

Abstract
Human ether-a-go-go-related gene (hERG) channels conduct delayed rectifier K+ current. However, little information is available on physiological situations affecting hERG channel protein and function. In the present study we examined the effects of intermittent hypoxia (IH), which is a hallmark manifestation of sleep apnea, on hERG channel protein and function. Experiments were performed on SH-SY5Y neuroblastoma cells, which express hERG protein. Cells were exposed to IH consisting of alternating cycles of 30 s of hypoxia (1.5% O2) and 5 min of 20% O2. IH decreased hERG protein expression in a stimulus-dependent manner. A similar reduction in hERG protein was also seen in adrenal medullary chromaffin cells from IH-exposed neonatal rats. The decreased hERG protein was associated with attenuated hERG K+ current. IH-evoked hERG protein degradation was not due to reduced transcription or increased proteosome/lysomal degradation. Rather it was mediated by calcium-activated calpain proteases. Both COOH- and NH2-terminal sequences of the hERG protein were the targets of calpain-dependent degradation. IH increased reactive oxygen species (ROS) levels, intracellular Ca2+ concentration ([Ca2+]i), calpain enzyme activity, and hERG protein degradation, and all these effects were prevented by manganese-(111)-tetrakis-(1-methyl-4-pyridyl)-porphyrin pentachloride, a membrane-permeable ROS scavenger. These results demonstrate that activation of calpains by ROS-dependent elevation of [Ca2+]i mediates hERG protein degradation by IH.
Keywords: sleep apnea, oxidative stress, arrhythmia, apnea of prematurity, adrenal medullary chromaffin cells
human ether-a-go-go-related gene (hERG) channels conduct delayed rectifier K+ current (23). The hERG K+ channel has been implicated in a variety of cellular functions including the maintenance of resting membrane potential in excitable cells (2). Altered hERG channel function has dire physiological consequences. For instance, mutations in hERG channel protein underlie long-QT syndrome (LQT2), a cardiac repolarization disorder that predisposes to cardiac arrhythmia (5, 23). However, little information is available on physiological situations affecting hERG channel protein expression and function.
Hypoxia (i.e., decreased O2 availability) is a pervasive physiological stimulus that impacts a variety of cellular functions including ion channel function (11, 15). However, people living at sea level most often experience intermittent hypoxia (IH) as a consequence of sleep apnea (i.e., brief, repetitive cessation of breathing) (22). IH, mimicking O2 profiles encountered during sleep apnea, affects various cellular functions including activation of early genes (31), hypoxia-inducible factor (HIF)-1α (33), and voltage-gated Ca2+ channels (14). Sleep apnea patients and IH-exposed rodents exhibit cardiovascular abnormalities, including hypertension and cardiac arrhythmias (18, 30). Given that hERG K+ channels are implicated in cardiac arrhythmia (5, 23), we examined the effects of IH on hERG channel protein and function in neuroblastoma (SH-SY5Y) cells, which express high levels of endogenous hERG protein (2, 3). Our results demonstrate that IH decreases hERG protein and hERG-mediated K+ current and these effects are mediated by reactive oxygen species (ROS)-dependent activation of Ca2+-activated calpain proteases.1
MATERIALS AND METHODS
Exposure of cell cultures and rats to intermittent hypoxia.
Neuroblastoma (SH-SY5Y) cells were obtained from ATCC (no. CRL-2266). Cells were grown in DMEM medium supplemented with 10% FBS, 100 U/ml penicillin, and 100 mg/ml streptomycin and maintained at 37°C in 10% CO2. Cell cultures were exposed to IH 60 cycles (IH60) or 120 cycles (IH120) with each cycle consisting 1.5% O2 for 30 s followed by 20% O2 for 5 min at 37°C as described previously (33). Ambient O2 levels in the IH chamber were monitored by an O2 analyzer (Alpha Omega Instruments). Cells were pretreated for 30 min with either drug or vehicle in all experiments involving drug treatment.
Studies were performed on Sprague-Dawley rats of both sexes. Experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Chicago. Rat pups (ages P0–P10) along with their dams were exposed to IH between 9:00 AM and 5:00 PM for 5 days as described previously (19, 25, 26). Control experiments were performed on age-matched rat pups exposed to alternating cycles of room air instead of hypoxia.
hERG NH2-terminal; and/or COOH-terminal deletion plasmid constructs and transfections.
pcDNA3-full-length [wild-type (WT)] HA-tagged hERG plasmid was used as a template for PCR and cloned into pCRII-T/A vector (Invitrogen). The amino (NH2) terminus deletion construct pcDNA3-HA-ΔN300 was generated by amplifying ∼300-bp NH2-terminal flanking regions using primers HindIII-hERG-ΔN-F (5′-GCG GCG AAG CTT GCC ACC ATG TAT CCA TAC GAT GTA CCT GAC TAC GCA CGC CAC GCC AGC ACC GGG GCC ATG CAC-3′) and BstE2-hERG-ΔN-R (5′-CAG GAG CTG GGT GAC CTT CTC AGT GAC ATT GTG GGT TCG C-3′). The NH2/carboxy (COOH)-terminal deletion construct pcDNA3-HA-ΔN300/ΔC679 was generated by amplifying ∼900-bp COOH-terminal flanking regions using the primers hERG-CtD-XhoI-Stop Codon-F (5′-CGC CTC GAG TAG ATG AAT AAT ACT TCC AGC ACG CCT GGT CCT AC-3′) and hERG-CtD-XbaI-R (5′-GCG TCT AGA GTT GGA CAC TCC TGA GAA GGC GCC-3′). The PCR product was cloned into pCRII-T/A vector. This plasmid was cut by XhoI and XbaI and then ligated into pcDNA3-HA-hERG predigested by XhoI and XbaI. All plasmid DNA sequences were verified by DNA sequencing at the core facility of the University of Chicago. SH-SY5Y cells were transiently transfected with different plasmid constructs using TransT-2020 transfection reagent (Mirus Bio, Madison, WI) according to the manufacturer's instructions.
Immunoblot assay.
SH-SY5Y cells or adrenal medullary tissues were solubilized at 4°C in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100) containing a protease inhibitor cocktail (Sigma, St. Louis, MO). Protein concentrations were determined, and equal amounts of proteins were separated on a 6% SDS-PAGE gel and transferred to polyvinylidene difluoride membrane (PVDF). Membranes were blocked in 5% milk overnight and immunoblotted with specific primary antibodies, followed by horseradish peroxidase-conjugated secondary antibody. ECL (GE Healthcare) was used to develop the blots. Image densities were quantified using ImageJ (National Institutes of Health) by integrating pixel densities of individual protein bands.
Immunocytochemistry.
Neuroblastoma cells coated on coverslips were fixed with 4% formalin, blocked, and permeabilized in 5% BSA and 0.1% Triton X-100 for 30 min at room temperature. Rat pups were anesthetized with urethane (1.2 g/kg ip), and perfused transcardially with ice-cold heparinized PBS followed by 4% paraformaldehyde. Adrenal glands were harvested and mounted in OCT compound (Tissue Tek; VWR Scientific), and 8-μm sections were cut and stored at −80°C till further analysis. Sections were blocked with 20% normal goat serum in PBS with 0.2% Triton X-100 for 30 min. Permeabilized cells or adrenal gland tissues were incubated with anti-hERG antibody (1:2,000; Alamone Labs) for 2 h at 37°C, washed with PBS, and incubated with Alexa Fluor conjugated secondary antibody (1:250; Molecular Probes) for 1 h at 37°C. Cells were mounted in DAPI containing media and analyzed using a Nikon fluorescence microscope.
Real-time RT-PCR assay.
SH-SY5Y cells exposed to normoxia or IH were used to generate total RNA followed by first-strand cDNA. Aliquots of cDNA were used in quantitative real-time reverse transcription polymerase chain reaction (rt RT-PCR) with rat-specific primers detecting either hERG or18S using SYBR green as a fluorogenic binding dye as described previously (17). Relative mRNA level was calculated based on the comparative threshold method known as the 2−[ΔΔ]Ct method, where [ΔΔ]Ct = ΔCt (IH) − ΔCt (normoxia). The ΔCt values of both normoxia and IH are normalized to an internal standard (18S RNA).
Measurements of hERG K+ currents.
hERG currents were monitored in SH-SY5Y cells at room temperature by whole cell configuration of the patch-clamp technique as described previously (15). hERG currents were recorded using patch pipettes filled with solution containing (in mM): 100 K+-aspartate, 20 KCl, 2 MgCl2, 1 CaCl2, 10 EGTA, and 10 HEPES (pH 7.2). The extracellular solution was the following (in mM): 140 NaCl, 5 KCl, 1 MgCl2, 1.8 CaCl2, 10 HEPES, and 10 glucose (pH 7.4). Currents were elicited from a holding potential of −90 mV with depolarizing pulses to +50 mV in 10-mV step range. The current density was analyzed after maximal activation of hERG currents in a 2-s prepulse. Maximal tail current amplitudes measured at −90 mV were normalized to cell capacitance to compute hERG current densities.
Measurements of calpain activity.
Calpain enzyme activity was measured using Innozyme Calpain1/2 Kit (no. CBA054; Calbiochem) as described previously (17). Briefly, following exposure to IH, cells were lysed in 50 mM Tris·Cl (pH 7.4), 0.5 mM EDTA, 0.05% β-mercaptoethanol, 1 mm PMSF, and protease inhibitor cocktail (Sigma). Calpain activity was measured using purified calpain as standard. Enzyme activity was normalized to protein content and expressed as percentage of normoxic controls.
Measurements of intracellular Ca2+ concentration.
Intracellular calcium levels were monitored in SH-SY5Y cells using fura-2 AM as described previously (13). Background fluorescence was subtracted from signals. Image intensity at 340 nm was divided by 380-nm image intensity to obtain the ratiometric image. Ratios were converted to free intracellular Ca2+ concentration ([Ca2+]i) using calibration curves constructed in vitro by adding fura-2 (50 μM, free acid) to solutions containing known concentrations of Ca2+ (0–2,000 nM).
Measurement of malondialdehyde levels and aconitase enzyme activity.
Malondialdehyde (MDA) levels were determined as described previously (20) and expressed as nanomoles per milligrams of protein. Mitochondrial and cytosol fractions were isolated by differential centrifugation and aconitase activity was determined using aconitase assay kit (no. 705502; Cayman Chemical) as described previously (20). Protein concentration was estimated in the cell extracts using Bio-Rad protein assay kit. The enzyme activities were expressed as nanomoles per milligram of protein per minute.
Data analysis.
Data are expressed as means ± SE from three to five independent experiments. Statistical analysis was performed by one-way ANOVA, and P values <0.05 were considered as significant.
RESULTS
IH reduces hERG protein expression.
Previous studies showed that cell cultures exposed to 60 cycles of IH (IH60) activates early genes (31) and HIF-1-mediated transcriptional activity (33). Based on these studies, we chose to treat SH-SY5Y cells with IH60 and hERG protein expression was analyzed by immunoblot assay. Cells treated with IH60 showed ∼40% reduction in hERG protein compared with normoxia-treated control cells (Fig. 1A). Increasing the IH exposure to 120 cycles of IH (IH120) further decreased hERG protein by 80% (Fig. 1A), suggesting that IH decreases hERG protein in a stimulus dependent manner.
Fig. 1.
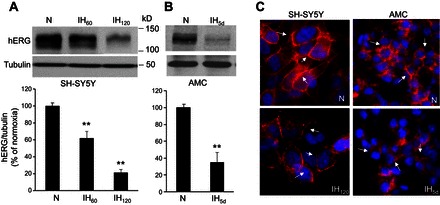
Intermittent hypoxia (IH) downregulates human ether-a-go-go-related gene (hERG) protein expression. A: representative immunoblot of hERG protein in SH-SY5Y cells exposed to either normoxia (N) or 60 and 120 cycles of IH (IH60 and IH120; top). Densitometric analysis (means ± SE; n = 5 independent experiments) of hERG protein normalized to tubulin and expressed as percentage of normoxia (bottom). B: representative immunoblot of hERG protein in adrenal medullary chromaffin cells (AMC) harvested from rat pups exposed to either normoxia or 5 days of IH (IH5d: top). Densitometric analysis (means ± SE; Normoxia and IH; n = 8 rat pups each) of hERG protein normalized to tubulin and expressed as percentage of normoxia (bottom). C: immunolocalization of hERG (red) in SH-SY5Y cells and AMC exposed to normoxia or IH. Blue: DAPI staining of nuclei. Arrows denote membrane localization of hERG. **P < 0.01, compared with normoxia.
Catecholamine secretion from adrenal medullary chromaffin cells (AMC) is augmented in rat pups exposed to 5 days of IH (25, 27). Since neonatal rat AMC express hERG protein (10), we investigated whether 5 days of IH affects hERG protein. As shown in Fig. 1B, hERG protein decreased by 65 ± 12% in IH-treated AMC compared with control tissue derived from rats reared under normoxia.
Immunocytochemical analysis showed localization of hERG-like protein primarily in the plasma membrane of both SH-SY5Y and AMC. The membrane expression of hERG decreased following IH exposure in both cell types (Fig. 1C). Based on these results, all further experiments were performed on SH-SY5Y cells to further assess the effects of IH on hERG K+ current and to delineate the mechanisms underlying hERG protein degradation.
IH reduces hERG-mediated K+ current.
To assess the effect of IH on hERG K+ channel function, K+ currents were recorded in SH-SY5Y cells in the presence and absence of 2 μM E-4031, a potent and selective blocker of hERG K+ channels (29). E-4031 blocked 70 ± 4% of the K+ current (n = 15 cells; P < 0.01), suggesting hERG channel mediates the majority of the observed K+ current. IH progressively decreased the hERG K+ current in a stimulus-dependent manner (Fig. 2A). Normalizing the current values to cell capacitance showed that hERG K+ current density decreased significantly with IH60 and IH120 compared with control cells exposed to normoxia (Fig. 2B).
Fig. 2.
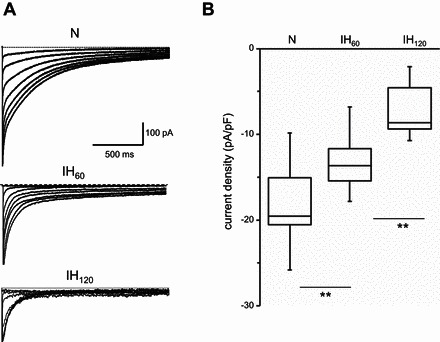
Effect of IH on hERG K+ conductance. A: representative whole cell currents recorded from SH-SY5Y cells grown in normoxia and in cells exposed to IH60 and IH120. B: HERG K+ current density was calculated by normalizing average peak tail current amplitude measured at −90 mV to cell capacitance (pA/pF) from cells exposed to normoxia (n = 15 cells), IH60 (n = 11 cells), and IH120 (n = 17 cells). To give a measure of data dispersion, data are presented in box charts, whiskers making the 5th and 95th percentile and the box determining the 25th and 75th percentile. Median is represented by the line in the boxes. **P < 0.01.
Mechanisms underlying decreased hERG protein by IH.
To assess the mechanisms underlying decreased hERG protein expression by IH, experiments were performed with SH-SY5Y cells exposed to IH120, which produced maximal changes in hERG protein expression (Fig. 1A) and hERG K+ current (Fig. 2, A and B). We first analyzed hERG mRNA expression by real-time quantitative PCR. hERG mRNA levels increased in IH120-exposed cells compared with control cells (Fig. 3A), suggesting that transcriptional changes are unlikely to account for down regulation of hERG protein by IH. Given that hERG protein can be degraded by either proteasome or lysosome (4, 9), we tested the effects of MG-132, a proteosomal inhibitor or baflomycin, a lysosomal inhibitor on hERG protein degradation by IH. Neither MG-132 nor baflomycin were able to prevent degradation of hERG protein by IH (Fig. 3B).
Fig. 3.
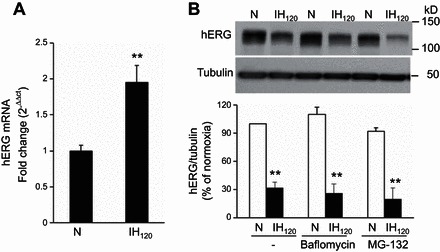
Effect of IH on hERG mRNA and the effects of proteosomal and lysomal inhibitors on IH-induced degradation of hERG protein. A: quantitative real-time PCR analysis of hERG mRNA levels in SH-SY5Y cells exposed to normoxia or IH120. The data are means ± SE from 4 individual experiments. B: representative immunoblot of hERG protein in cells exposed to normoxia and IH120 in the presence of baflomycin (1 μM) and MG-132 (5 μM), inhibitors of lysome and proteosome, respectively (top). Densitometric analysis of hERG protein normalized to tubulin expressed as percentage of normoxia (bottom). Data are means ± SE from 3 individual experiments. **P < 0.01, compared with normoxia.
ROS-dependent Ca2+ signaling mediates hERG degradation by IH.
IH activates ROS signaling in cell cultures and in intact animals (21). We therefore examined whether ROS signaling contributes to IH-induced hERG protein degradation. To this end, we first determined ROS levels in IH120-treated SH-SY5Y cells by two approaches: one by measuring MDA levels, which represents oxidized lipids (20), and the other by determining the aconitase enzyme activity, an established biochemical marker of ROS (20), in cytosol and mitochondrial fractions. IH-exposed cells showed increased MDA levels and decreased aconitase enzyme activity in the cytosolic and mitochondrial fractions (Fig. 4, A and B), demonstrating increased ROS levels. MnTmPyP, a membrane permeable ROS scavenger, prevented increased ROS levels and hERG protein degradation by IH (Fig. 4, A–C).
Fig. 4.
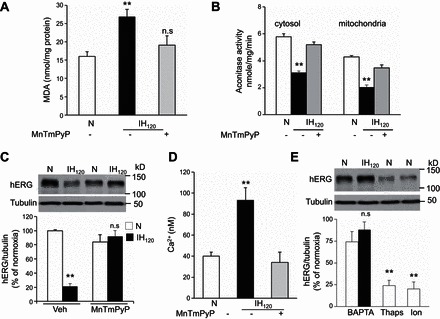
Reactive oxygen species (ROS)-dependent elevation of intracellular Ca2+ concentration ([Ca2+]i) is necessary for IH-induced hERG degradation. Malondialdehyde (MDA) levels (A) and cytosolic and mitochondrial aconitase activity (B) were monitored as indexes of ROS generation in SH-SY5Y cells exposed to normoxia or IH120 with and without MnTmPyP (50 μM), a membrane permeable ROS scavenger. Data shown are means ± SE from 4 independent experiments. C: effect of MnTmPyP on hERG protein in cells exposed to normoxia or IH120 (IH). Representative immunoblot (top) and densitometric analysis (bottom; means ± SE from 3 individual experiments). D: basal [Ca2+]i levels in cells exposed to either normoxia or IH120 with and without MnTmPyP. Data shown are means ± SE from 3 independent experiments. E: BAPTA-AM (10 μM), a chelator of Ca2+, prevents IH120-induced hERG protein degradation. Ionomycin (Ion; 1.4 μM), a Ca2+ ionophore, and thapsagargin (Thaps; 1.5 μM), which elevates [Ca2+]i, degrade hERG protein in cells exposed to normoxia. Representative immunoblot (top) and densitometric analysis (bottom; means ± SE from 3 individual experiments). **P < 0.01; n.s = not significant, P > 0.05, compared with normoxia.
We then investigated how ROS contributes to IH-induced hERG degradation. We previously reported that ROS generated by IH increases [Ca2+]i levels in rat pheochromocytoma PC12 cells (17). To determine whether Ca2+ signaling contributes to hERG protein degradation, we first monitored [Ca2+]i levels in IH120-treated SH-SY5Y cells. Basal [Ca2+]i levels were elevated in IH-treated cells, and this effect was absent following treatment with MnTmPyP, a ROS scavenger (Fig. 4D). BAPTA-AM, a chelator of Ca2+ prevented hERG degradation by IH, whereas thapsagargin and ionomycin, a Ca2+ ionophore, which elevate [Ca2+]i decreased hERG protein levels under normoxia, mimicking the effects of IH (Fig. 4E).
Calpain proteases mediate IH-induced hERG degradation.
Elevation of [Ca2+]i activates several downstream effector molecules including Ca2+-activated proteases, calpains (8). To determine the role of calpains in IH-induced hERG protein degradation, calpain enzyme activity was determined in IH120-treated SH-SY5Y cells. As shown in Fig. 5A, calpain activity was elevated in IH-treated cells. N-Acetyl-Leu-Leu-Methional (ALLM), a potent inhibitor of calpains, prevented IH-evoked calpain activation and hERG degradation and restored hERG-mediated K+ currents (Fig. 5, A–D).
Fig. 5.
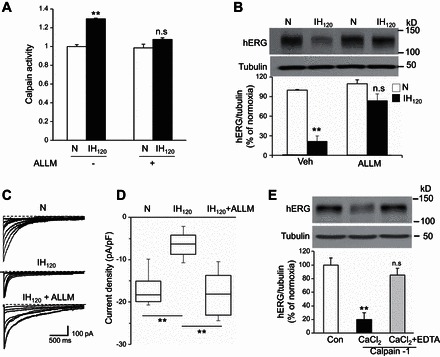
Calpains mediate IH-induced hERG degradation. A: calpain activity was determined in SH-SY5Y cells exposed to either normoxia (N) or to IH120. Calpain activity increased in cells exposed to IH120 which was inhibited by the calpain inhibitor N-Acetyl-Leu-Leu-Methional (ALLM; 10 μM; means ± SE from 8 independent experiments). B: representative immunoblot of hERG in cells exposed to normoxia or IH120 with and without pretreatment with ALLM (10 μM; top). Densitometric analysis (bottom; means ± SE from 3 experiments). C and D: representative hERG currents (C) and hERG K+ current density (D) in normoxia, IH120, and IH120 + ALLM-treated cells. Current density in D are represented by box charts from cells exposed to normoxia (n = 13 cells) and IH120 with (n = 11 cells) and without (n = 12 cells) pretreatment with ALLM (10 μM). Current density was derived by dividing the peak tail currents measured at −90 mV by the cell capacitance. E: representative hERG immunoblot (top) in SH-SY5Y cell lysates incubated for 15 min with purified calpain-1 (3 mg/ml) in presence of 1 mM CaCl2, with and without 2 mM EDTA. Densitometric analysis (bottom; means ± SE from 3 individual experiments). **P < 0.01; n.s = not significant, P > 0.05 compared with normoxia.
To further establish that hERG is a substrate for calpains, cell lysates from normoxic cells were incubated with purified calpain-1 in presence of CaCl2 with and without EDTA, a chelator of Ca2+ ions. hERG protein was completely degraded when cell lysates were incubated as brief as 15 min with calpain-1 and CaCl2, and addition of EDTA prevented this effect (Fig. 5E).
Identification of calpain binding sites in hERG protein.
Calpain cleavage prediction using multiple Kernel learning (6) from Calpain Modulatory Proteolysis Data Base (CaMPDB) identified potential calpain binding sites in the hERG protein both at the NH2 terminus containing a PER-ARNT-SIM (PAS) domain and the COOH terminus containing a cyclic nucleotide binding (cNBD) domain (28). The contributions of NH2 and COOH terminus to IH-induced hERG degradation were determined. To this end, SH-SY5Y cells were transfected with HA-tagged hERG plasmids with deletions of either NH2 or COOH terminus (Fig. 6A) and then were exposed to IH120. Control experiments were performed on cells transfected with full length WT plasmid. hERG protein, synthesized in the endoplasmic reticulum, as an immature core glycosylated (cg) protein, is exported to the Golgi apparatus for glycosylation and subsequent insertion into the plasma membrane as a fully glycosylated (fg) mature protein (34). Immunoblot analysis of cells transfected with WT hERG (Fig. 6A, plasmid 1) revealed two bands corresponding to cg and fg forms under normoxia (Fig. 6B). IH-exposed cells showed 81 ± 2% reduction in the fg form, and 48 ± 2% reduction in the cg form (Fig. 6, B and C, lane 1). The effects of IH on cells expressing NH2-terminal deletion of 300 amino acids including the PAS domain (Fig. 6A, plasmid 2) were similar to those seen in cells expressing WT hERG (Fig. 6, B and C, lane 2). In cells expressing COOH-terminal deletion with intact cNBD (Fig. 6A, plasmid 3), IH exposure resulted in near absence of the fg form (90 ± 2% reduction) with modest reduction (39 ± 1%) in the cg form (Fig. 6, B and C, lane 3). Combined deletion of NH2 or COOH terminus either with cNBD (Fig. 6A, plasmid 4) or without cNBD (Fig. 6A, plasmid 5) showed no detectable expression of the fg form under normoxia, and the reduction of cg form by IH was comparable with WT hERG (Fig. 6, B and C, lanes 4 and 5).
Fig. 6.
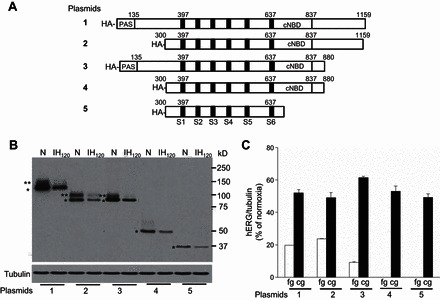
Contribution of NH2 terminus and COOH terminus of hERG in IH-induced hERG degradation by calpains. A: schematic diagram showing the full-length wild-type (WT; 1); NH2 terminus (2); COOH terminus (3); and both NH2- and COOH-terminal (4 and 5) deleted plasmids with HA-tag. B: representative immunoblot showing hERG protein expression in SH-SY5Y cells with forced expression of WT hERG (1) and NH2 (2) and COOH (3) terminus and NH2- and COOH (4 and 5)-terminal truncated constructs exposed to either normoxia or IH120. The recombinant hERG proteins were detected with anti-HA antibody (Acris Antibodies; AP23352PU-N). Tubulin protein expression was used as a loading control. **Full glycosylated (fg) form of hERG protein. *Core glycosylated (cg) form of hERG protein. C: densitometric analysis (means ± SE from 3 individual experiments) of fg and cg forms of hERG protein normalized to tubulin and expressed as percentage of normoxia.
DISCUSSION
The results of the present study demonstrate that IH degrades hERG protein in SH-SY5Y neuroblastoma cell cultures. Similar degradation of hERG protein was also seen in AMC from IH-treated neonatal rats, suggesting that the effects of IH are not cell selective and can be seen in intact rodent models of IH. The finding that IH exposure reduces hERG-mediated K+ current further demonstrates the functional importance of hERG protein degradation by IH.
Analysis of the underlying mechanisms revealed that the reduced hERG protein was not due to transcriptional downregulation, because IH increased hERG mRNA levels. The immature core glycosylated protein (cg) of hERG protein is degraded by proteasomes with a half-life of 4 h (9), and the fully glycosylated mature protein (fg) is degraded by the lysosomes with a half-life of 10–12 h (4, 7). Although cg and fg forms were not always distinguishable by Western blot assay in native SH-SYY5 neuroblastoma cells, immunocytochemical analysis showed that IH selectively degrades the membrane fg form of hERG. However, both MG-132 and baflomycin were unable to prevent the reduced hERG protein expression caused by IH, demonstrating that neither proteasome nor lysosome contribute to hERG protein degradation by IH.
The following observations demonstrate that ROS signaling mediates hERG protein degradation by IH: 1) ROS levels were elevated in IH-exposed neuroblastoma cells as evidenced by increased MDA levels as well as decreased aconitase activity in the cytosolic and membrane fractions; and 2) MnTmPyP, a membrane permeable ROS scavenger, prevented IH-induced ROS generation as well as the degradation of hERG protein. Previous studies reported that IH increases ROS levels by activating several prooxidant enzymes including NADPH oxidases 2 and 4 (32), xanthine oxidase (16), and mitochondrial electron transport chain at the complex I (12) as well as by decreasing antioxidant enzymes such as superoxide dismutase-2 (17). Further studies are needed to determine which of these ROS-generating sources contribute to hERG degradation by IH.
How might ROS contribute to hERG degradation by IH? We found that 1) IH-exposed cells exhibit elevated [Ca2+]i and ROS scavenger prevents this response; and 2) BAPTA-AM, a membrane permeant Ca2+ chelator, prevents hERG degradation by IH, whereas thapsagargin or ionomycin, which increase [Ca2+]i, decrease hERG protein levels in neuroblastoma cells exposed to normoxia, mimicking the effects of IH. These findings demonstrate that ROS-dependent [Ca2+]i elevation is essential for hERG degradation by IH.
The novel finding of the present study is the demonstration that Ca2+-activated proteases, calpains, mediate hERG protein degradation by IH. Calpain enzyme activity increased in IH-exposed cells, and ALLM, a potent inhibitor of calpains, not only blocked IH-induced calpain enzyme activity but also prevented the degradation of hERG protein. More importantly, the calpain inhibitor restored the hERG-mediated K+ currents in IH-exposed cells. The finding that hERG is degraded in presence of exogenous calapin and Ca2+, which could be prevented by EDTA, a Ca2+ chelator, further establishes that hERG protein is one of the substrates for calpains during IH. While both NH2- and COOH-terminal regions of the hERG protein contain putative calpain binding sites, COOH-terminal deletion was ineffective in preventing IH-induced fg degradation, whereas NH2-terminal deletion resulted in modest degradation of fg form of hERG in IH-exposed cells. Cells with forced expression of combined NH2- and COOH-terminal deletion constructs showed only the cg form of hERG, which was relatively resistant to IH-induced degradation. We did not measure K+ currents in cells treated with various NH2- and COOH-terminal deletion constructs for two reasons: 1) cells transfected with these constructs expressed predominantly the cg form and very little or none of the fg form, which is the main hERG form expressed in the membrane and important for K+ currents; and 2) these experiments were performed with forced expression of HA-tagged constructs in neuroblastoma cells expressing native hERG protein, which complicates the interpretation of results. Despite these limitations, our observations suggest that calpain-mediated hERG protein degradation by IH is complex and likely involves multiple calpain binding sites. Nonetheless, collectively these findings demonstrate a hitherto uncharacterized role for calpains in regulating hERG protein expression.
Although hERG is an important K+ channel in many cell types, arguably it is most important for regulating the heartbeat. A recent study reported that obstructive sleep apnea (OSA) patients with congenital long-QT syndrome (LQTS) exhibit increased QT prolongation and sudden cardiac death (24). Given that hERG is implicated in long-QT syndrome (24), it would be of interest to assess whether downregulation of hERG protein by IH contributes to heart rate abnormalities associated with OSA. Recurrent apnea is also a major clinical problem in infants born preterm (1). Neonatal rats exposed to IH exhibit markedly augmented catecholamine secretion from AMC (25). hERG protein is expressed in neonatal AMC, and the hERG-mediated K+ current is important for catecholamine secretion (10). Given that IH increases calpain enzyme activity in the adrenal medulla (17), it would be interesting to determine whether calpain-mediated hERG degradation contributes to IH-induced augmented catecholamine secretion from neonatal AMC.
GRANTS
This research is supported by National Heart, Lung, and Blood Institute Grant P01-HL-90554.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: N.W., H.S.K., S.A.K., G.A., and V.V.M. performed experiments; N.W., S.A.K., G.A., V.V.M., and J.N. analyzed data; N.R.P. and J.N. edited and revised manuscript; J.N. conception and design of research; J.N. interpreted results of experiments; J.N. prepared figures; J.N. drafted manuscript; J.N. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Drs. Ganesh K. Kumar and Aaron P. Fox for critical reading of the manuscript.
Footnotes
This article is the topic of an Editorial Focus by Ramon J. Ayon, Haiyang Tang, and Jason X.-J. Yuan (2a).
REFERENCES
- 1.Abu-Shaweesh JM, Martin RJ. Neonatal apnea: what's new? Pediatr Pulmonol 43: 937–944, 2008. [DOI] [PubMed] [Google Scholar]
- 2.Arcangeli A, Bianchi L, Becchetti A, Faravelli L, Coronnello M, Mini E, Olivotto M, Wanke E. A novel inward-rectifying K+ current with a cell-cycle dependence governs the resting potential of mammalian neuroblastoma cells. J Physiol 489: 455–471, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2a.Ayon RJ, Tang H, Yuan JX. Gasping for answers. Focus on “Calpain activation by ROS mediates human ether-a-go-go-related gene protein degradation by intermittent hypoxia.” Am J Physiol Cell Physiol. doi: 10.1152/ajpcell.00017.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bianchi L, Wible B, Arcangeli A, Taglialatela M, Morra F, Castaldo P, Crociani O, Rosati B, Faravelli L, Olivotto M, Wanke E. herg encodes a K+ current highly conserved in tumors of different histogenesis: a selective advantage for cancer cells? Cancer Res 58: 815–822, 1998. [PubMed] [Google Scholar]
- 4.Chapman H, Ramström C, Korhonen L, Laine M, Wann KT, Lindholm D, Pasternack M, Törnquist K. Downregulation of the HERG (KCNH2) K(+) channel by ceramide: evidence for ubiquitin-mediated lysosomal degradation. J Cell Sci 118: 5325–5334, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80: 795–803, 1995. [DOI] [PubMed] [Google Scholar]
- 6.DuVerle DA, Ono Y, Sorimachi H, Mamitsuka H. Calpain cleavage prediction using multiple kernel learning. PLoS One 6: e19035, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ficker E, Dennis AT, Wang L, Brown AM. Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ Res 92: e87–100, 2003. [DOI] [PubMed] [Google Scholar]
- 8.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev 83: 731–801, 2003. [DOI] [PubMed] [Google Scholar]
- 9.Gong Q, Keeney DR, Molinari M, Zhou Z. Degradation of trafficking-defective long QT syndrome type II mutant channels by the ubiquitin-proteasome pathway. J Biol Chem 280: 19419–19425, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Gullo F, Ales E, Rosati B, Lecchi M, Masi A, Guasti L, Cano-Abad MF, Arcangeli A, Lopez MG, Wanke E. ERG K+ channel blockade enhances firing and epinephrine secretion in rat chromaffin cells: the missing link to LQT2-related sudden death? FASEB J 17: 330–332, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Kemp PJ, Peers C. Oxygen sensing by ion channels. Essays Biochem 43: 77–90, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Khan SA, Nanduri J, Yuan G, Kinsman B, Kumar GK, Joseph J, Kalyanaraman B, Prabhakar NR. NADPH oxidase 2 mediates intermittent hypoxia-induced mitochondrial complex I inhibition: relevance to blood pressure changes in rats. Antioxid Redox Signal 14: 533–542, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Makarenko VV, Nanduri J, Raghuraman G, Fox AP, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. Endogenous H2S is required for hypoxic sensing by carotid body glomus cells. Am J Physiol Cell Physiol 303: C916–C923, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Makarenko VV, Peng YJ, Yuan G, Fox AP, Kumar GK, Nanduri J, Prabhakar NR. CaV3.2 T-type Ca2+ channels in H2S-mediated hypoxic response of the carotid body. Am J Physiol Cell Physiol 308: C146–C154, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nanduri J, Bergson P, Wang N, Ficker E, Prabhakar NR. Hypoxia inhibits maturation and trafficking of hERG K(+) channel protein: role of Hsp90 and ROS. Biochem Biophys Res Commun 388: 212–216, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nanduri J, Vaddi DR, Khan SA, Wang N, Makerenko V, Prabhakar NR. Xanthine oxidase mediates hypoxia-inducible factor-2α degradation by intermittent hypoxia. PLoS One 8: e75838, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nanduri J, Wang N, Yuan G, Khan SA, Souvannakitti D, Peng YJ, Kumar GK, Garcia JA, Prabhakar NR. Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc Natl Acad Sci USA 106: 1199–1204, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D'Agostino RB, Newman AB, Lebowitz MD, Pickering TG. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA 283: 1829–1836, 2000. [DOI] [PubMed] [Google Scholar]
- 19.Pawar A, Peng YJ, Jacono FJ, Prabhakar NR. Comparative analysis of neonatal and adult rat carotid body responses to chronic intermittent hypoxia. J Appl Physiol 104: 1287–1294, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng YJ, Nanduri J, Khan SA, Yuan G, Wang N, Kinsman B, Vaddi DR, Kumar GK, Garcia JA, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 2α (HIF-2α) heterozygous-null mice exhibit exaggerated carotid body sensitivity to hypoxia, breathing instability, and hypertension. Proc Natl Acad Sci USA 108: 3065–3070, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prabhakar NR, Kumar GK, Nanduri J, Semenza GL. ROS signaling in systemic and cellular responses to chronic intermittent hypoxia. Antioxid Redox Signal 9: 1397–1403, 2007. [DOI] [PubMed] [Google Scholar]
- 22.Prabhakar NR, Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev 92: 967–1003, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81: 299–307, 1995. [DOI] [PubMed] [Google Scholar]
- 24.Shamsuzzaman AS, Somers VK, Knilans TK, Ackerman MJ, Wang Y, Amin RS. Obstructive sleep apnea in patients with congenital long QT syndrome: implications for increased risk of sudden cardiac death. Sleep 38: 1113–1119, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Souvannakitti D, Kumar GK, Fox A, Prabhakar NR. Neonatal intermittent hypoxia leads to long-lasting facilitation of acute hypoxia-evoked catecholamine secretion from rat chromaffin cells. J Neurophysiol 101: 2837–2846, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Souvannakitti D, Kuri B, Yuan G, Pawar A, Kumar GK, Smith C, Fox AP, Prabhakar NR. Neonatal intermittent hypoxia impairs neuronal nicotinic receptor expression and function in adrenal chromaffin cells. Am J Physiol Cell Physiol 299: C381–C388, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Souvannakitti D, Nanduri J, Yuan G, Kumar GK, Fox AP, Prabhakar NR. NADPH oxidase-dependent regulation of T-type Ca2+ channels and ryanodine receptors mediate the augmented exocytosis of catecholamines from intermittent hypoxia-treated neonatal rat chromaffin cells. J Neurosci 30: 10763–10772, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K(+) channels: structure, function, and clinical significance. Physiol Rev 92: 1393–1478, 2012. [DOI] [PubMed] [Google Scholar]
- 29.Wang S, Morales MJ, Liu S, Strauss HC, Rasmusson RL. Modulation of HERG affinity for E-4031 by [K+]o and C-type inactivation. FEBS Lett 417: 43–47, 1997. [DOI] [PubMed] [Google Scholar]
- 30.Young T, Finn L, Peppard PE, Szklo-Coxe M, Austin D, Nieto FJ, Stubbs R, Hla KM. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin sleep cohort. Sleep 31: 1071–1078, 2008. [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan G, Adhikary G, McCormick AA, Holcroft JJ, Kumar GK, Prabhakar NR. Role of oxidative stress in intermittent hypoxia-induced immediate early gene activation in rat PC12 cells. J Physiol 557: 773–783, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol 226: 2925–2933, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan G, Nanduri J, Bhasker CR, Semenza GL, Prabhakar NR. Ca2+/calmodulin kinase-dependent activation of hypoxia inducible factor 1 transcriptional activity in cells subjected to intermittent hypoxia. J Biol Chem 280: 4321–4328, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Zhou Z, Gong Q, January CT. Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J Biol Chem 274: 31123–31126, 1999. [DOI] [PubMed] [Google Scholar]