horiuchi M, Mori M, Suhara W, Okabe Y, Fukuhara Y, Terasawa H, Akira S, Fujita T, Inagaki F. 2003. X-ray crystal structure of IRF-3 and its functional implications. Nat Struct Biol 10: 922–927. [DOI] [PubMed] [Google Scholar]
- Thompson BJ, Sahai E. 2015. MST kinases in development and disease. J Cell Biol 210: 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, et al. 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11: 997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman M, Xu W, Stempak JM, Milgrom R, Bernstein CN, Griffiths AM, Greenberg GR, Steinhart AH, Silverberg MS. 2011. Distinct and overlapping genetic loci in Crohn's disease and ulcerative colitis: correlations with pathogenesis. Inflamm Bowel Dis 17: 1936–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr MC, Holder MV, Gailite I, Saunders RE, Maile TM, Ciirdaeva E, Instrell R, Jiang M, Howell M, Rossner MJ, Tapon N. 2013. Salt-inducible kinases regulate growth through the Hippo signalling pathway in Drosophila. Nat Cell Biol 15: 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wies E, Wang MK, Maharaj NP, Chen K, Zhou S, Finberg RW, Gack MU. 2013. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 38: 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DS, Hansen M. 2015. LC3 is a novel substrate for the mammalian Hippo kinases, STK3/STK4. Autophagy 11: 856–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Chen ZJ. 2014. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol 32: 461–488. [DOI] [PubMed] [Google Scholar]
- Xiong F, Ma W, Hiscock TW, Mosaliganti KR, Tentner AR, Brakke KA, Rannou N, Gelas A, Souhait L, Swinburne IA, et al. 2014. Interplay of cell shape and division orientation promotes robust morphogenesis of developing epithelia. Cell 159: 415–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Derynck R. 2010. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol Cell 37: 551–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Bailey-Bucktrout S, Xi Y, Xu D, Du D, Zhang Q, Xiang W, Liu J, Melton A, Sheppard D, et al. 2014. Innate antiviral host defense attenuates TGF-β function through IRF3-mediated suppression of Smad signaling. Mol Cell 56: 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. 1998. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J 17: 1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Onomoto K, Jogi M, Akaboshi T, Fujita T. 2015. Viral RNA detection by RIG-I-like receptors. Curr Opin Immunol 32: 48–53. [DOI] [PubMed] [Google Scholar]
- Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, Chen ZJ. 2010. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 141: 315–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. 2011. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol 12: 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Lei Q, Guan KL. 2010. The Hippo–YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev 24: 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Medoff BD, Chen L, Li L, Zhang XF, Praskova M, Liu M, Landry A, Blumberg RS, Boussiotis VA, et al. 2008. The Nore1B/Mst1 complex restrains antigen receptor-induced proliferation of naive T cells. Proc Natl Acad Sci 105: 20321–20326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, Bardeesy N. 2009. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16: 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.