

Single-molecule microscopy has the potential to link repeatable discrete molecular behaviors to changes in cellular behavior. The approach used here finds characteristic changes in integrin density and mobility that are linked to local cellular protrusion. Mutants show that the density changes can be separated from mobility changes.
Abstract
Recent advances in light microscopy permit visualization of the behavior of individual molecules within dense macromolecular ensembles in live cells. It is now conceptually possible to relate the dynamic organization of molecular machinery to cellular function. However, inherent heterogeneities, as well as disparities between spatial and temporal scales, pose substantial challenges in deriving such a relationship. New approaches are required to link discrete single-molecule behavior with continuous cellular-level processes. Here we combined intercalated molecular and cellular imaging with a computational framework to detect reproducible transient changes in the behavior of individual molecules that are linked to cellular behaviors. Applying our approach to integrin transmembrane receptors revealed a spatial density gradient underlying characteristic molecular density increases and mobility decreases, indicating the subsequent onset of local protrusive activity. Integrin mutants further revealed that these density and mobility transients are separable and depend on different binding domains within the integrin cytoplasmic tail. Our approach provides a generalizable paradigm for dissecting dynamic spatiotemporal molecular behaviors linked to local cellular events.
INTRODUCTION
The organization of macromolecular assemblies such as focal adhesions, neurological synapses, and immunological synapses is essential for regulating cellular mobility and signaling. Recent advances in light microscopy permit the development of cytoarchitectural blueprints of these assemblies with single-molecule resolution in fixed cells (Shroff et al., 2007; Dani et al., 2010; Kanchanawong et al., 2010; Rossy et al., 2013). However, difficulties in analyzing the fast movement of many individual molecules within a high-density field have yielded only the rapid movement of sparse subsets of molecules (Manley et al., 2008; Rossier et al., 2012) or the slow evolution of dense macromolecular complexes (Shroff et al., 2008). Because dynamic molecular organization is likely to imbue cellular-level function (Scannevin and Huganir, 2000), we sought to develop a paradigm that could capture a high density of fast-moving molecules with the aim of identifying characteristic transient changes in molecular behavior that could be linked to cellular behaviors.
Identifying characteristic molecular behaviors that can be linked to cellular behaviors is challenging; it requires imaging many molecules over large enough areas of the cell fast enough to capture molecular behavior and long enough to capture inherently slower cell behaviors. We decided to use photoactivatible fluorophores (PAFPs) to provide a high molecular density (Shroff et al., 2007; Shtengel et al., 2009). We modulated the limited photon output of the PAFPs by optimizing excitation parameters to extend fluorophore lifetime and implementing a method for calculating the diffusion coefficient for all but the shortest molecular trajectories, analyzing 70% of the localized molecules, an ∼20-fold improvement over single-particle tracking PALM (sptPALM) (Manley et al., 2008). We then calculated the density and mobility of individual molecules in regions that are the size of the smallest spot that is resolvable with conventional light microscopy—information that was previously either temporally collapsed into low-frequency, single live-cell PALM images (Shroff et al., 2008) or undetectable in low-density sptPALM images (Manley et al., 2008). Finally, we linked the characteristic dynamic behaviors of the individual molecules located within these diffraction-limited spots to changes in cellular activity derived from intercalated conventional microscopy images.
Using this approach, we uncovered characteristic local changes in integrin density and mobility just before protrusion. Integrin molecules outside of adhesions exhibit transient behaviors that are detectable with superresolution microscopy, despite having been previously reported as randomly diffusing when visualized with conventional microscopy (Duband et al., 1988; Wehrle-Haller, 2007; Rossier et al., 2012). We also found that the integrin density and mobility changes in preparation for protrusion are separable and depend on different domains of the cytoplasmic tail. These data suggest a basis for future studies to dissect the interactions that modulate single-molecule behavior and initiate local changes in cellular function.
RESULTS AND DISCUSSION
To characterize dense, dynamic molecular behaviors and link them to cell behavior, we intercalated fast, live-cell, single-molecule imaging at 40 Hz with conventional cellular imaging at 0.1 Hz. Both imaging schemes used total internal reflection fluorescence microscopy to focus on the ventral surface of the cell. The slower, conventional cellular imaging monitored either untagged green fluorescent protein (EFGP) to identify the cell edge or Emerald-paxillin to identify focal adhesions. The faster, single-molecule imaging monitored integrin heterodimers that were expressed in CHO cells (CHO-K1) as αv tagged with the monomeric photoactivatible fluorescent protein mEos2 and untagged β3. We used genetic expression in CHO-K1 cells because they do not express endogenous αv or β3 (Xu et al., 2011). Transfection of human integrins into the hamster background permitted the use of human specific antibodies to verify receptor expression (Materials and Methods), and expression levels were quantified in individual cells by counting the number of integrin molecules on the cell surface. Stochastically photoconverted molecules were initially localized and tracked (Figure 1, a and b; Jaqaman et al., 2008; Shroff et al., 2008), with the single-molecule data combined in 10-s intervals to create either live-cell PALM or single-molecule trajectory time-lapse movies. Still frames from the movies are shown in Figure 1, c–f. Although these data confirm our earlier finding that integrin is locally positioned along the leading edge by actin polymerization (Galbraith et al., 2007), they also suggest the existence of additional heterogeneities in integrin localization and movement outside of adhesions.
FIGURE 1:
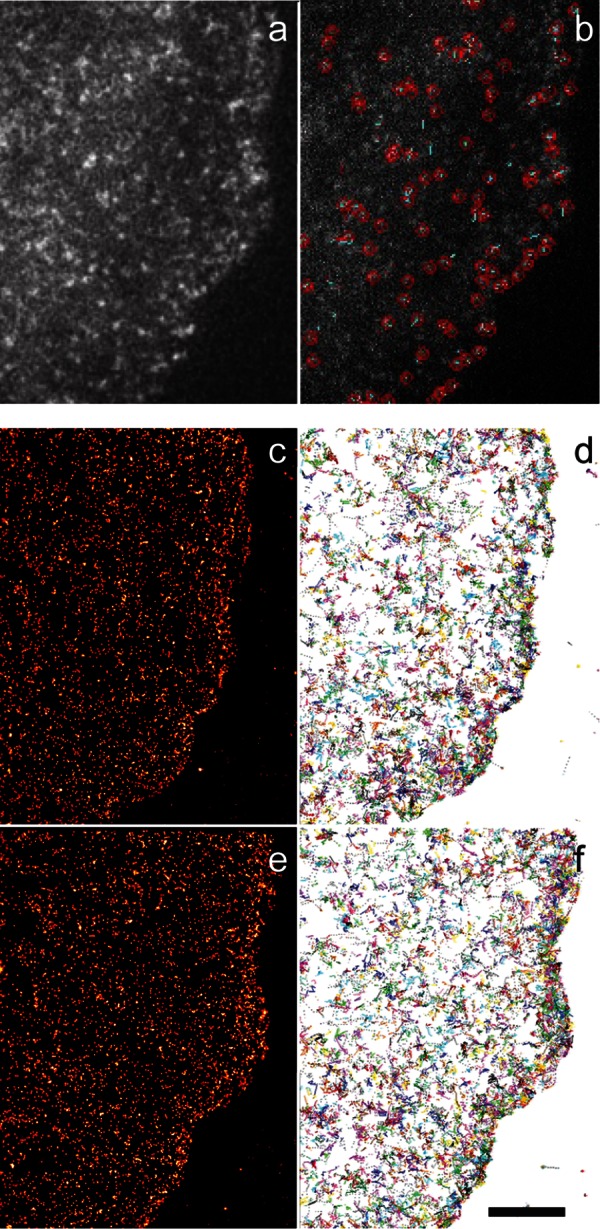
Single-molecule analysis detects heterogeneities in integrin distributions and trajectories in protruding cells. (a, b) mEos2-αvβ3 single-molecule image (a) with localization (red circles) and trajectories (b). (c, f) Time-lapse images created by accumulating 10 s of single- molecule images of localized molecules (c, e) whose trajectories (d, f) lasted at least five frames. Intensity and diameter of molecules in c and e indicate localization precision. Bar, 5 μm.
Quantification of dynamic molecular behavior in dense fields
To quantify the localization of the heterogeneities, we calculated the number of neighbors surrounding each molecule. We detected spatially discrete variations in concentration outside of adhesions, larger regions of higher density indicative of adhesions within the cell body, and a spatial gradient between the leading edge and the cell body (Figure 2a). We initially quantified movement heterogeneities by calculating the diffusion coefficient of the molecules using moment-scaling spectrum analysis (MSS; Ewers et al., 2005; Jaqaman et al., 2011), but the requirement of trajectories lasting at least 20 frames in length meant that we were not able to calculate the mobility of enough molecules to capture the spatial information content seen in our molecular density maps (Figure 2, a and b). Therefore we implemented a diffusion analysis based on frame-to-frame (F2F) mean square displacements with the diffusion coefficient, D, calculated as (Wieser et al., 2008)
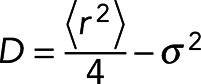 (1) (1)
|
FIGURE 2:

Molecular mobility calculated from short F2F trajectory analysis has spatial heterogeneities similar to molecular density. (a) Number of neighbors per αvβ3-integrin molecule detected during a 6-min experiment. (b) Diffusion coefficients calculated via MSS analysis requiring trajectories lasting ≥0.5 s (20 frames). (c) Map of diffusion coefficients calculated via F2F analysis requiring trajectories lasting ≥0.125 s (5 frames). (d) Four exponentials, indicative of four diffusion modes, are required to fit the distribution of square frame-to-frame displacements (>900,000 displacements in 3 min). (e) Adhesion complexes identified by image segmentation of paxillin. (f) Paxillin intensity correlates with diffusion modes. Notches on boxplots indicate 95% confidence interval around the median, and daggers indicate outlier points. (g) Fraction of each diffusion mode inside vs. outside of adhesions in e. Mean ± SD, p < 0.005 by χ2. (h) Integrin diffusion modes of molecules in a. Seventeen cells in f and g. Bars, 5 μm.
where <r2> is the F2F mean square displacement and σ is the positional uncertainty (calculated from the molecular localization). With this analysis, we were able to calculate the diffusion coefficients from trajectories as short as five frames, making it possible for us analyze enough molecules to create mobility maps that exhibit spatial heterogeneities similar to those identified in integrin density maps (Figure 2c).
To validate our F2F diffusion analysis, we compared diffusion coefficients obtained with MSS and F2F where both were applicable (trajectories >20 frames) and found good agreement (Supplemental Figure S1). To confirm that the observed heterogeneity in diffusion coefficients was not a consequence of analyzing short trajectories, we also examined the distribution of frame-to-frame displacements. A single-diffusion mode leads to a monoexponential distribution of F2F square displacements (Wieser et al., 2008); however, our data required four exponentials, representing four modes of diffusion, to fully fit the distribution (Figure 2d and Supplemental Figure S2). The three slower modes have diffusion coefficients similar to those previously reported (Rossier et al., 2012), whereas the fourth mode has a mean diffusion coefficient of 1 μm2/s, which is the speed of free diffusion in a membrane. To further confirm the validity of the short-trajectory analysis technique, we analyzed integrin diffusion modes inside and outside of adhesions. As expected, integrins exhibited slower diffusion modes within adhesions and faster diffusion modes outside of adhesions that were >1.1 μm2 as identified by paxillin localization (Rossier et al., 2012; Figure 2, e–h). A heterogeneous mixture of integrin diffusion modes was also identified along the leading edge in rapidly remodeling regions of elevated paxillin concentration, which are indicative of close contacts (Izzard and Lochner, 1980; Lee and Jacobson, 1997). Taken together, these data indicate that our experimental and analytical approach is capable of capturing previously undetected transient receptor diffusion mode heterogeneity outside of established adhesions.
Linking transient modulations in molecular behavior to changes in cellular function
We then added a framework to investigate whether the heterogeneities, which were more pronounced near the leading edge (Figure 1, c–f), were linked to cell protrusion activity. We performed the same intercalated imaging described earlier (Figure 3a), using images of untagged GFP to create a mask that identified the cell edge (Figure 3b). The cell mask was divided into sampling windows that were propagated from one frame to the next (Vilela et al., 2013; Cai et al., 2014), and localized molecules were assigned to the sampling windows based on their mean position and existence time (Figure 3c). Of importance, because the sampling windows were the size of a diffraction-limited spot (220 nm × 220 nm), we were able to quantify the dynamic behavior of multiple individual integrin receptors within an area that would yield only a single intensity readout from the ensemble of molecules if conventional fluorescence microscopy were used. Finally, we synchronized our integrin density and mobility analysis to the onset of protrusion by placing the origin of our newly defined coordinate system at the time and location of protrusion onset and grouping windows according to their distance and time relative to the origin (Machacek et al., 2009; Lee et al., 2015). These alignment and synchronization procedures allowed us to compare spatial and temporal molecular information as a function of morphological variation within an individual cell and across multiple cells (Figures 3c and 4a). Although we developed this framework to examine integrin behavior in relation to the onset of protrusion, it is readily generalizable to different alignment and synchronization schemes to analyze other molecular variations associated with changes in cell behaviors.
FIGURE 3:

Multiscale live-cell imaging and analysis framework to connect molecular density and mobility to local cell protrusion. (a) Multiscale microscopy intercalates high-speed, single-molecule, superresolution imaging with slower, diffraction-level imaging of the cell. (b) Data are extracted to construct single-molecule trajectories and track the cell edge or segment adhesions. (c) Molecular properties are connected to cell protrusive activity.
FIGURE 4:
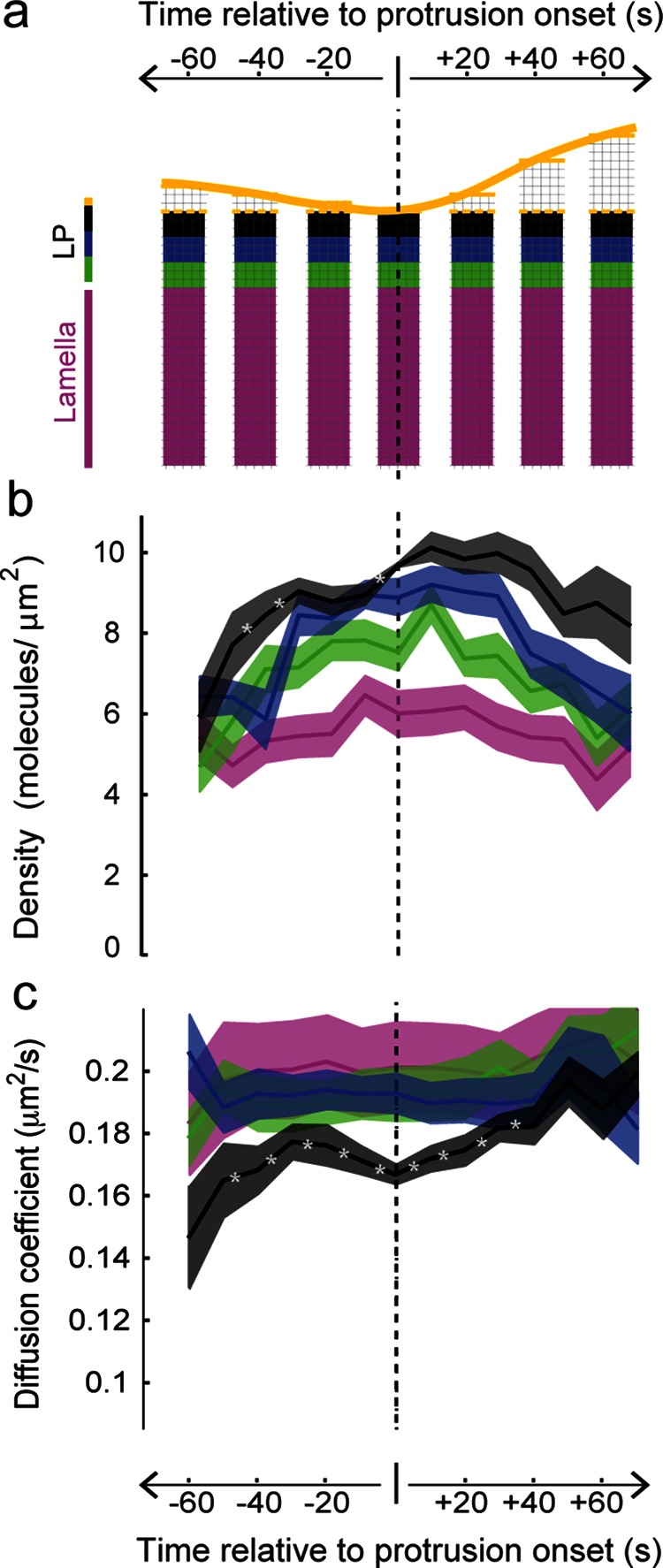
Spatial and temporal changes in receptor density and mobility precede local cellular protrusion. (a) Molecular information in diffraction-limited molecular sampling windows (thin lines) plotted in a fixed coordinate system with the origin set as the time and location of protrusion onset. Yellow line and colored bands indicate edge and distance from the cell edge, respectively. LP, lamellipodium. (b) αvβ3 molecular density grouped according to part a exhibits a significant spatial gradient between −50 and +70 s (with 90% confidence for black > blue > green > magenta). Darker line indicates mean, and outer edges indicate ±1 SD. Asterisk indicates temporal change with 90% confidence (Dorn et al., 2005). (c) Integrin mobility at the protrusion onset location (black) has a significant increase and then decrease before protrusion onset, followed by a significant increase afterward. Sixteen cells.
Changes in integrin density initiate before local cellular protrusion
We discovered that the integrin density, as well as the prominent front-to-back spatial gradient, increased in preparation of cell protrusion. The increase was attenuated with distance from the cell edge, and the density returned toward baseline values after protrusion onset (Figure 4b). Although diffraction-limited studies do suggest a higher integrin density at the leading edge (Supplemental Movie S1; Kiosses et al., 2001; Galbraith et al., 2007), they do not have sufficient spatial sensitivity to detect the variations in density across the narrow (1 to 2 μm wide) lamellipodium that are quantified here using single-molecule data. Control single-molecule experiments using the membrane marker farnesyl did not exhibit a density peak surrounding protrusion or a density spatial gradient, indicating that the integrin density increase before protrusion onset is not a reflection of nonspecific changes in membrane behavior associated with the onset of protrusion (Supplemental Figure S3). In addition, neither the randomization of integrin single-molecule behavior nor simulated integrin behavior that was not coupled to edge activity exhibited a spatiotemporal density modulation (Supplemental Figure S3). Thus changes in integrin density reflect a specific adhesion receptor redistribution that initiates before local areas of the cell protrude.
Changes in integrin mobility initiate before protrusion at the protrusion onset location
Our single-molecule analysis also detected nonuniformity in integrin mobility across the surface of the cell (Figure 2h). Using the mean diffusion coefficient of all molecules within a sampling window, we determined that mobility was slowest at the leading edge, where density was highest (Figure 4c). We also found that mobility near the front of the cell, at the protrusion onset location, increased before decreasing starting 30 s prior to protrusion. Mobility then increased during protrusion, approaching the lamellar values as the cell edge advanced forward (Figure 4c). No such molecular mobility change in association with protrusion was observed in experiments performed with the membrane marker farnesyl (Supplemental Figure S3). As with density, experimental data randomization and simulations without coupling between single-molecule behavior and cell edge activity abolished the integrin spatiotemporal mobility trends (Supplemental Figure S3). The combined increase in integrin density and decrease in mobility before protrusion onset support the hypothesis that adhesions initiate before protrusion in order to provide a physical anchor point for the advancing edge (Lee et al., 2015). Of importance, the change in integrin density and mobility in local regions before the onset of protrusion indicate that we have identified a characteristic transient change in the organization and speed of individual molecules that precede a local cellular behavior.
Changes in integrin density and mobility before protrusion onset depend on different domains of the cytoplasmic tail
To gain insight into the mechanisms underlying the changes in density and mobility, we analyzed several integrin mutants. We first tested two truncation mutants, β3-Δ724 and β3-Δ717 (O’Toole et al., 1994; Hughes et al., 1995; Huttenlocher et al., 1996). The latter mutant is locked in a high-affinity state, which facilitates binding to the extracellular matrix independently of cytoplasmic activators (Hughes et al., 1995). Although both mutants did have a slight density increase before protrusion onset, similar to the membrane marker farnesyl (Supplemental Figure S3), neither mutant demonstrated the prominent spatial density gradient or the temporal density peak surrounding protrusion onset seen with (wild type [WT]) β3 (Figure 5, a and c). These mutants also did not exhibit the mobility modulation surrounding protrusion onset observed with (WT) β3 (Figure 5, b and d). The absence of a density gradient and the lack of density and mobility changes before protrusion onset indicate that these characteristic behaviors cannot be rescued by simply increasing integrin affinity for matrix; they require the integrin cytoplasmic tail.
FIGURE 5:

Density and mobility changes surrounding protrusion onset depend on separate binding domains on the integrin cytoplasmic tail. (a, b) High-affinity αvβ3-717Δ truncation mutants do not have a density gradient or a mobility modulation surrounding protrusion onset. (c, d) Lower-affinity αvβ3-724Δ also does not have a density gradient or a mobility modulation surrounding protrusion onset. (e, f) αvβ3-Y747A integrins with a NPXY point mutation do have a spatial density gradient similar to WT integrins but do not have a mobility modulation (Figure 4). The spatial gradient is significant between −20 and +70 s; black > blue > green > magenta. LP, asterisk, and bands as in Figure 4. Nine cells for αvβ3-717Δ, 10 for αvβ3-724Δ, and 4 for αvβ3-Y747A.
We next investigated an integrin mutant that has a complete cytoplasmic tail but has a point mutation that inhibits binding to the NPXY domain and cannot bind talin, β3-Y747A (Calderwood et al., 2002). We found that β3-Y747A had spatiotemporal variations in density that were similar to (WT) β3 (Figure 5e) but did not exhibit the mobility changes surrounding protrusion onset that were observed with (WT) β3 (Figure 5f). These data indicate that the integrin density gradient and the change in density that initiate before protrusion onset require interaction with a site on the cytoplasmic tail outside of the NPXY domain, and these data further suggest that the NPXY is involved in regulating integrin mobility. Of importance, these data identify an underlying separation in the dynamic molecular positioning and speed before the initiation of a cellular event, suggesting that density and mobility regulation depend on interactions with different domains on the β-integrin cytoplasmic tail.
The finding of a separation between control of integrin position and speed is consistent with biochemical (Ma et al., 2008; Harburger et al., 2009) and cross-variance analyses (Bachir et al., 2014), which suggest that integrins assemble adhesions through a sequential series of molecular interactions. Because the NPXY domain appears to be necessary for mobility modulation, our data support both the model of kindlin binding to the β cytoplasmic tail before talin binding (Bachir et al., 2014) and the model of α-actinin positioning integrin at the leading edge (Choi et al., 2008). Our studies also suggest that separating control of molecular position from speed may act as a functional mechanism to establish the order of molecular binding events to the cytoplasmic tail and integrin activation. Thus we have laid the foundation for future studies that use the dynamic modulation of molecular spatial and temporal behaviors to dissect the hierarchy of adhesion formation.
To our knowledge, our study is the first to identify characteristic transient changes in single-molecule organization and mobility that consistently precede a change in cell behavior. It is an important step toward using characteristic dynamic molecular patterns to understand cell behaviors such as the seemingly random variations in cell edge protrusion first observed almost a half-century ago (Abercrombie et al., 1970). Earlier studies aimed at analyzing integrin behavior sampled molecular information too sparsely (Rossier et al., 2012), lacked the spatial and temporal resolution to capture transient heterogeneities (Digman et al., 2009; Lee et al., 2015), or analyzed only a very small region of the cell (Mueller et al., 2013). These limitations led to the current consensus model in which integrins outside of adhesion complexes undergo predominantly fast random diffusion that appears uniform across the cell surface (Duband et al., 1988; Wehrle-Haller, 2007; Rossier et al., 2012). By characterizing the behavior of individual molecules within a spot that is at the limit of resolution of a conventional light microscope, our approach captures subtle dynamic characteristic variations in molecular organization and mobility linked to cellular behavior that would likely be disregarded as signal noise with other approaches. Moreover, by synchronizing spatial and temporal molecular behaviors with changes in cellular behavior, we identified an approach for dissecting sequential molecular interactions that generate a cellular output. Finally, although we demonstrated the usefulness of our approach using integrin transmembrane receptors, it is readily generalizable to any transmembrane receptor or similar signaling system, as well as to the reorganization of molecules within macromolecular assemblies, such as adhesion complexes and neurological and immunological synapses.
MATERIALS AND METHODS
Cell culture and transfection
CHO-K1 cells (American Type Culture Collection [ATCC]) were grown in DMEM-F12 supplemented with 10% fetal bovine serum. Cells were transfected with DNA using a Nucleofector II and T kit (Lonza) transfection reagents following manufacturer’s protocols. To verify coexpression of human integrin heterodimers for WT and mutants, cells were immunolabeled with an antibody specific for human αvβ3 (clone LM609). All DNA was prepared using a Plasmid Midi Kit (Qiagen). Approximately 24 h after transfection, cells were trypsinized and replated on 25-mm-diameter #1.5 RCA-1 cleaned cover glass that had been coated overnight with 5 μg/ml human plasma fibronectin. Cells were allowed to attach and spread for approximately 30 min prior to imaging. Coverslips were mounted into Attoflour cell chambers (Invitrogen) and maintained at 37°C in media containing 25 mM HEPES.
Multiscale light microscopy
Imaging was performed using total internal reflection fluorescence (TIRF) illumination on an Olympus IX71 with a 60X 1.49 NA objective. The TIRF illumination consisted of three laser lines (405, 488, 561 nm) (Coherent) that were combined to yield a common beam diameter on a separate optics breadboard (Thor Labs). The combined beam was introduced into the TIRF illumination port of the microscope through free space. To stochastically excite a subpopulation of the mEos2 labeled molecules, a low level of 405-nm activation light (5 μW at the back aperture) was combined with the 561-nm excitation light (2.5 mW at the back aperture). Every 400th frame, or 10 s, the excitation light was switched using an acousto-optic tunable filter (AOTF; AA Opto-Electronic) to 488 nm (100 μW at the back aperture) to collect a single image of either unconjugated EGFP or Emerald-Paxillin (Fig. 3). These data were used to identify the cell edge (unconjugated EGFP) or adhesion complexes (Emerald-Paxillin) and create a context for the single-molecule data. All images were magnified to ×144 before being acquired with an Andor 897 electron-multiplying charge-coupled device camera at an exposure time of 25 ms. The recorded image had a final magnification of 111 nm/pixel. Cells were imaged continuously for a minimum of 6 min and a maximum of 30 min. In all cases cells did not exhibit any deleterious effects due to imaging as indicated by normal cell morphology and motility at the end of the experiment.
Plasmid construction
All photoactivatable fluorescent protein expression vectors were constructed using C1 and N1 (Clontech-style) cloning vectors. To generate the fusion constructs, an EGFP variant (mEmerald) was first used to characterize the localization of the protein target. The mEos2 fluorescent protein was amplified with a 5′ primer encoding an AgeI site and a 3′ primer encoding a NotI site (N1) or a BspEI site (C1) for insertion into the appropriate cloning vector backbone. The resulting PCR products were sequentially digested, gel purified, and ligated into similarly treated EGFP-C1 and EGFP-N1 vector backbones, yielding mEos2-C1 and mEos2-N1 cloning vectors. Following sequence verification, the fusion vector and mEos2-C1 or mEos2-N1 were digested with the appropriate enzymes and ligated to produce the mEos2 expression vectors.
To construct the human alpha V integrin (NM_002210.4) fusion, the following primers were used to create a 25–amino acid linker separating the integrin from the fluorescent protein:
KpnI forward: GCG AGG TAC CTC GGC GAT GGC TTT TCC GCC GCG GCG ACG G
AgeI reverse: TCG CAC CGG TGG GGA TCC CGG GCC CGC GGT ACC GTC GAC TGC AGA ATT CGA AGC TTG AGC TCG AGA TCC CGG AGT TTC TGA GTT TCC TTC
The PCR product and mEmerald-N1 were digested with the appropriate enzymes, purified, and ligated to yield mEmerald-integrin-alpha-V-25.
The farnesyl expression vector was constructed using the 20–amino acid (KLNPPDESGPGCMSCKCVLS) farnesylation signal from c-Ha-Ras (NM_001130442.1). To produce the farnesyl fusion, pEGFP-farnesyl was sequentially digested with AgeI and BspEI. The resulting product was purified and ligated to a similarly treated mEos2-C1 cloning vector to yield mEos2-farnesyl-5.
The chicken paxillin (NM_204984.1) protein contains an alternate splice site. We used a paxillin construct with T132S and M133L mutations, eliminating the Kozak sequence responsible for the splicing. To generate a 22–amino acid linker separating paxillin from the fluorescent protein, the following primers were used to amplify paxillin:
NheI forward: GTC AGA TCC GCT AGC GCC ACC ATG GAC GAC CTC GAT GCC TTA CTG GCA GAC C
BamHI reverse: CCT ATG TAC GGA TCC GCG CTA CCA CTG GCT GCG CTT GCT CCA CCG CTG CTA CAG AAG AGT TTG AGA AAG CAG TTC TGA CAG TAG GGC TTG
The PCR product and mEmerald-N1 were digested, purified, and ligated to yield mEmerald-paxillin-22.
To construct the mouse talin1 (NM_011602.5) fusion, the following primers were used to amplify the N-terminal 4021 nucleotides of and create EcoRI and XmaI restriction sites:
EcoRI forward: GAC GGT AGA ATT CTA GCC ACC ATG GTT GCG CTT TCG CTG AAG ATT AGC ATT GG
XmaI reverse: TCC GTC ACT GCC CGG GCA GCC GCA GCC AGC TGA CTC TTG AGG TTG GGA GAA G
The resulting PCR product was ligated into a similarly cut mEmerald-N1 cloning vector. Next the following primers were used to amplify the C-terminal 3608 nucleotides of mouse talin1 and create XmaI and AgeI restriction sites:
XmaI forward: GAT TCA TCG CCC GGG CAG TGA CGG ACA GCA TCA ACC AGC TC
AgeI reverse: GGT GGC GAC CGG TGA TCG GGA CGC GGA GCC TGC GCC GGA CCC TTG CCC GGA CCC GGA CCC AGC GCC GTG CTC GTC TCG AAG CTC TGA AGG C
This PCR product was ligated into the mEmerald-N1 vector containing the EcoRI and XmaI insert, yielding a fusion of mEmerald and mouse talin1 separated by a 22–amino acid linker. This vector along with mEos2-N1 was digested using AgeI and NotI and ligated to produce mEos2-Talin-22.
To verify localization, mEmerald and mEos2 fusion proteins were transfected in HeLa cells (CCL2 line; ATCC) using Effectene (QIAGEN) and ∼1 μg vector. Transfected cells were grown on coverslips in DMEM/F12, fixed after 48 h, and mounted with Gelvatol.
F2F diffusion analysis
Diffusion mode decomposition.
For two-dimensional diffusion, the distribution of square frame-to-frame displacements, r2, is (Wieser et al., 2008)
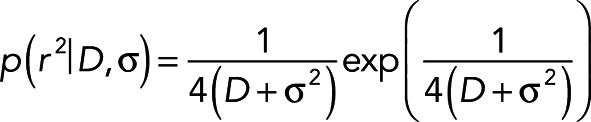 (2) (2)
|
where D is the diffusion coefficient and σ is the localization precision. If there are multiple diffusion modes N with diffusion coefficients D1…DN and fractions f1…fN, then the distribution of square frame-to-frame displacements follows the superposition of the individual probabilities in Eq. 2:
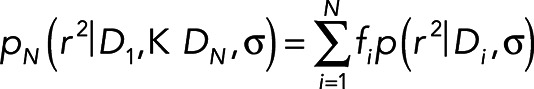 (3) (3)
|
To determine the number of diffusion modes in our data and the corresponding diffusion coefficients and fractions, Eq. 3 was fitted to the experimentally observed distribution of r2 in each cell (typically ∼900,000 points) with increasing values of N (1, 2, …), and each fit was compared to the previous one using the F-test (Sheskin, 2011). The addition of modes was stopped when the F-test failed (p-value > 0.05). As a negative control, monoexponential distributions with the same number of data points and average value as the experimental data were subjected to the decomposition analysis and overwhelmingly found only one mode (Supplemental Figure S2).
Trajectory diffusion mode classification.
Diffusion mode dividers for trajectory classification into the four modes were determined via simulations. Specifically, 1000 trajectories in each of modes 1–4, at each trajectory lifetime (5–20 frames, in steps of 1 frame) and at each localization precision (0–55 nm, in steps of 5 nm) were simulated (a total of 1000 × 4 × 16 × 11 = 710,400 trajectories). From this, the diffusion coefficient value maximally distinguishing between adjacent modes for a given trajectory lifetime and localization precision was determined and taken as the “diffusion mode divider” (Supplemental Figure S2). Since the divider values plateaued at 15–20 frames, the dividers at 20 frames were used for trajectories longer than 20 frames. The ability to distinguish between adjacent modes ranged between 60 and 100%, increasing with trajectory length and improved localization precision (Supplemental Figure S2).
Cell edge detection and tracking
The cell edge was detected using a combination of intensity value, intensity gradient (calculated via a steerable filter [ Jacob and Unser, 2004]), and single-molecule density gradient information. The edge was then tracked as described previously (Cai et al., 2014).
Adhesion segmentation
The paxillin images were first band-pass filtered to eliminate noise (Gaussian kernel sigma of 1 pixel) and then background subtracted (Gaussian kernel sigma of 10 pixels). The segmentation threshold was then taken as the 95th percentile of the band-pass-filtered intensity inside the cell mask and applied to the band-pass-filtered image. Segmented areas smaller than 10 pixels were discarded.
Mask division into edge activity-based sampling areas
Mask division into sampling areas was achieved via a three-step process.
Step 1: Initial 2 pixel × 2 pixel windows.
For all frames, the mask was divided into a series of 2-pixel strips, propagating from the cell edge to the cell interior. In the first frame, the cell edge was divided into 2-pixel segments. These segments were propagated from one frame to the next, using the cell edge frame-to-frame displacement vectors calculated via edge tracking (described above). The edge segments were combined with the first 2-pixel strip in each frame to generate windows at the cell edge for all frames. In each frame the windows were then propagated in space from one strip to the next, such that each window was associated with the edge segment closest to it.
To preserve window identity and distance from the edge, window length parallel to the cell edge could deviate from the target length of 2 pixels (the length perpendicular to the edge was fixed at 2 pixels). As a result, the windows could in some cases turn into virtual windows, which served the purpose of placeholders to maintain window identity (Machacek et al., 2009; Vilela et al., 2013). Nevertheless, we refer to these initial windows as 2 pixel × 2 pixel windows.
Step 2: Characterization of edge activity.
Periods of protrusion, retraction, and pause were identified for each 2-pixel edge segment as follows: 1) Individual frame-to-frame displacements normal to the local cell edge with magnitude >σedge (σedge = edge position SD = 1 pixel) were classified as protrusion if positive or retraction if negative. 2) A group of consecutive normal displacements with magnitude >0.5 × σedge were classified as protrusion if all were positive or as retraction if all were negative, with the rationale that persistence compensated for smaller individual displacements. 3) Individual normal displacements with magnitude <0.5 × σedge surrounded by displacements that were both classified as protrusion or both classified as retraction were given the classification of the surrounding displacements. 4) Normal displacements not satisfying conditions 1, 2, or 3 were classified as pause. Consecutive displacements of the same type were then grouped into periods of that movement type.
Step 3: Final activity-based sampling areas.
The initial 2 pixel × 2 pixel windows were combined into larger sampling areas based on time and location relative to protrusion onset in the following ways. All fixed areas were sampled at all times.
Protrusion onset location: The fixed area of the windows in the first three 2-pixel strips sampled 0–0.667 μm from the cell edge at protrusion onset.
Bands 2 and 3: Bands 2 and 3 were generated by grouping, respectively, strips 4–6 and 7–9 at protrusion onset, that is, 0.667–1.332 and 1.332–2 μm from the cell edge at protrusion onset. Together, the protrusion onset location and bands 2 and 3 covered the lamellipodium (0–2 μm) of the cell at protrusion onset.
Lamella: The lamella band was generated by grouping strips 10–30, sampling 2–6.6 μm from the cell edge at protrusion onset.
Calculation of single-molecule properties in sampling windows per cell
To increase sample size, areas within a cell with the same spatiotemporal relation to protrusion onset were grouped together. Areas prior to protrusion onset were grouped with no distinction between retraction and pause. Each event contributed a number of time points equal to its duration (e.g., a protrusion lasting 30 s would yield measurements at 0, 10, 20, and 30 s, while a retraction/pause lasting 20 s would yield measurements at –10 and –20 s). This grouping yielded a sample of molecule densities (sample size = number of 2 pixel × 2 pixel windows in group) and a sample of molecule diffusion coefficients (sample size = number of trajectories in group), from which the mean and SD of each property were calculated. Only measurements with sample size ≥5 were included in the calculation.
Combining single-molecule properties from multiple cells
Measurements from multiple cells were combined as the weighted average and SD of individual cell measurements. The weight of each individual cell measurement was inversely proportional to its square SE (= variance/sample size). Only measurements with sample size ≥5 were included in the average.
To tackle cell-to-cell heterogeneity when averaging, the single-molecule properties in each cell were first normalized by their value at the time and location of protrusion onset. Consequently, the mean and SD of the normalized single-molecule property and the normalization constant were calculated across cells. The average single-molecule property from multiple cells was taken as the product of the two, and the associated SD was calculated via error propagation of their two SDs.
Special considerations for density calculation
Windows in the first or last frame of a video had only a one-sided association with single-molecule trajectories. To compensate for this, single-molecule densities in these windows were multiplied by 2.
Supplementary Material
Acknowledgments
We thank H. Elliott and G. Danuser for cell mask and windowing software, J. R. Galbraith for circuit assistance, and M. Ginsberg and D. Calderwood for constructs. u-Track is available at www.utsouthwestern.edu/labs/jaqaman/software. Funding was provided by the National Institutes of Health Intramural Director’s Challenge Award (C.G.G. and J.A.G.) and Super-resolution Initiative (C.G.G.), the OHSU Knight Cancer Institute (C.G.G. and J.A.G.), the intramural program of the National Institute of Neurological Disorders and Stroke/National Institutes of Health (J.A.G.), National Institutes of Health Grant P50 GM068762 (K.J.; P. Sorger, PI), and the UT Southwestern Endowed Scholars Program (K.J.).
Abbreviations used:
- EGFP
enhanced green fluorescent protein
- F2F
frame-to-frame
- LP
lamellipodium
- MSS
momentum-scaling spectrum
- PAFPs
photoactivatible fluorophores
- PALM
photoactivation localization microscopy
- sptPALM
single-particle tracking PALM
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-01-0018) on March 23, 2016.
REFERENCES
- Abercrombie M, Heaysman JE, Pegrum SM. The locomotion of fibroblasts in culture. I. Movements of the leading edge. Exp Cell Res. 1970;59:393–398. doi: 10.1016/0014-4827(70)90646-4. [DOI] [PubMed] [Google Scholar]
- Bachir AI, Zareno J, Moissoglu K, Plow EF, Gratton E, Horwitz AR. Integrin-associated complexes form hierarchically with variable stoichiometry in nascent adhesions. Curr Biol. 2014;24:1845–1853. doi: 10.1016/j.cub.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Chen SC, Prasad M, He L, Wang X, Choesmel-Cadamuro V, Sawyer JK, Danuser G, Montell DJ. Mechanical feedback through E-cadherin promotes direction sensing during collective cell migration. Cell. 2014;157:1146–1159. doi: 10.1016/j.cell.2014.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood DA, Yan B, de Pereda JM, Alvarez BG, Fujioka Y, Liddington RC, Ginsberg MH. The phosphotyrosine binding-like domain of talin activates integrins. J Biol Chem. 2002;277:21749–21758. doi: 10.1074/jbc.M111996200. [DOI] [PubMed] [Google Scholar]
- Choi CK, Vicente-Manzanares M, Zareno J, Whitmore LA, Mogilner A, Horwitz AR. Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat Cell Biol. 2008;10:1039–1050. doi: 10.1038/ncb1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani A, Huang B, Bergan J, Dulac C, Zhuang X. Superresolution imaging of chemical synapses in the brain. Neuron. 2010;68:843–856. doi: 10.1016/j.neuron.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digman MA, Wiseman PW, Horwitz AR, Gratton E. Detecting protein complexes in living cells from laser scanning confocal image sequences by the cross correlation raster image spectroscopy method. Biophys J. 2009;96:707–716. doi: 10.1016/j.bpj.2008.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn JF, Jaqaman K, Rines DR, Jelson GS, Sorger PK, Danuser G. Yeast kinetochore microtubule dynamics analyzed by high-resolution three-dimensional microscopy. Biophys J. 2005;89:2835–2854. doi: 10.1529/biophysj.104.058461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duband JL, Nuckolls GH, Ishihara A, Hasegawa T, Yamada KM, Thiery JP, Jacobson K. Fibronectin receptor exhibits high lateral mobility in embryonic locomoting cells but is immobile in focal contacts and fibrillar streaks in stationary cells. J Cell Biol. 1988;107:1385–1396. doi: 10.1083/jcb.107.4.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewers H, Smith AE, Sbalzarini IF, Lilie H, Koumoutsakos P, Helenius A. Single-particle tracking of murine polyoma virus-like particles on live cells and artificial membranes. Proc Natl Acad Sci USA. 2005;102:15110–15115. doi: 10.1073/pnas.0504407102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith CG, Yamada KM, Galbraith JA. Polymerizing actin fibers position integrins primed to probe for adhesion sites. Science. 2007;315:992–995. doi: 10.1126/science.1137904. [DOI] [PubMed] [Google Scholar]
- Harburger DS, Bouaouina M, Calderwood DA. Kindlin-1 and -2 directly bind the C-terminal region of beta integrin cytoplasmic tails and exert integrin-specific activation effects. J Biol Chem. 2009;284:11485–11497. doi: 10.1074/jbc.M809233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes PE, O’Toole TE, Ylanne J, Shattil SJ, Ginsberg MH. The conserved membrane-proximal region of an integrin cytoplasmic domain specifies ligand binding affinity. J Biol Chem. 1995;270:12411–12417. doi: 10.1074/jbc.270.21.12411. [DOI] [PubMed] [Google Scholar]
- Huttenlocher A, Ginsberg MH, Horwitz AF. Modulation of cell migration by integrin-mediated cytoskeletal linkages and ligand-binding affinity. J Cell Biol. 1996;134:1551–1562. doi: 10.1083/jcb.134.6.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzard CS, Lochner LR. Formation of cell-to-substrate contacts during fibroblast motility: an interference-reflexion study. J Cell Sci. 1980;42:81–116. doi: 10.1242/jcs.42.1.81. [DOI] [PubMed] [Google Scholar]
- Jacob M, Unser M. Design of steerable filters for feature detection using canny-like criteria. IEEE Trans Pattern Anal Mach Intell. 2004;26:1007–1019. doi: 10.1109/TPAMI.2004.44. [DOI] [PubMed] [Google Scholar]
- Jaqaman K, Kuwata H, Touret N, Collins R, Trimble WS, Danuser G, Grinstein S. Cytoskeletal control of CD36 diffusion promotes its receptor and signaling function. Cell. 2011;146:593–606. doi: 10.1016/j.cell.2011.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, Waterman CM. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468:580–584. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiosses WB, Shattil SJ, Pampori N, Schwartz MA. Rac recruits high-affinity integrin alphavbeta3 to lamellipodia in endothelial cell migration. Nat Cell Biol. 2001;3:316–320. doi: 10.1038/35060120. [DOI] [PubMed] [Google Scholar]
- Lee J, Jacobson K. The composition and dynamics of cell-substratum adhesions in locomoting fish keratocytes. J Cell Sci. 1997;110:2833–2844. doi: 10.1242/jcs.110.22.2833. [DOI] [PubMed] [Google Scholar]
- Lee K, Elliott HL, Oak Y, Zee CT, Groisman A, Tytell JD, Danuser G. Functional hierarchy of redundant actin assembly factors revealed by fine-grained registration of intrinsic image fluctuations. Cell Syst. 2015;1:37–50. doi: 10.1016/j.cels.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YQ, Qin J, Wu C, Plow EF. Kindlin-2 (Mig-2): a co-activator of beta3 integrins. J Cell Biol. 2008;181:439–446. doi: 10.1083/jcb.200710196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461:99–103. doi: 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley S, Gillette JM, Patterson GH, Shroff H, Hess HF, Betzig E, Lippincott-Schwartz J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat Methods. 2008;5:155–157. doi: 10.1038/nmeth.1176. [DOI] [PubMed] [Google Scholar]
- Mueller V, Honigmann A, Ringemann C, Medda R, Schwarzmann G, Eggeling C. FCS in STED microscopy: studying the nanoscale of lipid membrane dynamics. Methods Enzymol. 2013;519:1–38. doi: 10.1016/B978-0-12-405539-1.00001-4. [DOI] [PubMed] [Google Scholar]
- O’Toole TE, Katagiri Y, Faull RJ, Peter K, Tamura R, Quaranta V, Loftus JC, Shattil SJ, Ginsberg MH. Integrin cytoplasmic domains mediate inside-out signal transduction. J Cell Biol. 1994;124:1047–1059. doi: 10.1083/jcb.124.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossier O, Octeau V, Sibarita JB, Leduc C, Tessier B, Nair D, Gatterdam V, Destaing O, Albiges-Rizo C, Tampe R, et al. Integrins beta1 and beta3 exhibit distinct dynamic nanoscale organizations inside focal adhesions. Nat Cell Biol. 2012;14:1057–1067. doi: 10.1038/ncb2588. [DOI] [PubMed] [Google Scholar]
- Rossy J, Pageon SV, Davis DM, Gaus K. Super-resolution microscopy of the immunological synapse. Curr Opin Immunol. 2013;25:307–312. doi: 10.1016/j.coi.2013.04.002. [DOI] [PubMed] [Google Scholar]
- Scannevin RH, Huganir RL. Postsynaptic organization and regulation of excitatory synapses. Nat Rev Neurosci. 2000;1:133–141. doi: 10.1038/35039075. [DOI] [PubMed] [Google Scholar]
- Sheskin DJ. Handbook of Parametric and Nonparametric Statistical Procedures, 5th ed. Boca Raton, FL: Chapman and Hall/CRC; 2011. [Google Scholar]
- Shroff H, Galbraith CG, Galbraith JA, Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat Methods. 2008;5:417–423. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff H, Galbraith CG, Galbraith JA, White H, Gillette J, Olenych S, Davidson MW, Betzig E. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc Natl Acad Sci USA. 2007;104:20308–20313. doi: 10.1073/pnas.0710517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtengel G, Galbraith JA, Galbraith CG, Lippincott-Schwartz J, Gillette JM, Manley S, Sougrat R, Waterman CM, Kanchanawong P, Davidson MW, et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc Natl Acad Sci USA. 2009;106:3125–3130. doi: 10.1073/pnas.0813131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilela M, Halidi N, Besson S, Elliott H, Hahn K, Tytell J, Danuser G. Fluctuation analysis of activity biosensor images for the study of information flow in signaling pathways. Methods Enzymol. 2013;519:253–276. doi: 10.1016/B978-0-12-405539-1.00009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrle-Haller B. Analysis of integrin dynamics by fluorescence recovery after photobleaching. Methods Mol Biol. 2007;370:173–202. doi: 10.1007/978-1-59745-353-0_13. [DOI] [PubMed] [Google Scholar]
- Wieser S, Axmann M, Schutz GJ. Versatile analysis of single-molecule tracking data by comprehensive testing against Monte Carlo simulations. Biophys J. 2008;95:5988–6001. doi: 10.1529/biophysj.108.141655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Nagarajan H, Lewis NE, Pan S, Cai Z, Liu X, Chen W, Xie M, Wang W, Hammond S, et al. The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat Biotechnol. 2011;29:735–741. doi: 10.1038/nbt.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.