

Abstract
Phylogenetic inference artifacts can occur when sequence evolution deviates from assumptions made by the models used to analyze them. The combination of strong model assumption violations and highly heterogeneous lineage evolutionary rates can become problematic in phylogenetic inference, and lead to the well-described long-branch attraction (LBA) artifact. Here, we define an objective criterion for assessing lineage evolutionary rate heterogeneity among predefined lineages: the result of a likelihood ratio test between a model in which the lineages evolve at the same rate (homogeneous model) and a model in which different lineage rates are allowed (heterogeneous model). We implement this criterion in the algorithm Locus Specific Sequence Subsampling (LS³), aimed at reducing the effects of LBA in multi-gene datasets. For each gene, LS³ sequentially removes the fastest-evolving taxon of the ingroup and tests for lineage rate homogeneity until all lineages have uniform evolutionary rates. The sequences excluded from the homogeneously evolving taxon subset are flagged as potentially problematic. The software implementation provides the user with the possibility to remove the flagged sequences for generating a new concatenated alignment. We tested LS³ with simulations and two real datasets containing LBA artifacts: a nucleotide dataset regarding the position of Glires within mammals and an amino-acid dataset concerning the position of nematodes within bilaterians. The initially incorrect phylogenies were corrected in all cases upon removing data flagged by LS³.
Keywords: long branch attraction, phylogenomics, rate heterogeneity, artifacts, Glires, Nematoda.
Introduction
In recent years, new sequencing technologies have enabled phylogenetic studies to move from the analysis of single genes to the analysis of hundreds of genes, thus marking the advent of phylogenomics. The benefits derived from the production of large DNA sequence datasets include the reduction of stochastic errors and single-gene biases in phylogenetic inferences (Soltis et al. 2004; Philippe et al. 2005a). As a result, the analysis of large datasets is often considered valuable for the recovery of statistically well-supported and “true” phylogenies.
However, a number of studies have suggested that analyzing large datasets under optimal models of sequence evolution does not guarantee robust phylogenetic inferences (Ho and Jermiin 2004; Philippe et al. 2005b; Rodríguez-Ezpeleta et al. 2007; Salichos and Rokas 2013). In particular, the misleading effects of certain biases may increase along with the size of a dataset (e.g., Sullivan and Swofford 1997; Lartillot and Philippe 2004). One such bias is long-branch attraction (LBA), which was first described in the framework of maximum parsimony (MP) by Felsenstein (1978) and results in the clustering of taxa with high evolutionary rates (long branches) regardless of the phylogenetic relatedness. This incorrect arrangement occurs because converging characters in fast-evolving taxa are interpreted as synapomorphic (Felsenstein 1978; Bergsten 2005). This artifact has also been reported when probabilistic phylogenetic inference methods have been used, such as the maximum-likelihood (ML) method (Kück et al. 2012; Parks and Goldman 2014) and Bayesian inference method (Lartillot et al. 2007), although these methods tend to be less sensitive to LBA artifacts relative to nonprobabilistic methods such as MP or distance methods (distance measures based on probabilistic models can be used).
LBA falls within the class of systematic errors (Sullivan and Swofford 1997) that occur when models do not accurately describe the processes that generate the data. In phylogenetics, systematic errors may appear when the actual evolutionary process violates the sequence evolution assumptions made by the model when analyzing the data (Yang and Rannala 2012). Errors of this type are of major concern in phylogenomics because additional data with a consistent bias can exacerbate errors (Rodríguez-Ezpeleta et al. 2007) and obscure phylogenetic signals.
Several studies have successfully mitigated problems related to LBA by using more complex sequence evolution models and improving the model fit to the data, which reduces the model assumption violations. For example, Sullivan and Swofford (1997) recovered the monophyly of Rodentia by considering heterogeneous evolutionary rates among sites using a gamma distribution (Yang 1994). In addition, Lartillot et al. (2007) corrected the artifactual paraphyly of Ecdysozoa by analyzing their sequence data under a site-heterogeneous mixture model (Lartillot and Philippe 2004). Authors have resorted to optimizing datasets when LBA artifacts could not be further mitigated by improvements to the model. One method for optimizing a dataset is to partition the data using certain criteria and then analyze each partition independently. For example, data partitions have been analyzed according to site evolutionary rates to clarify conflicting signals related to fast-evolving sites (Brinkmann and Philippe 1999; Sperling et al. 2009). An alternative approach is to remove data that are deemed unfit for analysis, such as data from fast-evolving sites (e.g., Pisani 2004; Rodríguez-Ezpeleta et al. 2007; Goremykin et al. 2013), fast-evolving genes from multi-gene datasets (Brinkmann et al. 2005), and fast-evolving taxa (e.g., Aguinaldo et al. 1997; Stefanović et al. 2004).
However, only the latter approach specifically focuses on the heterogeneous rates of evolution across lineages, an important condition required for LBA to occur in a phylogenetic reconstruction. Eliminating fast-evolving taxa decreases the heterogeneity of evolutionary rates among the lineages in a dataset, which reduces the possibility of generating LBA artifacts during phylogenetic inferences. Nevertheless, data removal leads to missing data in the dataset, and the effects of these data gaps have been debated (e.g., Lemmon et al. 2009; Roure et al. 2013). In the studies by Aguinaldo et al. (1997) and Stefanović et al. (2004), the deleted taxa were selected subjectively. To systematically and reproducibly remove sequence information from multi-gene and phylogenomic datasets, objective criteria are required to indicate whether removing particular taxa will decrease LBA artifacts. In addition, these criteria must provide information on whether removing a taxon was effective in reducing the heterogeneity of lineage evolutionary rates.
Here, we present one such criterion that was employed in a sequence data exploration algorithm that we developed called “Locus Specific Sequence Subsampling” (LS³), which aims to reduce LBA artifacts in phylogenetic inferences. This algorithm extends and refines the approach proposed by Brinkmann et al. (2005) and automatically identifies, for any given gene, a subset of taxon sequences with homogeneous evolutionary rates and flags as potentially problematic the sequences with excessively high rates of evolution. In a multi-gene context, identifying these taxon sequences is performed independently for each gene, taking into account gene-specific evolutionary patterns. With this information, the user can decide to remove the sequences flagged by LS³ and analyze a subset of the dataset in which lineage rate homogeneity has been enforced. The criterion used in LS³ derives from a likelihood ratio test (LRT) and hence can be applied to both nucleotide and amino-acid datasets.
To assess whether the new LS3 method effectively identifies data that can lead to LBA, we tested the LS3 method with three types of data: (i) simulated data of nucleotides; (ii) a dataset of biological nucleotide sequences; and (iii) a dataset of biological amino acid sequences. In the first case, we simulated gene evolution leading to LBA by introducing highly heterogeneous rates of evolution among lineages. Upon analyzing 10,000 such datasets, the LS³ method effectively flagged all of the introduced fast-evolving taxon sequences in virtually all the alignments. Next, we tested the effectiveness of LS³ in two biological multi-gene datasets with well-documented LBA artifacts. The first biological case we addressed was a nucleotide dataset that we assembled with the aim of reproducing the artifactual paraphyly of Glires (rodents and lagomorphs), in the tree of placental mammals as inferred by D’Erchia et al. (1996) and corrected by Sullivan and Swofford (1997). The second biological case we analyzed was the amino-acid dataset from Philippe et al. (2005b) leading to the incorrect paraphyly of Ecdysozoa (Aguinaldo et al. 1997; Philippe et al. 2005b). This same dataset was used by Lartillot et al. (2007) to demonstrate the efficiency of their CAT site-heterogeneous mixture model of protein evolution for correcting LBA artifact. In our study, both real cases initially led to a wrong phylogeny with high bootstrap supports, resulting from LBA artifacts. On both cases we were able to recover the correct phylogeny upon removing the sequence information flagged by LS³ as potentially problematic.
New Approaches
Assessing the Heterogeneity of Evolutionary Rates among Lineages
Taxa with long branches are a result of ancient divergence times (with poor taxon sampling), fast evolutionary rates, or a combination of both (Felsenstein 2004). The relative contribution of these two factors can be evaluated using ad hoc information, such as well-established phylogenetic relationships and/or well-documented time calibration points, which can help account for different evolutionary rates among lineages. However, a number of phylogenetic questions cannot be resolved because information on these factors is unavailable, which increases the difficulty of determining whether the inferred phylogenies suffer from LBA artifacts. Our approach aims to address whether errors of phylogenetic inference are caused by unequal evolutionary rates among lineages.
To explain the LS3 procedure, we will consider three monophyletic lineages (“ingroup lineages”) with unknown interrelationships and one or more distantly related lineages as outgroups (fig. 1). The ingroup lineages contain species that evolved at different evolutionary rates, and the mean lineage evolutionary rates differ among the three lineages. To test whether differences in the evolutionary rates among the three lineages are significant, we perform an LRT (Felsenstein 1981) between two models: (1) a model that assumes a single evolutionary rate for all three ingroup lineages (“homogeneous model”) and (2) a model that allows each ingroup lineage to have its own evolutionary rate (“heterogeneous model”). If the P-value resulting from the LRT is >0.05, then the homogeneous model cannot be rejected because the difference in evolutionary rates among lineages is small. However, if the P-value resulting from the LRT is ≤0.05, then the heterogeneous model provides a significantly improved explanation of the data relative to the homogeneous model and the hypothesis that all ingroup lineages have a homogeneous rate of evolution must be rejected.
Fig. 1.
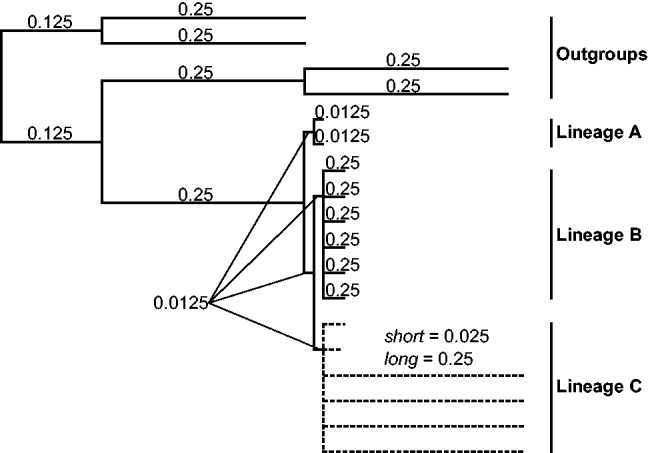
Guide tree used to produce the simulated gene evolution datasets. Two long-branch assemblages (the outgroups and the Lineage C) are placed on both extremes of the tree. Fifty different cases of this general topology were considered, and they differed according to the taxa in Lineage C to which the long and short branches were assigned. These 50 cases represented all possible combinations of two to four fast-evolving taxa in the Lineage C, with all other branch lengths equal. For each of the 50 cases, 200 simulations were performed, and a total of 10,000 trees were produced. Branch length units are substitutions per site.
By using an LRT and its associated P-value as criteria to evaluate differences in evolutionary rates among ingroup lineages, the lineage rate homogeneity of virtually any dataset can be assessed. This can be applied to automatically test several subsets of taxa of the same gene to identify a subset in which the evolutionary rates among lineages are not significantly different. Once such a subset is identified, the sequences that disrupted the lineage rate homogeneity are flagged as potentially problematic.
LS³: Locus Specific Sequence Subsampling Algorithm
The LS³ algorithm employs the LRT to compare the homogeneous versus the heterogeneous models of lineage evolutionary rate to remove the fast-evolving sequences in successive subsamples of taxa in a gene. The likelihood associated with the sequence data under each model is calculated based on a rooted tree given by the user in which the three ingroup lineages of interest are each monophyletic, but all emerge from a single basal polytomy (step 1 in fig. 2). The polytomy in the input tree for the likelihood calculation reduces the possible misleading effect that may result from using a wrong branching order in the LRT analysis, as this interrelationship represents the question to be solved. Then, LS³ performs the following steps: (i) This algorithm employs the LRT (as explained in the previous section) to determine whether the homogeneous model can be accepted or rejected for a particular gene (step 3 in fig. 2). (ii) If rejected, the sequence displaying the longest sum of branch lengths beginning from the polytomic node is removed (step 5 in fig. 2). (iii) Using the reduced dataset, a new LRT is performed (return arrow in fig. 2). If the homogeneous model is retained, the algorithm stops. Otherwise, a new sequence is removed according to step (ii), and the process is iterated until the homogeneous model is not rejected in a subset of taxa. As the taxon sequence subsampling progresses, the input tree required for the LRT is modified according to the taxon sequence sample, and its branch lengths are optimized again via ML methods.
Fig. 2.
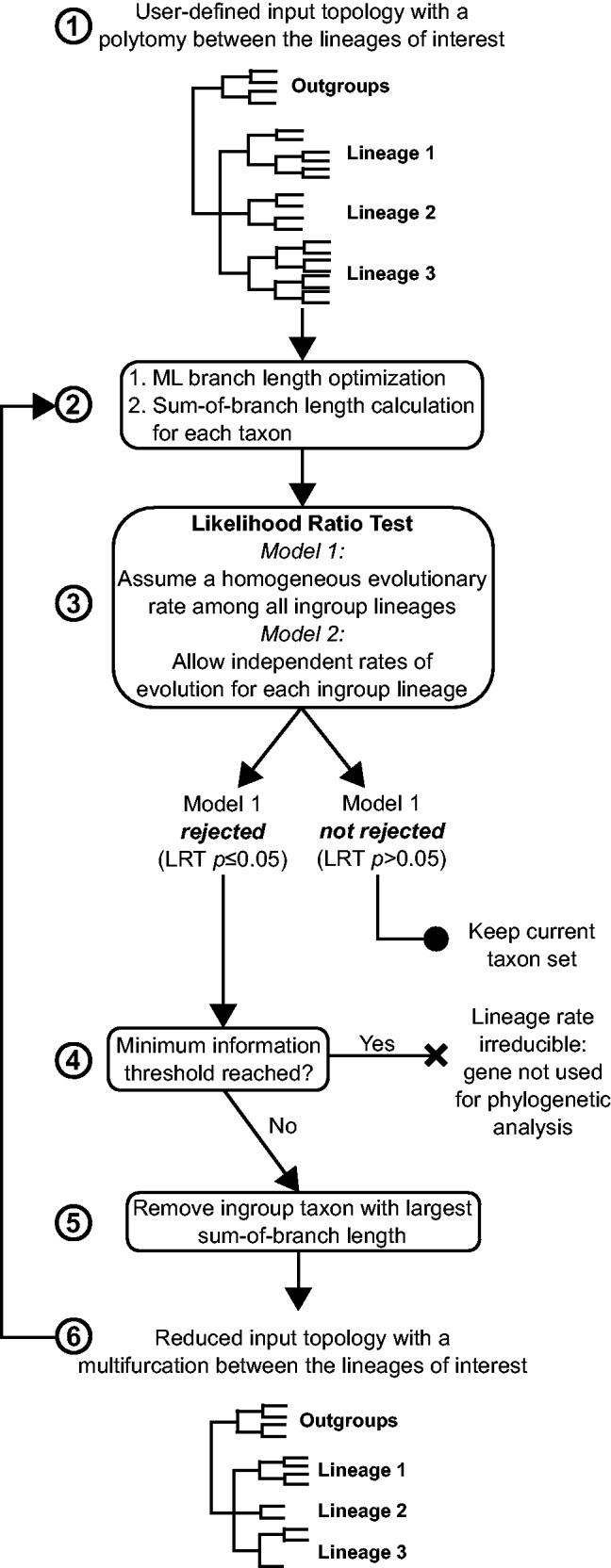
Flow chart representation of the LS³ algorithm. The input tree with the gene sequence alignment passes through a ML branch optimization step. The input tree contains a multifurcation that consists of the three lineages of interest (Lineage 1, Lineage 2, and Lineage 3). The sum of branch lengths for each ingroup taxon is calculated based on this tree (steps 1 and 2). Next, an LRT is performed between a model that assumes homogeneous evolutionary rates among ingroup lineages and a model that allows for a local rate of evolution for each of the three lineages (step 3). If the homogeneous evolutionary rate model is accepted for a set of taxon sequences, these sequences are maintained for further analysis. If the homogeneous evolutionary rate model is rejected, the fastest-evolving sequence is flagged and removed and the LRT test is repeated (steps 4–6). A minimum taxon sequence threshold is defined, and after reaching this threshold, sequences cannot be removed (in this study, this threshold is defined as only two species remaining per lineage, step 5). If a gene dataset reaches this threshold and the homogeneous model is rejected, the gene is flagged as unfit for phylogenetic analysis and can be removed by the user.
The removal of the fast evolving taxa progresses until a user-defined stopping point is reached after which no more sequences can be removed from a gene because it would render the dataset inadequate for phylogenetic analysis. We suggest it to be when each lineage is represented by only two taxon sequences. If homogeneous lineage rates are never attained on a given gene by the time it reaches this stopping point, the entire gene is flagged as unfit for analysis due to its persistent lineage rate heterogeneity.
After the LS³ algorithm is applied to each gene of a multi-gene or phylogenomic dataset, an additional script produces a table for each gene showing the sequences flagged as potentially problematic by the LS³ method. Because sequences are flagged according to their branch lengths relative to the other lineages, paralogous or markedly misaligned sequences will also be flagged by the LS³ algorithm. If the user decides to explore a multi-gene dataset without flagged sequences and genes (i.e., enforcing gene-by-gene lineage rate homogeneity), we include a script that generates these datasets. Eliminating the flagged data of potential problematic sequences increases the overall quality of the dataset taking into account the specific biases and mode of evolution of each gene. A fast-evolving taxon sequence that was flagged in one gene may not be flagged in another gene because the taxa in the latter dataset may have evolved at a more homogeneous rate. Therefore, taxa that were problematic in certain genes may still be placed in the phylogeny even after removing the sequences flagged by the LS³ algorithm. This approach contrasts with the more common practice of completely removing taxa from the entire dataset when bias is suspected.
Results
Validation of LS3 through Simulations
To assess the LS3 method when the true phylogeny is known, we applied the LS3 algorithm to simulated datasets that produce LBA artifacts. We simulated the evolution of 2,000 bp genes with INDELible v1.03 (Fletcher and Yang 2009) using an input tree designed to generate LBA artifacts. The input tree had markedly divergent (long-branched) outgroups and three ingroup lineages. One of the two ingroup lineages furthest from the outgroups contained long terminal branches (fig. 1). This setting facilitated the generation of an artifactual nonphylogenetic signal that incorrectly located the fast-evolving crown lineage closer to the root of the tree toward the highly divergent outgroups. To include variation in the datasets, we produced 50 different arrangements of long-branched taxa in the fast-evolving lineage (Lineage C in fig. 1), representing all of the possible combinations of two to four fast-evolving taxa in the six taxon fast-evolving lineage (supplementary fig. S1, Supplementary Material online). The final dataset was composed of 200 simulated genes for each of the 50 long-branch arrangement cases, thus totaling 10,000 genes.
For 9,775 genes (97.75%), the LS3 method successfully flagged as potentially problematic all of the fast-evolving taxon sequences that were simulated in the fast-evolving lineage. The remaining 225 genes (2.25%) were flagged as unfit for analysis because with only two species remaining per ingroup lineage, significant evolutionary rate heterogeneity remained among the lineages. In 277 of the cases in which all of the fast-evolving sequences were correctly flagged (2.83% of the 9,775), additional taxa that were not simulated as fast-evolving were removed because they contributed significantly to evolutionary rate heterogeneity among the lineages, which was likely caused by case-specific variations in the simulation processes.
We constructed ML trees for each of the 10,000 simulated genes to examine whether the simulated long branches had an effect on the phylogenetic inferences. Although all of the inferences were conducted under the same model and model parameters used to simulate the data, 6,705 (67.05%) datasets produced an incorrect LBA-affected topology. A total of 3,211 (32.11%) datasets produced the correct topology, and the remaining 84 (0.84%) datasets produced a variety of incorrect arrangements. Consistent with our rationale, when the taxon sequences flagged by the LS³ method were removed from these datasets, 8,446 inferences produced the correct tree (86.40% of the 9,775). The remaining 1,329 cases (13.60% of the 9,775) produced a variety of incorrect arrangements presumably due to the lack of phylogenetic signal to solve the short internal branches. This suspicion was confirmed by analyzing these remaining 1,329 genes concatenated (total alignment size of 2,658,000 bp), which produced a correct phylogeny with full bootstrap supports for all correct relationships (data not shown).
To examine the effect of introducing fast-evolving sequences within a multi-gene context, we produced 100 multi-gene alignments that were each composed of 100 simulated genes randomly chosen from the pool of 10,000. The ML trees for these 200,000 bp multi-gene alignments showed that the bias introduced by the fast-evolving species was sufficient to generate LBA-affected phylogenies. We tested whether removing the sequences and genes flagged by the LS3 method would result in more robust inferences and found that misleading information was reduced and the correct phylogenetic signal was recovered. When analyzing the full simulated data, 59 out of the 100 multi-gene datasets (59%) resulted in the correct topology; however, the bootstrap support for the correct grouping of lineages B and C was low ( = 68.17% ± 13.22). The remaining 41 multi-gene datasets produced incorrect topologies in which the fast-evolving Lineage C was incorrectly displaced toward the outgroups (albeit with little support for the resulting incorrect grouping of lineages A and B; = 35.22% ± 14.42), thus indicating that concatenating these genes did not entirely correct their initial bias. However, when the sequence data flagged by the LS³ method was removed, all 100 multi-gene datasets generated the correct topology. Moreover, the bootstrap support for the correct placement of the fast-evolving lineage was 100% in all datasets, thus reflecting a systematic recovery of the phylogenetic signal. The mean percent of missing data in the datasets in which the LS³-flagged data had been removed remained relatively low ( = 17.0% ± 0.50 for the full datasets, = 50.3% ± 1.38 for the fast-evolving lineage).
Based on these assays of simulated data we confirmed that the LS3 method could efficiently and automatically flag potentially problematic fast-evolving sequences that disrupt lineage evolutionary rate homogeneity in a gene-specific manner. We further observed that the exclusion of LS³-flagged data resulted in a reduction of misleading signals and a corresponding increase of the true phylogenetic signal.
Biological Datasets—Position of Glires within Placental Mammals
We further assessed the efficiency of the LS³ method by analyzing two actual cases of evolutionary relationships that were once problematic to solve because of LBA but for which a reasonable solution is currently available. The first case addresses the historically problematic recognition of Glires (grouping rodents, rabbits, and hares) as a monophyletic lineage and the challenging position of this group within the mammalian tree (currently accepted to be the sister group of Primates, which together form the clade Euarchontoglires). In early molecular phylogenies, Glires was recovered as a paraphyletic assemblage (e.g., D’Erchia et al. 1996); however, this relationship was subsequently explained by systematic errors and LBA artifacts due to several fast-evolving Glires species (e.g., Sullivan and Swofford 1997; Goremykin et al. 2010).
We assembled a multi-gene dataset with high amounts of LBA artifacts by selecting, from all mammalian single-copy orthologous genes (n = 679) as provided in OrthoDB (Waterhouse et al. 2013), the genes that individually led to the nonmonophyly of Glires and the inaccurate placement of its members within the mammalian tree. We excluded representatives from both Afrotheria and Xenarthra to avoid unnecessary controversial results because the positioning of these groups is still debated (Nikolaev et al. 2007; Song et al. 2012; Akanni et al. 2014; Moran et al. 2015). The subselection of LBA-affected genes resulted in 57 genes that amounted to a total of 135,394 bp (list of gene accession numbers are in supplementary table S1, Supplementary Material online). Concatenating these 57 genes produced a dataset referred to as the “only LBA” (OL) dataset, which was expected to produce strong LBA biases. Indeed, the resulting ML inference produced an incorrect tree, with Lagomorpha (rabbits and hares) diverging first within the analyzed placentals, followed by Rodentia (hence rendering Glires paraphyletic) and the incorrect grouping of Laurasiatheria sister to Primates. The incorrect grouping of laurasiatherians and primates had 93% bootstrap support, and the incorrect grouping of laurasiatherians, primates, and rodents had 92% bootstrap support (topology in fig. 3A).
Fig. 3.

ML phylogenies and missing data plots obtained with (A) the unprocessed OL + 2 dataset, and (B) the OLLS³+2 dataset, for which data flagged by LS³ has been removed. Bootstrap support values <100 are presented in black, and support values for the same node in the ML trees obtained with the OL and OLLS³ dataset, which had the same topological arrangement of the three lineages of interest, are presented in red. Primates are colored green, Laurasiatherians are colored red, and Glires are colored dark blue. When using the unprocessed OL + 2 dataset (A), the Euarchontoglires group (Primates and Glires) is not recovered as monophyletic and Glires is recovered as paraphyletic. Upon removing the LS³-flagged sequences and genes from the dataset, the correct topology is recovered (B). Note that in (B), the bootstrap support from the OLLS³ dataset (in red) is much lower compared with that of the OLLS³+2 dataset (corresponding number in black). The missing data plots show the percent of total missing data by species (orange) and the percent of genes in which the species was not represented in the dataset (green). Asterisks (*) mark species that were eliminated from the OLLS³ dataset because they disrupted the lineage rate homogeneity in all genes. The scientific names of the species are as follows: Gibbon = Nomascus leucogenys; Human = Homo sapiens; Macaque = Macaca mulatta; Marmoset = Callithrix jacchus; Mouse lemur = Microcebus murinus; Cow = Bos taurus; Pig = Sus scrofa; Dog = Canis lupus familiaris; Microbat = Myotis lucifugus; Horse = Equus caballus; Chinese hamster = Cricetulus griseus; Mouse = Mus musculus; Kangaroo rat = Dipodomys ordii; Squirrel = Ictidomys tridecemlineatus; Guinea pig = Cavia porcellus; Naked mole-rat = Heterocephalus glaber; Pika = Ochotona princeps; Rabbit = Oryctolagus cuniculus; Opossum = Monodelphis domestica; and Platypus = Ornithorhynchus anatinus.
We examined this multi-gene dataset using the LS3 method and considered Laurasiatheria, Glires and Primates to be three ingroup lineages with unknown relationships. The LS³ analysis flagged as potentially problematic 44 genes out of the initial 57 genes (77% of genes, an expected result considering the high amount of bias contained in this dataset) and 81 sequences contained within the remaining 13 genes (35%; supplementary table S2, Supplementary Material online). We removed the LS³-flagged data to produce the OLLS³ dataset, which contained a total of 28,230 bp and had 30% missing data. In addition, three species, pika (Ochotona princeps), mouse (Mus musculus), and Chinese hamster (Cricetulus griseus), produced consistent nonconformity with the lineage rate homogeneity; thus, their data were removed from all 57 genes. Compared with the OL dataset (fig. 3, and supplementary fig. S2A and B, Supplementary Material online), the reduced but clean OLLS³ dataset generated an ML inference that successfully recovered the correct topology (fig. 3, and supplementary fig. S2A and B, Supplementary Material online). However, the bootstrap support for the correct groupings was low (57% for the monophyly of Glires and 55% for the monophyly of Euarchontoglires, fig. 3B). Excluding the aforementioned three species from the unprocessed OL dataset was not sufficient to correct the topology (data not shown).
We hypothesized that the phylogenetic signal contained in the reduced OLLS³ dataset was most likely too low to produce high bootstrap supports; therefore, we tested whether adding two genes that produced the correct species tree would increase the bootstrap support. The LS3 analysis indicated that the first gene did not include any flagged taxa, whereas the second gene required the removal of a single taxon to reach a homogeneous rate of evolution across the ingroup lineages. The two additional LS³-processed genes (totaling 3,114 bp) were added to the OLLS³ dataset, thus producing the OLLS³ + 2 dataset (supplementary table S3, Supplementary Material online). With this slightly extended dataset, the correct phylogeny was recovered with high bootstrap support: 90% for the monophyly of Glires and 89% for the monophyly of Euarchontoglires (fig. 3B). However, when the same two genes were added to the unprocessed OL dataset to form the OL + 2 dataset, the topology remained incorrect and had high support values (fig. 3A). Similar results were obtained when adding other gene pairs that initially produced the correct species tree (data not shown).
Biological Datasets—Position of Nematoda within Bilateria
To test the performance of the LS³ method with amino-acid data, we used a second biological case study on the placement of the fast-evolving Nematoda within the animal tree of life using the protein dataset published by Philippe et al. (2005b). This dataset consists of 146 proteins (alignment length: 35,371 aa positions) and includes sequences from nematodes, arthropods, and deuterostomes as well as a large set of sequences belonging to the distant Fungi outgroup. This dataset contains a strong signal that incorrectly displaces the fast-evolving Nematoda toward the base of the bilaterian animals and the Fungi outgroup, thus impeding the monophyletic recovery of Ecdysozoa (formed by nematodes and arthropods). The full dataset resulted in an ML phylogeny that incorrectly placed Nematoda as the first diverging lineage of the bilaterian ingroup and sister to a clade formed by Deuterostomia and Arthropoda (the “coelomates” clade) with 76% bootstrap support (fig. 4A). We processed the data with the LS3 method using Deuterostomia, Arthropoda and Nematoda as the three ingroup lineages with unknown interrelationships. A subsample of taxon sequences with homogeneous evolutionary rates was found in 98 out of the initial 146 proteins (67%), whereas the remaining 61 genes were flagged as potentially misleading because of their persistent lineage rate heterogeneity (supplementary table S4, Supplementary Material online). Removing the flagged sequences and genes produced an alignment of 23,011 aa positions and 32% missing data. Compared with the full dataset, this filtered dataset resulted in a correct ML phylogeny and generated the monophyly of Ecdysozoa with 73% bootstrap support (fig. 4B).
Fig. 4.

ML phylogenies and missing data plots obtained for the 35,371 amino acid alignments from Philippe et al. (2005b) before (A) and after (B) removing the sequences and genes flagged by the LS³ algorithm. Nematoda is colored in red, Deuterostomia is in blue, and Arthropoda is in yellow. When the full dataset is analyzed, Ecdysozoa (arthropods and nematodes) is not recovered, and an incorrect “coelomate” clade is produced with substantial bootstrap support (support value in red). However, the dataset without the LS³-flagged data recovers Ecdysozoa with relatively high bootstrap support (support value in green). The missing data plots show the percent of total missing data by species (orange) and the percent of genes in which the species was not represented in the dataset (green). Only bootstrap support values <100 are shown.
Discussion
Currently, no automated data filtering process is available to mitigate LBA artifacts by directly reducing the evolutionary rate heterogeneity among taxa. In this study, we showed that an LRT can be employed to assess lineage evolutionary rate heterogeneity in different taxon subsets of a gene to recover lineages with homogeneous evolutionary rates. This was accomplished by comparing the likelihood scores of a model in which all lineages are assumed to evolve at the same rate with the scores of a model that allows for local lineage rates. We then presented a taxon sequence subselection algorithm (LS³) developed by our group that uses this test to identify in any given gene a subset of taxon sequences that evolved at a homogeneous rate. We hypothesized that when LBA-related errors are suspected, flagging potentially problematic sequences and genes in multi-gene datasets through the LS³ algorithm and subsequently removing the flagged data would result in a corrected phylogenetic tree. By testing the LS3 method on simulated and biological datasets that produced incorrect topologies due to LBA, we successfully recovered the correct topologies in all cases when the sequence data that had been flagged by the LS³ algorithm was excluded.
As our knowledge of molecular evolutionary processes increases, more realistic models of sequence evolution that can provide more robust data analyses will be developed. However, the analysis of data with highly heterogeneous evolutionary rates among taxa can still lead to artifactual phylogenies even under the best available models of sequence evolution. Probabilistic phylogenetic methods have been shown to be relatively robust to artifacts despite high heterogeneity in rates of evolution across taxa (Bergsten 2005); however, it is not possible to tell when this robustness fails. Consequently, subselecting the data that will reduce the bias in phylogeney construction is a safe strategy. Data subselection has been suggested by Philippe et al. (2005a) in order to increase the phylogenetic to nonphylogenetic signal ratio. More recently, Salichos and Rokas (2013) vouched for the subselection of genes with high amounts of signal for phylogenomic analyses and for the use of conditional combination approaches, instead of “total evidence” approaches. Moreover, the amount of sequence data available nowadays is high, and this amount is increasing as sequencing technologies improve and their costs decrease. This enables the use of data exploration techniques such as LS³ to identify subsamples of a larger dataset that can be better interpreted, with a high chance that enough phylogenetic signal will finally remain to recover the “true” tree even after the removal of the potentially problematic information.
Nevertheless, excluding data raises concerns because of the fraction of valuable phylogenetic information this excluded data may contain. In this study, the amount of excluded data in the analysis of the monophyly and position of Glires within the mammalian tree does not reflect a real situation because the dataset was assembled with the intention of inflating LBA artifacts. However, the amino-acid alignment from Philippe et al. (2005b) was not assembled for that purpose and reflects a realistic situation. The LS³ method identified 98 out of 146 (67%) proteins as suitable for the final analysis. In the concatenated protein dataset in which the LS³-flagged sequences and genes were removed, missing data represented 32% of the matrix, whereas missing data represented 10% of the original dataset. Thus, the final quantity of excluded data was 38% of the genes and 22% of the sequences in the remaining genes (missing data plots in fig. 4, supplementary table S5, Supplementary Material online). These exclusions were not detrimental to the question addressed with this dataset; however, the effects of missing data on phylogenetic inferences are disputed (e.g., Lemmon et al. 2009; Roure et al. 2013).
Because of its efficiency at detecting and flagging fast-evolving sequences in a dataset, the LS³ method may also be effective for automatically identifying paralogous, misaligned, and mislabeled sequences, which can help to increase the general quality of multi-gene datasets. This expectation is based on the fact that such problematic sequences will appear as long branches within the predetermined ingroup lineages and thus be detected by the LS³ method. We tested this conjecture using an alignment from Zhou et al. (2010) that included a gene duplication deep in the eukaryote tree and intentionally changed the actual orthologous sequence using a paralogous copy in one species and then in two species. In both tests, the LS³ algorithm successfully flagged the paralogous sequences (data not shown).
LS3 Limitations
One limitation of the LS³ method as currently implemented is that the three ingroup lineages, whose interrelationships remain to be resolved, must be defined a priori. The number of ingroup lineages to be compared can be increased without issue; however, the monophyly of each of the compared lineages must be known. In addition, the guide tree provided by the user for the LRT step must also be rooted, otherwise the lineage evolutionary rate calculations will lack an evolutionary context. We believe that the majority of the current and future cases of dubious relationships in phylogenetics can be reduced to the positioning of a lineage relative to two other lineages plus an outgroup with three branching alternatives (a “three-taxon” situation). For example, if a phylogenetic tree has several questionable (or unexpected) lineage positions, the problematic areas in the tree can be analyzed by first solving a subtree that reduces the problem to the three-taxon situation. If a given problematic area of a tree contains more than three competing branching alternatives due to, for instance, the presence of four or more lineages whose interrelationships are debated, four or more lineages can be considered under the LS3 algorithm without any issues. However, considering additional lineages may result in less precise assessments because more internal branches collapse when forcing the multifurcation at the base of the ingroup lineages during the LS3 procedure.
A possible outcome of removing LS³-flagged data is that, if a given species evolves faster than the rest in a consistent manner across all genes, and is flagged on all gene datasets, it will be absent from the final phylogenetic tree. In this case, the result of removing LS³-flagged sequences would resemble—for the particular species—the outcome of the classical taxon removal approach, in which a taxon is removed from the entire multi-gene dataset (as in Stefanović et al. 2004). However, the rate of evolution of protein-coding genes in a genome is known to have a spread distribution (e.g., Montoya-Burgos 2011). Thus, with greater numbers of loci available, then the probability that there will be genes in which this species displays an average rate of evolution is higher, enabling it to be placed in the final phylogeny. However, we recommend removing LS³-flagged data in cases where the main question to be resolved is the positioning of a multi-species lineage within the phylogeny. In this case, for an entire lineage to be absent in the final dataset, then all of the members should be flagged for all of the genes. This situation becomes less probable as the number of genes increases.
In this study, reducing our datasets to sequences that evolved at homogeneous rates resulted in the inference of correct ML trees and in the reduction of LBA artifacts. Their identification and flagging enable the exploration of the signal in the dataset by comparing the phylogeny obtained with and without the flagged sequences, which can help uncover the true evolutionary signal. As sequence data production accelerates, we must develop in parallel well-grounded, fast and automated data assessment methods able to identify potentially misleading signal in order to take informed decisions about the phylogenetic information contained in the data. In this way, we will hopefully be able to solve the most elusive splits in the tree of life.
Materials and Methods
Automatic Implementation of LS³
We implemented the LS³ algorithm in a pipeline (available at http://genev.unige.ch/en/users/Juan-Montoya/unit, last accessed March 1, 2016) with the following dependencies: PAML v4.6 (Yang 2007) and R (R Core Team 2014), with the R packages “ape” (Paradis et al. 2004) and “adephylo” (Jombart and Dray 2010). The input for the LS³ pipeline is an initial alignment in PHYLIP format, a tree including all taxa with a multifurcation between the three ingroup lineages of interest; and a table that assigns each taxon either to the ingroup lineage they belong to (“Clade1,” “Clade2,” or “Clade3”) or to the outgroups (“Ogs”). Next, the LS³ algorithm is run until the user-defined minimal taxa threshold is reached (for this study, the threshold was when only two species remained per declared ingroup lineage). At each iteration, output files with general information and all of the values of the LRT (relative rates of evolution, log-likelihood values, P-value, etc.) are produced. In certain cases, PAML may not converge systematically to the best parameter estimations; therefore, we ran the LS³ algorithm several times on each gene until the minimal taxa threshold was reached. Once the data exploration step was finished, and the correct parameter estimations were confirmed, another script (LS3_tabs.sh) developed by our group summarized the data into a table that plotted all of the taxon sequences for all of the unflagged genes and categorized them as flagged (“0”), unflagged (“1”), or not present (“NA”). Should the user decide to remove the flagged taxon sequences, we developed a script (serial_trimmer.sh) to select the first LS³ iteration for each gene that reached lineage rate homogeneity (showing an LRT P-value ≥0.05) and output an alignment with the corresponding taxon sequences subset. These subsets can then be analyzed gene-by-gene and/or concatenated to construct a multi-gene phylogeny based on the datasets without the LS³-flagged data. We did not include this last script embedded in the LS³ pipeline because we consider running the LS³ subsampling steps several times on the same dataset important to verify the consistency among the results of the analysis with PAML. Note that our scripts run the LS³ algorithm until the minimum taxa threshold is reached and do not have a stopping point defined by the LRT P-value of 0.05. This feature permits the exploration of gene dataset subsamples that produced LRT P-values higher than the classical 0.05 threshold. In these cases, evolutionary rate heterogeneity among lineages is expected to be even lower because the null hypothesis of a single evolutionary rate among all ingroup lineages will be more easily rejected.
Production and Analysis of the Simulated Datasets
The gene evolution simulations were performed using INDELible v1.03 (Fletcher and Yang 2009). Each simulated gene was 2,000-bp long, and the taxonomic samples included 14 ingroup taxa organized into three ingroup lineages: Lineage A (two taxa), Lineage B (six taxa), and Lineage C (six taxa) and four outgroup taxa, with the following interrelationships (outgroup(A,(B,C))) (fig. 1). To introduce variation to the simulation process, we produced 50 guide trees, and they each differed in the number and identity of fast-evolving taxa present in the fast-evolving Lineage C (supplementary fig. S1, Supplementary Material online). These 50 cases represent all possible combinations of two to four fast-evolving taxa in Lineage C. To produce the dataset with 10,000 simulated genes, we performed 200 simulations for each of the 50 guide trees under the Jukes–Cantor (JC) model of sequence evolution, including eight gamma rate categories (α = 1.0) and a 0.3 proportion of invariable sites (I). The simple JC model was selected to produce simulated data of low complexity to reduce the number of misleading signals. Gene phylogenies were obtained for each of the 10,000 simulated genes using PhyML v3.0 (Guindon and Gascuel 2003) using the same model and parameter values as in the simulation.
We screened all 10,000 resulting topologies to quantify the number of correct and incorrect topologies before and after eliminating the LS³-flagged sequences. A topology was deemed to be correct if the bifurcation (B,C),(A,outgroup) was present, conversely, a topology was considered incorrect if the bifurcation (A,B),(C,outgroup) was present. To assess the impact of the flagged sequences in a multi-gene phylogeny context, we concatenated 100 sets of 100 simulated genes selected at random from the 10,000 simulated genes and analyzed each dataset before and after removing the LS³-flagged sequences. ML analyses were performed with PhyML using the same model and parameter values as in the simulation as well as 200 bootstrap replicates to assess the statistical support for the correct and incorrect bipartitions. All of the simulated datasets are available online at http://genev.unige.ch/en/users/Juan-Montoya/unit, last accessed March 1, 2016.
Production of the Biological Datasets
The mammalian multi-gene dataset was constructed by extracting all of the single-copy 1:1 orthologous genes from mammalian species available in OrthoDB (Waterhouse et al. 2013). We then used a reduced taxa set that included platypus (Ornithorhynchus anatinus) and opossum (Monodelphis domestica) as outgroups, five Laurasiatheria species, eight Glires species, and five Primates species (fig. 3). We then automatically aligned all of the genes with TranslatorX (Abascal et al. 2010) using default parameters, and ambiguously aligned fragments were removed with Gblocks (Talavera and Castresana 2007) using default parameters with the following exceptions: b3 = 10, b4 = 5, and b5 = a. Next, we performed ML inferences for every gene alignment >800 bp in size. For the OL dataset, we only selected genes leading to an ML tree that incorrectly placed Primates as the sister group to Laurasiatheria. Thus, all cases in which Glires were incorrectly placed toward the outgroup (as predicted by LBA) were selected. The two genes added to produce the OLLS³+2 dataset were selected by applying the LS³ algorithm to all of the genes that had initially resulted in a correct tree and choosing the two genes for which the least number of taxa were flagged, thus indicating an overall low lineage rate heterogeneity. Other genes that had initially resulted in a correct tree were also tested for the production of alternative OLLS³+2 datasets. All of the aforementioned datasets are available upon request.
The amino-acid dataset for the bilaterian case (Philippe et al. 2005b) was received directly from the authors.
Phylogenetic Analyses of the Biological Datasets
For the biological cases, all of the phylogenetic analyses of single-gene nucleotide data were performed with RAxML 7.4.8 (Stamatakis 2006) using the GTR + G+I model of evolution with four gamma rate categories and ten independent inferences. The concatenated nucleotide datasets were analyzed with RAxML using the GTR + G+I model of sequence evolution with four gamma rate categories and 100 independent inferences. RAxML was used to analyze the amino acid multi-gene datasets using the WAG + G+F model, which is the same model used by Lartillot et al. (2007), with four gamma rate categories and 100 independent inferences. Both the nucleotide and amino-acid concatenated datasets were partitioned according to genes, thus enabling independent estimations of the parameters of the evolutionary model for each gene. Node support values were assessed by analyzing 1,000 bootstrap replicates using the same RAxML parameters applied to infer the phylogeny.
Supplementary Material
Supplementary figures S1 and S2, and tables S1–S5 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors are very grateful for the constructive comments and feedback received from Nicolas Galtier, Manolo Gouy, Michel Milinkovitch, Raymond Moran, Davide Pisani, Omar Rota-Stabelli, and Ziheng Yang. We also acknowledge Hervé Philippe and Xiaofan Zhou for kindly providing us with their datasets. This study was supported by the Swiss National Science Foundation (Grant number 31003A_141233 to J.I.M.B.), the Geneva Canton, the Kinesis Foundation of Puerto Rico, the Excellence Master Fellowship of the University of Geneva, the iGE3 PhD Salary Award, and the Donation Claraz of Switzerland.
References
- Abascal F, Zardoya R, Telford MJ. 2010. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38:W7–W13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguinaldo A, Turbeville J, Linford L, Rivera MC, Garey JR, Raff RA, Lake JA. 1997. Evidence for a clade of nematodes, arthropods and other moulting animals. Nature 387:489–493. [DOI] [PubMed] [Google Scholar]
- Akanni WA, Creevey CJ, Wilkinson M, Pisani D. 2014. L.U.St: a tool for approximated maximum likelihood supertree reconstruction. BMC Bioinformatics 15:183.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsten J. 2005. A review of long-branch attraction. Cladistics 21:163–193. [DOI] [PubMed] [Google Scholar]
- Brinkmann H, Philippe H. 1999. Archaea sister group of Bacteria? Indications from tree reconstruction artifacts in ancient phylogenies. Mol Biol Evol. 16:817–825. [DOI] [PubMed] [Google Scholar]
- Brinkmann H, van der Giezen M, Zhou Y, Poncelin de Raucourt G, Philippe H. 2005. An empirical assessment of long-branch attraction artefacts in deep eukaryotic phylogenomics. Syst Biol. 54:743–757. [DOI] [PubMed] [Google Scholar]
- D’Erchia A, Gissi C, Pesole G, Saccone C, Arnason U. 1996. The guinea-pig is not a rodent. Nature 381:597–600. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 1978. Cases in which parsimony or compatibility methods will be positively misleading. Syst Zool. 27:401–410. [Google Scholar]
- Felsenstein J. 1981. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 17:368–376. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 2004. Inferring phylogenies. Sunderland (MA): Sinauer Associates, Inc. [Google Scholar]
- Fletcher W, Yang Z. 2009. INDELible: a flexible simulator of biological sequence evolution. Mol Biol Evol. 26:1879–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goremykin VV, Nikiforova SV, Biggs PJ, Zhong B, Delange P, Martin W, Woetzel S, Atherton R, McLenachan P, Lockhart PJ. 2013. The evolutionary root of flowering plants. Syst Biol. 62:50–61. [DOI] [PubMed] [Google Scholar]
- Goremykin VV, Nikiforova SV, Bininda-Emonds ORP. 2010. Automated removal of noisy data in phylogenomic analyses. J Mol Evol. 71:319–331. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 52(5): 696–704. [DOI] [PubMed] [Google Scholar]
- Ho S, Jermiin L. 2004. Tracing the decay of the historical signal in biological sequence data. Syst Biol. 53(49): 623–637. [DOI] [PubMed] [Google Scholar]
- Jombart T, Dray S. 2010. Adephylo: new tools for investigating the phylogenetic signal in biological traits. Bioinformatics 26(15): 1907–1909. [DOI] [PubMed] [Google Scholar]
- Kück P, Mayer C, Wägele J-W, Misof B. 2012. Long branch effects distort maximum likelihood phylogenies in simulations despite selection of the correct model. PLoS One 7:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartillot N, Brinkmann H, Philippe H. 2007. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol Biol. 7 (1 Suppl):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartillot N, Philippe H. 2004. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol. 21:1095–1109. [DOI] [PubMed] [Google Scholar]
- Lemmon AR, Brown JM, Stanger-Hall K, Lemmon EM. 2009. The effect of ambiguous data on phylogenetic estimates obtained by maximum likelihood and Bayesian inference. Syst Biol. 58:130–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya-Burgos JI. 2011. Patterns of positive selection and neutral evolution in the protein-coding genes of Tetraodon and Takifugu. PloS One 6(9): e24800.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran R, Morgan C, O’Connell M. 2015. A guide to phylogenetic reconstruction using heterogeneous models—a case study from the root of the placental mammal tree. Computation 3:177–196. [Google Scholar]
- Nikolaev S, Montoya-Burgos JI, Margulies EH, Rougemont J, Nyffeler B, Antonarakis SE. 2007. Early history of mammals is elucidated with the ENCODE multiple species sequencing data. PLoS Genet. 3:e2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290. [DOI] [PubMed] [Google Scholar]
- Parks S, Goldman N. 2014. Maximum likelihood inference of small trees in the presence of long branches. Syst Biol. 63(5): 798–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe H, Delsuc F, Brinkmann H, Lartillot N. 2005a. Phylogenomics. Annu Rev Ecol Evol Syst. 36:541–562. [Google Scholar]
- Philippe H, Lartillot N, Brinkmann H. 2005b. Multigene analyses of bilaterian animals corroborate the monophyly of Ecdysozoa, Lophotrochozoa, and Protostomia. Mol Biol Evol. 22:1246–1253. [DOI] [PubMed] [Google Scholar]
- Philippe H, Zhou Y, Brinkmann H, Rodrigue N, Delsuc F. 2005c. Heterotachy and long-branch attraction in phylogenetics. BMC Evol Biol. 5:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani D. 2004. Identifying and removing fast-evolving sites using compatibility analysis: an example from the Arthropoda. Syst Biol. 53:978–989. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2014. R: a language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing. [Google Scholar]
- Rodríguez-Ezpeleta N, Brinkmann H, Roure B, Lartillot N, Lang BF, Philippe H. 2007. Detecting and overcoming systematic errors in genome-scale phylogenies. Syst Biol. 56:389–399. [DOI] [PubMed] [Google Scholar]
- Roure B, Baurain D, Philippe H. 2013. Impact of missing data on phylogenies inferred from empirical phylogenomic data sets. Mol Biol Evol. 30:197–214. [DOI] [PubMed] [Google Scholar]
- Salichos L, Rokas A. 2013. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 497:327–331. [DOI] [PubMed] [Google Scholar]
- Soltis DE, Albert V, Savolainen V, Hilu K, Qiu Y-L, Chase MW, Farris JS, Stefanović S, Rice DW, Palmer JD, et al. 2004. Genome-scale data, angiosperm relationships, and “ending incongruence”: a cautionary tale in phylogenetics. Trends Plant Sci. 9:477–483. [DOI] [PubMed] [Google Scholar]
- Song S, Liu L, Edwards SV, Wu S. 2012. Resolving conflict in eutherian mammal phylogeny using phylogenomics and the multispecies coalescent model. Proc Natl Acad Sci U S A. 109:14942–14947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling EA, Peterson K, Pisani D. 2009. Phylogenetic-signal dissection of nuclear housekeeping genes supports the paraphyly of sponges and the monophyly of eumetazoa. Mol Biol Evol. 26(10):2261–2274. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. [DOI] [PubMed] [Google Scholar]
- Stefanović S, Rice DW, Palmer JD. 2004. Long branch attraction, taxon sampling, and the earliest angiosperms: amborella or monocots? BMC Evol Biol. 4:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan J, Swofford D. 1997. Are guinea pigs rodents? The importance of adequate models in molecular phylogenetics. J Mamm Evol. 4:77–86. [Google Scholar]
- Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 56:564–577. [DOI] [PubMed] [Google Scholar]
- Waterhouse RM, Tegenfeldt F, Li J, Zdobnov EM, Kriventseva EV. 2013. OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 41:D358–D365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 1994. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol. 39:306–314. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24(8): 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang Z, Rannala B. 2012. Molecular phylogenetics: principles and practice. Nat Rev Genet. 13:303–314. [DOI] [PubMed] [Google Scholar]
- Zhou X, Lin Z, Ma H. 2010. Phylogenetic detection of numerous gene duplications shared by animals, fungi and plants. Genome Biol. 11:R38.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.