

Abstract
Alkyl carboxylic acids are ubiquitous in all facets of chemical science, from natural products to polymers and represent an ideal starting material with which to forge new connections. This study demonstrates how the same activating principles used for decades to make simple C–N (amide) bonds from carboxylic acids with loss of water can be employed to make C–C bonds through coupling with dialkylzinc reagents and loss of carbon dioxide. This disconnection strategy benefits from the use of a simple, inexpensive nickel catalyst and exhibits a remarkably broad scope across a range of substrates (>70 examples).
The heart of chemical synthesis relies on forging new C–C bonds with the evolution and advancement of the field being easily correlated to new developments on this front. For example, pioneering work on the cross-coupling of halogenated aromatic or vinylic (sp2) systems (Heck, Suzuki, Negishi, Stille) has transformed the practice of organic synthesis.(1) Similarly, a general and practical approach to C(sp3)–C(sp3) variants, would have the potential to open up new vistas in retrosynthetic analysis. Indeed, such transformations have been on organic chemists' wish list for well over a century.(2,3) Historically, alkyl-alkyl transition metal-catalyzed cross-coupling reactions have been difficult to accomplish, but examples can be traced to the early work of Kharasch in the 1950s,(4) followed by Noller,(5,6) Kochi and Tamura (7,8) in the 1960s to more recent work from the groups of Suzuki, (9) Fu,(10) Knochel,(11) Kambe,(12) Oshima,(13) and many others.(14) Thus far, the vast majority of approaches to this problem have involved the coupling of alkyl halides (or related species) to organometallic reagents.(15–18) However, the limited availability, perceived instability, and frequent toxicity of alkyl halides has perhaps prevented the area of alkyl cross-coupling from blossoming. If one only considers convenience, stability, and availability as desired attributes in a functional group for such a coupling, the carboxylic acid reigns supreme (Figure 1A). Alkyl carboxylic acids are ubiquitous in every aspect of chemistry and can be readily found in medicines, materials, natural products, and in the pages of commercial chemical supplier catalogs. They are a stable functional group, non-toxic, and eminently diversifiable owing to the field of combinatorial chemistry where they are the "workhorse" building block. Although certain carboxylic acids have already been demonstrated to engage in cross-coupling reactions,(19) the use of alkyl carboxylic acids in alkyl-alkyl cross-coupling remains elusive.
Figure 1. Development of a Ni-catalyzed decarboxylative alkyl-alkyl cross-coupling.

(A) Activation of carboxylic acids in organic synthesis. (B) Potential side-products. (C) Ni-catalyzed cross-coupling of redox-active esters and alkylzinc reagents.
Carboxylic acids can be primed for reaction by a process known as activation (such as formation of an active ester, –OA*), dating back to the classic work of Sheehan in the synthesis of penicillin.(20) Once activated, a gateway opens to access a myriad of related functional groups such as amides, ketones, esters or alcohols via addition of a nucleophile or alternative oxidation states by the formal addition of hydrogen. In this Report, a broadly useful transform, able to forge C(sp3)–C(sp3) bonds via this age-old activation process, is presented.
We recently reported a Ni-catalyzed decarboxylative cross-coupling of alkyl carboxylic acids with arylzinc reagents to forge C(sp3)–C(sp2) bonds by repurposing activating methods more typically associated with amide-bond formation.(21, 22) Certain active esters (e.g. HOAt (aza –N-hydroxybenzotriazole), HOBt (N-hydroxybenzotriazole), NHPI (N-hydroxyphathalimide) TCNHPI (tetrachloro N-hydroxyphthalimide)) can accept an electron to trigger an ensuing cascade of events that liberates CO2 from the parent alkyl group (Alk1); such esters (23) are termed redox-active.(21, 22) The application of this chemistry to sp3–sp3 C–C bond formation poses a number of substantial challenges, with potentially unproductive pathways far outnumbering the desired reaction (Figure 1B).(15–18) For example, β-hydride elimination from the alkyl metal intermediates, dimerization of the organometallic reagent, reduction of the electrophile, and proto-demetallation are problems which also historically plague traditional C(sp3)–C(sp3) cross-coupling reactions. With a redox-active ester as an electrophile, oxidative addition of low valent Ni into the activated C–O bond could result in formation of an acyl-Ni complex, which could reductively eliminate and ultimately result in undesired ketone byproducts. These fundamental challenges notwithstanding, described below is a straightforward solution to this problem.
Dialkylzinc reagents were chosen for the organometallic coupling partner due to their ease of preparation from the parent alkyl halide, high functional group tolerance, and their propensity for facile transmetallation. Over the years, mono and dialkylzinc reagents have been shown to be viable cross-coupling partners in Negishi cross-coupling using alkyl halides and related species using Ni-based catalysts.(16,18) Thus, an exploration of the coupling of redox-active esters 1a and 1b (Figure 1C) with diethylzinc (2) was undertaken. An exhaustive screen of ligands, including bipy (L1, entry 1 – 2) and di-tBubipy (L2, entries 3 and 4) showed that the electron-deficient TCNHPI ester 1b with L2 afforded the desired product in 84% isolated yield (entry 4). TCNHPI can be easily prepared from tetrachlorophthalic anhydride, an industrial non-toxic flame retardant (ca. $48/kg from VWR)(24) and it has recently been commercialized by Aldrich (catalog # ALD00564). Additional screening of ligands based on phenanthroline (L3 – L5, entries 5 – 7) and terpyridine (L6 and L7, entries 8 and 9) did not improve the yield and were in many cases detrimental. When the reaction was performed in the absence of NiCl2·glyme, the desired product was not formed (entry 10).
With an optimized set of conditions in hand, the scope of this new reaction was explored and found to be remarkably broad. First, a range of dialkylzinc reagents (sixteen, Figure 2a) was explored with piperidine esters 1a and 1b. With the exception of dibenzylzinc reagents, all primary dialkylzinc reagents explored were viable in the cross coupling. From dimethylzinc (4) and simple alkyl chains (3, 5, 6, 9, 10) to derivatives harboring olefins (7–8), alkynes (11–12), acetals (16), ethers (14–15), and even alkyl halides (13) were tolerated. The reaction was easily run on a gram scale (as exemplified by 3) and could even be employed to produce isotopically labeled piperidine 4-13C. In addition to 3, this example highlights that a lower loading of dialkylzinc reagent (0.5 equiv) could be employed when a highly valuable alkyl group is involved. Secondary alkylzinc reagents such as cyclopropylzinc could also be accommodated as exemplified by 17.
Figure 2. Scope of the Ni-catalyzed decarboxylative alkyl-alkyl cross-coupling.

Standard conditions: redox-active ester (1 equiv.), dialkylzinc reagent (2 equiv.), NiCl2·glyme (20 mol%), L2 (40 mol%), THF:DMF, 25 ºC, 8–14 h. THF=tetrahydrofuran; DMF=dimethylformamide.
Next, the scope of secondary alkyl carboxylic acids (fifteen, Figure 2b) was explored. Cyclic (18, 19, 21), heterocyclic (20, 22, 26–28, 31), bridging (25), indane (24), acyclic (29), and even fluorinated (23, 30) alkyl carboxylic acids were all viable coupling partners. Substrates 30 through 32 are particularly striking and open the door to access a vast array of fluorinated building blocks and optically pure tartrate-derived materials. Many of these products would be either inconvenient or chemically intractable to access from the corresponding alkyl halide starting materials (27, 28, 30, 31 and 32).
Primary alkyl carboxylic acids, representing some of the most inexpensive organic materials available, could also be readily employed. Of the eight examples depicted in Figure 2c, most notable are the use of mono-methyladipic acid (34, 2.5 billion kg of adipic acid are produced per year),(25) a pyridine-containing substrate (36), and a polyfluorinated acid (40). Tertiary alkyl carboxylic acids (Figure 2d) could also be employed to generate quaternary centers. Although limited in scope, bridgehead systems such as adamantane (41 and 42) or bicyclo[2.2.2]octane (43) smoothly participated in the cross-coupling. The preparation of such bridged-systems is traditionally performed via a 3-step Wittig/hydrogenation sequence from the parent aldehyde.(26) Although pivalic acid (44) did not couple in this context, its failure led to a distinct type of coupling (vide infra).
Simple primary and secondary α-oxyacids could also be employed (Figure 2e, 45–49), representing a practical route to form ethers that would not be possible using alkyl halides. The classic Williamson ether synthesis is still the staple transform for constructing ethers but in many cases the SN2 reaction is either sluggish or unworkable due to steric hindrance. This cross coupling opens a distinct disconnection strategy for ether synthesis by using easily obtained α oxyacids (either commercial or derived via alkylation with bromoacetic acid) as progenitors for a virtually limitless array of new ethers.
Redox-active esters enable the cross-coupling of alkylzinc reagents with high chemoselectivity (Figure 2f) even in complex contexts, reminiscent of that exhibited in classic amide-bond formation. For example, a variety of sensitive fatty acids reacted smoothly to give the corresponding alkyl chains (50, 57 and 58) without olefin isomerization or epoxide opening. Naturally occurring carboxylic acids such as biotin, cholic acid and dehydrocholic acid were also amenable to alkylation (54–56). Pharmaceuticals such as pregabalin, 2,4-D, cetirizine and atorvastatin smoothly reacted with alkylzinc reagents to afford good yields of the alkyl coupling products (51–53 and 59).
In perhaps the most impressive feat of chemoselectivity for this methodology, the cross-coupling could be conducted on solid phase in the context of peptide synthesis. On-resin coupling of dialkylzinc reagents to proteinogenic amino acids (both side-chain and α-carboxylic acids) facilitates the late-stage introduction of designer amino acids, which would otherwise require de novo synthesis. Valuable synthetic handles including alkenes (60 and 62) and alkyl ethers (63) could be readily incorporated into resin-bound substrates (Figure 3), and the installation of an aliphatic side-chain (61) provides a facile approach to the modulation of peptide lipophilicity.(27) Double activation of a peptide substrate bearing both Asp and Glu residues also enables the simultaneous introduction of multiple non-proteinogenic side-chains (64 and 65). This convenient approach to diversely functionalized peptides provides a tool for the construction of therapeutic peptide leads, stapled peptides (28,29) and for applications in bioconjugation.(30)
Figure 3. Ni-catalyzed decarboxylative alkyl-alkyl coupling in solid phase synthesis.

† See Supplementary Materials for experimental details.
Returning to the limitations posed by tertiary systems, mechanistic studies involving 5-exo-dig cyclization, racemization and ring-strain opening experiments clearly point to the radical nature of this reaction (see Figures S1 – S3). We reasoned that the steric limitations encountered could be overcome by engaging these reactive species with a radical trap that could subsequently combine with an organozinc reagent. Remarkably, such a conjunctive cross-coupling could be realized as shown in Figure 4.(31) By using benzylacrylate as a radical trap and phenylzinc as a coupling partner, thirteen different tertiary alkyl carboxylic acids could be smoothly engaged in a three-component cross coupling to generate useful building blocks that would be extremely difficult to access in any other way (66–78). This reaction is scalable (as exemplified by 76), generates 2 new C–C bonds, and is a rare example of a multicomponent cross coupling reaction that forms quaternary centers in high yield. In addition to the broad functional group tolerance (heteroatom containing substrates, Boc (tert-butoxycarbonyl), TBS (tert-butyldimethylsilyl), MOM (methoxymethyl) groups), this reaction features the use of electronically different radical precursors, such as α-heteroatom acids or electronically unbiased and neutral alkyl acids such as pivalic acid.
Figure 4. Scope of the Ni-catalyzed three-component conjunctive cross-coupling.
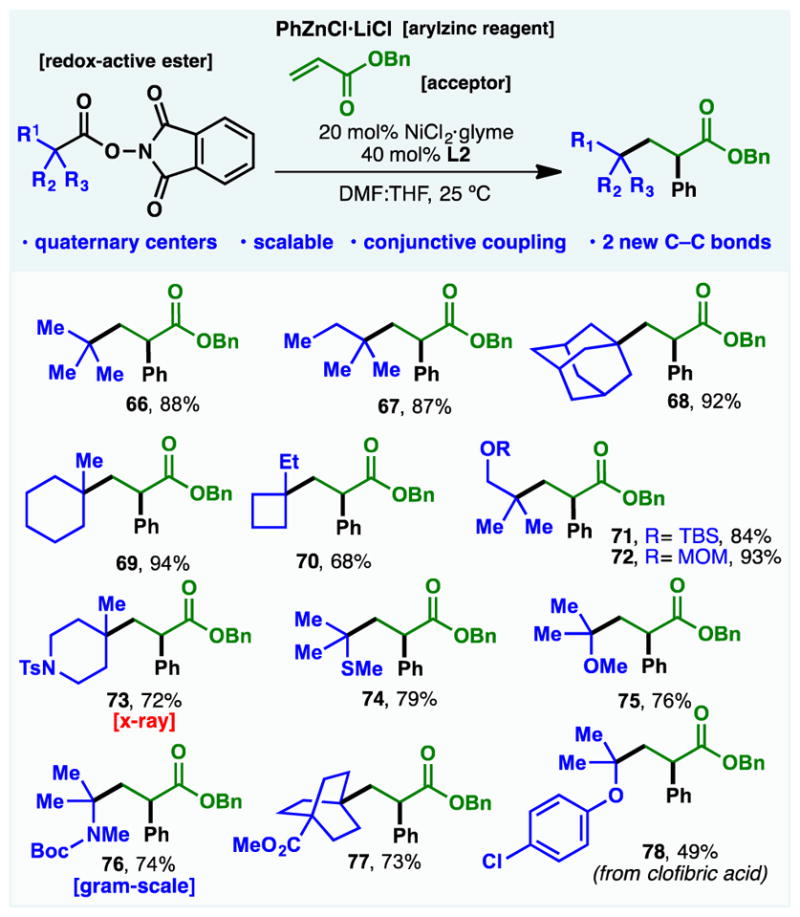
Standard conditions: redox-active ester (1 equiv.), acceptor (2.5 equiv.), PhZnCl·LiCl complex (3 equiv.), NiCl2·glyme (20 mol%), L2 (40 mol%), THF:DMF, 25 ºC, 8 h. THF=tetrahydrofuran; DMF=dimethylformamide. All the products shown were obtained in a racemic form.
As with any methodology there are obvious drawbacks. For example, the atom-economy is low due to the use of an activating group. We do note, however, that such considerations are ignored in the enormous field of peptide chemistry where expensive coupling agents are regularly employed to make a simple amide bond.(32) The activating agents utilized herein, NHPI ($19.5/mol) and TCNHPI (vide supra), are both commercially available and derived from cheap, readily available materials and open a gateway of reactivity heretofore inaccessible. The use of two equivalents of the dialkylzinc reagent is another disadvantage in cases where the zinc-derived fragment is valuable. However, as shown in 3 and 4-13C, the reaction proceeds with workable yields when simply equimolar amounts of alkylzinc reagents and 5 mol% catalyst were employed.
The data presented herein suggests that the advantages associated with this method far outweigh its limitations. Without exception, the carboxylic acids employed represent the most inexpensive sources of these carbon frameworks; in fact all of these were commercially available. In contrast, in the cases where an alkyl halide would be chemically stable, only a handful are commercially available. Conceptually, carboxylic acids can perhaps be considered as Nature's version of a boronic acid. For the past seven decades these ubiquitous functional groups have usually been dehydrated with incorporation of a nucleophile (such as an amine to make an amide). This method extends the native diversification of this functional group,(33) to allow for the addition of a new carbon framework via extrusion of CO2. As such it is anticipated that this technique will greatly expand planning options in retrosynthetic analysis.
Supplementary Material
Acknowledgments
Financial support for this work was provided by Bristol-Myers Squibb, Catalan Government (Postdoctoral Fellowship to J.C.), Department of Defense (NDSEG fellowship to J.T.E.), NIH (F32GM117816 Postdoctoral Fellowship to L.R.M.) China Scholarship Council (postdoctoral fellowship to C.L.) and NIGMS (GM106210). We thank Dr. D.-H. Huang and Dr. L. Pasternack for assistance with NMR spectroscopy; Dr. M. Collins for providing samples of atorvastatin, cetirizine and pregabalin; Mr. Riley Mills for experimental assitance; Dr. A. Rheingold, Dr. C. E. Moore, and Dr. M. A. Galella for x-ray crystallographic analysis; and Dr. K. Chen for helpful discussions. Metrical parameters for the structures of 11 and 73 are available free of charge from the Cambridge Crystallographic Data Center under reference numbers CCDC-1457710 and CCDC-1457711 respectively.
References and Notes
- 1.Diederich F, Stang PJ. Metal-catalyzed Cross-coupling Reactions. Wiley-VCH; New York: 1998. [Google Scholar]
- 2.Jolibois P. Compt Rend Acad Sciences. 1912;155:213. [Google Scholar]
- 3.Späth E. Monatsch. 1913;34:1965. [Google Scholar]
- 4.Kharasch MS, Hambling JK, Rudy TP. J Org Chem. 1959;24:303. [Google Scholar]
- 5.Parker VD, Noller CR. J Am Chem Soc. 1964;86:1110. [Google Scholar]
- 6.Parker VD, Noller CR. J Am Chem Soc. 1964;86:1112. [Google Scholar]
- 7.Tamura M, Kochi J. J Am Chem Soc. 1970;93:1483. [Google Scholar]
- 8.Tamura M, Kochi J. J Am Chem Soc. 1970;93:1485. [Google Scholar]
- 9.Ishiyama T, Abe S, Miyaura N, Suzuki A. Chem Lett. 1992;21:691. [Google Scholar]
- 10.Netherton MR, Dai C, Neuschütz K, Fu GC. J Am Chem Soc. 2001;123:10099. doi: 10.1021/ja011306o. [DOI] [PubMed] [Google Scholar]
- 11.Devasagayaraj A, Stüdemann T, Knochel P. Angew Chem Int Ed. 1995;34:2723. [Google Scholar]
- 12.Terao J, Watanabe H, Ikumi A, Kuniyasu H, Kambe N. J Am Chem Soc. 2002;124:4222. doi: 10.1021/ja025828v. [DOI] [PubMed] [Google Scholar]
- 13.Tsuji T, Yorimitsu H, Oshima K. Angew Chem Int Ed. 2002;41:4137. doi: 10.1002/1521-3773(20021104)41:21<4137::AID-ANIE4137>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 14.Urkalan KB, Sigman MS. J Am Chem Soc. 2009;131:18042. doi: 10.1021/ja908545b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Netherton MR, Fu GC. Adv Synth Catal. 2004;346:1525. [Google Scholar]
- 16.Frisch AC, Beller M. Angew Chem Int Ed. 2005;44:674. doi: 10.1002/anie.200461432. [DOI] [PubMed] [Google Scholar]
- 17.Rudolph A, Lautens M. Angew Chem Int Ed. 2009;48:2656. doi: 10.1002/anie.200803611. [DOI] [PubMed] [Google Scholar]
- 18.Jana R, Pathak TP, Sigman MS. Chem Rev. 2011;111:1417. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodriguez N, Goossen LJ. Chem Soc Rev. 2011;40:5030. doi: 10.1039/c1cs15093f. [DOI] [PubMed] [Google Scholar]; Sheehan JC. The Untold Story of Penicillin. Cambridge: MIT Press; 1982. The Enchanted Ring. [Google Scholar]
- 20.Cornella J, et al. J Am Chem Soc. 2016;138:2174. doi: 10.1021/jacs.6b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huihui KMM, et al. J Am Chem Soc. 2016 doi: 10.1021/jacs.6b01533. [DOI] [Google Scholar]
- 22.Okada K, Okamoto K, Oda M. J Am Chem Soc. 1988;110:8736. [Google Scholar]
- 23.Data retrieved from https://us.vwr.com/store/ on the 6th of April of 2016.
- 24.Musser MT. Adipic Acid. Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH; Weinheim: 2000. [Google Scholar]
- 25.Waddell ST, et al. 20040133011. PCT Intl Appl US. 2004
- 26.Arnott JA, Planey SL. Expert Opin Drug Discov. 2012;7:863. doi: 10.1517/17460441.2012.714363. [DOI] [PubMed] [Google Scholar]
- 27.Kritzer JA. Nat Chem Biol. 2010;6:566. doi: 10.1038/nchembio.407. [DOI] [PubMed] [Google Scholar]
- 28.Lau YH, de Andrade P, Wu Y, Spring DR. Chem Soc Rev. 2015;44:91. doi: 10.1039/c4cs00246f. [DOI] [PubMed] [Google Scholar]
- 29.Boutureira O, Bernardes GJL. Chem Rev. 2015;115:2174. doi: 10.1021/cr500399p. [DOI] [PubMed] [Google Scholar]
- 30.Zhang L, et al. Science. 2016;351:70. [Google Scholar]
- 31.Ishihara Y, Montero A, Baran PS. The Portable Chemist’s Consultant. Apple Publishing Group; Cupertino, CA: 2013. [Google Scholar]
- 32.Ogliaruso MA, Wolfe JF. Synthesis of Carboxylic Acids, Esters and their Derivatives. John Wiley; Chichester, England: 1991. [Google Scholar]
- 33.Nakamura T, Yorimitsu H, Shinokubo H, Oshima K. Synlett. 1999;1415 Ksdjc. [Google Scholar]
- 34.Bowry VW, Ingold KU. J Am Chem Soc. 1992;114:4992. Sdcnksjdnc. [Google Scholar]
- 35.Beckwith ALJ, Ingold KU. Free Radical Rearrangements. In: de Mayo P, editor. Rearrangements in Ground and Excited States. Vol. 1. Academic Press; NY: 1980. pp. 161–310. [Google Scholar]
- 36.Sugamoto K, Matsushita Y, Kameda Y, Suzuki M, Matsui T. Synth Comm. 2005;35:67. [Google Scholar]
- 37.Seebach D, Behrendt L, Felix D. Angew Chem Int Ed Engl. 1991;30:1008. [Google Scholar]
- 38.Piller FM, Appukkuttan P, Gavryushin A, Helm M, Knochel P. Angew Chem Int Ed. 2008;47:6802. doi: 10.1002/anie.200801968. [DOI] [PubMed] [Google Scholar]
- 39.Piller FM, Metzger A, Schade MA, Haag BA, Gavryushin A, Knochel P. Chem Eur J. 2009;15:7192. doi: 10.1002/chem.200900575. [DOI] [PubMed] [Google Scholar]
- 40.Hettche F, Hoffmann RW. New J Chem. 2003;27:172. [Google Scholar]
- 41.Nyfeler E, Reinaud P. Org Lett. 2008;10:985. doi: 10.1021/ol702832x. [DOI] [PubMed] [Google Scholar]
- 42.Yeager AR, Finney NS. Bioorg Med Chem. 2004;12:6451. doi: 10.1016/j.bmc.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 43.Linney ID, McDonald IM. 2001085704. PCT Int Appl. 2001
- 44.Ali A, et al. 2007070173. PCT Int Appl. 2007
- 45.Bertram LS, Fyfe MCT, Procter MJ, Williams GM. 2007116229. PCT Int Appl. 2007
- 46.Tochtrop GP, DeKoster GT, Cistola DP, Covey DF. Bioorg Med Chem Lett. 2002;12:433. doi: 10.1016/s0960-894x(01)00763-6. [DOI] [PubMed] [Google Scholar]
- 47.Medivir UK Ltd, Peptimmune, Inc. 2006/64286 A1. WO. 2006
- 48.Dowdy SF, Meade BR, Gogoi K. 2015/238516 A1. US. 2015
- 49.Clark R, et al. 2011/145747 A1. WO. 2011
- 50.Roham and Haas Company. 213718 B1. EP. 1991
- 51.Flynn DL, et al. 2014/275016 A1. US. 2014
- 52.Guerlavais V, et al. J Med Chem. 2003;46:1191. doi: 10.1021/jm020985q. [DOI] [PubMed] [Google Scholar]
- 53.Lackner GL, Quasdorf KW, Overman LE. J Am Chem Soc. 2013;135:15342. doi: 10.1021/ja408971t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brittain DEA, et al. Angew Chem Int Ed. 2005;44:2732. doi: 10.1002/anie.200500174. [DOI] [PubMed] [Google Scholar]
- 55.Yang Y, Fu X, Chen J, Zhai H. Angew Chem Int Ed. 2012;51:9825. doi: 10.1002/anie.201203176. [DOI] [PubMed] [Google Scholar]
- 56.Fleming I, et al. J Chem Soc, Perkin Trans 1. 1995;4:317. [Google Scholar]
- 57.Ackermann L, Kapdi AR, Schulzke C. Org Lett. 2010;12:2298. doi: 10.1021/ol100658y. [DOI] [PubMed] [Google Scholar]
- 58.Zhou J, Fu GC. J Am Chem Soc. 2003;125:12527. doi: 10.1021/ja0363258. [DOI] [PubMed] [Google Scholar]
- 59.Connolly T, et al. Org Lett. 2014;16:4444. doi: 10.1021/ol5019739. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.