

Abstract
Fatty acids (FA) modify DNA methylation in vitro, but limited information is available on whether corresponding associations exist in vivo and reflect any short-term effect of the diet. Associations between global DNA methylation and FAs were sought in blood from lactating infants (LI; n = 49) and adult males (AMM; n = 12) equally distributed across the three conventional BMI classes. AMM provided multiple samples at 2-hour intervals during 8 hours after either a single Western diet-representative meal (post-prandial samples) or no meal (fasting samples). Lipid/glucose profile, HDAC4 promoter and PDK4 5’UTR methylation were determined in AMM. Multiple regression analysis revealed that global (in LI) and both global and PDK4-specific DNA methylation (in AMM) were positively associated with eicosapentaenoic and arachidonic acid. HDAC4 methylation was inversely associated with arachidonic acid post-prandially in AMM. Global DNA methylation did not show any defined within-day pattern that would suggest a short-term response to the diet. Nonetheless, global DNA methylation was higher in normal weight subjects both post-prandially and in fasting and coincided with higher polyunsaturated relative to monounsaturated and saturated FAs. We show for the first time strong associations of DNA methylation with specific FAs in two human cohorts of distinct age, diet and postnatal development stage.
The consumption of high-fat diets and alterations in lipid metabolism are associated with metabolic diseases and cancer1,2,3. In vitro data indicate that very low density lipoproteins (VLDL) and specific fatty acids (FAs) can change DNA methylation patterns4,5,6,7. These data, together with the known association of global DNA hypomethylation with cancer (reviewed by Kulis and Esteller8), metabolic syndrome9 and the post-rupture atherosclerotic lesion10, and of DNA hypermethylation with early-stage atherosclerosis11,12, suggest that dietary lipids may exert pathological effects at least in part by imposing pathological DNA methylation profiles. If so, associations should be detectable between DNA methylation and specific FAs, yet the topic has received little attention to date.
Another pending issue is whether such associations reflect short- or long-term responses to the diet. Mid- and long-term high-fat dietary supplementation can change DNA methylation profiles in mammals13,14. Epigenetic short-term effects of the diet could be equally relevant, as the frequent transitions from pre-prandial/fasting to post-prandial states that are experienced by humans, may result in corresponding frequent oscillations between fasting- and post-prandial-specific epigenetic marks in at least selected loci in the genome. If so, it is conceivable that any error-prone diet-driven oscillations may lock selected loci in a pathogenic chromatin state. For example, locking in a fasting-specific state may exacerbate a pre-existing thrifty genotype15.
In the present work, we assessed whether any association exists between whole peripheral blood DNA methylation and specific FAs and lipids in two distinct human cohorts - lactating infants and adult men - and whether those associations are rapidly modified in the postprandial state.
Results
Associations between DNA methylation and FAs in the lactating infant (LI) cohort
We first asked whether any association existed between global DNA methylation and specific FAs in whole peripheral blood. To that end, we interrogated a cohort of 49 Mexican lactating infants of both sexes. A description of the lactating infant cohort is presented in Supplementary Table 1. Sex, age at sampling, weight at birth, normalized weight gain - i.e. difference between the weight at sampling and at birth divided by the birth-to-sampling time (days) - whole peripheral blood FAs and global DNA methylation were the available variables for that cohort. When grouped by degree of saturation and percent-normalized, FAs showed a non-significant tendency to associate inversely (saturated and monounsaturated FAs or SFAs and MUFAs, respectively) or positively (polyunsaturated FAs or PUFAs) with DNA methylation (Supplementary Table 2.1). A multiple regression analysis adjusting for the above mentioned variables only revealed significant positive associations of global DNA methylation with normalized (% of all FAs) n-3 PUFA eicosapentaenoic acid (EPA; C20:5) and the n-6 PUFA arachidonic acid (AA; C20:4) among all detected FAs, with a noticeably stronger (higher Beta) association with EPA (Table 1). Although the estimation of the effects was not the primary aim of the study, the adjusted R2 indicates that EPA and AA explain ~17% of the variation in DNA methylation in the presented model. Furthermore, although DNA methylation changes with age16, we failed to detected any corresponding association between these two parameters in the LI cohort (Supplementary Table 2.1). However, a handful of FAs were associated with age but not with sex (Supplementary Table 3). Notably, age accounted for nearly half of the variability of the sum of the SFA and MUFA C22:0 and C14:1 (p = 0.0015, adjusted R2 = 0.49) and the latter FAs were not associated with DNA methylation (p = 0.184, adjusted R2 = 0.04).
Table 1. Significant associations of FAs with global DNA methylation (dependent variable) in the LI cohort.
| Variable | Beta | t | p | adjusted R2 |
|---|---|---|---|---|
| C20:4 (AA) | 0.0848 | 2.1084 | 0.0420 | 0.1683 |
| C20:5 (EPA) | 0.9867 | 2.3512 | 0.0384 |
Beta values indicate the change in % 5mdC per one-point increase in percent FA. Non-significant associations are shown in Supplementary Table 2.1.
DNA methylation trend across the sampling period in adult men (AMM)
The observation that global DNA methylation was associated with specific FAs in LI, prompted us to ask whether those associations were reproducible in an entirely distinct cohort and whether they were dynamically responsive to the diet. These questions were addressed in the AMM subjects, for which a complete metabolic and anthropometric profile was obtained, in addition to global and gene-specific DNA methylation data.
A total of 12 men entered and completed the study. A description of the AMM subjects is presented in Supplementary Table 4. The AMM subjects underwent a repeated blood draw procedure across an 8-hour period (see Methods), which is schematically shown in Fig. 1. Four subjects represented each of the normal weight, overweight and obese groups (BMI < 25, 25–30 and >30, respectively). The comparison between BMI groups showed expected significant differences in body weight, waist and hip circumference (WC and HC, respectively), waist-to-height ratio (WHtR) and systolic blood pressure (SBP). Meanwhile, age, height, waist-to-hip ratio (WHR) and diastolic blood pressure (DPB) showed no differences (Supplementary Table 4).
Figure 1. AMM subject sampling scheme.
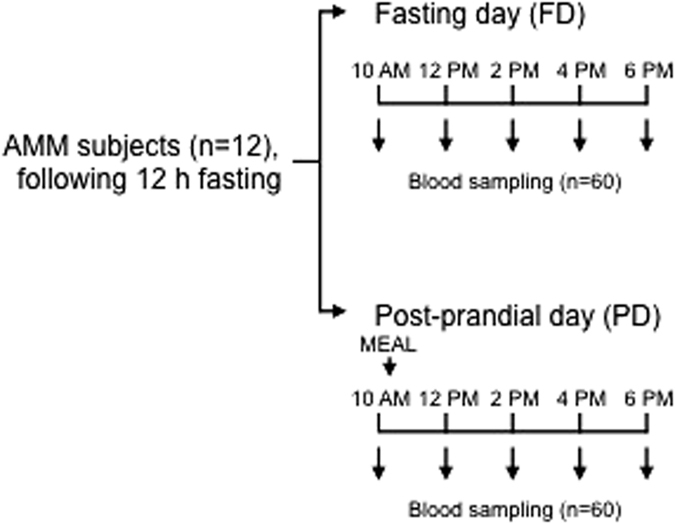
Five consecutive, same-day blood samples were obtained from a total of 12 subjects in two prandial conditions: fasting (FD) and post-prandial (PD). The 10 AM blood sample represented the baseline for each day. A total of 120 samples were obtained and analyzed (60 per prandial condition).
We hypothesized that total blood FA-dependent and/or lipid-dependent trends in global DNA methylation - i.e. an increase or decrease relative to 10 AM sample baseline, whether transient or stable throughout the 8 h sampling period - would be observed. However, a highly variable and not identifiable DNA methylation pattern across the sampling period was observed in the PD or FD sets - i.e. no significant difference between the 10AM baseline and any other time point or between the maximum and minimum values within any of the 6 sets (3 PD, 3 FD) could be detected (ANOVA and Scheffé's post hoc test; Fig. 2 upper panels). Noticeably, the variability (SD) in the 5 average values (one for each time point) of the normal weight subjects was 3–5-fold lower compared to the other two BMI classes in both PD and FD (p < 0.001 in all comparisons with the normal weight set; Bartlett’s test).
Figure 2. DNA methylation trends across the sampling period in AMM.

Average and SD for n = 4 subjects per time point are shown. Open circles: normal weight; solid squares: overweight; open squares: obese. The arrows indicate the approximate time of the meal consumption in PD.
Associations between DNA methylation and metabolic parameters in AMM
Based on the lack of any time-dependent global DNA methylation pattern and with the aim of optimising measurement accuracy, we treated the 5 PD or FD time-point data for each AMM subject as repeated measurements. The analysis of such subject-averaged data revealed an interplay between global DNA methylation, post-prandial state and BMI. First, global DNA methylation was significantly higher in PD relative to FD when comparing normal weight or obese AMM of the two sets (Fig. 3). Furthermore, within FD or PD, global DNA methylation was significantly lower in overweight and obese, compared to normal weight subjects. Weak (p = 0.04) differences between overweight and obese subjects were observed and only in the FD set. The extreme global DNA methylation differences and levels shown in Fig. 3 and in the results below, although counterintuitive have been reported in several studies (see for example17,18,19). As for metabolic parameters, an initial approach by Pearson’s correlation analysis revealed that VLDL and TG were significantly and inversely correlated with global DNA methylation in both the FD and PD sets (Supplementary Table 5). To account for a possible delayed response, we compared global DNA methylation at 6 PM against metabolic variables at the 12 PM blood draw, but correlations were generally weaker (Supplementary Table 6).
Figure 3. Global DNA methylation in FD, PD and BMI classes.
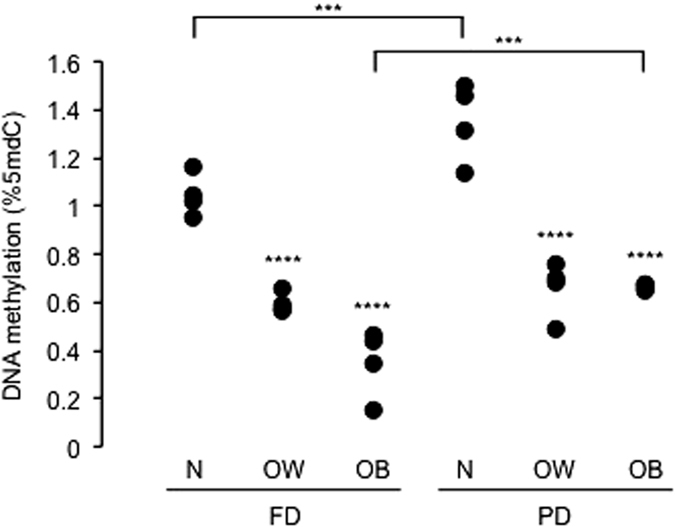
Datapoints are the four subjects’ averages of 5 repeated measurements in each condition. Asterisks above data points indicate the significance of the comparison with normal weight subjects within the FD or PD set. Asterisks above the horizontal lines indicate the significance of the comparison between FD and PD for the same BMI group. N, OW and OB indicate normal weight, overweight and obese subjects, respectively. Significance levels: ***p < 0.001; ****p < 10−4; ANOVA and Scheffé's test.
Although the correlation with total FAs was not significant, the above results suggested that specific components of the VLDL FA pool could be associated with global DNA methylation. To explore the potential interplay between FAs and DNA methylation, we examined the distribution of FAs across sample groups, according to the degree of FA saturation. In order to take the data variability into account, we analysed the complete data point set (n = 5 in each time point) for each subject in either FD or PD, i.e. n = 20 observations per BMI group. Normalized (% of total FAs) FA data were used. When plotted by global DNA methylation, a clear stratification of FA types could be observed despite the fact that data were not subject-averaged, which suggested specific associations with both global DNA methylation and BMI (Fig. 4). Both SFAs and MUFAs showed an inverse trend relative to DNA methylation, while the opposite was observed for PUFAs. Concomitantly, the data suggested an increase of SFAs and MUFAs, and a decrease of PUFAs, respectively, with BMI. PUFAs were clearly stratified by BMI, particularly in the PD set.
Figure 4. FA type distribution according to BMI and global DNA methylation in AMM.

Green, blue and red: normal weight, overweight and obese subjects. n = 20 datapoints for each BMI class are shown, corresponding to the 5 repeated measurements for each of the 4 subjects in each BMI class.
Based on the above data, we undertook a multiple linear regression analysis to assess the association of DNA methylation with normalized (% of total) FAs while adjusting for anthropometric and metabolic variables. The analysis was performed in subject-averaged values (n = 12 for either FD or PD). Global DNA methylation was significantly associated with PUFAs, but not SFAs or MUFAs, in PD and in FD after adjusting as indicated in the Methods section (Table 2). Multiple linear regression analysis within PUFAs revealed a significant positive association of global DNA methylation with EPA in both FD and PD. A significant although weaker positive association with AA was detected but only in PD. Global DNA methylation was inversely associated with BMI in FD and PD. An additional positive association with glucose was detected in PD. Generally, the multiple regression models, including FAs and metabolic variables, accounted for a higher portion of global DNA methylation variation (60–70%) in AMM compared to LI (compare Table 2 and Table 1).
Table 2. Significant associations with global DNA methylation (dependent variable) in the AMM subjects.
| Sample | Variable | Beta | t | p | adjusted R2 |
|---|---|---|---|---|---|
| PD | PUFAs | 0.0214 | 3.7356 | 0.0047 | 0.4815¶ |
| BMI | −0.1611 | −3.1200 | 0.0268 | 0.7437§ | |
| glucose | 0.0849 | 3.2093 | 0.0237 | ||
| C20:4 (AA) | 0.1118 | 4.2941 | 0.0013 | ||
| C20:5 (EPA) | 0.7000 | 6.1371 | 0.0001 | ||
| FD | PUFAs | 0.0146 | 2.5549 | 0.0309 | 0.4273¶ |
| BMI | −0.0862 | −2.9481 | 0.0163 | 0.6235§ | |
| C20:5 (EPA) | 1.1339 | 4.2449 | 0.0017 |
¶SFA/MUFA/PUFA model.
§Individual PUFA model. Beta values indicate the change in % 5mdC per one-point increase in FA (%), BMI or glucose (mg/dl). Non-significant associations are shown in Supplementary Table 2.4.
Akin to the LI cohort, we also tested the associations between FAs and age in AMM. A number of significant positive associations (which included both AA and EPA) were consistent in both FD and PD, but none overlapped with those identified in the LI cohort (Supplementary Table 7).
Associations with gene-specific DNA methylation
Next, we probed for any trend or associations between FAs and the histone deacetylase 4 (HDAC4) promoter or pyruvate dehydrogenase kinase 4 (PDK4) 5′UTR methylation across sampling time in the AMM subjects. These particular genes and genic regions were chosen based on previous evidence showing an inverse correlation between expression and the methylation state of these regions20,21,22 (Silva-Martínez et al., in press). Furthermore, HDAC4 and PDK4 show opposite trends in methylation and expression in obese individuals20,21,22 and PDK4 was the most up-regulated gene in response to the VLDL-rich lipoprotein-induced DNA hypermethylation in the human monocytic THP-1 cell line5.
For HDAC4, we analysed average methylation data for 2 CpGs located in the distal promoter region, one of which showed a ~40% decrease in methylation between 1 and 100 μM AA-stimulated human cultured THP-1 monocytes that was associated with an increase in expression (Silva-Martínez et al., in press). In the case of PDK4, we analysed average methylation data of five CpGs that mapped within the 5′UTR region that shows an inverse association with expression when methylated in both a CG and non-CG context (Supplementary Figure 1)21. Importantly, traditional bisulfite sequencing of this region in two AMM subjects in PD and FD (n = 20) demonstrated a concordance between the average of bisulfite-sequenced CpGs (n = 35) or non-CpG (n = 83) site methylation and the average methylation of the five pyrosequenced CpGs (Pearson’s r = 0.72, p = 3.4 × 10−3 and r = 0.51, p = 0.022, respectively), indicating that average methylation data of the latter is representative of the 5′UTR region.
The average methylation data of the 2 (HDAC4) and 5 (PDK4) CpGs sequenced were: 0.62 + 0.17% (FD) and 0.67 + 0.25% (PD) for HDAC4 and 2.70 + 0.69% (FD) and 3.63 + 2.87% (PD) for PDK4. Similarly to global DNA methylation, no significant trend with time could be observed for methylation in either gene (Fig. 2 middle and lower panels). However, with respect to variability, PDK4 showed the opposite pattern to global DNA methylation, i.e. a ~2-fold higher variability in the normal weight set, compared to overweight and obese (p < 0.001 in both FD and PD, Bartlett’s test). Gene-specific differences in methylation levels were also observed. HDAC4 methylation was lower in normal weight PD compared to overweight and obese (p = 0.0006 and p = 5.94 × 10−5, respectively; ANOVA followed by the Scheffé's post hoc test). The opposite trend, i.e. higher methylation in the normal weight set, was observed in FD for PDK4, although only compared to the obese set (p = 0.047). The paired differences between the same BMI class sample in FD and PD were not significant. The data therefore hinted at inverse and positive relationships of HDAC4 and PDK4 methylation, respectively, with PUFAs, given the relative enrichment of the latter in the normal weight set. Indeed, multiple regression analysis revealed that HDAC4 methylation in PD and PDK4 methylation in FD was inversely and positively associated with AA, respectively (Tables 3 and 4). PDK4 methylation showed a positive association with EPA in both FD and PD (Table 4). Similar to global DNA methylation, the association with EPA was stronger than with AA. The FA models accounted for 25–70% of the respective DNA methylation variation. Notably, glucose accounted for ~85% of HDAC4 methylation variation in FD. Examples of the methylation trend at the two loci in two representative samples with extreme AA levels are shown in Supplementary Figure 1. Despite the obvious stratification by BMI class (Fig. 2), only a marginal association of HDAC4 methylation with BMI was detected in our multiple regression model in PD (Supplementary Table 2.5).
Table 3. Significant associations with HDAC4 promoter methylation in the AMM subjects.
| Sample | Variable | Beta | t | p | adjusted R2 |
|---|---|---|---|---|---|
| PD | C20:4 (AA) | −0.0664 | −2.0042 | 0.0464 | 0.2524 |
| FD | glucose | 0.0397 | 4.7465 | 0.0177 | 0.8581 |
Beta values indicate the change in % 5mdC per one-point increase in percent FA. Non-significant associations are shown in Supplementary Table 2.5.
Table 4. Significant associations with PDK4 5′UTR methylation (dependent variable) in the AMM subjects.
| Sample | Variable | Beta | t | p | adjusted R2 |
|---|---|---|---|---|---|
| PD | C20:5 (EPA) | 4.1432 | 2.2777 | 0.0499 | 0.3140 |
| FD | PUFAs | 0.0373 | 2.7347 | 0.0291 | 0.6990¶ |
| C20:4 (AA) | 0.2411 | 2.9776 | 0.0155 | 0.5828§ | |
| C20:5 (EPA) | 1.6523 | 2.6578 | 0.0261 |
¶SFA/MUFA/PUFA model.
§Individual PUFA model. Beta values indicate the change in % 5mdC per one-point increase in percent FA. Non-significant associations are shown in Supplementary Table 2.8.
Associations between FAs, weight or BMI
In order to better understand the interplay between FAs, DNA methylation and the metabolism, we asked whether any specific associations of FAs with body weight existed in LI or AMM. In LI, three FAs were inversely associated with the weight at birth or the normalized weight gain (Supplementary Table 8). In all cases, the portion of weight variance accounted for by these associations was ~17% or less. In AMM, significant associations were observed for the SFA C16:0 (palmitic acid) and the MUFA C16:1 (palmitoleic acid) and for the SFA C18:0 (stearic acid) in FD (Supplementary Table 9). None of these FAs was significantly associated with global or gene-specific DNA methylation (Tables 1 and 2). In summary, associations with weight involved different FAs in LI, PD and FD, and the same FA (C16:0) showed inconsistent associations between LI and AMM.
Discussion
Although the majority of DNA methylation profiles which are established during early development, are faithfully inherited following replication, several evidences show that the DNA methylation state of specific genes or repetitive sequences is altered, albeit modestly, in response to metabolic changes such as exercise23,24,25, alterations in body weight following RYGB22,26,27,28 and dietary intervention13,14,29,30,31,32,33. Similarly, our data show that variation in DNA methylation between two different metabolic states, postprandial and fasting, occurs both in lean, overweight and obese individuals. As for post-prandial time-dependent global or gene-specific DNA methylation trends, our survey did not clearly show any consistently identifiable profile. Thus, the data do not support the occurrence of diet-driven, short-term intra-individual global DNA methylation oscillations in an 8-hour time window. Rather, the significant increase in global DNA methylation in the PD sets at least in the two extreme BMI groups compared to FD, suggests dietary effects that are subtle and diluted over time.
We detect an inverse association between BMI and global DNA methylation, which includes potential methylation changes in high copy number repetitive elements, such as LINE and Alu. Several studies have used the methylation state of such repeated elements to probe for associations between BMI and DNA methylation, but in the majority of cases no significant associations or trend, was uncovered (reviewed by van Dijk an colleagues34). Similarly, genome-wide obesity-relevant studies of DNA methylation using methylation arrays show either a net increase26 or decrease in methylation35,36,37,38 of specific genes in obese relative to lean individuals. While the underlying heterogeneity of these results is not understood, several factors such as gender and race/ethnicity39,40,41,42, folate and homocysteine levels41,43, aging and nutritional intake44 are all known to affect methylation. Notwithstanding these variables, our results show that EPA and AA are significantly associated with DNA methylation in two cohorts of distinct diet and post-natal developmental stage, independent of BMI and sex. The consistency and relevance of those associations are further evidenced by the fact that global DNA methylation was measured by two entirely different methods (the MethylFlash™ ELISA-based system and an HPLC-based technique) in different laboratories and sample types - i.e. whole blood in LI and PBMCs in AMM. Although we also uncovered associations of specific FAs with age and weight, none were consistent across cohorts or related to DNA methylation. Thus our data do not support the idea that specific fatty acids mediate age-related changes in DNA methylation.
Collectively, our data show that whole blood levels of selected PUFAs, which are lower in obese individuals, are important determinants of both global and gene-specific methylation profiles. To our knowledge this is a little explored topic, the only related report describing an inverse association between PUFAs and BMI in healthy obese individuals45. Interestingly, a recently published DNA methylome study in twins revealed that only twins discordant for both BMI and elevated liver fat differed significantly in leukocyte DNA methylation, with the heavier twin showing decreased levels of methylation in addition to lower levels of the PUFA linoleic acid46. These data point to an important association between liver lipid metabolism and blood methylation profiles. Indeed, the above discussed discrepancies between BMI and DNA methylation may in part be explained by the observations that elevated liver fat content, a characteristic of non alcoholic dependant fatty liver disease, is present in many, but not all obese individuals and disproportionately in Hispanics, predominantly of Mexican origin47.
The present analysis included two genes, HDAC4 and PDK4, both of which are known to be important regulators of glucose and fatty acid metabolism23,26,48. In particular, we found that the opposite associations of HDAC4 and PDK4 methylation with PUFAs are consistent with the previously published methylation and expression trend of these two genes with BMI20,22. However, we have recently shown that AA and the MUFA oleic acid have distinct, wide-spread, albeit small effects on a large number of genes in a monocytic THP-1 cell line, some of which are associated with changes in gene expression (Silva-Martínez et al., in press). We predict that some of the target genes identified in the above described study will show BMI-dependant changes in methylation and expression, although the effects may be subtle. In line with this view, DHA and EPA intake Yup’ik Alaskan natives, measured using red blood cell δ15N, identified several biologically relevant CpGs sites that showed an robust association between methylation and DHA and EPA intake, of which the majority showed increased levels of methylation49. Furthermore, dietary supplementation with n-3 PUFA in pregnant women is associated with a 1% increase in infant LINE-1 methylation and alterations in promoter levels of immune response genes IFNγ and IL1330. Similarly, n-3 PUFA or fish oil supplementation in adult humans also lead to modest alterations in promoter regions of genes involved in PUFA metabolism31 or genes previously known to be regulated by n-3 FA, the latter of which included PDK432.
The associations uncovered here may reflect a direct effect of FAs on the DNA methylome. In support of this view, our recent work identified PPAR-alpha and SIRT1 as mediators of AA-induced DNA hypermethylation in cultured monocytes (Silva-Martínez et al., in press). In addition, functional annotation analysis of the top 150 genes (ranked by the number of differentially methylated CpGs between the 100 and 1 μm AA concentrations/number of probes present in Illumina’s Infinium HumanMethylation450 BeadChip platform for a given gene) revealed a significant enrichment for the G protein-coupled receptor signalling pathway that are known to be activated by specific fatty acids50. On the other hand, a complex metabolic network links the availability of methyl group donors, lipid metabolism genes and FA synthesis, in which the hepatic metabolic activity plays a central role (reviewed by da Silva and colleagues)46,51,52. Interestingly, hyperhomocysteinaemia, an imbalance of the folate cycle that often results in DNA hypomethylation, is associated with low levels of AA in mice53. These studies suggest that the positive association between PUFAs and DNA methylation reflects a balanced cellular metabolism in which the correct methyl donor availability maintains physiological levels of DNA methylation and the correct expression of AA-generating enzymes such as FADS2. To our knowledge, no information is available on the opposite mechanism, i.e. a direct effect of FAs on methyl donor availability.
We also document a complex interplay between BMI, high-fat diet (PD set) and DNA methylation. On the one hand, global DNA methylation decreases with BMI in both PD and FD sets, yet an increase is observed in PD relative to FD. It is tempting to speculate that the data reflect opposite short-term and long-term effects of high fat diet. This view is supported by the documented induction of DNA hypermethylation by short-term high-fat diet in humans and mice, and by a 24- exposure of cultured cells to lipoproteins or selected FAs4,5,13,54 (and Silva-Martínez et al., in press) whereas a 6 year-long high-fat diet induced the opposite response in leukocytes in a primate model55. We speculate that the transient BMI-independent increase in DNA methylation observed in PD relative to FD reflects a genomic response to an acute increase in nutrient-activated pathways relevant to glucose and lipid metabolism such as the described pathways involving G-protein coupled signalling and PPARs. Oppositely, the reduced levels of DNA methylation characteristic of overweight and obese individuals may be related to structural changes in liver morphology and/or lipid metabolism following prolonged hyperlipidaemia.
Both global and gene-specific DNA methylation was more strongly associated with EPA than with AA. Yet, EPA circulating levels were 2–3-fold lower than other PUFAs such as docosahexaenoic acid in our samples, in accordance with previously reported data56, thus underlining EPA’s significant biological importance. EPA and AA are products of essential FA metabolism, therefore are potentially relevant to understand the epigenetic effects of dietary components in human health and disease. Both FAs are involved in inflammation resolution57. The n-3 PUFA EPA has been long regarded as a protective FA, particularly in the light of the favourable cardiometabolic effects of fish oil (see Kromhout and De Goede for a recent review58). A growing literature documents the regulation of specific signalling pathways and gene-specific methylation by EPA in vitro59,60. At chromatin level, EPA decreases the expression of polycomb group proteins that alter histone posttranslational histone modifications at specific loci61. As for AA, this PUFA has complex biological functions, ranging from the participation in inflammation resolution mentioned above, to being a precursor of inflammatory mediators. The impact of AA on cardiovascular health and other inflammation-related disorders has not yet been conclusively established62. We showed that AA induces global DNA hypermethylation in human THP-1 monocytes (Silva-Martínez et al., in press), in accordance with the positive association detected in the present study.
In summary, we demonstrate for the first time a strong association of DNA methylation with EPA and AA in two populations of different age and developmental stage. In addition, this study replicates in human subjects in vivo data previously obtained in cell lines, underlining the significance of the latter models to mechanistically link EPA and AA to DNA hypermethylation. Further work is needed to correct important caveat in this study, such as the relatively small sample size and missing information on potentially diet-dependent blood cell type-specific DNA methylation profiles.
Methods
LI cohort
Infants were recruited at the public paediatric centre “Secretaría de Salud UIMAPS San Felipe de Jesús”, in the city of León, state of Guanajuato, in central Mexico, which provides routine infant health checks. Inclusion criteria were exclusive breast-feeding or sporadic use of formula milk (reported in 4 out of 49 infants), absence of any mother’s or infant’s chronic or acute diseases, normal birth weight, uncomplicated pregnancy and delivery. The requirement for predominant breast-feeding aimed at increasing sample homogeneity. All participating mothers gave their informed consent to the procedure and the ethical committee of the hosting institution approved the protocol, which included a presentation of the main results of the study to the mothers and medical personnel. The methods were carried out in accordance with the approved guidelines. The LI cohort and AMM (see below) procedures were conducted in accordance with the WMA’s 2013 Declaration of Helsinki guidelines.
AMM group participants
Volunteers were recruited among the caretaker personnel of the University of Guanajuato in central Mexico and selected according to metabolic profiling prior to the project. The inclusion criteria were: male sex; age >25 years; normal fasting glucose and total cholesterol (TC) (<100 mg/dL and <250 mg/dL, respectively); normal blood pressure (BP; range: 90–139/60–89 mmHg). Subjects were grouped according to body mass index (BMI) in normal, overweight and obese (n = 4 in each group). The inclusion of males only was aimed at simplifying the statistical analysis and increasing the chances of detecting any effects in the light of the documented sexual dimorphism of obesity and cardiovascular risk63,64. All participants gave their informed consent to the procedure and the Ethical Committee of the Department of Medical Sciences, University of Guanajuato, approved the protocol. The methods were carried out in accordance with the approved guidelines.
Blood samples
In the LI cohort, 500 μL of arm venous blood were drawn in an EDTA-coated tube and immediately frozen. In the AMM group, ~10 mL peripheral blood were obtained in two different days apart by at least one week. On both days, the subject was asked to fast for 12 h and to abstain from exercise in the 48 h period prior to the first blood draw. On either day, venous blood was drawn 5 times at 2 h intervals starting at 10 AM. On one day (post-prandial day; PD data set), the subject consumed a single standard meal representative of the Western diet (one each of McDonald’s McChicken, medium-size fries, medium-size coke) immediately after the first blood draw at 10 AM. The 10 AM blood draw data are therefore considered as basal for that day. On the other day (fasting day; FD dataset), the subject maintained fasting throughout the blood draw period and was offered a small meal after the last (6 PM) blood draw. The rationale for including a complete fasting sample set was that it could allow accounting for circadian rhythm-related65 and blood cell type composition-related effects. Blood cell-type composition effects would be attenuated in intra-subject comparisons - i.e. between PD and FD - by the similar habits the subjects were required to adhere to in the 48 h previous to sampling. Blood cell-type composition may well contribute to inter-individual differences in FA profiles, global DNA methylation levels or other parameters, but correcting for that variable would mask genuine markers of prandial or metabolic state, whatever their underlying biological origin. Water was allowed ad libitum throughout the procedure in all cases. A schematic diagram of the procedure is shown in Fig. 1. Peripheral blood mononuclear cells (PBMCs) were obtained from ~5 mL EDTA-anticoagulated blood by centrifugation in Ficoll-Paque (GE Healthcare Life Sciences) according to the manufacturer’s recommendations. PBMCs and the remaining whole blood samples were stored at −80 °C in 200 μL aliquots until processed.
Blood lipid and FA determination
Standard enzymatic-colorimetric methods (Spinreact) were used to determine triglycerides (TG), high-density lipoproteins (HDL) and TC levels according to the manufacturer’s instructions. Low-density lipoproteins (LDL) and VLDL levels were determined using the Friedewald equation (LDL = TC-HDL-(TG/5 = VLDL)66.
Whole blood total (i.e. esterified and free) FAs were determined in LI and AMM with two different methods by two different research groups in different laboratories at two different time points. For AMM, 500 μL whole blood aliquots were frozen-lyophilized. One mL 0.5 M NaOH in methanol and 10 μL methyl-nonadecanoic acid (5 mg/mL) as an internal standard, were added and incubated at 90 °C for 1 h. After cooling to room temperature, 1 mL BF3 in methanol was added and incubated at 90 °C for 30 min and cooled again at room temperature. After transferring to a clean vial, 2 mL deionized water and 4 mL hexane were added, the organic phase was collected and the solvent evaporated with a stream of nitrogen. Dried sample were resuspended in 400 μL isooctane to be analysed by GC/IEMS. Samples were analysed in a Gas Chromatograph (Agilent Technologies 7890A) using a capillary column Zebron ZB-WAX (30 m × 250 μm × 0.25 μm) coupled to an Electron Impact Mass Spectrometer (Agilent Technologies 5975C). Oven temperature program was follows: initial temperature 50 °C during 3 min, then increasing at a rate of 10 °C/min to 250 °C and maintained for 20 min. Injector temperature of 220 °C was maintained constant. Helium 2 ml/min was use as carrier gas. Sample was injected with a 10:1 split. MSchem software was used to collect m/z data and NIST MS Search Software to compare the mass spectra and retention time vs. NIST 2011 database (National Institute of Standards and Technology Mass Spectra Database U.S. Department of Commerce) for the identification of the fatty acids methyl esters. For LI, FAs were determined as described above but using gas chromatography with flame ionization detector using a HP-88 capillary column (100 m × 0.25 mm, film 0.20 μm, Agilent Technologies, USA). Methyl tridecanoate was used as the internal standard, five point calibration was performed using a FAMEs standard mix containing esters of C8-C22 FAs (Supelco reference number 18920) with addition of AA. The identification of FAs in blood extracts was based on their retention times.
Global DNA methylation assays
Whole blood (0.2 mL) or PBMC DNA was extracted by using the DNeasy Blood & Tissue Kit (QIAGEN) according to the manufacturer’s instructions and was quantified using SYBR Green I (Sigma-Aldrich). Total 5-methyldeoxycytosine (5mdC) was determined with the MethylFlash™ Methylated DNA Quantification Kit, colorimetric (Epigentek) according to the manufacturer’s instructions or by a HPLC-based method67 for AMM or LI, respectively.
AA-stimulated human THP-1 monocytes
THP-1 cells were stimulated with 1, 10 or 100 μM BSA-conjugated AA for 24 h as described (Silva-Martínez et al., in press). Genome-wide methylation data were obtained with the Infinium HumanMethylation450 BeadChip platform (Illumina) and are publicly available in NCBI’s Gene Expression Omnibus68 with the accession number GSE67331.
Gene-specific DNA methylation and expression
DNA methylation in the 5′UTR of pyruvate dehydrogenase kinase, isozyme 4 (PDK4) and the histone deacetylase 4 (HDAC4) distal promoter was measured by pyrosequencing in bisulfite-treated DNA (EZ DNA Methylation™, Zymo Research), using the PyroMark Q96 ID (Qiagen) according to the manufacturer’s instructions. The following primers designed with the PyroMark Assay Design software were used for PDK4 pyrosequencing (5′ to 3′): GGTGGGAAGATTTGAATTTGAA and AAAACTACTCRAAACAAAACCTAATTCC (PCR primers; R indicates A or G); GGTATTTTTAAATTTTAGTTTAGG (sequencing primer). The corresponding primers for HDAC4 were: TGGGAGGTTTGTGTTGAGTT and CACCACCAAAAAAATAACCACTA (PCR primers); TGGTTGTTAGTAGGTG (sequencing primer). Supplementary Figure 1 schematically shows the sequenced loci and primer positions. For profiling the PDK4 5′UTR by traditional bisulfite-modified DNA sequencing, the primers AAATGTTTTATTTTTYGGGGTA (forward; Y indicates C or T) and AACTCCTCCTATTTAAAACT (reverse) were used. Fifteen-twenty clones were sequenced per sample. HDAC4 expression was assessed by reverse transcription-PCR with the primers GTGGGTTTCAACGTCAACATG and GTGACACGGGAAAGTTTCTTG.
Statistical analysis
Groups were compared by ANOVA followed by Scheffé’s post hoc test, while the Wilcoxon test was used in paired comparisons between experimental days. Correlations were measured with the Pearson’s r test. Associations between DNA methylation and FAs were assessed by best fit multiple regression analysis with global or gene-specific DNA methylation as dependent variable. In the case of the LI cohort, percent-normalized total FAs, sex, age at sampling, weight at birth, birth-to-sampling weight change/day (i.e. normalized weight gain) were the predictors. In the case of AMM, predictor variables were age, BMI, lipoproteins, TG, cholesterol, total FAs, percent-normalized individual FAs or grouped by degree of saturation and BP. Multiple regression analysis was then repeated with the individual members of the FA saturation class that was significantly associated in the first analysis and all other significantly associated variables as predictors. Dependent variables for all other multiple regression models are indicated in the Results section. Differences in variability (SD) between groups were tested by the Bartlett’s test. Tests were performed with the STATISTICA software (StatSoft) or with the StatPlus (AnalystSoft) MS Excel plug-in.
Additional Information
How to cite this article: de la Rocha, C. et al. Associations between whole peripheral blood fatty acids and DNA methylation in humans. Sci. Rep. 6, 25867; doi: 10.1038/srep25867 (2016).
Supplementary Material
Acknowledgments
We thank the mothers of the infants included in the study for their generosity and the Medical and Nurse staff at “Secretaría de Salud UIMAPS San Felipe de Jesús”, León, Gto., Mexico for their assistance, and the Clinical Laboratory of the Department of Medical Sciences, University of Guanajuato personnel for assistance in plasma lipoprotein and glucose determination. We also thank Victoria G. Reyes-Sánchez for assistance. This work was funded by the Council for Science and Technology of the State of Guanajuato (CONCyTEG), Mexico grant no. 08-03-K662-020-A01 to G.L., University of Guanajuato’s intramural research grant no. 114/08 to S.Z., Mexican Council for Science and Technology (CONACyT) “Ciencia Básica” grant no. 83401 to G.L. and by the CONACyT Sabbatical Fellowships no. 166209 to G.L. and no. 166058 to S.Z. CONACyT Studentships supported C.d.l.R., J.E.P.-M., S.Z.-D.L., B.C.-P. and F.E.T.-F.
Footnotes
Author Contributions C.d.l.R., J.E.P.-M. and S.Z.-D.L. performed the majority of blood sample gathering, molecular biology and data management/analysis work. J.E.P.-M. wrote the initial manuscript draft. B.C.-P. and F.E.T.-F. performed part of the molecular biology work. D.R.-R. and Y.A.-C. coordinated the laboratory work. J.M.-T., E.R.-C., K.W. and K.W. performed FA determination. K.W. and K.W. performed HPLC-based global DNA methylation analysis. F.J.C. and M.E. performed pyrosequencing. R.H.-R. contributed intellectually to the study design. S.Z. participated in designing the study and writing the manuscript, and performed part of the global DNA methylation analysis. G.L. designed and supervised the study and wrote the final manuscript version.
References
- Waqar A. B. et al. High-fat diet without excess calories induces metabolic disorders and enhances atherosclerosis in rabbits. Atherosclerosis 213, 148–55 (2010). [DOI] [PubMed] [Google Scholar]
- Tang F.-Y., Pai M.-H. & Chiang E.-P. I. Consumption of high-fat diet induces tumor progression and epithelial-mesenchymal transition of colorectal cancer in a mouse xenograft model. J. Nutr. Biochem. 23, 1302–13 (2012). [DOI] [PubMed] [Google Scholar]
- Zhang F. & Du G. Dysregulated lipid metabolism in cancer. World J. Biol. Chem. 3, 167–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund G. et al. DNA methylation polymorphisms precede any histological sign of atherosclerosis in mice lacking apolipoprotein E. J. Biol. Chem. 279, 29147–29154 (2004). [DOI] [PubMed] [Google Scholar]
- Rangel-Salazar R. et al. Human native lipoprotein-induced de novo DNA methylation is associated with repression of inflammatory genes in THP-1 macrophages. BMC Genomics 12, 582 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrès R. et al. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab. 10, 189–98 (2009). [DOI] [PubMed] [Google Scholar]
- Hall E. et al. Effects of palmitate on genome-wide mRNA expression and DNA methylation patterns in human pancreatic islets. BMC Med. 12, 103 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulis M. & Esteller M. DNA methylation and cancer. Adv. Genet. 70, 27–56 (2010). [DOI] [PubMed] [Google Scholar]
- Luttmer R. et al. Metabolic syndrome components are associated with DNA hypomethylation. Obes. Res. Clin. Pract. 7, e106–e115 (2013). [DOI] [PubMed] [Google Scholar]
- Zaina S. et al. DNA Methylation Dynamics in Human Carotid Plaques After Cerebrovascular Events. Arterioscler. Thromb. Vasc. Biol. 35, 1835–1842 (2015). [DOI] [PubMed] [Google Scholar]
- Zaina S. et al. DNA methylation map of human atherosclerosis. Circ. Cardiovasc. Genet. 7, 692–700 (2014). [DOI] [PubMed] [Google Scholar]
- Valencia-Morales M. D. P. et al. The DNA methylation drift of the atherosclerotic aorta increases with lesion progression. BMC Med. Genomics 8, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen S. C. et al. Effects of short-term high-fat overfeeding on genome-wide DNA methylation in the skeletal muscle of healthy young men. Diabetologia 55, 3341–9 (2012). [DOI] [PubMed] [Google Scholar]
- Brøns C. et al. Deoxyribonucleic Acid Methylation and Gene Expression of PPARGC1A in Human Muscle Is Influenced by High-Fat Overfeeding in a Birth-Weight-Dependent Manner. J. Clin. Endocrinol. Metab. 95, 3048–3056 (2010). [DOI] [PubMed] [Google Scholar]
- Neel J. Diabetes mellitus: a ‘thrifty’ genotype rendered detrimental by ‘progress’? Am. J. Hum. Genet. 14, 353–362 (1962). [PMC free article] [PubMed] [Google Scholar]
- Heyn H. et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. USA 109, 10522–10527 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A.-P. et al. D2HGDH regulates alpha-ketoglutarate levels and dioxygenase function by modulating IDH2. Nat. Commun. 6, 7768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervouet E. et al. The autophagy GABARAPL1 gene is epigenetically regulated in breast cancer models. BMC Cancer 15, 729 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose R. et al. Tet3 mediates stable glucocorticoid-induced alterations in DNA methylation and Dnmt3a/Dkk1 expression in neural progenitors. Cell Death Dis. 6, e1793 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Farha M. et al. Proteomics analysis of human obesity reveals the epigenetic factor HDAC4 as a potential target for obesity. Plos One 8, e75342 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni S. S. et al. Mitochondrial regulators of fatty acid metabolism reflect metabolic dysfunction in type 2 diabetes mellitus. Metabolism. 61, 175–185 (2012). [DOI] [PubMed] [Google Scholar]
- Kirchner H. et al. Altered promoter methylation of PDK4, IL1 B, IL6, and TNF after Roux-en Y gastric bypass. Surg. Obes. Relat. Dis. 10, 671–678 (2014). [DOI] [PubMed] [Google Scholar]
- Barrès R. et al. Acute Exercise Remodels Promoter Methylation in Human Skeletal Muscle. Cell Metab. 15, 405–411 (2012). [DOI] [PubMed] [Google Scholar]
- Rönn T. et al. A six months exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue. Plos Genet. 9, e1003572 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm M. E. et al. An integrative analysis reveals coordinated reprogramming of the epigenome and the transcriptome in human skeletal muscle after training. Epigenetics 9, 1557–1569 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres R. et al. Weight loss after gastric bypass surgery in human obesity remodels promoter methylation. Cell Rep. 3, 1020–1027 (2013). [DOI] [PubMed] [Google Scholar]
- Nilsson E. K. et al. Roux-en Y gastric bypass surgery induces genome-wide promoter-specific changes in DNA methylation in whole blood of obese patients. Plos One 10, e0115186 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton M. C. et al. An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss. Genome Biol. 16, 8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milagro F. I. et al. A dual epigenomic approach for the search of obesity biomarkers: DNA methylation in relation to diet-induced weight loss. FASEB J. 25, 1378–1389 (2011). [DOI] [PubMed] [Google Scholar]
- Lee H.-S. et al. Modulation of DNA methylation states and infant immune system by dietary supplementation with ω-3 PUFA during pregnancy in an intervention study. Am. J. Clin. Nutr. 98, 480–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoile S. P. et al. Supplementation with N-3 long-chain polyunsaturated fatty acids or olive oil in men and women with renal disease induces differential changes in the DNA methylation of FADS2 and ELOVL5 in peripheral blood mononuclear cells. Plos One 9, e109896 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- do Amaral C. L., Milagro F. I., Curi R. & Martínez J. A. DNA methylation pattern in overweight women under an energy-restricted diet supplemented with fish oil. Biomed Res. Int. 2014, 675021 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Cruzata L. et al. Dietary modifications, weight loss, and changes in metabolic markers affect global DNA methylation in Hispanic, African American, and Afro-Caribbean breast cancer survivors. J. Nutr. 145, 783–790 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk S. J., Molloy P. L., Varinli H., Morrison J. L. & Muhlhausler B. S. Epigenetics and human obesity. Int. J. Obes. (Lond). 39, 85–97 (2015). [DOI] [PubMed] [Google Scholar]
- Wang X. et al. Obesity related methylation changes in DNA of peripheral blood leukocytes. BMC Med. 8, 87 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almén M. S. et al. Genome wide analysis reveals association of a FTO gene variant with epigenetic changes. Genomics 99, 132–137 (2012). [DOI] [PubMed] [Google Scholar]
- Xu X. et al. A genome-wide methylation study on obesity: differential variability and differential methylation. Epigenetics 8, 522–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlman I. et al. The fat cell epigenetic signature in post-obese women is characterized by global hypomethylation and differential DNA methylation of adipogenesis genes. Int. J. Obes. (Lond) 39, 910–919 (2015). [DOI] [PubMed] [Google Scholar]
- Zhang F. F. et al. Significant differences in global genomic DNA methylation by gender and race/ethnicity in peripheral blood. Epigenetics 6, 623–629 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. et al. DNA methylation as a biomarker for cardiovascular disease risk. Plos One 5, e9692 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng W. et al. Dietary intake, plasma homocysteine, and repetitive element DNA methylation in the Multi-Ethnic Study of Atherosclerosis (MESA). Nutr. Metab. Cardiovasc. Dis. 24, 614–622 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Rocha J. L. et al. LINE-1 methylation is positively associated with healthier lifestyle but inversely related to body fat mass in healthy young individuals. Epigenetics 1–12 (2016) doi: 10.1080/15592294.2015.1135286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird J. K. et al. Obesity is associated with increased red blood cell folate despite lower dietary intakes and serum concentrations. J. Nutr. 145, 79–86 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes M. V. M. et al. Age-related changes in the global DNA methylation profile of leukocytes are linked to nutrition but are not associated with the MTHFR C677T genotype or to functional capacities. Plos One 7, e52570 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondanelli M. et al. Effects of two-months balanced diet in metabolically healthy obesity: lipid correlations with gender and BMI-related differences. Lipids Health Dis. 14, 139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollikainen M. et al. Genome-wide blood DNA methylation alterations at regulatory elements and heterochromatic regions in monozygotic twins discordant for obesity and liver fat. Clin. Epigenetics 7, 39 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.-J. & Fallon M. B. Gender and racial differences in nonalcoholic fatty liver disease. World J. Hepatol. 6, 274–283 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova M. M. et al. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 145, 607–621 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslibekyan S. et al. DNA methylation patterns are associated with n-3 fatty acid intake in Yup’ik people. J. Nutr. 144, 425–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukdar S., Olefsky J. M. & Osborn O. Targeting GPR120 and other fatty acid-sensing GPCRs ameliorates insulin resistance and inflammatory diseases. Trends Pharmacol. Sci. 32, 543–550 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva R. P., Kelly K. B., Al Rajabi A. & Jacobs R. L. Novel insights on interactions between folate and lipid metabolism. Biofactors 40, 277–283 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R. & Sanyal A. J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 10, 686–690 (2013). [DOI] [PubMed] [Google Scholar]
- Devlin A. M. et al. Hypermethylation of Fads2 and altered hepatic fatty acid and phospholipid metabolism in mice with hyperhomocysteinemia. J. Biol. Chem. 282, 37082–37090 (2007). [DOI] [PubMed] [Google Scholar]
- Yoo T. et al. Hypermethylation of repetitive DNA elements in livers of mice fed an atherogenic diet. Nutrition 28, 127–130 (2012). [DOI] [PubMed] [Google Scholar]
- Pheiffer C., Dias S., Muller C. & Louw J. Decreased global DNA methylation in the white blood cells of high fat diet fed vervet monkeys (Chlorocebus aethiops). J. Physiol. Biochem. 70, 725–33 (2014). [DOI] [PubMed] [Google Scholar]
- Sun Q. et al. Blood concentrations of individual long-chain n-3 fatty acids and risk of nonfatal myocardial infarction. Am. J. Clin. Nutr. 88, 216–223 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire M. O. & Van Dyke T. E. Natural resolution of inflammation. Periodontol. 2000 63, 149–164 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kromhout D. & de Goede J. Update on cardiometabolic health effects of ω-3 fatty acids. Curr. Opin. Lipidol. 25, 85–90 (2014). [DOI] [PubMed] [Google Scholar]
- Ceccarelli V. et al. Eicosapentaenoic acid demethylates a single CpG that mediates expression of tumor suppressor CCAAT/enhancer-binding protein delta in U937 leukemia cells. J. Biol. Chem. 286, 27092–27102 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli V. et al. Eicosapentaenoic acid activates RAS/ERK/C/EBPβ pathway through H-Ras intron 1 CpG island demethylation in U937 leukemia cells. Plos One 9, e85025 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri M., Bommi P. V., Sahasrabuddhe A. A., Khandekar J. D. & Dimri G. P. Dietary omega-3 polyunsaturated fatty acids suppress expression of EZH2 in breast cancer cells. Carcinogenesis 31, 489–495 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai M. et al. Arachidonic Acid and Cerebral Ischemia Risk: A Systematic Review of Observational Studies. Cerebrovasc. Dis. Extra 4, 198–211 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer B. F. & Clegg D. J. The sexual dimorphism of obesity. Mol. Cell. Endocrinol. 402, 113–119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedungadi T. P. & Clegg D. J. Sexual dimorphism in body fat distribution and risk for cardiovascular diseases. J. Cardiovasc. Transl. Res. 2, 321–327 (2009). [DOI] [PubMed] [Google Scholar]
- Bönsch D. et al. Daily variations of homocysteine concentration may influence methylation of DNA in normal healthy individuals. Chronobiol. Int. 24, 315–326 (2007). [DOI] [PubMed] [Google Scholar]
- Warnick G. R., Knopp R. H., Fitzpatrick V. & Branson L. Estimating low-density lipoprotein cholesterol by the Friedewald equation is adequate for classifying patients on the basis of nationally recommended cutpoints. Clin. Chem. 36, 15–9 (1990). [PubMed] [Google Scholar]
- Magaña A. A. et al. High-performance liquid chromatography determination of 5-methyl-2′-deoxycytidine, 2′-deoxycytidine, and other deoxynucleosides and nucleosides in DNA digests. Anal. Biochem. 374, 378–385 (2008). [DOI] [PubMed] [Google Scholar]
- Edgar R., Domrachev M. & Lash A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.