

This is the first study that has directly measured mitochondrial respiration in the cerebral arteries of insulin-resistant rats. Our results show that severe mitochondrial dysfunction is not necessary for the development of IR and that oxidative stress plays a significant role in vascular dysfunction during IR.
Keywords: middle cerebral arteries, insulin resistance, mitochondria, mitochondrial respiration, reactive oxygen species, cerebral vessels, cerebral microvessels
Abstract
Little is known about mitochondrial functioning in the cerebral vasculature during insulin resistance (IR). We examined mitochondrial respiration in isolated cerebral arteries of male Zucker obese (ZO) rats and phenotypically normal Zucker lean (ZL) rats using the Seahorse XFe24 analyzer. We investigated mitochondrial morphology in cerebral blood vessels as well as mitochondrial and nonmitochondrial protein expression levels in cerebral arteries and microvessels. We also measured reactive oxygen species (ROS) levels in cerebral microvessels. Under basal conditions, the mitochondrial respiration components (nonmitochondrial respiration, basal respiration, ATP production, proton leak, and spare respiratory capacity) showed similar levels among the ZL and ZO groups with the exception of maximal respiration, which was higher in the ZO group. We examined the role of nitric oxide by measuring mitochondrial respiration following inhibition of nitric oxide synthase with Nω-nitro-l-arginine methyl ester (l-NAME) and mitochondrial activation after administration of diazoxide (DZ). Both ZL and ZO groups showed similar responses to these stimuli with minor variations. l-NAME significantly increased the proton leak, and DZ decreased nonmitochondrial respiration in the ZL group. Other components were not affected. Mitochondrial morphology and distribution within vascular smooth muscle and endothelium as well as mitochondrial protein levels were similar in the arteries and microvessels of both groups. Endothelial nitric oxide synthase (eNOS) and ROS levels were increased in cerebral microvessels of the ZO. Our study suggests that mitochondrial function is not significantly altered in the cerebral vasculature of young ZO rats, but increased ROS production might be due to increased eNOS in the cerebral microcirculation during IR.
NEW & NOTEWORTHY
This is the first study that has directly measured mitochondrial respiration in the cerebral arteries of insulin-resistant rats. Our results show that severe mitochondrial dysfunction is not necessary for the development of IR and that oxidative stress plays a significant role in vascular dysfunction during IR.
insulin resistance (IR), with the contributing factors of central obesity, genetic factors, and a sedentary lifestyle, is a primary feature in the development of the metabolic syndrome (1). Impaired insulin metabolism can lead to various metabolic and vasculature abnormalities resulting in clinical consequences, such as hypertension, atherosclerosis, dyslipidemia, diabetes, cerebrovascular dysfunction, and/or stroke (2, 32). Although the incidence of IR is growing dramatically, the mechanisms of its pathological effects on cerebral blood vessels as well as the underlying etiology of IR have not been completely elucidated. Impaired mitochondrial function and mitochondrial content have been suggested as underlying causes of IR (31, 39), whereas other reports have not found altered mitochondrial respiratory capacity (18, 33) or mitochondrial content (18, 26) in insulin-resistant humans. Our laboratory has shown that mitochondrial-dependent vasodilatory responses of major cerebral arteries are diminished in Zucker obese (ZO) rats (27, 28). On the other hand, we have also shown that local cortical microcirculatory responses to pharmacological blood flow stimulators, such as bradykinin, N-methyl-d-aspartate (NMDA), bicuculline, and physiologically induced hypercapnia, were not significantly altered in insulin-resistant ZO rats compared with phenotypically normal, Zucker lean (ZL) controls (24).
Therefore, we further investigated mitochondrial energetics in the cerebral vasculature during IR using young ZO rats with normal levels of glucose and blood pressure but with elevated plasma insulin levels (16, 17). We hypothesized that mitochondrial respiration would be reduced in large cerebral arteries of young insulin-resistant ZO rats as would mitochondrial and nonmitochondrial protein expression associated with mitochondrial function. We also examined the effects of the nonselective nitric oxide (NO) synthase (NOS) inhibitor Nω-nitro-l-arginine methyl ester (l-NAME) and diazoxide (DZ), a mitochondrial ATP-sensitive potassium (mitoKATP) channel opener, on mitochondrial respiration. l-NAME was used to test the role of NO, and DZ was used because of its known depolarizing effect on mitochondria (47). We used freshly isolated cerebral vessels to determine levels of mitochondrial and related nonmitochondrial proteins. In freshly isolated cerebral microvessels of ZO and ZL rats, we measured and compared superoxide (O2·−) levels to determine oxidative stress levels in the cerebral microcirculation during IR. Finally, we assessed for the first time the morphology of large cerebral arteries and microvessels using electron microscopy to determine whether there are any differences between ZO and ZL rats.
MATERIALS AND METHODS
The procedures and protocols involving animals were approved by the Institutional Animal Care and Use Committee (IACUC) of Tulane University School of Medicine and comply with the National Institutes of Health (NIH) guidelines. Animal care was provided by the Department of Comparative Medicine. Animals were given standard food and tap water ad libitum. We used 10–12-wk-old male ZO and ZL rats from Charles River Laboratories (Wilmington, MA) (n = 34 each).
ZO rat model.
ZO rats have been widely used as a model of IR. These rats have a leptin receptor mutation (fa/fa, homozygous for the mutation), which leads to hyperphagia with subsequent obesity and IR. ZL rats (Fa/fa, heterozygous for the mutation) were used as genetically appropriate controls (6). We and other laboratories have shown that the metabolic profile of ZO rats is similar to the human condition (16, 17, 19, 36). At 10–12 wk of age, ZO rats develop hyperinsulinemia, hypercholesterolemia, hypertriglyceridemia, and impaired glucose tolerance but retain normal levels of glucose and arterial blood pressure (27).
Isolation of cerebral arteries and microvessels.
Animals were deeply anesthetized using 5% isoflurane (VetOne, Boise, ID) and decapitated. Brains were removed immediately and placed in ice-cold PBS. All of the following procedures were carried out on ice or at 4°C. Anterior, middle cerebral, and basilar arteries were isolated, cleaned, and used for mitochondrial respiration measurements and for Western blotting. Brain microvessel isolation was then performed as previously described (42, 43, 49) with minor modifications. Cerebellum, brain stem, choroid plexus, and large superficial arteries and meninges were all removed. The remaining cortex was homogenized using a hand-held glass homogenizer and centrifuged at 1,000 g for 10 min. A three-step process yielded the final microvessels for the experiments: 1) the supernatant was discarded, and the pellet was resuspended in 10 ml of 17.5% dextran (64–76 kDa, Sigma-Aldrich, St. Louis, MO), passed through 300-μm nylon mesh, and then centrifuged at 4,400 g for 15 min; 2) the microvessel pellet was transferred to a clean tube, and the supernatant was resuspended and centrifuged as before to increase the yield of microvessels; and 3) the two resulting microvessel pellets were collected, resuspended, and centrifuged again, as described previously. The resulting microvessel pellet was collected in 1% BSA and passed through 70-μm nylon mesh. Captured microvessels were washed with ice-cold PBS, collected, and used for Western blotting and determination of superoxide levels. Purity of the microvessels was constantly monitored and evaluated via immunohistochemistry (Fig. 1).
Fig. 1.

Immunohistochemical staining of cerebral microvessels. Cerebral microvessels were stained with antibodies specific for different cells that could be present in the sample to evaluate the purity of the microvessel sample. Images are from frozen sections of the microvessel pellet and cut into 10-μm sections, stained, and visualized with fluorescence microscopy. Antibodies that were used are: anti-glial fibrillary acidic protein (GFAP), as a marker of glial cells (A); antineuronal nuclei, as a neuronal marker (E); and anti-von Willebrand factor (vWF), an endothelial cell marker (B, F). C and G: DAPI was used as a nuclear stain. D and H represent merged images. All images in this figure were viewed in a ×10 magnification.
Immunohistochemical evaluation of the microvessel samples.
Immunohistochemistry experiments were done essentially as described by Toth et al. (44). Microvessel pellets were embedded in the Tissue-Tek optimal cutting temperature compound (Sakura Finitek, Torrance, CA), and 10-μm-thick cryostat sections were prepared and fixed in acetone. After being blocked with normal goat serum (Sigma-Aldrich), samples were stained with antibodies specific for different cells that could be present in the sample to evaluate the purity of the microvessel sample. The antibodies used in these experiments were antiglial fibrillary acidic protein at 1:200 dilution (no. G4546, Sigma-Aldrich), as a marker of glial cells; antineuronal nuclei at 1:1,000 dilution (no. ABN90, Sigma-Aldrich), as a neuronal marker; and anti-von Willebrand factor (vWF) at 1:200 dilution (no. ab6994; Abcam, Cambridge, MA), as an endothelial cell marker. After incubation with primary antibodies, sections were washed with PBS and incubated with adequate, fluorescence-conjugated, secondary antibodies. Sections were then visualized via fluorescence microscopy (Fig. 1).
Electron microscopy.
For characterization of large arteries, rats were euthanized with anesthesia and perfused with a PBS solution containing 2% glutaraldehyde and 3% formaldehyde. Arteries were removed and kept in the perfusion solution for 1 h. The microvessel pellets were fixed in a PBS solution containing 2% glutaraldehyde and 3% formaldehyde. Arteries and microvessels were postfixed in 1% osmium tetroxide and embedded in Spurr's resin. Ultrathin sections (80–90 nm) were mounted on formvar-coated copper grids (200 mesh), air dried, and stained with uranyl acetate and lead citrate (at 7 min and 7 min, respectively). The sections were put on grids and viewed at a magnification of ×11,000 using a FEI Tecnai BioTwin 120 keV TEM with a digital imaging setup (Wake Forest University Health Sciences, Winston-Salem, NC) (Fig. 2).
Fig. 2.

Electron microscopy of middle cerebral arteries and microvessels from Zucker obese (ZO) and lean (ZL) rats. The basic features of mitochondrial morphology and location are similar in arteries from ZO (A) and ZL rats (B) and conform in general terms to the Western blot and oxygen consumption rate (OCR) data. Representative sections show heavy investment of mitochondria in cerebral vascular endothelium and vascular smooth muscle (VSM) in both types of rats. To illustrate the spatial arrangement of mitochondria especially in the VSM cells, we used a longitudinal cross section of the artery from an insulin-resistant (IR) rat (A) and a cross-sectional view in the middle cerebral artery from the Zucker lean rat (B). Mitochondria are usually present singly and dispersed throughout in the endothelial cells. Mitochondria in VSM are much larger and of different configurations than in endothelium. Typically, mitochondria in VSM are relatively large and numerous and are present in fields or clusters with sarcoplasmic reticulum (SR) and are not dispersed evenly throughout the VSM. This characteristic can be seen clearly by comparing the longitudinal and cross sections of the arteries. Representative sections of microvessels from ZO (C) and ZL (D) rats are shown. As described in materials and methods, harvested microvessels are composed of arterioles and capillaries (not shown) with little contamination of nonvascular cells except for occasional erythrocytes. Similar to arteries, the VSM of the arterioles contains extensive mitochondrial fields intermixed with SR in both strains of rats. In the arterioles, the mitochondria in VSM retain relatively normal configuration, and the mitochondria in endothelium also appear intact in ZO and ZL rats. However, the mitochondria in the endothelium of the capillaries in both strains are often swollen in appearance or are difficult to visualize (images not shown), suggesting that mitochondrial morphology is better preserved during the harvesting procedure in the more muscular blood vessels. Magnification was ×11,000 in each section. IEL, internal elastic lamina; M, mitochondrion or mitochondria; EEL, external elastic lamina.
Mitochondrial respiration measurement.
The Seahorse Bioscience XFe24 extracellular flux analyzer was used to measure mitochondrial oxygen consumption rate (OCR) as an indicator of mitochondrial respiration (5, 20, 22). For these measurements, we used freshly isolated cerebral arteries as described previously (40). Briefly, arteries were placed into wells of the XF24 islet capture microplate (no. 101122-100; Seahorse Bioscience, Billerica, MA), and wells were filled with 525 μl XF assay medium (no. 102365-100, Seahorse Bioscience) containing 5.0 mM/l glucose and 2 mM/l pyruvate and maintained at 37°C for 20 min in a non-CO2 incubator before the measurement. Oxygen and hydrogen ion-sensitive fluorophores of the Seahorse XFe24 analyzer then repeatedly measured the oxygen and hydrogen concentration in the medium surrounding the arteries, thus calculating OCR. The assay protocols consisted of three cycles of baseline measurements followed by five cycles for each treatment (Fig. 3). To test the effect of DZ on mitochondrial respiration, plates were loaded with 250 μM/l DZ or an equivalent amount of dimethyl sulfoxide (DMSO) as vehicle. The plates were then exposed sequentially to 2 μM/l oligomycin, 1 μM/l carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone, and 1.5 μM/l antimycin plus 1.5 μM/l rotenone to evaluate different components of mitochondrial respiration. The same approach was used to test the effect of l-NAME at 100 μmol/l using deionized water as a control treatment (untreated group). We had four sample groups each of ZO and ZL rats: 1) untreated group, 2) l-NAME-treated group, 3) DMSO-treated group, and 4) DZ-treated group. The OCR data were normalized to arterial protein concentration as determined by Pierce BCA protein assay kit (Thermo Scientific, Rockford, IL) and expressed in picomoles per microgram per minute. After data normalization, a series of calculations were performed using OCR data to provide information about specific segments of mitochondrial (basal respiration, proton leak, maximal respiration, and spare respiratory capacity) and nonmitochondrial respiration.
Fig. 3.
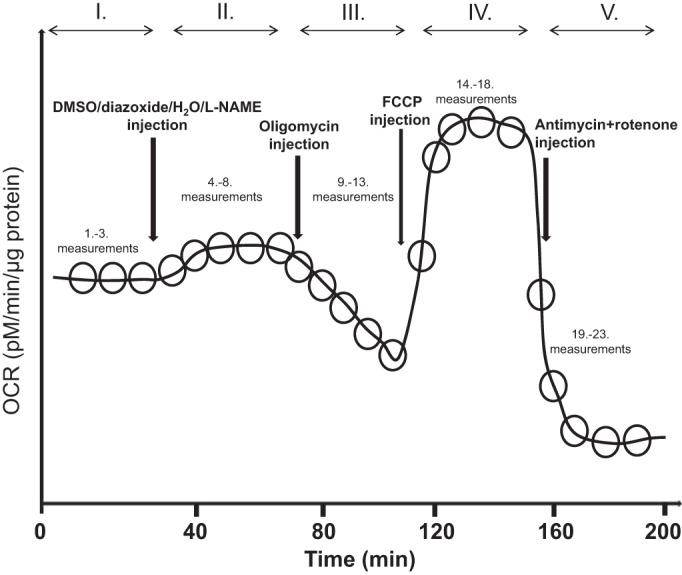
Seahorse experimental design shown through a schematic of the Seahorse experiment protocol. Time points of different drug administration are marked with vertical arrows and resulting changes in OCR, expressed in pM/μg protein, are shown. Roman numerals show different measurement cycles. FCCP, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone; l-NAME, Nω-nitro-l-arginine methyl ester.
Electron spin resonance studies.
We used electron spin resonance (ESR) spectroscopy to measure the levels of reactive oxygen species (ROS), specifically superoxide, in the cerebral microvessels of ZO and ZL rats. Freshly isolated microvessels were used to measure ROS production using the spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethyl-pyrrolidine (CMH), as described previously (28). Deferoxamine (25 μmol/l) and diethyldithiocarbamate (DETC; 2.5 μmol/l) were dissolved under nitrogen gas bubbling in ice-cold modified Krebs-Hepes (KH) buffer containing (in mmol/l) 99.01 NaCl, 4.69 KCl, 1.87 CaCl2, 1.20 MgSO4, 25 NaHCO3, 1.03 K2HPO4, 20 sodium-HEPES, and 11.1 d-glucose, at pH 7.35. Microvessel samples were placed in a microtube containing 300 μl of freshly made 200 μmol/l CMH solution of KH buffer containing DETC and deferoxamine. Samples were incubated at 37°C for 60 min. Afterward, the needle end of a 1-ml syringe barrel was cut, the plunger was retracted from the cut end, and microvessels with CMH containing KH buffer were placed into the syringe barrel and frozen in liquid nitrogen. When the sample was ready to be measured, the syringe was warmed gently between the palms of the hands, and the syringe plunger was used to push the frozen column of microvessels out of the syringe barrel directly into a finger Dewar (Noxygen Science Transfer & Diagnostics, Elzach, Germany) containing liquid nitrogen. Subsequently, the ESR spectra were obtained by placing the finger Dewar into the measuring cavity of a benchtop X-band EMX series ESR spectrometer (Bruker Biospin, Karlsruhe, Germany) using a high-sensitivity SHQ microwave cavity. Time-dependent formation of ROS was determined using the following ESR settings: center field, 1.99 g; microwave power, 20 mW; modulation amplitude, 2 G; sweep time, 10 s; number of scans, 10; field sweep, 60 G. The amplitude measurements of the ESR spectra were normalized to microvessel protein concentrations and expressed in arbitrary units; n represents the number of experiments that include microvessels isolated from two rat brains.
Western blot analysis.
Proteins were harvested as described previously (28, 40). Briefly, cerebral arteries and microvessels were homogenized in ice-cold NP40 lysis buffer (Invitrogen, Frederick, MD) supplemented with proteinase inhibitor cocktail (cat. no. P8340, Sigma-Aldrich) and phosphatase inhibitor cocktail (cat. No. P2850, Sigma-Aldrich). Samples were centrifuged, and the supernatant was used for further analysis. The protein concentration was determined using Pierce BCA protein assay (Thermo Scientific). Protein samples were separated by gel electrophoresis on a 4–20% SDS-PAGE gradient gel, and proteins were transferred onto a polyvinylidene difluoride membrane. Membranes were blocked with casein blocking buffer (no. 92740200; Li-Cor, Lincoln, NE) for 60 min at room temperature. Membranes were then washed with Tris-buffered saline and 0.1% Tween-20 (TBST) (Sigma-Aldrich) and incubated overnight at 4°C with primary antibodies in casein-blocking buffer. The following primary antibodies for mitochondrial proteins were used: anti-Complex II Fp subunit I at 1:1,000 dilution (70 KDa, no. 459200; Invitrogen); anti-Complex III Subunit I core at 1:1,000 dilution (53 KDa, no. 459140; Invitrogen); ATP synthase Complex V subunit-α at 1:500 dilution (50 KDa, no. 459240; Invitrogen); anti-voltage-dependence anion channel (VDAC) at 1:1,000 dilution to detect the endogenous levels of total VDAC (32 KDa, no. 4866S; Cell Signaling Technology, Danvers, MA); and total dynamin-related protein-1 (DRP-1) at 1:1,000 dilution (no. 611112; BD Transduction, San Jose, CA). Antibodies for primary nonmitochondrial proteins were antimanganese superoxide dismutase (MnSOD) aa114-220 at 1:5,000 (25 KDa, no. 611581; BD Transduction Laboratories); total endothelial nitric oxide synthase (eNOS) at 1:500 dilution (140 KDa, no. 610297; BD Transduction Laboratories); and the loading control β-actin at 1:5,000 dilution (42 KDa, no. A5441; Sigma-Aldrich). After incubation with the primary antibody, membranes were washed and incubated for 90 min at room temperature in 1% BSA-TBST with secondary goat anti-rabbit IgG at 1:2,500 dilution (no. 7074S, Cell Signaling Technology) or goat anti-mouse IgG at 1:5000 dilution (no. 7076P2, Cell Signaling Technology). The final reaction was visualized using chemiluminescence (LumiGLO, Gaithersburg, MD) and autoradiography. Immunobands were scanned, and the optical density of each band was quantified and normalized to the intensity of the corresponding β-actin band using ImageJ software.
Data analysis and statistics.
Results were expressed as means ± SE; n indicates the number of independent measurements. Data were analyzed using unpaired t-test and one-way ANOVA with Tukey's post hoc test. P < 0.05 was considered statistically significant.
RESULTS
Electron microscopy.
The basic features of mitochondrial morphology and location are similar in middle cerebral arteries from ZO and ZL rats (Fig. 2, A and B, respectively) and microvessels from ZO and ZL rats (Fig. 2, C and D, respectively). Representative sections of middle cerebral arteries show heavy investment of mitochondria in cerebral vascular endothelium and vascular smooth muscle (VSM) in both types of rats. Usually, mitochondria are present singly and dispersed throughout the endothelial cells. However, mitochondria in VSM are much larger and display different configurations than in endothelium. Additionally, mitochondria in VSM are relatively large and numerous and characteristically are present in fields or clusters together with sarcoplasmic reticulum (SR) and are not dispersed evenly throughout the VSM. Harvested microvessels, prepared as described in materials and methods, are composed of arterioles and capillaries with little contamination of nonvascular cells except for occasional erythrocytes. Similar to arteries, the VSM of arterioles contains extensive mitochondrial fields intermixed with SR in both strains of rats. In the arterioles, the VSM mitochondria retain relatively normal configuration, and the endothelial mitochondria also appear intact in ZO and ZL rats. However, the endothelial mitochondria of the capillaries in both strains are often swollen in appearance or are difficult to visualize (not shown), suggesting that mitochondrial morphology is better preserved during the harvesting procedure in the more muscular blood vessels. Although we did not perform a qualitative analysis, visual observation indicated that microvessels of ZO (Fig. 2C) had more vacuoles in the endothelium than ZL microvessels (Fig. 2D).
Mitochondrial respiration measurements.
Oxygen consumption rates for different segments of mitochondrial and nonmitochondrial respiration are shown in Figs. 4 and 5. Maximal respiration was significantly higher in the untreated ZO group (375 ± 6.5 pM/μg protein per min) compared with the untreated ZL group (334.6 ± 6.66 pM/μg protein per min; n = 15, P < 0.05) (Fig. 5D). Other components of mitochondrial (basal respiration, ATP production, spare capacity, and proton leak) and nonmitochondrial respiration did not show a statistically significant difference among the untreated groups (Fig. 5, A–F). The l-NAME treatment significantly increased the proton leak in the ZL group (65.24 ± 8.5 pM/μg protein per min; 94.94 ± 9.4 pM/μg protein per min; n = 15, P < 0.05) but not in the ZO group [68.95 ± 7.4 pM/μg protein per min; 61.43 ± 9.45 pM/μg protein per min; n = 15, P = not significant (NS)] (Fig. 5F). The l-NAME treatment had no significant effect on the other components of mitochondrial and nonmitochondrial respiration (Fig. 5, A–F). In the DMSO-treated group, basal respiration (173.6 ± 12.7 pM/μg protein per min; 134.5 ± 11.03 pM/μg protein per min; n = 17, P < 0.05) and proton leak (122 ± 11.07 pM/μg protein per min; 86.5 ± 10.47 pM/μg protein per min; n = 17, P < 0.05) were significantly increased in ZO compared with ZL rats (Fig. 5, H and L). Similar to observations from the untreated group, DMSO had no significant effect on other mitochondrial and nonmitochondrial respiration components of the ZL and ZO groups (Fig. 5, G–L). Treatment with DZ significantly decreased nonmitochondrial respiration in the ZL (73.15 ± 6.89 pM/μg protein per min; 53.24 ± 5.81 pM/μg protein per min; n = 16, P < 0.05) but not in the ZO groups (67.48 ± 6.43 pM/μg protein per min; 65.43 ± 12.85 pM/μg protein per min; n = 16, P = NS) (Fig. 5G). The DZ treatment did not significantly alter other mitochondrial respiration components in either ZL or ZO groups (Fig. 5, H–L).
Fig. 4.

Mitochondrial respiration profile of major cerebral arteries from ZO and ZL rats. A: effects of l-NAME treatment on mitochondrial respiration in cerebral arteries of ZL and ZO rats. B: effects of treatment with diazoxide (DZ) on mitochondrial respiration of ZL and ZO groups. Data are expressed as means ± SE; n = 16–17 per group.
Fig. 5.

Mitochondrial respiration segments and the effect of l-NAME and DZ on each individual segment. Mitochondrial bioenergetic parameters shown are as follows: nonmitochondrial respiration (A, G), basal respiration (B, H), ATP production (C, I), maximal respiration (D, J), spare respiratory capacity (E, K), and proton leak (F, L). Data are expressed as means ± SE. Sample sizes are the same as in Fig. 4. *P < 0.05 (untreated samples, ZO vs. ZL), #P < 0.05 (l-NAME-treated ZL vs. untreated ZL), †P < 0.05 (DMSO-treated ZO vs. ZL), ‡P < 0.05 (DZ-treated ZL vs. DMSO-treated ZL).
Protein expression.
Levels of mitochondrial proteins (Complex II, Complex III, Complex V, VDAC, DRP-1, and MnSOD) were not significantly different in cerebral arteries and microvessels of ZO compared with ZL (Figs. 6 and 7). Levels of eNOS were significantly higher in microvessels of ZO compared with ZL (132.7 ± 20.84%; 76.67 ± 10.54%, n = 8, P < 0.05).
Fig. 6.

Mitochondrial and nonmitochondrial protein expression in major cerebral arteries of ZL and ZO rats. Representative Western blots and summary data: Complex II, 70 kDa (A); Complex III, 53 kDa (B); Complex V, 50 kDa (C); voltage-dependence anion channel (VDAC), 32 kDa (D); dynamin-related protein-1 (DRP-1), 80 kDa (E); and manganese superoxide dismutase (MnSOD), 24 kDa (F). L = lean, O = obese. Data are expressed as means ± SE (n = 8–12 per group).
Fig. 7.

Mitochondrial and nonmitochondrial protein expression in cerebral microvessels of ZL and ZO rats. Representative Western blots and summary data: Complex II, 70 kDa (A); Complex III, 53 kDa (B); Complex V, 50 kDa (C); VDAC, 32 kDa (D); DRP-1, 80 kDa (E); MnSOD, 24 kDa (F). Data are expressed as means ± SE (n = 8 - 12 per group).
ROS production in cerebral microvessels.
A characteristic ESR signal with characteristic spectrum (28) was detected in microvessels incubated with CMH. The magnitude of this signal (normalized arbitrary units) was significantly increased in the freshly isolated cerebral microvessels of the ZO (1,397.33 ± 317.86) compared with the ZL group (560.6 ± 63.43, n = 7 each, P < 0.05) (Fig. 8).
Fig. 8.

Superoxide production is significantly higher in cerebral microvessels of ZO compared with ZL rats; n = 7 per group, *P < 0.05 (ZO vs. ZL).
DISCUSSION
We have characterized for the first time the mitochondrial fine structure, mitochondrial protein mass, and functional mitochondrial dynamics of cerebral arteries and microvessels of ZL and ZO rats. The major findings of this study are the following: 1) mitochondrial morphological profiles in endothelium and VSM are similar in ZL and ZO arteries and extend to the level of arterioles and capillaries; 2) differences in mitochondrial respiration between ZL and ZO cerebral arteries are modest; 3) levels of mitochondrial proteins were similar between ZL and ZO groups both for major arteries and for microvessels; 4) the eNOS levels were significantly higher in the cerebral microvessels of the ZO compared with the ZL group; and 5) the mitochondrial ROS production was significantly increased in ZO cerebral microvessels compared with ZL. Collectively, these findings indicate that subtle differences, which might set the stage for more extensive changes during prolonged IR, are present in mitochondrial dynamics in the early stages of the metabolic syndrome.
Although the fine structure of large cerebral arteries in normal rats has been examined previously (41), we are unaware of any examination of mitochondrial profiles in cerebral arteries or microvessels of IR rats (11, 30, 40). Similar to our previous studies in male, non-IR rats (40), we found that mitochondria in endothelial cells are smaller and more oval in appearance than mitochondria in VSM and that mitochondria are dispersed throughout the endothelium. In contrast, mitochondria are normally localized in discrete fields running down the longitudinal axis rather than evenly distributed in VSM cells in both normal ZO and ZL rats. Additionally, mitochondria in these fields are interspersed with SR, cellular structures associated with calcium release and storage (11, 13, 14). We and others have shown that mitochondrial activation in VSM of large cerebral arteries leads to the generation of calcium sparks from the SR, which promotes vasorelaxation (11, 30, 47). A new finding is that the mitochondrial profile seen in large surface arteries is also found in parenchymal arterioles in our microvessel preparation. Thus it seems likely that the functional relationship between mitochondria and SR in VSM is present at all levels of the precapillary cerebral circulation. Nonetheless, we have found that the mitochondrial-SR relationship, with respect to mitochondrial depolarization and the generation of calcium sparks to diazoxide and BMS-191095, are reduced in large cerebral arteries of IR rats (15, 16, 27). Additionally, because we could rapidly correct vascular dysfunction in cerebral arteries from young, IR animals with a ROS scavenger or a PKC inhibitor, it appears that obvious and irreversible changes in vascular structures have not yet occurred at this stage of the metabolic syndrome. The only difference in vascular morphology relates to the presence of a large number of vacuoles in endothelial cells of the microvessels from the ZO animals, which might be related to enhanced superoxide anion generation.
Different aspects of mitochondrial (basal respiration, ATP production, spare respiratory capacity, and proton leak) and nonmitochondrial respiration showed similar results in untreated ZL and ZO cerebral arteries except for maximal respiration, which was significantly higher in the ZO compared with the ZL group. Although there are opposing reports regarding the correlation between mitochondrial dysfunction and IR (18, 26, 31, 33, 39), our data are similar to that found by other laboratories regarding ex vivo measurements of mitochondrial respiration in different samples (3, 4, 8, 12, 23). Our previous data showed impaired vasodilation of cerebral arteries from ZO rats mediated by mitochondrial depolarization via DZ (27). Therefore, we examined mitochondrial respiration in cerebral arteries of ZO rats. Our data suggest that mitochondrial respiration is not significantly altered in cerebral arteries of young ZO rats. Increased maximal mitochondrial respiration levels support this claim, suggesting that the mechanisms necessary to increase respiration are preserved in young ZO rats and possibly are more functional than in the ZL group.
We also treated arteries with the nonselective NOS inhibitor, l-NAME, to examine the influence of NO in the mitochondrial respiration of these arteries. NO is an inhibitor of the mitochondrial respiratory chain, and as such it binds to cytochrome oxidase at the oxygen-binding site, causing reversible inhibition of cytochrome oxidase, thus reducing ATP production. It can also act through production of reactive nitrogen species, causing slow and weak but irreversible inhibition of different mitochondrial components (7). Therefore, we would have expected an increase in mitochondrial respiration after the inhibition of NOS, as has been reported previously (40). However, there was no significant effect of l-NAME treatment on the components of the mitochondrial respiration in the ZL and ZO groups with the exception of significantly increased proton leak in the ZL group. During conditions with normal oxygen levels, very little NO reaches cytochrome oxidase in mitochondria because it is scavenged by oxymyoglobin for nitrate and metmyoglobin production (7). Morley and Mattammal (34) showed that levels of NOS are significantly lower in ZO compared with ZL rats. Therefore, we can also project that there are certain variabilities in NOS and NO levels between ZL rats and other typically used strains. In contrast, we reported increased eNOS levels in cerebral arteries from ZO rats (16, 27). However, despite these increased levels, NO bioavailability was found to be diminished (27, 29). We went on to demonstrate that eNOS in ZO arteries partly exists in an uncoupled state, contributing to increased ROS levels (29).
Our laboratory previously demonstrated the importance of mitoKATP channels in preconditioning and regulation of vascular tone (9, 10, 28, 30, 41). In the present study, treatment with DZ did not significantly affect any of the segments of mitochondrial respiration in either the ZL or ZO group. This finding is consistent with some of the previous findings (37, 40). However, DZ treatment significantly reduced nonmitochondrial respiration but only in the ZL group, which suggests the existence of differences in other segments of cellular respiration in these groups. MitoKATP channel openers may have different effects on mitochondrial respiration under different energetic conditions (38). This could explain the different tendencies in response to DZ, although without statistical significance. During this experiment, we also found increased proton leak and basal respiration of vehicle (DMSO)-treated ZO compared with ZL samples. Considering the multiple possible effects of DMSO, such as inhibition of inflammation, reduction in free radical formation, and increase in membrane transport (25), we suggest that any of these effects may be more noticeable in ZO than in ZL arteries.
Expression of mitochondrial (Complex II, Complex III, Complex V, VDAC, MnSOD, and DRP-1) proteins was not significantly different among the major cerebral artery groups. This finding is consistent with mitochondrial respiration measurements and corresponds with studies that found no correlation between IR and mitochondrial dysfunction (3, 4, 18, 23, 26, 33). Mitochondrial protein expression levels were similar in microvessel samples of the ZL and ZO groups.
We also measured ROS production in cerebral microvessels and found that superoxide levels were significantly higher in ZO compared with ZL rats. Higher eNOS levels and ROS production in ZO rats were observed previously in major cerebral arteries in which they were considered to be a possible compensatory mechanism to increased oxidative stress (16, 27); however, it is also feasible that uncoupled eNOS is the source of elevated superoxide levels (45, 48). Furthermore, the presence of relatively larger amounts of vacuoles in the endothelial cells is consistent with enhanced superoxide levels in microvessels from the ZO rats.
Type 2 diabetes and metabolic syndrome in general are enormous public health problems. With tens of millions of people affected by type 2 diabetes, it represents a heavy burden for both patients as well as the health system (21). Considering the complications of type 2 diabetes, attributable to increased oxidative stress such as diabetic neuropathy (35, 46) in which the ROS levels are increased in the initial stage of the disease, early antioxidative treatment could be beneficial in slowing the disease, specifically its complications.
There were a few limitations to our study. We did not have a sufficient microvessel mass for reproducible determinations of OCR, but we hope to overcome this limitation in future studies. In addition, we performed our experiments on young, male ZO and ZL rats and thus do not know how sex, aging, or a greater expression of the metabolic syndrome affects mitochondrial dynamics of large cerebral arteries and microvessels. Fisher-Wellman et al. (18) argued that mitochondrial dysfunction needs to be presented early in development to be considered a possible cause of IR. Also, the initial NO levels in the examined cerebral arteries were unknown, and therefore the interpretation of NOS inhibition is limited. Possible changes in mitochondrial function that may occur later in the development of the metabolic syndrome and their eventual influence on cerebral vasculature are yet to be investigated. Even though our present study and some of the previous studies from our laboratory show that uncoupled eNOS may be the source of increased ROS production, additional tests are required to fully understand underlying mechanisms.
In summary, we have for the first time directly measured mitochondrial respiration and determined levels of mitochondrial proteins in the large cerebral arteries and microvessels of insulin-resistant rats. Our results show that severe mitochondrial dysfunction is not present in the early stages of IR and that oxidative stress, possibly arising from uncoupling eNOS, might play a significant role in vascular dysfunction during IR.
GRANTS
This work was supported by National Institutes of Health grants (HL-077731 and HL093554 to D. Busija), Louisiana Board of Regents Support Fund-Research Competitiveness Subprogram [LEQSF (2014-17)-RD-A-11 to P. Katakam], American Heart Association National Center NRCP Scientist Development Grant (14SDG20490359 to P. Katakam), and American Heart Association Post-Doctoral Fellowship Grant (15POST23040005 to I. Rutkai). This research was supported in whole or in part by the Louisiana Board of Regents Endowed Chairs for Eminent Scholars program.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: I.M. and D.W.B. conception and design of research; I.M., I.R., T.T., V.N.S., and P.V.G.K. performed experiments; I.M. and D.W.B. analyzed data; I.M. and D.W.B. interpreted results of experiments; I.M. and D.W.B. prepared figures; I.M. drafted manuscript; I.M., I.R., T.T., V.N.S., P.V.G.K., and D.W.B. edited and revised manuscript; I.M., I.R., T.T., V.N.S., P.V.G.K., and D.W.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Nancy Busija, M.A. for editorial assistance. We thank Dana Liu for technical help. We also thank Kenneth Grant of the Cellular Imaging Shared Resource at Wake Forest University Health Sciences for assistance with electron microscopy.
REFERENCES
- 1.Alberti KG, Zimmet P, Shaw J. Metabolic syndrome–a new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet Med 23: 469–480, 2006. [DOI] [PubMed] [Google Scholar]
- 2.Arenillas JF, Moro MA, Davalos A. The metabolic syndrome and stroke: potential treatment approaches. Stroke 38: 2196–2203, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM. Increased malonyl-CoA levels in muscle from obese and type 2 diabetic subjects lead to decreased fatty acid oxidation and increased lipogenesis; thiazolidinedione treatment reverses these defects. Diabetes 55: 2277–2285, 2006. [DOI] [PubMed] [Google Scholar]
- 4.Boushel R, Gnaiger E, Schjerling P, Skovbro M, Kraunsoe R, Dela F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia 50: 790–796, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J 435: 297–312, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bray GA. The Zucker-fatty rat: a review. Fed Proc 36: 148–153, 1977. [PubMed] [Google Scholar]
- 7.Brown GC, Borutaite V. Nitric oxide and mitochondrial respiration in the heart. Cardiovasc Res 75: 283–290, 2007. [DOI] [PubMed] [Google Scholar]
- 8.Bugger H, Abel ED. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin Sci 114: 195–210, 2008. [DOI] [PubMed] [Google Scholar]
- 9.Busija DW, Katakam PV. Mitochondrial mechanisms in cerebral vascular control: shared signaling pathways with preconditioning. J Vasc Res 51: 175–189, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Busija DW, Lacza Z, Rajapakse N, Shimizu K, Kis B, Bari F, Domoki F, Horiguchi T. Targeting mitochondrial ATP-sensitive potassium channels—a novel approach to neuroprotection. Brain Res Brain Res Rev 46: 282–294, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Cheranov SY, Jaggar JH. Mitochondrial modulation of Ca2+ sparks and transient KCa currents in smooth muscle cells of rat cerebral arteries. J Physiol 556: 755–771, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choo HJ, Kim JH, Kwon OB, Lee CS, Mun JY, Han SS, Yoon YS, Yoon G, Choi KM, Ko YG. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 49: 784–791, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Dai J, Kuo KH, Leo JM, van Breemen C, Lee CH. Rearrangement of the close contact between the mitochondria and the sarcoplasmic reticulum in airway smooth muscle. Cell Calcium 37: 333–340, 2005. [DOI] [PubMed] [Google Scholar]
- 14.Dorn GW 2nd, Scorrano L. Two close, too close: sarcoplasmic reticulum-mitochondrial crosstalk and cardiomyocyte fate. Circ Res 107: 689–699, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erdos B, Simandle SA, Snipes JA, Miller AW, Busija DW. Potassium channel dysfunction in cerebral arteries of insulin-resistant rats is mediated by reactive oxygen species. Stroke 35: 964–969, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Erdos B, Snipes JA, Miller AW, Busija DW. Cerebrovascular dysfunction in Zucker obese rats is mediated by oxidative stress and protein kinase C. Diabetes 53: 1352–1359, 2004. [DOI] [PubMed] [Google Scholar]
- 17.Erdos B, Snipes JA, Tulbert CD, Katakam P, Miller AW, Busija DW. Rosuvastatin improves cerebrovascular function in Zucker obese rats by inhibiting NAD(P)H oxidase-dependent superoxide production. Am J Physiol Heart Circ Physiol 290: H1264–H1270, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Fisher-Wellman KH, Weber TM, Cathey BL, Brophy PM, Gilliam LA, Kane CL, Maples JM, Gavin TP, Houmard JA, Neufer PD. Mitochondrial respiratory capacity and content are normal in young insulin-resistant obese humans. Diabetes 63: 132–141, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frisbee JC. Hypertension-independent microvascular rarefaction in the obese Zucker rat model of the metabolic syndrome. Microcirculation 12: 383–392, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Gerencser AA, Chinopoulos C, Birket MJ, Jastroch M, Vitelli C, Nicholls DG, Brand MD. Quantitative measurement of mitochondrial membrane potential in cultured cells: calcium-induced de- and hyperpolarization of neuronal mitochondria. J Physiol 590: 2845–2871, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract 103: 137–149, 2014. [DOI] [PubMed] [Google Scholar]
- 22.Hill BG, Benavides GA, Lancaster JR Jr, Ballinger S, Dell'Italia L, Jianhua Z, and Darley-Usmar VM. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem 393: 1485–1512, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holloway GP, Thrush AB, Heigenhauser GJ, Tandon NN, Dyck DJ, Bonen A, Spriet LL. Skeletal muscle mitochondrial FAT/CD36 content and palmitate oxidation are not decreased in obese women. Am J Physiol Endocrinol Metab 292: E1782–E1789, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Institoris A, Lenti L, Domoki F, Wappler E, Gaspar T, Katakam PV, Bari F, Busija DW. Cerebral microcirculatory responses of insulin-resistant rats are preserved to physiological and pharmacological stimuli. Microcirculation 19: 749–756, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacob SW, de la Torre JC. Pharmacology of dimethyl sulfoxide in cardiac and CNS damage. Pharmacol Rep 61: 225–235, 2009. [DOI] [PubMed] [Google Scholar]
- 26.Karakelides H, Irving BA, Short KR, O'Brien P, Nair KS. Age, obesity, and sex effects on insulin sensitivity and skeletal muscle mitochondrial function. Diabetes 59: 89–97, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katakam PV, Domoki F, Snipes JA, Busija AR, Jarajapu YP, Busija DW. Impaired mitochondria-dependent vasodilation in cerebral arteries of Zucker obese rats with insulin resistance. Am J Physiol Regul Integr Comp Physiol 296: R289–R298, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katakam PV, Gordon AO, Sure VN, Rutkai I, Busija DW. Diversity of mitochondria-dependent dilator mechanisms in vascular smooth muscle of cerebral arteries from normal and insulin-resistant rats. Am J Physiol Heart Circ Physiol 307: H493–H503, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katakam PV, Snipes JA, Steed MM, Busija DW. Insulin-induced generation of reactive oxygen species and uncoupling of nitric oxide synthase underlie the cerebrovascular insulin resistance in obese rats. J Cereb Blood Flow Metab 32: 792–804, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katakam PV, Wappler EA, Katz PS, Rutkai I, Institoris A, Domoki F, Gaspar T, Grovenburg SM, Snipes JA, Busija DW. Depolarization of mitochondria in endothelial cells promotes cerebral artery vasodilation by activation of nitric oxide synthase. Arterioscler Thromb Vasc Biol 33: 752–759, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51: 2944–2950, 2002. [DOI] [PubMed] [Google Scholar]
- 32.Kendall DM, Harmel AP. The metabolic syndrome, type 2 diabetes, and cardiovascular disease: understanding the role of insulin resistance. Am J Manag Care 8: S635–S653; quiz S654–S637, 2002. [PubMed] [Google Scholar]
- 33.Lefort N, Glancy B, Bowen B, Willis WT, Bailowitz Z, De Filippis EA, Brophy C, Meyer C, Hojlund K, Yi Z, Mandarino LJ. Increased reactive oxygen species production and lower abundance of complex I subunits and carnitine palmitoyltransferase 1B protein despite normal mitochondrial respiration in insulin-resistant human skeletal muscle. Diabetes 59: 2444–2452, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morley JE, Mattammal MB. Nitric oxide synthase levels in obese Zucker rats. Neurosci Lett 209: 137–139, 1996. [DOI] [PubMed] [Google Scholar]
- 35.Obrosova IG. How does glucose generate oxidative stress in peripheral nerve? Int Rev Neurobiol 50: 3–35, 2002. [DOI] [PubMed] [Google Scholar]
- 36.Oltman CL, Richou LL, Davidson EP, Coppey LJ, Lund DD, Yorek MA. Progression of coronary and mesenteric vascular dysfunction in Zucker obese and Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol 291: H1780–H1787, 2006. [DOI] [PubMed] [Google Scholar]
- 37.Ovide-Bordeaux S, Ventura-Clapier R, Veksler V. Do modulators of the mitochondrial K(ATP) channel change the function of mitochondria in situ? J Biol Chem 275: 37291–37295, 2000. [DOI] [PubMed] [Google Scholar]
- 38.Riess ML, Camara AK, Heinen A, Eells JT, Henry MM, Stowe DF. KATP channel openers have opposite effects on mitochondrial respiration under different energetic conditions. J Cardiovasc Pharmacol 51: 483–491, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 54: 8–14, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Rutkai I, Dutta S, Katakam PV, Busija DW. Dynamics of enhanced mitochondrial respiration in female compared with male rat cerebral arteries. Am J Physiol Heart Circ Physiol 309: H1490–H1500, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rutkai I, Katakam PV, Dutta S, Busija DW. Sustained mitochondrial functioning in cerebral arteries after transient ischemic stress in the rat: a potential target for therapies. Am J Physiol Heart Circ Physiol 307: H958–H966, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silbergeld DL, Ali-Osman F. Isolation and characterization of microvessels from normal brain and brain tumors. J Neurooncol 11: 49–55, 1991. [DOI] [PubMed] [Google Scholar]
- 43.Stirone C, Duckles SP, Krause DN, Procaccio V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol 68: 959–965, 2005. [DOI] [PubMed] [Google Scholar]
- 44.Toth A, Czikora A, Pasztor ET, Dienes B, Bai P, Csernoch L, Rutkai I, Csato V, Manyine IS, Porszasz R, Edes I, Papp Z, Boczan J. Vanilloid receptor-1 (TRPV1) expression and function in the vasculature of the rat. J Histochem Cytochem 62: 129–144, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA Jr. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci USA 95: 9220–9225, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vincent AM, Russell JW, Low P, Feldman EL. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocrine Rev 25: 612–628, 2004. [DOI] [PubMed] [Google Scholar]
- 47.Xi Q, Cheranov SY, Jaggar JH. Mitochondria-derived reactive oxygen species dilate cerebral arteries by activating Ca2+ sparks. Circ Res 97: 354–362, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem 273: 25804–25808, 1998. [DOI] [PubMed] [Google Scholar]
- 49.Yousif S, Marie-Claire C, Roux F, Scherrmann JM, Decleves X. Expression of drug transporters at the blood-brain barrier using an optimized isolated rat brain microvessel strategy. Brain Res 1134: 1–11, 2007. [DOI] [PubMed] [Google Scholar]