

Abstract
An increase in cytosolic free Ca2+ concentration ([Ca2+]cyt) in pulmonary arterial smooth muscle cells (PASMC) is a major trigger for pulmonary vasoconstriction and a critical stimulation for PASMC proliferation and migration. Previously, we demonstrated that expression and function of calcium sensing receptors (CaSR) in PASMC from patients with idiopathic pulmonary arterial hypertension (IPAH) and animals with experimental pulmonary hypertension (PH) were greater than in PASMC from normal subjects and control animals. However, the mechanisms by which CaSR triggers Ca2+ influx in PASMC and the implication of CaSR in the development of PH remain elusive. Here, we report that CaSR functionally interacts with TRPC6 to regulate [Ca2+]cyt in PASMC. Downregulation of CaSR or TRPC6 with siRNA inhibited Ca2+-induced [Ca2+]cyt increase in IPAH-PASMC (in which CaSR is upregulated), whereas overexpression of CaSR or TRPC6 enhanced Ca2+-induced [Ca2+]cyt increase in normal PASMC (in which CaSR expression level is low). The upregulated CaSR in IPAH-PASMC was also associated with enhanced Akt phosphorylation, whereas blockade of CaSR in IPAH-PASMC attenuated cell proliferation. In in vivo experiments, deletion of the CaSR gene in mice (casr−/−) significantly inhibited the development and progression of experimental PH and markedly attenuated acute hypoxia-induced pulmonary vasoconstriction. These data indicate that functional interaction of upregulated CaSR and upregulated TRPC6 in PASMC from IPAH patients and animals with experimental PH may play an important role in the development and progression of sustained pulmonary vasoconstriction and pulmonary vascular remodeling. Blockade or downregulation of CaSR and/or TRPC6 with siRNA or miRNA may be a novel therapeutic strategy to develop new drugs for patients with pulmonary arterial hypertension.
Keywords: G protein-coupled receptor, ionic ligand, hypoxia-induced pulmonary hypertension
excessive proliferation of pulmonary arterial smooth muscle cells (PASMC) is an important contributor to the concentric pulmonary arterial remodeling, a major cause of the elevated pulmonary vascular resistance (PVR) and pulmonary arterial pressure (PAP) in patients with pulmonary arterial hypertension (PAH) and animals with experimental pulmonary hypertension (PH). Many intracellular signaling cascades are involved in the regulation of cell proliferation and migration; both are involved in the development and progression of pulmonary vascular remodeling (34, 55). Among various membrane receptors and ion channels functionally expressed in the plasma membrane of PASMC, G protein-coupled receptors (GPCR) are a large family of membrane receptors sensing extracellular stimuli such as growth factors, angiogenic factors, vasoconstrictors, hormones, and inflammatory cytokines (34, 38). Activation of GPCR by mitogenic and vasoconstrictive factors, as well as upregulated GPCR and GPCR ligands, has been implicated in the development and progression of pulmonary hypertension in patients and experimental animals (61).
Calcium-sensing receptor (CaSR) is a GPCR that can be activated by extracellular cations (e.g., Ca2+, Mg2+, and Sr+), polyamines (e.g., spermine, spermidine), protamine, polyarginine, aromatic amino acids, aminoglycoside antibiotics (e.g., neomycin), β-amyloids, and calcimimetics (e.g., cinacalcet or Sensipar) (5, 7, 10, 12, 21, 35, 55). CaSR consists of three molecular domains: an extracellular domain (ECD) or extracellular ligand-binding domain, a transmembrane domain, and a cytosolic domain (23). Different agonists bind to the different domains of CaSR, and extracellular Ca2+ binds to the ECD of CaSR or, more precisely, to the inside pocket of two lobes in the ECD that are separated by a hinge region (30, 50). Calcimimetics bind to the transmembrane domain, and amino acids bind to the ECD, regions very close to Ca2+-binding site (28, 49).
For a healthy adult, the free Ca2+ concentration ([Ca2+]) in the serum ranges from 4.8 to 5.7 mg/dl (or 1.2 to 1.43 mM), whereas extracellular free [Ca2+] around pulmonary vascular smooth muscle and endothelial cells is similar to the concentration in the blood and ranges from 1.6 to 2.0 mM. In human embryonic kidney (HEK), cells transfected with the wild-type (WT) CaSR gene had a threshold for a response to extracellular [Ca2+] at 1.5–2.0 mM, an EC50 extracellular [Ca2+] is about 3.5–4.5 mM (46). Our previous studies showed that the EC50 of extracellular Ca2+-induced increase in cytosolic free Ca2+ concentration ([Ca2+]cyt) in PASMC from patients with idiopathic pulmonary arterial hypertension (IPAH) was 1.22 mM (63). These observations indicate that the pulmonary vasculature is normally exposed to a range of extracellular [Ca2+] that can sufficiently activate CaSR expressed on the surface membrane of pulmonary vascular smooth muscle and endothelial cells.
Activation of CaSR sets into motion a complex series of intracellular Ca2+ signaling events that cause PASMC contraction, migration, and proliferation. Like other GPCRs, CaSR activation increases the synthesis of inositol 1,4,5-phosphate (IP3) and diacylglycerol (DAG) via G protein-stimulated phospholipase C (PLC). IP3 binds to the IP3 receptor on the SR membrane and releases Ca2+ from the SR to the cytosol. Depletion of Ca2+ from the SR of PASMC induces Ca2+ entry through store-operated Ca2+ channels (SOC), also referred to as store-operated Ca2+ entry (SOCE). DAG directly activates receptor-operated Ca2+ channels (ROC) in the plasma membrane; the Ca2+ entry through ROC is termed receptor-operated Ca2+ entry (ROCE). In addition to increasing [Ca2+]cyt via ROCE and SOCE, extracellular Ca2+-induced activation of CaSR can also activate other signal transduction pathways [e.g., Akt/mammalian target of rapamycin (mTOR)] to induce cell proliferation (7, 25, 35).
CaSR is widely expressed in parathyroid glands, bones, and kidney (27). However, under normal or physiological conditions, the expression level of CaSR in lungs for the pulmonary vasculature is very low (58, 63). In lung tissues and PASMC isolated from patients with IPAH and animals with experimental PH, we recently reported that mRNA and protein expression of CaSR was significantly greater than in lung tissues and PASMC isolated from normal subjects and control animals (58, 63). Furthermore, the upregulated CaSR in IPAH-PASMC (and PASMC isolated from animals with hypoxia-induced PH and monocrotaline-mediated PH) was associated with a significant enhancement of extracellular Ca2+-induced increase in [Ca2+]cyt due primarily to augmented SOCE compared with normal and control PASMC (58).
In this study, our aim was to examine whether genetic deletion of the CaSR gene inhibits the development and progression of pulmonary hypertension in mice and what cellular and molecular mechanisms are involved in the CaSR-mediated PASMC proliferation and migration. The results from this study conducted in CaSR gene knockout mice provide strong evidence that CaSR, a not well-characterized membrane receptor in the pulmonary vasculature, may play an important pathogenic role in pulmonary hypertension, whereas pharmacological blockers of CaSR or siRNAs and microRNAs that can downregulate CaSR may potentially be new therapeutic approaches for patients with PAH and PH associated with lung diseases and hypoxia.
MATERIALS AND METHODS
Experimental animals and hemodynamic measurements.
All animal experiments were approved by the University of Arizona Animal Care and Use Committee and were performed according to university guidelines that comply with national and international regulations. To develop the mouse model with experimental pulmonary hypertension, male C57BL/6N mice were exposed to hypoxia (10% O2) in a ventilated chamber (BioSpherix) for 4 wk. Following the hypoxic exposure, right ventricular pressure was measured by a catheter (Millar Instruments) and positioned in the right ventricle (RV) via the external jugular vein and a 1.4-French pressure transducer (SPF 1030; Millar Instruments). The right ventricular pressure was recorded and analyzed using the AcgKnowledge software (Biopac Systems). The right ventricular systolic pressure (RVSP) was used as a surrogate for pulmonary arterial systolic pressure. After hemodynamic measurement, the heart was excised and dissected to determine the ratio of the RV weight to the left ventricle (LV) and septum (S) weight [RV/(LV + S) ratio], i.e., the Fulton index, as a parameter of RV hypertrophy (RVH). Lung tissues were used for Western blotting analysis and real-time RT-PCR analysis as well as for morphometric analysis.
Histological and morphometric analyses.
The Aperio ImageScope system was used to measure vascular wall thickness of the small pulmonary arteries. Briefly, lung tissues were perfused with cold PBS solution to remove blood and then fixed in a 10% normalized formalin solution overnight. The fixed lung tissues were dehydrated, embedded into paraffin, and then sectioned. Slides were further processed for hematoxylin and eosin (H & E) staining. The H & E-stained lung tissue slides were scanned with the Aperio scanner. Using the Aperio ImageScope software (version 11), the external and internal diameters as well as external and internal areas for each pulmonary artery were measured. The pulmonary arterial wall thickness was calculated by the following equation: thickness = (external vessel area − internal vessel area) ÷ (external vessel area). The diameter measured for each pulmonary artery was used to categorize it into two groups: the pulmonary artery with a diameter of <50 μm and the pulmonary artery with a diameter between 50 and 100 μm.
Isolated perfused/ventilated mouse lung experiment.
The PAP was measured using the isolated perfused/ventilated mouse lung system as described previously (51, 58, 66). Briefly, mice were anesthetized and ventilated with a gas mixture of 21% O2-5% CO2 in N2 via a rodent ventilator (minivent type 845; Harvard Apparatus). A stainless-steel catheter was inserted into the main pulmonary artery (PA) after a right ventriculotomy was performed, and the PA and ascending aorta were tied together using a 6-0 black silk suture. PAP was measured using a pressure sensor (P75 type 379; Hugo Sachs Elektronik-Harvard Apparatus), which was connected to the PA catheter. The other end of the catheter was connected to a tube for PA perfusion. The pulmonary circulation was maintained in a closed circuit via a peristaltic pump (ISM 834; Isomatec). For data acquisition and data storage, Powerlab 8/30, Quad Bridge Amp, and LabChart (AD Instruments) were used. After basal PAP was stabilized for 40–60 min, the experiments were performed.
Cell culture.
Human PASMC from normal subjects (Lonza, Walkersville, MD) and IPAH patients were cultured in M199 medium (Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum (Invitrogen, Grand Island, NY), 25 mg/l d-valine (Sigma-Aldrich, St. Louis, MO), 100 IU/ml penicillin and 100 μg/ml streptomycin (Sigma-Aldrich, St. Louis, MO), and 20 μg/ml cell growth supplement (BD Biosciences, Franklin Lakes, NJ) in a humidified atmosphere at 37°C and 5% CO2. Use of human lung tissues and cells was approved by the University of Arizona Institutional Review Board. The cells at passage 5–8 were used for the experiments.
Electrophysiological recording.
Whole-cell cation currents were recorded and analyzed by the patch-clamp technique using an Axopatch-1D amplifier, an analog-digital converter (Digidata 1200), and pCLMP8 software (Molecular Devices/Axon Instruments, Foster City, CA), as described previously (64). The HEPES-buffered solution was used as the bath (extracellular) solution. The pipette (intracellular) solution contained (in mM) 120 CsOH, 120 aspartic acid, 0.6475 CaCl2, 4 MgCl2, 5 MgATP, 10 HEPES, and 10 EGTA (pCa = 8.0). The pH of the pipette solution was adjusted to 7.2 with 1 N CsOH. Single cells were clamped at a holding potential of 0 mV, and ascending ramp protocol from −100 to +100 mV for 250 ms was applied to cells every 15 s. Electrophysiological recordings were performed at room temperature (24–25°C).
Measurement of [Ca2+]cyt.
[Ca2+]cyt in PASMC was measured according to the previously described method (52). Briefly, PASMC were grown to passage 5–8 at 50–60% confluence on 25-mm-diameter circular glass coverslips. The cells were loaded with 4 μM fura-2 acetoxymethyl ester (fura 2-AM; Invitrogen/Molecular Probes, Eugene, OR) in HEPES-buffered solution for 60 min at room temperature. PASMC loaded with fura 2-AM were placed in a recording chamber mounted on the stage of an inverted fluorescence microscope (Eclipse Ti-E; Nikon, Tokyo, Japan) equipped with an objective lens (S Plan Fluor ×20/0.45 ELWD; Nikon), an EM-CCD camera (Evolve; Photometrics, Tucson, AZ), and the NIS Elements 3.2 software (Nikon) and superfused with HEPES-buffered solution for 30 min to wash out extracellular residual fura 2-AM and allow sufficient time for intracellular esterase to convert fura 2-AM to fura 2. The fluorescence intensity emitted at 520 nm in cells excited by illumination at 340 and 380 nm was measured with a region of interest at the rate of every 2 s. The HEPES-buffered bath solution had an ionic composition of 137 mM NaCl, 5.9 mM KCl, 2.2 mM CaCl2, 1.2 mM MgCl2, 14 mM d-glucose, and 10 mM HEPES. The pH was adjusted to 7.4 with 10 N NaOH. The Ca2+-free bath solution was prepared by replacing CaCl2 with equimolar MgCl2; 1 mM EGTA was added to chelate the residual Ca2+ in the solution. To determine [Ca2+]cyt from fura-2/fluorescence ratios, the intracellular minimal (Rmin) and maximal fluorescence ratios (Rmax) were determined as described previously (52). To determine Rmin, fura-2-loaded PASMC were perfused with a solution containing (in mM) 137 NaCl, 5.9 KCl, 1.2 MgCl2, 14 d-glucose, 10 HEPES, and 5 EGTA with 2 mM ionomycin (pH 7.40). An intracellular Rmax value was determined by perfusing PASMC with a similar solution, except that the extracellular concentration of CaCl2 was increased to 11 mM. The values of intracellular Rmin and Rmax were used to calculate [Ca2+]cyt according to the following formula: [Ca2+]cyt = Kd (R − Rmin)/{[Rmax (Rmin) R](Sf2/Sb2)} (38, 39), where Kd is the dissociation constant of fura-2 and Sf2 and Sb2 are the fluorescence intensities at ∼510 nm of the Ca2+-free and Ca2+-saturated indicator, respectively. All [Ca2+]cyt measurements were performed at 32°C.
Western blotting.
Total protein was isolated from PASMC, which were lysed in 1× RIPA buffer (Bio-Rad). Protein was loaded on an 8% acrylamide gel, transferred to an Immobilon-P transfer membrane (Millipore, Bedford, MA), and immunoblotted with anti-CaSR (1:1,000; Alomone Laboratories) and anti-transient receptor potential canonical (TRPC)6 monoclonal antibody (1:1,000; Alomone Laboratories). Signals were detected using a Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific). The protein levels were normalized to β-actin (Santa Cruz Biotechnology) and expressed in arbitrary units.
Transfection of CaSR cDNA.
Cultred PASMC were transiently transfected with 2 μg of human CaSR cDNA [constructed into pcDNA3.1(+); Invitrogen], 2 μg of human TRPC6 cDNA [constructed into pcDNA3.1(+)], 50 nM control siRNA (sc-37007; Santa Cruz Biotechnology), or 50 nM TRPC6-siRNA (s2440; Applied Biosystems/Ambion, Austin, TX) using an Amaxa Basic Nucleofector kit for primary smooth muscle cells (Lonza). Experiments using CaSR-/TRPC6- or siRNA-transfected cells were performed 48 h after electroporation.
Transfection of interfering RNA (siRNA).
PASMC were transiently transfected with control small-interfering RNA (siRNA) (10 μM, sc-37007; Santa Cruz Biotechnology), TRPC6 siRNA (10 μM; Santa Cruz Biotechnology) or TRPC6 (10 μM; Santa Cruz Biotechnology) using Xfect siRNA transfection reagent protocol (Clontech Laboratories). [Ca2+]cyt measurement and Western blot experiments using siRNA-transfected cells were performed 48–72 h after transfection.
Drugs and chemicals.
A hydrophobic compound, 1-oleoyl-2-acetyl-sn-glycerol (OAG), was dissolved in dimethyl sulfoxide (DMSO) at the concentration of 100 mM as a stock solution; aliquots of the stock solution were then diluted 1,000 times to make a final concentration of OAG at 100 μM for the experiment. Lathanum (La3+) was prepared as a concentrated stock solution in distilled water, whereas 2-aminoethoxydiphenyl borate (2-APB) was prepared as a stock solution in ethanol and cyclopiazonic acid and NPS-2143 in DMSO. All stock solutions (in water, ethanol, or DMSO) were aliquoted and kept frozen at −20°C until use. On the day of experiments, aliquots of the stock solutions were diluted 1,000–2,000 times in the HEPES-buffered bath solution to the final concentrations for each drug. The pH values of all solutions were measured after addition of the drugs and adjusted to 7.4. All drugs were from Sigma Chemical unless otherwise indicated.
Statistical analysis.
Pooled data are shown as means ± SE. The statistical significance between two groups was determined by Student's t-test. The statistical significance among groups was determined by Scheffé's test after one-way analysis of variance. Significant difference is expressed in the figures or figure legends as P < 0.05.
RESULTS
Activation of CaSR by extracellular stimuli induces increases in [Ca2+]cyt via Ca2+ influx through ROC and SOC. Transient receptor potential canonical (TRPC) channels have been demonstrated as participating in forming both ROC and SOC in PASMC (67, 69). Ca2+ currents through TRPC6 channels or an increase in [Ca2+]cyt due to Ca2+ influx through TRPC6 channels are sensitive to La3+, whereas extracellular application of La3+ significantly inhibited the extracellular Ca2+-induced increase in [Ca2+]cyt in IPAH-PASMC as well. The question is whether the enhanced CaSR-mediated Ca2+ influx in IPAH-PASMC is due, at least partially, to DAG-mediated activation of TRPC6 channels.
CaSR functionally interacts with TRPC6 to regulate [Ca2+]cyt in IPAH-PASMC.
Similarly to other types of GPCR, extracellular Ca2+-mediated CaSR activation increases synthesis of IP3 and DAG via Gqα proteins, leading to rises in [Ca2+]cyt. The extracellular Ca2+-induced increase in [Ca2+]cyt in PASMC from IPAH patients (in which CaSR expression is significantly upregulated) was often composed of two components: a transient increase due mainly to Ca2+ release or mobilization from the intracellular stores to the cytosol and a sustained or plateaued phase due obviously to Ca2+ influx through Ca2+-permeable channels in the plasma membrane. As shown in Fig. 1, the extracellular Ca2+-induced sustained or plateaued phase of increase in [Ca2+]cyt was significantly attenuated by La3+, a potent blocker of cation channels, in IPAH-PASMC (Fig. 1, A and B). These results indicate that activation of CaSR in IPAH-PASMC leads to the opening of cation channels that are sensitive to La3+.
Fig. 1.

Blockade of transient receptor potential canonical 6 (TRPC6) channels significantly inhibits extracellular Ca2+-induced increase in cytosolic free Ca2+ concentration ([Ca2+]cyt) in idiopathic pulmonary (IPAH)-pulmonary arterial smooth muscle cells (PASMC). A: representative images (left) and traces (middle) showing the changes of [Ca2+]cyt before, during, and after extracellular application of 2.2 mM Ca2+ (2.2Ca) in normal (Nor) and IPAH-PASMC. Lathanum (La3+) was added into the perfusate when 2.2Ca-induced increase in [Ca2+]cyt reached the plateau phase. Summarized data (n = 43 cells; right) showing the amplitude of 2.2Ca-induced transient and plateau increases in [Ca2+]cyt in IPAH-PASMC with (La) or without (control) extracellular La3+. **P < 0.01 vs. control. B: representative images (left) and tracing (middle) showing the 2.2Ca-induced plateau increase [Ca2+]cyt in IPAH-PASMC before and during application of 3 mM La3+ (La). Summarized data (right) showing the amplitude of 2.2Ca-induced plateau increases in [Ca2+]cyt in IPAH-PASMC before (control) and during (La) application of extracellular La3+. **P < 0.01 vs. control. C: representative traces showing the changes of [Ca2+]cyt before, during, and after extracellular application of 2.2Ca in IPAH-PASMC transfected with control (scrambled siRNA; left) and TRPC6 siRNA (right) Nor cells. D: summarized data (means ± SE; left) showing the amplitude of 2.2Ca-induced increase in [Ca2+]cyt in IPAH-PASMC transfected with control (Cont) or TRPC6 siRNA. **P < 0.01 vs. Cont siRNA. Histogram at right shows the distribution of the amplitudes of 2.2Ca-induced increases in [Ca2+]cyt in IPAH-PASMC transfected with control siRNA (top) or TRPC6 siRNA (bottom). Data were obtained from 29 to 57 cells.
It has been well demonstrated that La3+ is a potent blocker of TRPC channels (32, 73), especially TRPC6 channels. Extracellular application of La3+ significantly reduced whole-cell currents in HEK-293 cells transiently transfected with TRPC6 (32) and markedly attenuated increases in [Ca2+]cyt induced by cyclopiazonic acid- or triglyceride-induced passive Ca2+ depletion in COS cells cotransfected with TRPC6 and TRPC1 (22). Downregulation of TRPC6 in IPAH-PASMC with siRNA significantly inhibited extracellular Ca2+-induced increase in [Ca2+]cyt in IPAH-PASMC (Fig. 1, C and D). These data indicate that TRPC6 is necessary for the CaSR-mediated increase in [Ca2+]cyt in IPAH-PASMC.
To obtain further evidence for the involvement of TRPC6 channels in the CaSR-mediated [Ca2+]cyt increase in IPAH-PASMC, overexpression experiments of CaSR or TRPC6 were carried out in normal PASMC. In normal PASMC (in which CaSR expression level is very low), overexpression of CaSR or TRPC6 alone significantly enhanced the extracellular Ca2+-induced increase in [Ca2+]cyt compared with normal cells transfected with an empty vector [green fluorescent protein (GFP)] (Fig. 2A), whereas cotransfection of CaSR and TRPC6 together in normal PASMC further enhanced the extracellular Ca2+-induced increases in [Ca2+]cyt (Fig. 2A). The amplitude of extracellular Ca2+-induced increase in [Ca2+]cyt in normal PASMC transfected with both CaSR and TRPC6 was doubled compared with the amplitude in normal PASMC transfected with CaSR or TRPC6 alone (Fig. 2A, right).
Fig. 2.

Overexpression of calcium-sensing receptor (CaSR) and/or TRPC6 enhances the extracellular Ca2+-induced increases in [Ca2+]cyt and cation currents in normal PASMC. A: representative traces (left) showing the changes in [Ca2+]cyt before, during, and after application of 2.2 mM extracellular Ca2+ (2.2Ca) in normal PASMC transfected with an empty vector (black), the CaSR gene (CASR; green), the TRPC6 gene (TRPC6; blue), or the CaSR and TRPC6 genes (CASR + TRPC6). Summarized data (means ± SE; right) showing the amplitudes of 2.2Ca-induced transient (left) and plateau increases in [Ca2+]cyt in normal PASMC transfected with vector, CASR, TRPC6, or CASR + TRPC6. **P < 0.01 vs. vector. Data from 8 to 14 cells were obtained. B: representative whole-cell cation currents, induced by a ramp test pulse (from −100 to +100 mV) from a holding potential of 0 mV, are recorded in vector-transfected (left) and CASR + TRPC6-transfected (right) normal PASMC during superfusion with Ca2+-free solution (0Ca-control; blue), 2.2-mM Ca2+-containing solution (2.2Ca; red) or Ca2+-free solution (0Ca-washout; green). C: summarized data (means ± SE) showing the amplitudes of extracellular Ca2+-induced currents at −80 and +80 mV in vector- or CASR + TRPC6-transfected normal PASMC; the Ca2+-induced currents were obtained by subtracting the currents recorded in cells superfused with 2.2-mM Ca2+-containing solution from the currents recorded in cells superfused with Ca2+-free solution. **P < 0.01 vs. vector. D: representative currents (left) in vector-transfected IPAH-PASMC during superfusion with Ca2+-free solution (0Ca; blue), 2.2-mM Ca2+-containing solution (2.2Ca; red), or Ca2+-free solution (0Ca; green). Summarized data (means ± SE; right) showing the amplitudes of extracellular Ca2+-induced currents at −80 and +80 mV in vector-transfected IPAH-PASMC. E: representative traces (left) showing the changes in [Ca2+]cyt before, during, and after application of 1.8 mM extracellular Ca2+ (1.8Ca) in human ebryonic kidney (HEK)-293 cells with a green fluorescent protein (GFP) vector (GFP; black), CASR (green), or CASR + TRPC6. Summarized data (means ± SE; right) showing the amplitudes of 1.8Ca-induced increases in [Ca2+]cyt in HEK cells transfected with GFP, CASR, or CASR + TRPC6. **P < 0.01 vs. GFP. Data from 6 to 8 cells were obtained for each experiment.
Using patch clamp techniques, we also measured and compared whole-cell cation currents in normal PASMC transfected with an empty vector (Vector) in normal PASMC cotransfected with both CaSR and TRPC6 (CaSR + TRPC6) and in PASMC isolated from IPAH patients (IPAH-PASMC). As shown in Fig. 2B, the amplitude of whole-cell cation currents elicited by a voltage ramp (from −100 to +100 mV for 250 ms with a holding potential of 0 mV) in normal PASMC transfected with CaSR and TRPC6 (CaSR + TRPC6) was significantly greater than in normal PASMC transfected with empty vector. In normal PASMC, few currents were elicited despite the presence of extracellular Ca2+ (2.2 mM). In normal PASMC transfected with CaSR and TRPC6, we observed significantly enhanced currents in both the absence and presence of extracellular Ca2+ (Fig. 2, B and C). IPAH-PASMC exhibited large currents with a reversal potential of ∼0 mV even in the absence of extracellular Ca2+, yet these currents are enhanced with application of 2.2-mM extracellular Ca2+. In these experiments, cotransfection of CaSR and TRPC6 in normal PASMC increased the whole-cell cation currents to the level of currents detected in IPAH-PASMC.
Furthermore, in HEK-293 cells transfected with an empty vector (GFP), the application of extracellular Ca2+ caused a very small increase in [Ca2+]cyt (Fig. 2F). Overexpression of CaSR alone significantly enhanced the extracellular Ca2+-induced increase in [Ca2+]cyt, whereas overexpression of both CaSR and TRPC6 further enhanced the Ca2+-induced rise in [Ca2+]cyt in HEK-293 cells (Fig. 2F). These data indicate that TRPC6 is sufficient to enhance the CaSR-mediated increase in [Ca2+]cyt in normal PASMC.
As a GPCR, activated CaSR increases not only IP3 to open the Ca2+-release channels (IP3 receptors) on the sarcoplasmic reticulum (SR) membrane, inducing Ca2+ release, but also DAG to open ROC on the plasma membrane, causing Ca2+ influx. In vascular smooth muscle cells, it has been reported that TRPC6 is an important TRPC subunit that participates in forming homotetrameric or heterotetrameric channels to function as ROC (32). To examine whether DAG-mediated Ca2+ influx via ROC is enhanced in IPAH-PASMC, we used OAG, a membrane-permeable analog of DAG that directly activates ROC formed by TRPC3 and TRPC6 (26, 42, 65), to stimulate PASMC and compared the OAG-mediated changes in [Ca2+]cyt in normal and IPAH PASMC. As shown in Fig. 3, extracellular application of OAG had little effect on [Ca2+]cyt in normal PASMC but caused oscillatory increases in IPAH-PASMC (Fig. 3, A and B). The amplitude and frequency of OAG-induced oscillatory increases were both significantly enhanced in IPAH-PASMC compared with normal control cells (Fig. 3C). These data indicate that upregulated TRPC6 channels in IPAH-PASMC are functionally sensitive to DAG, a critical intracellular second messenger increased by CaSR activation, and may play an important role in mediating excessive extracellular Ca2+-induced increase in [Ca2+]cyt in IPAH-PASMC.
Fig. 3.

Diacylglycerol-induced increase in [Ca2+]cyt is significantly enhanced in IPAH-PASMC compared with normal PASMC. A and B: representative images (A) and traces (B) showing the changes in [Ca2+]cyt before and during extracellular application of 100-μM 1-oleoyl-2-acetyl-sn-glycerol (OAG), a membrane-permeable analog of diacylglycerol, in normal (top) and IPAH (bottom) PASMC bathed in a 2.2-mM Ca2+-containing solution. C: summarized data (means ± SE) showing %cells responding to OAG (left) and the amplitude of OAG-induced increase in [Ca2+]cyt in normal (Nor) and IPAH-PASMC. Data were obtained from 32 to 34 cells. *P < 0.05 and **P < 0.01 vs. Nor. D: histogram showing the distribution of the amplitudes of OAG-induced increases in [Ca2+]cyt in Nor (top) and IPAH (bottom) PASMC.
Taken together, these observations strongly suggest that CaSR is functionally coupled to La3+- and OAG-sensitive TRPC6 channels in IPAH-PASMC; the receptor-operated and/or store-operated Ca2+ entry via CaSR-mediated TRPC6 activation is an important signaling cascade leading to PASMC contraction, migration, and proliferation in IPAH patients.
In the next set of experiments, we aimed to examine whether the in vitro data showing that upregulated CaSR and TRPC6 functionally interact with each other to coordinately enhance the Ca2+ signaling cascade in PASMC from IPAH patients can be translated into in vivo experiments showing that upregulated CaSR may play a causal role in the development and progression of pulmonary hypertension.
Deletion of the CaSR gene significantly inhibits the development and progression of experimental pulmonary hypertension.
To test the hypothesis that upregulated CaSR, as shown in PASMC from IPAH patients, plays a potential causal role in the development and progression of pulmonary hypertension, we first compared pulmonary hemodynamics and structural changes in pulmonary arteries and right ventricle in wild-type (WT) and genetically engineered mice. Since global knockout of the CaSR gene was embryonically lethal, we used the double-knockout mice in which both the CaSR gene (casr) and the parathyroid hormone gene (pth) were knocked out (casr−/−/pth−/−) to investigate whether CaSR, via at least in part activation of TRPC6 channels, is involved in the development and progression of pulmonary vascular remodeling and right ventricular hypertrophy in vivo. The development of this model and the phenotypic characteristics as well as the diet requirements have been described previously by Kantham et al. (33).
The mice were first genotyped using PCR and confirmed the presence of one or two nonfunctional mutant alleles in the casr+/−/pth−/− and casr−/−/pth−/− mice, respectively (Fig. 4, A and B). Exposure of WT mice to hypoxia (10% O2) in a normobaric hypoxic chamber for 4 wk significantly increased right ventricular systolic pressure (RVSP), a surrogate marker for elevated pulmonary artery pressure (PAP), from 24.2 ± 2.6 to 41.9 ± 1.6 mmHg (P < 0.001) (Fig. 4, C–E). The increased RVSP in chronically hypoxic WT mice was closely associated with pulmonary vascular remodeling determined by a significant increase in pulmonary arterial wall thickness from both small (<50 μM) and medium-sized (50–100 μM) arteries (from 0.47 ± 0.03 to 0.64 ± 0.06 μm in vessels with diameter <50 μm, P < 0.05, and from 0.43 ± 0.03 to 0.73 ± 0.04 μm in vessels with diameter between 50 and 100 μm, P < 0.01) (Fig. 4, F and G). Furthermore, the increased RVSP and increased PA wall thickness in chronically hypoxic WT mice were associated with marked right ventricular hypertrophy (RVH), indicated by a significant increase in the Fulton Index [the ratio of RV/(LV + S)] (from 0.29 ± 0.01 in normoxic control mice to 0.46 ± 0.08 in hypoxic mice, P < 0.05) (Fig. 5A).
Fig. 4.

Deletion of the CaSR gene casr attenuates hypoxia-induced pulmonary hypertension and pulmonary vascular remodeling in mice. A: a wild-type (WT) mouse and a casr−/−/pth−/− mouse. B: genotyping characterization of WT, casr+/−/pth−/−, and casr−/−/pth−/− mice demonstrates the presence of only knockout (KO) alleles in the homozygous mice (casr−/−/pth−/−), whereas heterozygotes have both KO and WT alleles (casr+/−/pth−/−). C and D: representative records of right ventricular pressure (RVP; C) and summarized data (means ± SE) of peak right ventricular systolic pressure (RVSP; D) in normoxic (Nor; n = 6) and hypoxic (Hyp; n = 6) WT and casr−/−/pth−/− mice. E: summarized data (means ± SE) showing actual (left) and relative (%Nor; right) RVSP increases induced by chronic hypoxia in WT and casr−/− mice. *P < 0.05 vs. WT. F: hematoxylin and eosin images of small pulmonary arteries isolated from Nor and Hyp WT (top) and casr−/− (bottom) mice. G, top: summarized data (means ± SE) showing the medial thickness of pulmonary arteries (PA) with a diameter between 50 and 100 μm (left) or <50 μm (right) from WT mice or casr−/− mice under Nor or Hyp conditions. Bottom: relative changes (%WT-Nor) of PA wall thickness in Nor and Hyp WT mice or casr−/− mice. *P < 0.05 and **P < 0.01 vs. WT.
Fig. 5.
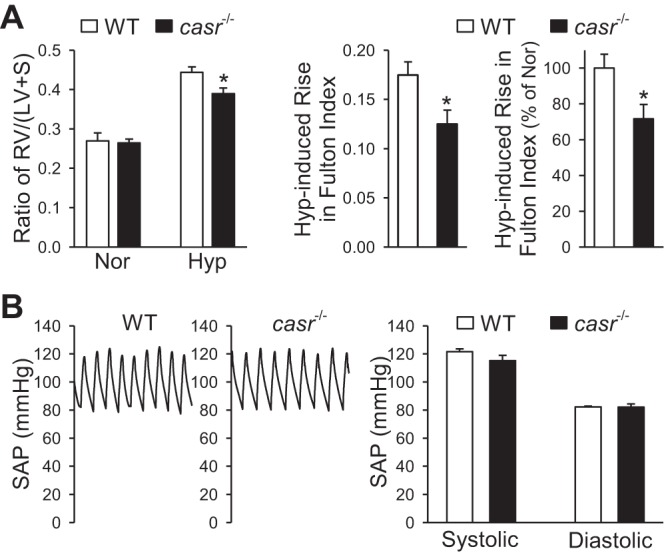
Deletion of the CaSR gene casr attenuates hypoxia-induced right ventricular hypertrophy but negligibly affected systemic arterial pressure (SAP) in mice. A: summarized data (means ± SE) showing averaged Fulton Index (left) and the ratio of RV/(LV + S) in normoxic (Nor) and hypoxic (Hyp) WT mice or casr−/− mice. The actual (middle) and relative (right) changes (%Nor) of the RV/(LV + S) ratio (Fulton Index) in WT or casr−/− mice under Nor and Hyp conditions are also shown. *P < 0.001 vs. WT. B: representative record of SAP (left) summarized data (means ± SE) of systolic and diastolic SAP (right) in WT and casr−/− mice under the Nor condition. No significant difference seen between WT and casr−/− mice.
In casr−/− mice (i.e., casr−/−/pth−/− mice), however, the hypoxia-mediated increases in RVSP, RV/(LV + S) ratio, and pulmonary arterial wall thickness were all significantly attenuated compared with WT mice. As shown in Fig. 4, the average RVSP in hypoxic casr−/− mice (33.3 ± 2.9 mmHg) was significantly lower than in hypoxic WT mice (41.9 ± 1.6 mmHg, P < 0.001), whereas RVSP in normoxic casr−/− mice (21.9 ± 1.3 mmHg) was comparable with RVSP in normoxic WT mice (24.2 ± 2.6 mmHg). The significantly lower RVSP in hypoxic casr−/− mice than in hypoxic WT mice was correlated with significantly reduced RVH as determined by Fulton's Index (Fig. 5A). In addition to RVSP and the Fulton Index, the wall thickness of small (diameter <50 μm) and medium-sized (diameter <100 μm) pulmonary arteries in hypoxic casr−/− mice was also significantly lower than in hypoxic WT mice. As shown in Figs. 4 and 5, deletion of casr caused >50% inhibition of hypoxia-mediated increases in RVSP (Fig. 4E), >65% inhibition of hypoxia-induced increase in PA wall thickness (Fig. 4G, bottom), and ∼30% inhibition of hypoxia-induced RVH (Fig. 5A, left).
The systemic arterial pressure (SAP) in casr−/− mice was comparable with WT mice. As shown in Fig. 5B, there was no significant difference of systolic and diastolic arterial pressure measured by a catheter positioned in carotid artery between the WT and casr−/− mice (Fig. 5B, right).
These data indicate that CaSR is functionally necessary for chronically hypoxic mice to develop pulmonary vascular remodeling and pulmonary hypertension, and downregulation of CaSR expression and/or inhibition of CaSR function may be a good target to develop novel drug therapy for hypoxia-induced pulmonary hypertension or pulmonary arterial hypertension.
Deletion of CaSR gene significantly inhibits acute hypoxic pulmonary vasoconstriction.
Hypoxic pulmonary vasoconstriction (HPV) is an important physiological mechanism that directs venous blood from poorly ventilated areas of the lungs to well-ventilated areas to assure maximal oxygenation by maintaining an optimal ventilation/perfusion ratio. Persistent hypoxia, however, causes sustained pulmonary vasoconstriction that may contribute to the elevated PVR and PAP in patients with PH associated with hypoxia and lung diseases and in residents living in high-altitude areas (40, 53).
To determine the functional relevance of CaSR and TRPC6 in the development of acute HPV, we compared high K+-induced increase in PAP and acute alveolar hypoxia-mediated increase in PAP in isolated perfused and ventilated lungs between WT and casr−/− or trp6−/− mice. A Trp6−/− mice breeding pair was initially obtained from National Institute of Environmental Health Sciences (Research Triangle Park, NC) and has been used in our previous studies (51).
Acute alveolar hypoxia (by ventilating 1% O2 gas into alveoli) reversibly increased PAP and reached its plateau within ∼5 min in WT mice (Fig. 6A, left, and B). Compared with the isolated perfused/ventilated lungs from WT mice, the acute hypoxia-induced increase in PAP or HPV was significantly inhibited in isolated perfused/ventilated lungs from casr+/−/pth−/− or casr−/−/pth−/− mice (Fig. 6A). These data demonstrate that CaSR is at least partially required for acute HPV in mice.
Fig. 6.

Deletion of the CaSR gene casr or the TRPC6 gene trpc6 attenuates acute hypoxia-induced pulmonary vasoconstriction. A, left: representative record of mean pulmonary arterial pressure (PAP) before, during, and after airway ventilation of hypoxic gas (Hyp; 1% O2) in isolated perfused/ventilated lungs from wild-type (WT), casr+/−, or casr−/− mice. A, right: summarized data (means ± SE) showing the increases in mean PAP induced by hypoxia challenges in isolated perfused/ventilated lungs from WT, casr+/−, or casr−/− mice. **P < 0.01 and ***P < 0.001 vs. WT. B: representative record of mean PAP before, during, and after superfusion of solutions with different K+ concentrations (10, 20, 30, 40, 60, 80, or 120 mM) and airway ventilation of Hyp (1% O2; shadowed) in isolated perfused/ventilated lungs from WT (top), trpc6−/− (middle), or casr−/− (bottom) mice. C: summarized data (means ± SE) showing the dose-response curves of high K+-induced increases in mean PAP in isolated perfused/ventilated lungs from WT, trpc6−/−, or casr−/− mice. No significant differences seen between WT and trpc6−/− or casr−/− mice. D, left: representative record mean PAP before, during, and after airway ventilation of Hyp in isolated perfused/ventilated lungs from WT, trpc6−/−, or casr−/− mice. D, right: summarized data (means ± SE) showing the amplitudes of hypoxia-induced increases in mean PAP in isolated perfused/ventilated lungs from WT, trpc6−/−, or casr−/− mice. ***P < 0.001 vs. WT.
Infusion of solutions with different concentrations of K+ (10, 20, 30, 40, 60, 80, and 120 mM) into the pulmonary artery caused a dose-dependent increase in PAP in isolated perfused/ventilated lungs from WT mice. The high K+-induced increase in PAP, due to high K+-mediated membrane depolarization, Ca2+ influx through voltage-dependent Ca2+ channels, and pulmonary vasoconstriction, was comparable in isolated perfused/ventilated lungs from WT and casr−/− or trp6−/− mice (Fig. 6, B and C); the dose-responsive curve of high K+-induced pulmonary vasoconstriction or increase in PAP in isolated perfused/ventilated lung from WT mice was actually superimposed with the dose-responses curves in isolated perfused/ventilated lung from either casr−/− or trpc6−/− mice (Fig. 6C).
We next compared acute HPV in isolated perfused/ventilated lung from WT and trpc6−/− mice. As shown in Fig. 6, B–D,similar attenuation of acute HPV was detected in trpc6−/− mice compared with isolated perfused/ventilated lungs from WT mice, which is in agreement with a prior study showing that acute HPV or acute alveolar hypoxia-induced increase in PAP in isolated perfused/ventilated lungs was significantly inhibited in trpc6−/− mice (51, 59). However, neither casr deficiency nor trpc6 knockout blocked the vasoconstrictor response to high K+ in isolated perfused/ventilated lungs (Fig. 6, B and C); deletion of casr or trpc6 significantly inhibited acute hypoxia-induced increases in PAP (Fig. 6D). These data suggest that attenuated acute HPV in casr−/− or trpc6−/− mice was not caused by a general defect in pulmonary arterial smooth muscle contractility or membrane depolarization-mediated Ca2+ influx through voltage-dependent Ca2+ channels in PASMC but appears to be more specific to the decrease in receptor-operated or store-operated Ca2+ entry in PASMC.
Deletion of CaSR gene significantly inhibits cell migration via reducing CaSR-associated ROCE in PASMC.
Concentric pulmonary vascular remodeling due to increased PASMC proliferation and migration is a major cause for the elevated PVR and PAP in patients with PAH and PH associated with hypoxia and lung diseases and in animals with experimental PH. In WT mice with chronic hypoxia-induced pulmonary hypertension (HPH), extracellular Ca2+-mediated increase in [Ca2+]cyt in freshly isolated PASMC was significantly enhanced compared with PASMC from normoxic WT mice (Fig. 7A). In PASMC freshly isolated from casr−/− mice, however, extracellular Ca2+-mediated increase in [Ca2+]cyt was no longer enhanced by hypoxia (Fig. 7A, bottom). These data indicate that chronic hypoxia-mediated enhancement of extracellular Ca2+-mediated [Ca2+]cyt increase in PASMC depends on the availability of CaSR or is due potentially and at least partially to upregulation of CaSR. Indeed, downregulation of CaSR with siRNA significantly inhibited PASMC proliferation (determined by BrdU uptake; Fig. 7B) and migration (Fig. 7, C and D). These data further demonstrate that CaSR is required for or involved in hypoxia-induced enhancement of Ca2+ signaling as well as PASMC proliferation and migration.
Fig. 7.

Deletion of the CaSR gene casr attenuates extracellular Ca2+-induced increase in [Ca2+]cyt in freshly isolated PASMC from chronically hypoxic mice and inhibits PASMC proliferation and migration. A: representative record of changes in [Ca2+]cyt before, during, and after extracellular application of 2.2 mM Ca2+ in freshly dissociated PASMC from WT and casr−/− mice exposed to normoxia (Nor; left) and hypoxia (Hyp; right) for 4 wk. Summarized data (means ± SE) showing the amplitude of extracellular Ca2+-induced increases in [Ca2+]cyt in PASMC isolated from WT and casr−/− under Nor and Hyp conditions. B: summarized data (means ± SE) showing BrdU incorporation in normal PASMC treated with control siRNA (Cont) and TRPC6 siRNA (siRNA) in Nor and Hyp. C: cell motility or migration was determined by a scratch wound assay. Representative images showing Nor and Hyp PASMC immediately (0 h) or 6 h after scratch with a sterile pipette in the presence of control siRNA (Scram-siRNA) or CaSR siRNA. D: summarized data (means ± SE) showing the no. of migrated cells into the gap measured at 6 h in Nor and Hyp PASMC treated with control siRNA (Scram-siRNA) or CaSR siRNA. *P < 0.05 vs. Nor; ***P < 0.001 vs. Scram-siRNA.
To further examine the potential role of upregulated CaSR in PASMC proliferation, we also conducted pharmacological experiments in vitro using normal and IPAH-PASMC. As shown in Fig. 8, protein expression level of CaSR in IPAH-PASMC was significantly greater than in normal PASMC (Fig. 8A; also see Ref. 63). The upregulated CaSR in IPAH-PASMC was associated with a basal increase in phosphorylated Akt (p-Akt) and an enhancement of extracellular Ca2+-mediated increase in p-Akt (Fig. 8B). Phosphorylation of Akt or activation of the Akt/mTOR signaling cascade is an important pathway for serum and growth factor-mediated cell proliferation (29). In the presence of 1.6 mM extracellular Ca2+ as well 10% serum and growth factors, pharmacological blockade of TRPC6 channels with a nonspecific TRPC channel inhibitor, 2-APB, significantly inhibited normal and IPAH-PASMC proliferation determined by the number of cells in the S phase (Fig. 8C). Blockade of CaSR by 1 and 2 μM NPS-2142, an allosteric blocker of CaSR (41), slightly inhibited normal PASMC proliferation but significantly inhibited IPAH-PASMC proliferation (Fig. 8C). These data indicate that upregulated CaSR in IPAH-PASMC is involved in promoting PASMC proliferation and migration due potentially to CaSR-mediated Akt phosphorylation and SOCE, which ultimately contribute to the development and progression of pulmonary vascular remodeling in patients with IPAH.
Fig. 8.

CaSR expression is upregulated and extracellular Ca2+-induced Akt phosphorylation and cell proliferation are enhanced in PASMC from IPAH patients. A: summarized data (means ± SE) showing CaSR protein expression level (determined by Western blot analysis) in normal and IPAH-PASMC. B: Western blot analysis of phosphorylated Akt (p-Akt) in normal and IPAH-PASMC treated with 0 or 0.5 mM extracellular Ca2+ for 5 or 13 min. C: summarized data (means ± SE) showing the no. of cells in the DNA synthesis (S) phase in normal and IPAH-PASMC in the absence (control) or presence of the TRPC channel blocker 2-aminoethoxydiphenyl borate (2-APB; 100 μM) or the CaSR blocker NPS-2143 (1 or 2 μM). **P < 0.01 and ***P < 0.001 vs. control in normal and IPAH PASMC.
DISCUSSION
CaSR is a class C GPCR that is activated by extracellular cations, polyamines, β-amyloids, antibiotics, and aromatic amino acids (6). Activation of CaSR sets into motion a complex series of downstream signaling cascades to cause cell contraction, migration, and proliferation. Previously, we reported that 1) mRNA and protein expression of CaSR is upregulated and extracellular Ca2+-induced increase in [Ca2+]cyt is enhanced (58, 63) and 2) mRNA and protein expression of TRPC6 is upregulated and store-operated Ca2+ entry enhanced (67, 69) in PASMC from IPAH patients compared with normal control PASMC. The results from this study indicate that 1) CaSR and TRPC6 functionally interact with each other to regulate [Ca2+]cyt in PASMC; 2) downregulation of TRPC6 with siRNA significantly inhibits extracellular Ca2+-induced increase in [Ca2+]cyt in IPAH-PASMC in which CaSR and TRPC6 are upregulated, whereas overexpression of TRPC6 significantly enhances extracellular Ca2+-induced increase in [Ca2+]cyt in normal PASMC in which CaSR and TRPC6 expression level is low; 3) downregulation of CaSR or TRPC6 with siRNA or pharmacological blockade of CaSR and TRPC6 significantly inhibit serum and growth factor-mediated PASMC proliferation in the presence of extracellular Ca2+; 4) deletion of the CaSR gene significantly inhibits the development and progression of chronic hypoxia-induced pulmonary vascular medial hypertrophy and pulmonary hypertension (HPH) in mice (casr−/−); and 5) knockout of casr in mice significantly inhibits acute hypoxia-induced pulmonary vasoconstriction in isolated perfused and ventilated lungs.
These data provide compelling evidence that upregulated CaSR functionally interacts with upregulated TRPC6 to coordinately enhance the Ca2+ signaling required for stimulating contraction, proliferation, and migration in PASMC from IPAH patients. Upregulation of CaSR and TRPC6 in PASMC may be an important pathogenic mechanism involved in the development of sustained pulmonary vasoconstriction and excessive pulmonary vascular remodeling. This is evident in our animal model of chronic hypoxia-induced PH, in which upregulation of both CaSR and TRPC6 in PASMC mimics the changes detected in PASMC isolated from IPAH subjects. Pharmacological blockers of CaSR or TRPC6 and siRNA/miRNA that specifically downregulate CaSR or TRPC6 expression should be considered to develop novel therapies for patients with IPAH and pulmonary hypertension associated with hypoxia and lung diseases.
CaSR-mediated enhancement of Ca2+ signaling and stimulation of Akt/mTOR cascade in PASMC.
The upregulated CaSR in lung tissues and PASMC not only occurs in IPAH patients but also takes place in mice and rats with experimental pulmonary hypertension (e.g., chronic hypoxia-induced pulmonary hypertension and monocrotaline-induced pulmonary hypertension). The subsequent enhancement of CaSR-mediated ROCE through upregulated TRPC6 in PASMC may be one of the important pathogenic mechanisms involved in stimulating PASMC contraction, migration, and proliferation and ultimately sustained pulmonary vasoconstriction and concentric pulmonary arterial wall thickening in patients with IPAH and animals with experimental PH.
The upregulated CaSR expression in IPAH-PASMC was also associated with a significant enhancement of basal line expression level of phosphorylated Akt (p-Akt) and a marked increase in extracellular Ca2+-mediated phosphorylation of Akt. The phosphatidylinositol 3-kinase (PI3K)/Akt1/mTOR pathway is involved in gene transcription and translation, protein synthesis, and cell proliferation. It is unclear whether CaSR-mediated phosphorylation of Akt is dependent of CaSR-mediated increase in [Ca2+]cyt via Ca2+ influx through TRPC6 in PASMC. Our previous studies showed that platelet-derived growth factor (PDGF) stimulates human PASMC proliferation via multiple intracellular signaling cascades, including the PI3K/Akt/mTOR pathway (43–45, 54). Abolishing increases in [Ca2+]cyt in PASMC by removal of extracellular Ca2+ (to eliminate Ca2+ influx) and chelation of cytosolic Ca2+ and intracellularly stored Ca2+ with BAPTA-AM (to eliminate Ca2+ mobilization) negligibly affected PDGF-mediated phosphorylation of Akt (43). These observations directed us to speculate that activation of upregulated CaSR in PASMC signals through at least the PLC/IP3/DAG/TRPC6 Ca2+ pathway and the PI3K/Akt/mTOR pathway to induce PASMC contraction, migration, and proliferation.
Multiple ligands of CaSR in the circulation and lungs.
CaSR can be activated by multiple ligands (6). The ligands of CaSR can be classified into three categories: 1) divalent and trivalent cations (i.e., Ca2+, Mg2+, Sr2+, and Gd3+) (48); 2) polyamines (i.e., spermine, spermidine); 3) aminoglycoside antibiotics (e.g., neomycin, streptomycin, tobramycin, kanamycin B, and paromomycin) (56); 4) aromatic amino acids (e.g., phenylalanine, tyrosine, and tryptophan); and 5) β-amyloids (14). Both the cationic ligands and polyamines are present in a large range of concentrations in the blood and the pulmonary vasculature. For example, the extracellular Ca2+ (and Mg2+) concentration is normally in the milimolar range (1–2 mM) that is much higher than the [Ca2+] of EC50 (∼1.22 mM) for extracellular Ca2+-induced increase in [Ca2+]cyt in IPAH-PASMC (58, 63). Using a transfection system (HEK-293 cells), it has been demonstrated that the extracellular Ca2+-induced response in HEK cells transfected with CaSR cDNA started at the [Ca2+] of 1.5–2.0 mM and a maximal response at 15 mM; the EC50 was around 3.0–4.5 mM of extracellular [Ca2+] (46, 49, 71). The greater sensitivity of extracellular Ca2+-induced increase in [Ca2+]cyt in IPAH-PASMC (∼1.22 mM) than in CaSR-transfected HEK cells (3.5–4.5 mM) was likely due to the presence of other CaSR activators that enhance the sensitivity of CaSR to extracellular Ca2+ in PASMC from patients with IPAH. Polyamines are normally present in the circulation and tissues in a very low concentration; however, polyamine levels (e.g., spermine and spermidine) can be increased significantly during tissue injury (10). Aromatic amino acids such as phenylalanine, histidine, and tryptophan are essential amino acids for mammals and humans that must be obtained from the daily diet (12). Therefore, the absorbed aromatic amino acids (phenylalanine, histidine, and tryptophan) can be accumulated in the extracellular space and activate CaSR in the plasma membrane of pulmonary vascular smooth muscle and endothelial cells, especially under the condition that CaSR is upregulated, for example, in PASMC in patients with IPAH.
As described above, most of the ligands or stimulators of CaSR are endogenously present in the circulation and in the lung tissues, and many are present in high concentrations (e.g., extracellular Ca2+ and Mg2+ concentrations are normally ∼1–2 mM). Therefore, CaSR on the plasma membrane of pulmonary vascular smooth muscle cells, (myo)fibroblasts, and endothelial cells are consistently exposed to a lot of ligands or stimulators; the change in the extracellular ligands' concentration and expression is thus not the critical rate-limiting step for the activation of CaSR and the CaSR-associated intracellular signaling cascades. It is the expression level of CaSR in PASMC and its downstream signaling pathway that control or determine the contribution of CaSR and its downstream signaling cascades in the development and progression of sustained pulmonary vasoconstriction and pulmonary vascular remodeling in patients with IPAH and animals with experimental PH.
The upregulated mRNA and protein expression of CaSR (58, 63), the subsequent enhancement of extracellular Ca2+-induced [Ca2+]cyt increase (via Ca2+ influx through TRPC6 channels) (11), and augmentation of CaSR-mediated activation of Akt (Fig. 8B) are major pathogenic causes for vasoconstriction and vascular remodeling observed in patients and animals with pulmonary hypertension. The upregulated TRPC6 further enhanced the CaSR-mediated ROCE and SOCE, resulting in sustained pulmonary vasoconstriction and excessive pulmonary vascular remodeling. CaSR is a unique GPCR that desensitizes very slowly (47), which allows the upregulated CaSR in PASMC to be activated persistently by high levels of circulating ligands and ligands accumulated in the lung tissues. Our animal model data are limited to using the animal model of HPH, and although it may be interesting to use other experimental models of PH (15), we found that this was a very reproducible model that simulated the changes in CaSR and TRPC6 in PASMC from human IPAH. Therefore, it remains unknown whether CaSR and/or TRPC6 is an important part in the development of severe PH in hypoxia/Sugen5416-induced PH murine models.
Transcriptional and posttranscriptional regulation of CaSR.
How CaSR (or TRPC6) is upregulated in PASMC from IPAH patients and animals with experimental pulmonary hypertension remains unknown. The human CaSR gene CASR can be transcriptionally regulated by transcription factors, posttranscriptionally regulated by microRNAs, and epigenetically regulated by DNA methylation (24). The promoter region of CASR contains numerous binding sites of transcription factors that are known to stimulate cell proliferation (13, 17, 20). A variety of transcription factors have been demonstrated to be upregulated in PASMC from patients with IPAH and animals with experimental PH (13, 20, 69, 70), which can directly bind to the promoter of CASR and may activate CASR transcription. A data-based analysis shows that there are many transcription factors that can directly bind to the promoter of CASR, including NF-κB, STAT, SP1, SMAD, octamer transcription factor 1, nuclear factor of activated T cells, cAMP response element-binding protein, GATA, and activator protein 1. These data direct us to speculate that the increased mRNA and protein expression of CaSR in IPAH-PASMC may be due to transcriptional upregulation induced by increased transcription factors (1, 13, 20, 69, 70). In addition to the transcription factors, many mitogenic, angiogenic, and fibrotic factors such as PDGF, VEGF, and TGFβ, along with inflammatory cytokines (e.g., TNFα, IL-1, and IL-6) (31, 57), have been demonstrated to be upregulated in PASMC and lung tissues from patients with IPAH and animals with experimental PH. These growth factors may be responsible for the upregulation of CaSR via increasing transcription factors that can bind directly to the promoter of CASR and enhance the transcription of CASR.
We also searched for the microRNAs (miRNA or miR) that can potentially regulate the CaSR mRNA and protein level by binding directly to the CaSR mRNA using the in silico prediction approach (provided by microrna.org) (4). Based on a stringency of the mirSVR score cutoff of −1.0 to select the top miRNA-mTNA binding pairs (3), there are more than 20 miRNAs with high likelihood to bind to the 3′-untranslated region (UTR) of the CaSR mRNA. These miRNAs bind to the 3′-UTR of the CaSR mRNA and decrease mRNA and/or protein level of CaSR by inhibiting protein translation and/or increasing mRNA degradation (2, 16). In addition, it has been demonstrated recently that miRNAs can also bind directly to the 5′-UTR of mRNA to inhibit translation and decrease gene expression level (39). Among the miRNAs that are predicted to bind to the CaSR mRNA, it has been demonstrated that miR-30, miR-92, and miR-126 are downregulated in lung tissues and PASMC from patients with IPAH or animals with experimental PH (8, 9, 60). One of the miRNAs that can bind directly to the 3′-UTR of the CaSR mRNA, miR-126, is actually significantly downregulated in PASMC from IPAH patients (data not shown). This miRNA can bind directly to the 3′-UTR of the CaSR mRNA and decrease the mRNA level of CaSR by increasing its degradation and decrease protein level of CaSR by inhibiting translation. It is thus possible that the downregulated miR-126 as well as miR-30 and miR-92 are involved in the upregulated CaSR expression and enhanced CaSR-mediated Ca2+ influx in PASMC from IPAH patients.
TRPC6 is involved in CaSR-mediated ROCE and SOCE in IPAH-PASMC.
Activation of CaSR by extracellular Ca2+ or other stimuli (e.g., spermine) increases synthesis of IP3 and DAG via Gqα proteins, leading to rises in [Ca2+]cyt. The extracellular Ca2+-induced increase in [Ca2+]cyt in PASMC from IPAH patients was often composed of two components: a transient increase due mainly to Ca2+ release or mobilization from the intracellular stores to the cytosol and a sustained or oscillatory phase due obviously to Ca2+ influx through Ca2+-permeable channels and/or inward Ca2+ transportation via Na+/Ca2+ exchangers in the plasma membrane (19, 36, 72). The data from this study demonstrate that 1) downregulation of TRPC6 with siRNA in IPAH-PASMC (in which CaSR expression is significantly upregulated) significantly inhibits the extracellular Ca2+-induced increase in [Ca2+]cyt and 2) overexpression of TRPC6 by transfecting TRPC6 alone or cotransfecting TRPC6 with CaSR in normal PASMC (in which CaSR expression level is low) significantly augments the extracellular Ca2+-induced increase in [Ca2+]cyt (Fig. 2). These data suggest that homotetrameric and/or heterotetrameric TRPC6 channels are required for CaSR-induced ROCE and SOCE, both contributing to the increase in [Ca2+]cyt in IPAH-PASMC. In other words, CaSR is functionally coupled to TRPC6 channels in IPAH-PASMC to induce increases in [Ca2+]cyt. It is not clear whether there is a similar functional interaction with other isomers of the TRPC family. TRPC1 has been shown to be an important in pulmonary vasoreactivity, yet it appears to have unique functions in the development and progression of PH in animal models (37, 62). Identifying other isoforms that can functionally couple with CaSR may be an important next step in defining this pathogenic mechanism and developing a novel therapy. In the present study, we have focused on TRPC6, as we have demonstrated previously that it contributes to forming both ROC and SOC in PASMC and is significantly upregulated in PASMC isolated from IPAH patients compared with PASMC isolated from normal subjects and patients without PH (18, 67, 68). Taken together, the data from this study suggest that the upregulated CaSR functionally interacts with TRPC6 channels via DAG (to induced ROCE) and IP3 (to indirectly induce SOCE) to coordinately enhance the Ca2+ signaling cascades, leading to sustained pulmonary vasoconstriction and pulmonary vascular remodeling by causing PASMC contraction and stimulating PASMC proliferation and migration.
Conclusion.
An increase in [Ca2+]cyt in PASMC is not only a major trigger for pulmonary vasoconstriction but also an important stimulus for PASMC migration and proliferation, with both contributing to developing concentric pulmonary arterial remodeling in patients with IPAH and animals with experimental PH. In this study, we identified a new potential pathogenic mechanism involving functional interaction of CaSR and TRPC6 in the development and progression of sustained pulmonary vasoconstriction and pulmonary vascular remodeling in patients with IPAH and animals with experimental PH. Specific blockers of CaSR and/or TRPC6 should be considered to develop novel drug therapies for patients with PAH and pulmonary hypertension associated with hypoxia and lung diseases. Combination therapy targeting both CaSR and TRPC6, along with other existing therapies, may be more effective in patients with severe PH.
GRANTS
This work was supported in part by grants from the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL-115014, HL-066012, and HL-125208).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.T., D.G., F.R., Z.I.K., S.M.B., J.G.G., A.M., and J.X.-J.Y. conception and design of research; H.T., A.Y., H.Y., S.S., D.R.F., J.C., Y.G., N.M.P., L.J.-P., D.G., and A.M. performed experiments; H.T., A.Y., H.Y., S.S., D.R.F., J.C., Y.G., N.M.P., T.Z., L.J.-P., R.J.A., A.A.D., D.G., F.R., Z.I.K., S.M.B., J.G.G., A.M., and J.X.-J.Y. interpreted results of experiments; H.T., A.Y., H.Y., S.S., D.R.F., J.C., Y.G., N.M.P., L.J.-P., R.J.A., A.M., and J.X.-J.Y. prepared figures; H.T. and J.X.-J.Y. drafted manuscript; H.T., D.R.F., Y.G., T.Z., R.J.A., A.A.D., F.R., Z.I.K., S.M.B., J.G.G., A.M., and J.X.-J.Y. edited and revised manuscript; H.T., A.Y., H.Y., S.S., D.R.F., J.C., Y.G., N.M.P., T.Z., L.J.-P., R.J.A., A.A.D., D.G., F.R., Z.I.K., S.M.B., J.G.G., A.M., and J.X.-J.Y. approved final version of manuscript; A.Y., H.Y., S.S., D.R.F., J.C., Y.G., N.M.P., T.Z., L.J.-P., R.J.A., A.A.D., F.R., Z.I.K., S.M.B., J.G.G., A.M., and J.X.-J.Y. analyzed data.
REFERENCES
- 1.Abud EM, Maylor J, Undem C, Punjabi A, Zaiman AL, Myers AC, Sylvester JT, Semenza GL, Shimoda LA. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc Natl Acad Sci USA 109: 1239–1244, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol 11: R90, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res 36: D149–D153, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bouschet T, Martin S, Henley JM. Regulation of calcium-sensing-receptor trafficking and cell-surface expression by GPCRs and RAMPs. Trends Pharmacol Sci 29: 633–639, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brennan SC, Thiem U, Roth S, Aggarwal A, Fetahu I, Tennakoon S, Gomes AR, Brandi ML, Bruggeman F, Mentaverri R, Riccardi D, Kallay E. Calcium sensing receptor signalling in physiology and cancer. Biochim Biophys Acta 1833: 1732–1744, 2013. [DOI] [PubMed] [Google Scholar]
- 7.Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev 81: 239–297, 2001. [DOI] [PubMed] [Google Scholar]
- 8.Caruso P, MacLean MR, Khanin R, McClure J, Soon E, Southgate M, MacDonald RA, Greig JA, Robertson KE, Masson R, Denby L, Dempsie Y, Long L, Morrell NW, Baker AH. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler Thromb Vasc Biol 30: 716–723, 2010. [DOI] [PubMed] [Google Scholar]
- 9.Chen T, Zhou G, Zhou Q, Tang H, Ibe JC, Cheng H, Gou D, Chen J, Yuan JX, Raj JU. Loss of microRNA-17∼92 in smooth muscle cells attenuates experimental pulmonary hypertension via induction of PDZ and LIM domain 5. Am J Respir Crit Care Med 191: 678–692, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng SX, Geibel JP, Hebert SC. Extracellular polyamines regulate fluid secretion in rat colonic crypts via the extracellular calcium-sensing receptor. Gastroenterology 126: 148–158, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Chow JY, Estrema C, Orneles T, Dong X, Barrett KE, Dong H. Calcium-sensing receptor modulates extracellular Ca2+ entry via TRPC-encoded receptor-operated channels in human aortic smooth muscle cells. Am J Physiol Cell Physiol 301: C461–C468, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conigrave AD, Quinn SJ, Brown EM. L-amino acid sensing by the extracellular Ca2+-sensing receptor. Proc Natl Acad Sci USA 97: 4814–4819, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crosswhite P, Sun Z. Molecular mechanisms of pulmonary arterial remodeling. Mol Med 20: 191–201, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dal Pra I, Armato U, Chioffi F, Pacchiana R, Whitfield JF, Chakravarthy B, Gui L, Chiarini A. The Aβ peptides-activated calcium-sensing receptor stimulates the production and secretion of vascular endothelial growth factor-A by normoxic adult human cortical astrocytes. Neuromolecular Med 16: 645–657, 2014. [DOI] [PubMed] [Google Scholar]
- 15.Das M, Fessel J, Tang H, West J. A process-based review of mouse models of pulmonary hypertension. Pulm Circ 2: 415–433, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Djuranovic S, Nahvi A, Green R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 336: 237–240, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fantozzi I, Zhang S, Platoshyn O, Remillard CV, Cowling RT, Yuan JX. Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 285: L1233–L1245, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez RA, Wan J, Song S, Smith KA, Gu Y, Tauseef M, Tang H, Makino A, Mehta D, Yuan JX. Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am J Physiol Cell Physiol 308: C581–C593, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Firth AL, Remillard CV, Platoshyn O, Fantozzi I, Ko EA, Yuan JX. Functional ion channels in human pulmonary artery smooth muscle cells: Voltage-dependent cation channels. Pulm Circ 1: 48–71, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Firth AL, Yao W, Remillard CV, Ogawa A, Yuan JX. Upregulation of Oct-4 isoforms in pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 298: L548–L557, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goltzman D, Hendy GN. The calcium-sensing receptor in bone—mechanistic and therapeutic insights. Nat Rev Endocrinol 11: 298–307, 2015. [DOI] [PubMed] [Google Scholar]
- 22.Gottlieb P, Folgering J, Maroto R, Raso A, Wood TG, Kurosky A, Bowman C, Bichet D, Patel A, Sachs F, Martinac B, Hamill OP, Honore E. Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflugers Arch 455: 1097–1103, 2008. [DOI] [PubMed] [Google Scholar]
- 23.Hendy GN, Canaff L, Cole DE. The CASR gene: alternative splicing and transcriptional control, and calcium-sensing receptor (CaSR) protein: structure and ligand binding sites. Best Pract Res Clin Endocrinol Metab 27: 285–301, 2013. [DOI] [PubMed] [Google Scholar]
- 24.Hendy GN, Guarnieri V, Canaff L. Calcium-sensing receptor and associated diseases. Prog Mol Biol Transl Sci 89: 31–95, 2009. [DOI] [PubMed] [Google Scholar]
- 25.Hofer AM, Brown EM. Extracellular calcium sensing and signalling. Nat Rev Mol Cell Biol 4: 530–538, 2003. [DOI] [PubMed] [Google Scholar]
- 26.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397: 259–263, 1999. [DOI] [PubMed] [Google Scholar]
- 27.Houillier P. Calcium-sensing in the kidney. Curr Opin Nephrol Hypertens 22: 566–571, 2013. [DOI] [PubMed] [Google Scholar]
- 28.Huang C, Miller RT. The calcium-sensing receptor and its interacting proteins. J Cell Mol Med 11: 923–934, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang K, Fingar DC. Growing knowledge of the mTOR signaling network. Semin Cell Dev Biol 36: 79–90, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang Y, Zhou Y, Yang W, Butters R, Lee HW, Li S, Castiblanco A, Brown EM, Yang JJ. Identification and dissection of (Ca2+)-binding sites in the extracellular domain of (Ca2+)-sensing receptor. J Biol Chem 282: 19000–19010, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med 151: 1628–1631, 1995. [DOI] [PubMed] [Google Scholar]
- 32.Inoue R, Okada T, Onoue H, Hara Y, Shimizu S, Naitoh S, Ito Y, Mori Y. The transient receptor potential protein homologue TRP6 is the essential component of vascular α1-adrenoceptor-activated Ca2+-permeable cation channel. Circ Res 88: 325–332, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Kantham L, Quinn SJ, Egbuna OI, Baxi K, Butters R, Pang JL, Pollak MR, Goltzman D, Brown EM. The calcium-sensing receptor (CaSR) defends against hypercalcemia independently of its regulation of parathyroid hormone secretion. Am J Physiol Endocrinol Metab 297: E915–E923, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuhr FK, Smith KA, Song MY, Levitan I, Yuan JX. New mechanisms of pulmonary arterial hypertension: role of Ca2+ signaling. Am J Physiol Heart Circ Physiol 302: H1546–H1562, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Magno AL, Ward BK, Ratajczak T. The calcium-sensing receptor: a molecular perspective. Endocr Rev 32: 3–30, 2011. [DOI] [PubMed] [Google Scholar]
- 36.Makino A, Firth AL, Yuan JX. Endothelial and smooth muscle cell ion channels in pulmonary vasoconstriction and vascular remodeling. Compr Physiol 1: 1555–1602, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malczyk M, Veith C, Fuchs B, Hofmann K, Storch U, Schermuly RT, Witzenrath M, Ahlbrecht K, Fecher-Trost C, Flockerzi V, Ghofrani HA, Grimminger F, Seeger W, Gudermann T, Dietrich A, Weissmann N. Classical transient receptor potential channel 1 in hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med 188: 1451–1459, 2013. [DOI] [PubMed] [Google Scholar]
- 38.Mandegar M, Remillard CV, Yuan JX. Ion channels in pulmonary arterial hypertension. Prog Cardiovasc Dis 45: 81–114, 2002. [DOI] [PubMed] [Google Scholar]
- 39.Meijer HA, Kong YW, Lu WT, Wilczynska A, Spriggs RV, Robinson SW, Godfrey JD, Willis AE, Bushell M. Translational repression and eIF4A2 activity are critical for microRNA-mediated gene regulation. Science 340: 82–85, 2013. [DOI] [PubMed] [Google Scholar]
- 40.Moudgil R, Michelakis ED, Archer SL. Hypoxic pulmonary vasoconstriction. J Appl Physiol (1985) 98: 390–403, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Nemeth EF, Delmar EG, Heaton WL, Miller MA, Lambert LD, Conklin RL, Gowen M, Gleason JG, Bhatnagar PK, Fox J. Calcilytic compounds: potent and selective Ca2+ receptor antagonists that stimulate secretion of parathyroid hormone. J Pharmacol Exp Ther 299: 323–331, 2001. [PubMed] [Google Scholar]
- 42.Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev 87: 165–217, 2007. [DOI] [PubMed] [Google Scholar]
- 43.Ogawa A, Firth AL, Ariyasu S, Yamadori I, Matsubara H, Song S, Fraidenburg DR, Yuan JX. Thrombin-mediated activation of Akt signaling contributes to pulmonary vascular remodeling in pulmonary hypertension. Physiol Rep 1: e00190, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogawa A, Firth AL, Smith KA, Maliakal MV, Yuan JX. PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 302: C405–C411, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ogawa A, Firth AL, Yao W, Madani MM, Kerr KM, Auger WR, Jamieson SW, Thistlethwaite PA, Yuan JX. Inhibition of mTOR attenuates store-operated Ca2+ entry in cells from endarterectomized tissues of patients with chronic thromboembolic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 297: L666–L676, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pearce SH, Bai M, Quinn SJ, Kifor O, Brown EM, Thakker RV. Functional characterization of calcium-sensing receptor mutations expressed in human embryonic kidney cells. J Clin Invest 98: 1860–1866, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ray K. Calcium-sensing receptor: Trafficking, endocytosis, recycling, and importance of interacting proteins. Prog Mol Biol Transl Sci 132: 127–150, 2015. [DOI] [PubMed] [Google Scholar]
- 48.Riccardi D, Park J, Lee WS, Gamba G, Brown EM, Hebert SC. Cloning and functional expression of a rat kidney extracellular calcium/polyvalent cation-sensing receptor. Proc Natl Acad Sci USA 92: 131–135, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rus R, Haag C, Bumke-Vogt C, Bahr V, Mayr B, Mohlig M, Schulze E, Frank-Raue K, Raue F, Schofl C. Novel inactivating mutations of the calcium-sensing receptor: the calcimimetic NPS R-568 improves signal transduction of mutant receptors. J Clin Endocrinol Metab 93: 4797–4803, 2008. [DOI] [PubMed] [Google Scholar]
- 50.Silve C, Petrel C, Leroy C, Bruel H, Mallet E, Rognan D, Ruat M. Delineating a Ca2+ binding pocket within the venus flytrap module of the human calcium-sensing receptor. J Biol Chem 280: 37917–37923, 2005. [DOI] [PubMed] [Google Scholar]
- 51.Smith KA, Voiriot G, Tang H, Fraidenburg DR, Song S, Yamamura H, Yamamura A, Guo Q, Wan J, Pohl NM, Tauseef M, Bodmer R, Ocorr K, Thistlethwaite PA, Haddad GG, Powell FL, Makino A, Mehta D, Yuan JX. Notch Activation of Ca(2+) Signaling in the Development of Hypoxic Pulmonary Vasoconstriction and Pulmonary Hypertension. Am J Respir Cell Mol Biol 53: 355–367, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song S, Yamamura A, Yamamura H, Ayon RJ, Smith KA, Tang H, Makino A, Yuan JX. Flow shear stress enhances intracellular Ca2+ signaling in pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension. Am J Physiol Cell Physiol 307: C373–C383, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev 92: 367–520, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tang H, Chen J, Fraidenburg DR, Song S, Sysol JR, Drennan AR, Offermanns S, Ye RD, Bonini MG, Minshall RD, Garcia JG, Machado RF, Makino A, Yuan JX. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L208–L220, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang H, Fernandez RA, Yuan JX. miRNA208/Mef2 and TNF-α in right ventricular dysfunction: the transition from hypertrophy to failure. Circ Res 116: 6–8, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thomsen AR, Hvidtfeldt M, Brauner-Osborne H. Biased agonism of the calcium-sensing receptor. Cell Calcium 51: 107–116, 2012. [DOI] [PubMed] [Google Scholar]
- 57.Voelkel NF, Tuder RM. Cellular and molecular mechanisms in the pathogenesis of severe pulmonary hypertension. Eur Respir J 8: 2129–2138, 1995. [DOI] [PubMed] [Google Scholar]
- 58.Wan J, Yamamura A, Zimnicka AM, Voiriot G, Smith KA, Tang H, Ayon RJ, Choudhury MS, Ko EA, Wang J, Wang C, Makino A, Yuan JX. Chronic hypoxia selectively enhances L- and T-type voltage-dependent Ca2+ channel activity in pulmonary artery by upregulating Cav1.2 and Cav3.2. Am J Physiol Lung Cell Mol Physiol 305: L154–L164, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weissmann N, Dietrich A, Fuchs B, Kalwa H, Ay M, Dumitrascu R, Olschewski A, Storch U, Mederos y Schnitzler M, Ghofrani HA, Schermuly RT, Pinkenburg O, Seeger W, Grimminger F, Gudermann T. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Natl Acad Sci USA 103: 19093–19098, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.White K, Loscalzo J, Chan SY. Holding our breath: The emerging and anticipated roles of microRNA in pulmonary hypertension. Pulm Circ 2: 278–290, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wright DB, Tripathi S, Sikarwar A, Santosh KT, Perez-Zoghbi J, Ojo OO, Irechukwu N, Ward JP, Schaafsma D. Regulation of GPCR-mediated smooth muscle contraction: implications for asthma and pulmonary hypertension. Pulm Pharmacol Ther 26: 121–131, 2013. [DOI] [PubMed] [Google Scholar]
- 62.Xia Y, Yang XR, Fu Z, Paudel O, Abramowitz J, Birnbaumer L, Sham JS. Classical transient receptor potential 1 and 6 contribute to hypoxic pulmonary hypertension through differential regulation of pulmonary vascular functions. Hypertension 63: 173–180, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yamamura A, Guo Q, Yamamura H, Zimnicka AM, Pohl NM, Smith KA, Fernandez RA, Zeifman A, Makino A, Dong H, Yuan JX. Enhanced Ca2+-sensing receptor function in idiopathic pulmonary arterial hypertension. Circ Res 111: 469–481, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamamura A, Yamamura H, Zeifman A, Yuan JX. Activity of Ca2+-activated Cl− channels contributes to regulating receptor- and store-operated Ca entry in human pulmonary artery smooth muscle cells. Pulm Circ 1: 269–279, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang XR, Lin MJ, Sham JS. Physiological functions of transient receptor potential channels in pulmonary arterial smooth muscle cells. Adv Exp Med Biol 661: 109–122, 2010. [DOI] [PubMed] [Google Scholar]
- 66.Yoo HY, Zeifman A, Ko EA, Smith KA, Chen J, Machado RF, Zhao YY, Minshall RD, Yuan JX. Optimization of isolated perfused/ventilated mouse lung to study hypoxic pulmonary vasoconstriction. Pulm Circ 3: 396–405, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci USA 101: 13861–13866, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu Y, Keller SH, Remillard CV, Safrina O, Nicholson A, Zhang SL, Jiang W, Vangala N, Landsberg JW, Wang JY, Thistlethwaite PA, Channick RN, Robbins IM, Loyd JE, Ghofrani HA, Grimminger F, Schermuly RT, Cahalan MD, Rubin LJ, Yuan JX. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 119: 2313–2322, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu Y, Sweeney M, Zhang S, Platoshyn O, Landsberg J, Rothman A, Yuan JX. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol 284: C316–C330, 2003. [DOI] [PubMed] [Google Scholar]
- 70.Zabini D, Crnkovic S, Xu H, Tscherner M, Ghanim B, Klepetko W, Olschewski A, Kwapiszewska G, Marsh LM. High-mobility group box-1 induces vascular remodelling processes via c-Jun activation. J Cell Mol Med 19: 1151–1161, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang C, Mulpuri N, Hannan FM, Nesbit MA, Thakker RV, Hamelberg D, Brown EM, Yang JJ. Role of Ca2+ and L-Phe in regulating functional cooperativity of disease-associated “toggle” calcium-sensing receptor mutations. PLoS One 9: e113622, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang S, Dong H, Rubin LJ, Yuan JX. Upregulation of Na+/Ca2+ exchanger contributes to the enhanced Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol 292: C2297–C2305, 2007. [DOI] [PubMed] [Google Scholar]
- 73.Zhu X, Jiang M, Peyton M, Boulay G, Hurst R, Stefani E, Birnbaumer L. trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell 85: 661–671, 1996. [DOI] [PubMed] [Google Scholar]