

Abstract

Fundamental research on nanoparticle (NP) interactions and development of next-generation biomedical NP applications relies on synthesis of monodisperse, functional, core–shell nanoparticles free of residual dispersants with truly homogeneous and controlled physical properties. Still, synthesis and purification of e.g. such superparamagnetic iron oxide NPs remain a challenge. Comparing the success of different methods is marred by the sensitivity of analysis methods to the purity of the product. We synthesize monodisperse, oleic acid (OA)-capped, Fe3O4 NPs in the superparamagnetic size range (3–10 nm). Ligand exchange of OA for poly(ethylene glycol) (PEG) was performed with the PEG irreversibly grafted to the NP surface by a nitrodopamine (NDA) anchor. Four different methods were investigated to remove excess ligands and residual OA: membrane centrifugation, dialysis, size exclusion chromatography, and precipitation combined with magnetic decantation. Infrared spectroscopy and thermogravimetric analysis were used to determine the purity of samples after each purification step. Importantly, only magnetic decantation yielded pure NPs at high yields with sufficient grafting density for biomedical applications (∼1 NDA-PEG(5 kDa)/nm2, irrespective of size). The purified NPs withstand challenging tests such as temperature cycling in serum and long-term storage in biological buffers. Dynamic light scattering, transmission electron microscopy, and small-angle X-ray scattering show stability over at least 4 months also in serum. The successful synthesis and purification route is compatible with any conceivable functionalization for biomedical or biomaterial applications of PEGylated Fe3O4 NPs.
Introduction
Superparamagnetic iron oxide nanoparticles (NPs), with core diameters of 3–15 nm, are used in a rapidly expanding number of applications in the biomedical field; the most common include cell labeling,1 hyperthermia,2 drug delivery,3 and as contrast agents for magnetic resonance imaging.4 For these applications, the iron oxide cores are coated with polymer, lipid, or other dispersants to enable dispersion of NPs in aqueous solutions containing biomolecules. Rapid aggregation and precipitation occur without a sterically stabilizing shell. The most common method to stabilize iron oxide NPs for biomedical applications has been to enwrap them in a weakly adsorbed shell of high molecular weight polymer (often dextran)5 or amphiphiles such as block copolymers or lipids.6 However, weakly adsorbed shells lead to low polymer densities on the particle surface that are further reduced with time under dilute conditions, resulting in low colloidal stability. Chemically grafted polymer shells are physically and chemically stable; they can also be made denser than physisorbed shells, and therefore they have received increasing attention.7
With recent improvements in the synthesis of NPs8 there has been a push toward more well-defined, core–shell NP architectures. Spherical iron oxide NPs can now be synthesized with monodisperse size (SD < 5%), but they are as-synthesized capped with a strongly adsorbed, hydrophobic shell of oleate that is difficult to replace;9 these NPs require stabilization with a shell of linear, end-grafted polymer dispersants of sufficient thickness and grafting density to ensure that the particles remain monodisperse during application. Defined core–shell architectures enable the prediction of all colloidal properties and can be used to define biological interactions through the attachment of organic ligands. The main advantage of grafting dispersants to the core is that the hydrodynamic size of the NPs, the stability of the shell, and the presentation of ligands can be precisely controlled, in contrast to for NPs with shells consisting of physisorbed high molecular weight dispersants; this critically determines NP performance in a biological fluid.10 Precise control over monodisperse size, purity, and colloidal interactions is crucial for NPs in biomedical applications to control biodistribution and self-assembly of drug delivery structures.7a
Achieving high dispersant grafting density on monodisperse iron oxide nanoparticles is challenging since the dispersant has to densely replace the (oleic acid) organic shell carried by the as-synthesized particles. In addition, a major problem for the synthesis and characterization of core–shell nanoparticles is the efficient separation of excess dispersants.7a,9a The PEG-dispersant density on nanoparticles is difficult to determine since continuous and complete removal of excess dispersants from nanoparticle dispersions can lead to particle aggregation and precipitation; alternatively, not all free ligands are removed, which leads to overestimation of the dispersant density on the nanoparticles. The actual stability and correct core–shell structure cannot be determined if excess dispersants are not removed. Efficient separation methods can also contribute to nanoparticle degradation and should be identified and avoided.
An irreversibly grafted shell of >2 chains/nm2 of PEG(5 kDa) on polydisperse and irregularly shaped single NPs was shown to be required for colloidal stability under application conditions.7a NP curvature facilitates polymer grafting by grafting-to of presynthesized polymer coils and chain grafting densities up to 2.5 chains/nm2 have been claimed on iron oxide NPs.11 Until recently, only ∼0.5 chains/nm2 of PEG(5 kDa) was reported grafted on spherical Fe3O4 NPs after ligand replacement by nitro-dihydrohyphenylalanine (nitro-DOPA)-PEG(5 kDa) on monodisperse nanoparticles synthesized using oleylamine as capping agent.12 Lak and colleagues recently synthesized large, 25 nm in diameter, Fe3O4 ferrimagnetic nanoparticles that were stabilized through ligand replacement with nitrodopamine (NDA)-PEG.13 Due to their size, these multidomain particles are optimized for applications where Brownian relaxation mechanisms are desired, while particles with weaker magnetic interactions and Néel relaxation as the dominant relaxation mechanism require iron oxide NPs smaller than ∼15 nm. These large particles form small aggregates in cell culture and PBS, which might be due to the stronger long-range core–core interactions that have to be screened by the grafted shell. In another recent study Davis et al. quantitatively addressed the problem of replacing oleic acid by ligand exchange on superparamagnetic iron oxide nanoparticles.14 Full ligand replacement was not shown, although the superiority of DOPA and nitro-DOPA anchor groups to displace oleic acid from the surface was demonstrated. These and other previous studies did not address the effect of purification from excess ligands on the results. The resultant colloidal stability of the core–shell nanoparticles under relevant conditions such as biological buffers and elevated temperature was also not investigated.
We present protocols to replace oleic acid for a dense spherical brush PEG-shell on monodisperse, monocrystalline, and spherical iron oxide nanoparticles in the superparamagnetic size range of 3–10 nm diameter at physiological temperatures. The colloidal stability in both phosphate buffered saline (PBS) and biofluids of these densely PEGylated core–shell particles significantly exceeds what has previously been described for particles synthesized by ligand exchange. Importantly, we also present an evaluation of different purification methods to separate PEGylated particles from free PEG, and we suggest a protocol for iterated solvent precipitation and magnetic extraction as most suitable to completely purify these nanoparticles at high yield. Only through this advancement is the high grafting density and colloidal stability conclusively demonstrated, with important implications for correct comparison of previous and future results on NP ligand density.
Materials and Methods
Materials
All chemicals (see Supporting Information for details) were purchased from Sigma-Aldrich, all solvents were from Roth, and PEG was from Jenkem and used as received without further purification. All NPs and nitrocatechol ligands originated from the same respective batch to ensure maximum reproducibility.
Methods
Transmission Electron Microscopy (TEM) and Analysis
TEM studies were performed on a FEI Tecnai G2 20 transmission electron microscope operating at 120 or 200 kV for high resolution imaging. Samples were prepared by dropping toluene dispersions (as-synthesized NPs) or aqueous dispersions (PEGylated NPs) onto a 300-mesh carbon-coated copper grid and subsequently evaporating the solvent in air. Size distributions were evaluated using the Pebbles15 software package with a local intensity-model fitting algorithm. Approximately 900 NPs were sampled for each size determination made by Pebbles.
Thermogravimetric Analysis (TGA) and Differential Scanning Calorimetry (DSC) Measurements
Thermograms were recorded on a Mettler-Toledo TGA/DSC 1 STAR system in the temperature range 25–650 °C with a ramp of 10 K/min in synthetic air stream of 80 mL/s in order to ensure complete combustion of ligands as NDA was found to polymerize by pyrolization under N2. 70 μL aluminum oxide crucibles were filled with 0.5–1.5 mg of sample, and the total organic content (TOC) was evaluated as the mass loss fraction at 500 °C by horizontal setting.
1H and 13C Nuclear Magnetic Resonance (NMR) Measurements
1H and 13C solution spectra were collected on a Bruker DPX operating at 300 MHz in CDCl3 and d6-DMSO as solvents using TMS as an internal standard.
Attenuated Total Reflection–Fourier Transform Infrared (ATR-FTIR) Measurements
Mid-IR powder spectra of the lyophilized samples were collected on a single reflection Bruker Tensor 37 FTIR spectrometer with Bruker Platinum Diamond single-reflection ATR equipment at a resolution of 4 cm–1 by averaging 32 scans.
Small-Angle X-ray Scattering (SAXS)
Samples were measured in type 0500 glass capillaries (Hilgenberg, Germany) with nominal diameter of 1 mm and wall thickness of 10 μm. Measurements were carried out using a Rigaku S-Max 3000 SAXS system equipped with a copper-target microfocus X-ray tube MicroMax-002+ (45 kV, 0.88 mA) with an energy of 8.05 keV, collimated through three pinholes (400, 200, and 700 μm) to achieve a beam diameter at the sample position of 210 μm (fwhm) and a Triton 200 2D multi wire gas-filled X-ray detector (200 mm diameter of active area, spatial resolution 200 μm). Data were acquired in the q-range from 0.01 to 0.26 Å–1 with a measurement time of 28.800 s for each scattering pattern at vacuum conditions better than 10–2 mbar. Subsequent data treatment included background correction based on the measured transmission and radial integration with the SaxsGui 2.8.03 software package.
Syntheses and Purification
Synthesis of Oleic Acid-Capped Iron Oxide Cores
Superparamagnetic oleic acid (OA)-stabilized magnetite nanoparticles were synthesized by thermal decomposition of an iron precursor according to a slightly modified heat-up procedure described by Hyeon et al.16 Briefly, for 9.6 nm NPs a mixture of 50 mL of dioctyl ether (Oct2O) and 7.04 mL of OA was heated to 100 °C under N2. 1 mL of iron pentacarbonyl (Fe(CO)5) was injected rapidly, and the reaction mixture was heated to 290 °C with a temperature ramp of 3 °C/min. After aging for 1 h the NP dispersion was allowed to cool to room temperature and precipitated thrice with ethanol (EtOH) from toluene in order to remove excess OA. The size was controlled by the Fe(CO)5:OA ratio; details can be found in the Supporting Information (Table S1). Four different core sizes were selected for the grafting and purification investigations.
Synthesis of Nitrocatechol Ligands
6-Nitrodopamine hemisulfate (NDA-HSO4) was synthesized according to literature with slight modifications.17 NDA-PEG(5 kDa) was synthesized by (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylaminomorpholinocarbenium hexafluorophosphate (COMU)-mediated peptide-coupling reactions9a,18 (see Supporting Information for experimental details).
Ligand Replacement
Typically, 1.3 g of wet (from EtOH washing) NPs (containing ∼0.11 g cores) were dispersed in 30 mL of DMF; the desired amount of NDA-PEG(5 kDa), usually a 10-fold excess (with respect to the grafting density of 1 NDA-PEG/nm2 expected from preliminary measurements of the maximum achievable grafting density by ligand replacement, e.g., 1.9 g NDA-PEG(5 kDa) for 9.6 nm NPs), was dissolved in DMF and mixed with the NPs. Note that only a low excess is needed for the ligand exchange, but to allow for fast ligand exchange and easier determination of the efficiency of the purification methods, a large excess was used. The dispersion was sonicated for 26 h at slightly elevated temperature (35 °C). Preliminary tests showed that 26 h was sufficient to replace OA, and longer sonication did not improve the exchange. Subsequently, the mixture was extracted thrice with n-hexane (30 mL each) in order to remove released oleic acid. Afterward, the solvent was evaporated; the PEGylated NPs were lyophilized for 24 h and stored as dry powder for further analysis and are referred to as “NPs raw” in the analysis. NPs were obtained as a light brown powder in 97% yield with respect to the amount of cores. All purification methods were applied after the extraction of the NPs with n-hexane. All NPs were characterized and analyzed before and after purification by TEM, TGA, IR, and DLS. Additionally, NPs purified by precipitation and magnetic decantation were analyzed by SAXS.
Purification Methods of NDA-PEG(5 kDa) Coated Iron Oxide NPs
Column Purification
0.3 g of the NPs after ligand replacement (NPs raw) was dissolved in 2 mL of H2O and sonicated for 15 s. Sephadex was swollen overnight in Milli-Q water at 40 °C and manually packed into a 25 × 2 cm column. The column was equilibrated with H2O and charged with the dispersed NPs. The length of the column was sufficient to split the NP sample in six to seven identifiable, differently colored fractions (Figure S1). The eluates were collected and freeze-dried for further analysis. For all NP sizes fraction II could be identified as the main fraction. For two NP sizes (9.6 and 7.1 nm) this main fraction was reapplied onto a fresh column, and the eluate was again characterized.
Dialysis Purification
The 1000 kDa molecular weight cutoff (MWCO) membranes were equilibrated for 10 min in 10% v/v EtOH and 30 min in H2O. 0.3 g of the raw NPs were dissolved in 5 mL of Milli-Q and dialyzed against 5 L of Milli-Q. To monitor the dialysis progress, equivalents of 0.5 mL were taken after 1, 2, 4, 6.5, and 24 h and freeze-dried. The 9.6 and 7.1 nm NPs were further dialyzed for 48 h and the small NPs (4.9 and 3.5 nm) for 70 h. The dispersions were lyophilized for further analysis by TEM, TGA, IR, and DLS.
Membrane Centrifugation
Typically, 0.15 g of raw NPs were dispersed in 2 mL of Milli-Q and loaded into a 50 kDa Amicon-Ultra-15 membrane centrifugation filter unit and centrifuged at 4500g for 15 min. The centrifuge effluent was collected, and the NPs were redispersed in 1 mL of Milli-Q. This procedure was repeated up to 15 times; equivalents were taken after 5, 7, and 11 centrifugation steps to track the removal of excess NDA-PEG. All dispersions and the collected effluents were freeze-dried for further analysis by TEM, TGA, and DLS.
Precipitation and Magnetic Decantation
Typically 0.3 g of the raw NPs (containing 0.019 g cores for 9.6 nm NPs) were dissolved in 10 mL of EtOH by sonication and slight heating with a hot gun to 60 °C. The clear dispersion was poured into a small beaker and mixed with 10 mL of ice-cold petrolether whereupon it got slightly turbid. The beaker was placed on a 5 × 5 cm 1 T magnet in the fridge at 4 °C, and the NPs were soaked from the cloudy mixture to the bottom of the beaker within a few minutes, dependent on the NP size. The supernatant was decanted by holding back the NPs with the magnet, collected, combined, and freeze-dried for TGA measurements. This procedure was repeated 7–9 times; after each step a small sample of NPs was kept and freeze-dried for TGA analysis to monitor the removal of excess ligands and impurities. The final products after the seventh step were freeze-dried and further analyzed. The NPs were obtained as dark brown powder in 96% yield with respect to the cores.
Investigation of Long-Term Stability
Freeze-dried samples of all purification methods were stored at room temperature without protection from light. After 6 months dry storage, suspensions of NPs in Milli-Q water and PBS of the NPs purified by dialysis (4.9 nm cores) and magnetic decantation (7.1 nm cores) were prepared (4 mg/mL) and characterized by TEM and DLS. Additionally, the samples purified by magnetic decantation were characterized by SAXS. The NP dispersions were stored for 4 months at 4 °C to prevent microbe growth and analyzed.
Additionally a sample of 7.1 nm cores purified by magnetic decantation was dissolved in PBS and water at 3 mg/mL; 10% bovine calf serum was added to investigate if the NPs aggregate upon nonspecific protein adsorption. DLS was measured over a temperature cycle from 20–70–20 °C in 5K steps with an equilibration time of 5 min at each temperature.
Results
Core Synthesis
Spherical, monodisperse, and single-crystalline superparamagnetic iron oxide nanoparticles were synthesized using a modified heat-up method introduced by Hyeon and co-workers.16 The NP size could be precisely tuned in the range of 3–10 nm without any further size selection process by varying the Fe(CO)5 to oleic acid molar ratio (see Table S1 in the Supporting Information for details). The obtained NPs were characterized by low- and high-resolution TEM, TGA, and ATR-FTIR. Figure 1 shows TEM micrographs of monodisperse 3.5 ± 0.3, 4.9 ± 0.4, 7.1 ± 0.4, and 9.6 ± 0.4 nm magnetite cores, calculated with Pebbles by evaluation of ∼900 NPs from multiple HR and LR-TEM micrographs of the respective batches. The HR-TEM (inset in Figure 1c) shows the single-crystallinity of the NPs with lattice spacing in the (311) direction of 0.26 nm. The ring diffraction pattern (inset Figure 1d) reveals the highly crystalline structure of the NPs. The ratio of d-spacings in the obtained pattern show good agreement with the JCPDS database numbers for maghemite or magnetite.19
Figure 1.

Transmission electron micrographs of as-synthesized monodisperse OA-capped Fe3O4 NPs: (a) 3.5 nm. (b) 4.9 nm. (c) 7.1 nm; inset: high resolution TEM image showing that the particles are single-crystalline. (d) 9.6 nm; inset: electron diffraction pattern of 9.6 nm NPs reveal the highly crystalline nature of the NPs.
Grafting of NDA-PEG and Purification of NDA-PEG Grafted Iron Oxide Nanoparticles
The grafting of NDA-PEG to the nanoparticle cores was performed in large excess of PEG in order to ensure fast and full ligand replacement and to provide an easily distinguished baseline for evaluation of different methods to remove unreacted dispersant. The separation of all excess free PEG-dispersant after grafting is challenging, as indicated by previous work;7a,11,20 we performed a thorough comparison and analysis of different methods that have been applied for core–shell nanoparticle preparation: cross-linked dextran size-exclusion column separation,11 dialysis,12,14,21 membrane centrifugation and precipitation with magnetic decantation. The different methods were compared on their ability to remove excess PEG to a stable value determined by TGA and their effect on nanoparticle stability. The TGA results with corresponding NDA-PEG(5 kDa) grafting density are summarized for all purification methods and core sizes in Table 1. The total organic content (TOC) was determined as the mass loss fraction up to 650 °C and converted to NDA-PEG(5 kDa) dispersant grafting density using the known molecular weight of NDA-PEG, the average iron oxide core area determined by TEM, and a core density of 5.17 g/cm3. The detailed results of each grafting method are presented in the sections below.
Table 1. TGA Results Converted into Nominal Grafting Densities ρgraft for Different Sizes and Purification Methods.
| raw |
column
purified (FR2) |
dialyzed
(48 h) |
membrane
centrifugation (11×) |
precipitation/magnetic
decantation (7×) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TOC [% w/w] | ρgraft [NDA-PEG(5 kDa)/nm2] | TOC [% w/w] | ρgraft [NDA-PEG(5 kDa)/nm2] | TOC [% w/w] | ρgraft [NDA-PEG(5 kDa)/nm2] | TOC [% w/w] | ρgraft [NDA-PEG(5 kDa)/nm2] | TOC [% w/w] | ρgraft [NDA-PEG(5 kDa)/nm2] | |
| 9.6 nm | 93.8 | 14.2a | 65.9 | 1.8a | 37.4 | 0.6 | 35.9 | 0.5 | 46.1 | 0.8 |
| 7.1 nm | 94.5 | 11.9a | 91.1 | 7.1a | 40.0 | 0.5 | 39.1 | 0.5 | 50.1 | 0.7 |
| 4.9 nm | 93.3 | 6.8a | 89.5 | 4.1a | 44.8 | 0.4 | 38.9 | 0.7 | 59.8 | 0.7 |
| 3.5 nm | 95.7 | 7.7a | 87.9 | 2.5a | 54.1 | 0.4 | 59.7 | 0.5 | 76.1 | 1.1 |
Samples contain amounts of free NDA-PEG(5 kDa).
Column
As described in the Materials and Methods section, the NPs were after column purification collected in 6–7 dark brown, red, orange, and yellow fractions. The same colors and amounts were observed for all NP core sizes (see Figure 2b). All samples passed the column without visible sticking to the dextran matrix, which demonstrates a high and homogeneous PEG shell density on the particles.11,20a Fraction 2 could be identified as the main fraction for all sizes; it had the largest amount of NPs with the smallest amount of NDA-PEG(5 kDa). Figure 2 shows the results of the column purification in terms of grafting density based on measurement of organic content by TGA. For two NP sizes (9.6 and 7.1 nm) the main fraction was dispersed in MQ and reapplied onto a fresh Sephadex column. For the 3.5 and 4.9 nm NPs the amounts contained in fraction 2 were too small for a second pass through the column. The reapplied samples passed the column as a single band with a very small fraction of particles that passed the column faster. The organic content of the reapplied fractions could be decreased from 66% to 49% (9.6 nm NPs) and 91% to 73% (7.1 nm NPs) by the second pass. For the 9.6 nm NPs this translates into a grafting density of 1.0 NDA-PEG(5 kDa)/nm2, while for 7.1 nm NPs a grafting density of ∼2 NDA-PEG(5 kDa)/nm2 is calculated. However, the yield of NPs obtained through the main fraction was low.
Figure 2.

(a) Total organic content of the column purification, measured by TGA and converted into NDA-PEG(5 kDa)/nm2 using the known molecular weight of NDA-PEG(5 kDa) and average core surface area. TOC includes large excesses of free PEG and Sephadex and was converted into dispersants/nm2 for comparison with the other methods but does therefore not reflect the actual grafting density of NDA-PEG. Fraction 2 could be identified as the main fraction. 2xFr2 is the main fraction 2 reapplied onto a fresh column. (b) Fractions of NPs after column purification dispersed in water (9.6 nm, upper) and freeze-dried (7.1 nm, lower).
Dialysis
Dialysis has to be performed with membranes having a pore size sufficiently large to allow diffusion of the dispersant through the pores; the MWCO of membranes is however typically given for equivalent protein or dextran sizes and cannot be used directly for comparison to highly solvated polymer molecular weights for which the radius of gyration is much bigger than for e.g. protein of the same molecular weight. A rule of thumb of choosing a pore size >5× that of the dialyzed molecule is often invoked. This was confirmed by attempting dialysis of dissolved PEG(5 kDa) (0.2 g/mL against 5 L of Milli-Q) in a membrane of 12–14 kDa MWCO, i.e., nominally 2–3 times the molecular weight of the NDA-PEG, which was used in previous publications studying grafting-to and ligand replacement of PEGylated NPs.14,21 After 24 h only 50% of the free PEG was removed, and only 60% had been removed after 5 days despite exchanging Milli-Q in the reservoir five times. Thus, choosing a too low MWCO results in remaining free dispersant and subsequent overestimation of the dispersant grafting density by TGA.11,14,22 We therefore changed to cellulose-based membranes with a MWCO of 1000 kDa, which nominally corresponds to a cutoff hydrodynamic size of ∼37 nm (assuming a scaling related to equivalent sizes of dextran). NPs with a hydrodynamic radius RH between 15 and 23 nm (for 7.1 nm cores) were still retained while free NDA-PEG(5 kDa) with an RH of ∼4 nm23 was presumably removed from the dispersion.
Figure 3a shows the removal of excess ligands by dialysis for all sizes of NPs. Large-core NPs (7.1 and 9.6 nm) dialyzed for 24 h yielded an average grafting density of ∼1 NDA-PEG(5 kDa)/nm2 determined by TGA. However, NPs also started to stick to the dialysis membrane after 24 h. TEM of large NPs (7.1 and 9.6 nm) extracted from the dialyzed dispersion after 24 h showed aggregated morphology (Figure 3b). The 24 h dialyzed samples could be redispersed in water but showed poor long-term stability. Visible aggregates were obtained after 1 week. Continued dialysis of the large cores led to an even lower grafting density (∼0.5 NDA-PEG(5 kDa)/nm2) and increased aggregation.
Figure 3.
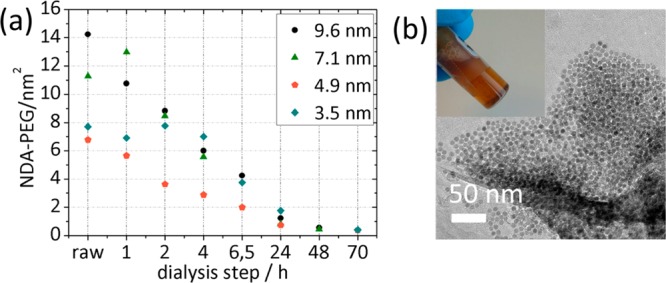
(a) Removal of excess ligands by dialysis with a 1000 kDa MWCO membrane. Total organic content was measured by TGA and converted into NDA-PEG(5 kDa)/nm2 using the known molecular weight of NDA-PEG(5 kDa) and average core surface area determined by TEM. After 24 h the surface coverage was ∼1 NDA-PEG(5 kDa)/nm2 independent of size, but core size-dependent aggregation and precipitation were observed. Dialysis for 70 h lowered the grafting density to 0.3 NDA-PEG(5 kDa)/nm2, resulting in colloidally unstable dispersions. (b) Typical TEM of nanoparticles with 9.6 nm core size that had visible aggregates (inset) grafted with NDA-PEG(5 kDa) after 24 h dialysis against water using a 1000 kDa MWCO cellulose membrane.
NPs with small core sizes (4.9 and 3.5 nm) had grafting densities of 0.8 and 1.7 NDA-PEG(5 kDa)/nm2, respectively, after 24 h dialysis and showed good colloidal stability. The small NPs were dialyzed also for 70 h, which resulted in lowering of the average grafting density to 0.3 NDA-PEG(5 kDa)/nm2.
During dialysis the NP suspension underwent visible color change from light brown to almost black (Figure S2 in the Supporting Information), indicating that as dialysis proceeds, surface-bound NDA-PEG is ripped off in addition to the initial removal of free NDA-PEG.
Membrane Centrifugation
In contrast to diffusion-driven dialysis, a MWCO of 50 kDa was sufficient to remove excess NDA-PEG. This can be ascribed to the convective flow and high forces induced by the centrifugation that deform the free polymer to flow through narrow pores. Figure 4 shows the results of the membrane centrifugation for the different NP sizes.
Figure 4.
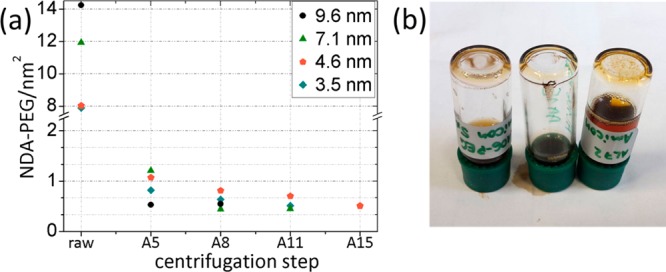
(a) Removal of excess ligands by repeated membrane centrifugation with Amicon 50 kDa centrifugation filter units. After 11 repetitions all sizes show an average grafting density around 0.5–0.7 NDA-PEG(5 kDa)/nm2. (b) More centrifugation steps lead to a decrease in grafting density and colloidal instability. Left to right: 4.6 nm NPs after 5, 4.6 nm NPs after 15, and 7.1 nm NPs after 11 centrifugation steps. All dispersions show visible aggregation.
For all sizes excess ligands could be removed with five centrifugation steps. The average grafting density at this point was measured to be ∼1 NDA-PEG(5 kDa)/nm2 by TGA, but the resuspended NPs were colloidally unstable, as can be seen by the visible aggregates sticking to the vial in Figure 4b.
In order to study if the grafting density stays constant, the centrifugation steps were repeated up to 15 times. As shown in Figure 4a, the grafting density did not stay constant but could be reduced to 0.4 NDA-PEG(5 kDa)/nm2. These NPs could be redispersed in Milli-Q but showed visible aggregation and precipitation after 2 days (Figure 4b). Analysis of the combined centrifugates showed a 99.8% organic composition, meaning that only ligands were removed from the dispersion and all cores were retained by the membrane. All NPs purified by this method showed strong aggregation and were therefore not analyzed further.
Precipitation and Magnetic Decantation
Both column separation and filtration (dialysis and membrane centrifugation) are established methods, but they failed to produce a high yield of colloidally stable nanoparticles for all sizes when the PEG-shell was grafted through ligand replacement. We therefore attempted a variation on precipitation that exploits the possibility to magnetize the nanoparticle cores. While hydrophilic, polymer-stabilized superparamagnetic nanoparticles cannot be pulled out of aqueous solution using a fixed magnet, such extraction is possible in solvent/nonsolvent mixtures in which the poor solubility leads to formation of small aggregates. By dispersing the particles in a solvent mixture of EtOH with an addition of petrolether, a cloudy suspension of poorly dispersed core–shell nanoparticles could be obtained. The poorly suspended NPs could be pulled to the container wall by application of a fixed magnet.
The decantation step has to be repeated several times due to the poor solubility also of the free PEG dispersant in the EtOH/petrolether. TGA was therefore performed after each decantation step to evaluate the stepwise reduction in dispersant concentration (Figure 5a). After the third precipitation and magnetic decantation step the grafting density stayed constant, interpreted as that no further dispersant removal is possible and that all remaining dispersants are strongly bound to the iron oxide core surface. The grafting density is within the measurement error independent of core size at ∼1 NDA-PEG(5 kDa)/nm2. Standard deviations were calculated based on results measured on 6–7 different samples for each core size. The combined supernatants were analyzed with TGA, and ∼1.5% inorganic material was found, which means that this purification method removes free NDA-PEG and no cores. After reaching the constant grafting density by precipitation and magnetic decantation the retained product included 98% of the cores initially added for ligand replacement independent of NP size.
Figure 5.
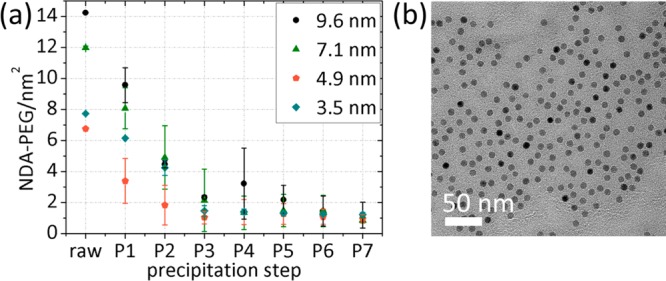
(a) Step-by-step removal of excess ligands by repeated precipitation/magnetic decantation. Total organic content was measured by TGA and converted into NDA-PEG(5 kDa)/nm2. After three precipitations with magnetic extraction the free dispersant is removed for all core sizes. A stable grafting density of ∼1 NDA-PEG(5 kDa)/nm2 independent of core size remains with a yield of 98% purified NPs. (b) Representative TEM micrograph of well-dispersed, NDA-PEG-grafted NPs with 9.6 nm core diameter. In comparison to OA-capped NPs it is possible to dry the NPs well dispersed and without order on the grid due to the steric repulsion of the added PEG shell (cf. Figure 1d). The morphology of the NP cores remains unchanged after ligand exchange.
Figure 5b shows a TEM micrograph of nicely dispersed core–shell PEGylated 9.6 nm NPs purified by precipitation/magnetic decantation. TEM micrographs of smaller cores purified by this method can be found in the Supporting Information (Figure S7). Compared to the OA-capped NPs (Figure 1d), the interparticle distance is increased due to the bulky NDA-PEG shell, and importantly an inspection of thousands of particles did not reveal any aggregated cores. This strongly suggests that the PEG shell was formed around individual cores and remained homogeneous. The morphology of the NP cores did not change after OA was replaced by NDA-PEG. A color change from light brown to dark brown could be observed, which is attributed to the removal of free PEG.
Characterization of Nanoparticle Shell Composition by IR and TGA
The difficulty of achieving complete exchange of OA for new dispersants previously reported by us9a,20a and others14 demonstrates the need to establish that the total organic content determined by TGA is traced exclusively to NDA-PEG. Significant amounts of other dispersants on the core surface might alter the physicochemical properties of the core–shell particles. ATR-FTIR analysis was performed to verify the composition of the dispersant shell after purification (Figure 6a; insets can be found in Figure S4). IR spectra were also recorded on the complete supernatant collected from the precipitation and magnetic decantation method. The presence of NDA-PEG on the NP surface could be confirmed by several marker bands below 1000 cm–1 as well as bands in the region from 1345 to 1240 cm–1 and from 1144 to 840 cm–1 that can be mainly attributed to ethylene glycol CH2 and C–O/C–C stretching vibrations, respectively.24
Figure 6.

(a) ATR-FTIR spectra of 7.1 nm iron oxide nanoparticles: (a) as-synthesized cores with OA ligands, (b) NDA-PEG-NPs after ligand exchange before purification, (c) PEG-NDA-NPs purified by precipitation and magnetic decantation, (d) PEG-NDA-NPs dialyzed 24 h, (e) PEG-NDA-NPs purified through Sephadex G75 column (Fr II), (f) Sephadex G75, (g) PEG-NDA, and (h) combined supernatants of all magnetic decantation steps. Labeling: (∗) 1960 cm–1 OH-bending from PEG-COOH, (+) 1740 cm–1 COOR or COOH, (△) 1702 cm–1 (C=O) free OA, (○) 1640 cm–1 amide CONH or NH2, (■) 1074–974 cm–1 sugar band, (ω) 965 cm–1 ((C=O)–OH) free OA; 680–400 cm–1 NP bands. (b) Representative TGA (solid lines) and DSC (dashed) graphs of 4.9 nm NPs measured in synthetic air: OA-capped NPs (orange), NDA-PEG-NPs “raw” after ligand exchange before purification (black), precipitation and magnetic decantation (green), dialyzed (light blue), membrane filtration with Amicon (dark cyan), column purified (dark yellow), and pure ligand NDA-PEG (navy).
The amide stretching vibrations at 1640 (CO) and 1550 cm–1 (NH)25 (Figure 6a (○)) that can be found in all spectra except for the OA-NP are a good indication of successful coupling of NDA to PEG and its presence at the NP surface; this peak is slightly larger for NPs purified by magnetic decantation compared to dialyzed NPs. A weak peak (Figure S4b (×)) at 1491 cm–1 fits monodeprotonated nitrocatechol26 and is attributed to surface-bound NDA-PEG. The band at 1728 cm–1 (Figure S4a (★)) arises from residual carboxylic carbonyl groups of free PEG(5 kDa) that has not been coupled to NDA and is found only in the nonpurified sample and the combined supernatants. A similar trend for the band at 1960 cm–1 (Figure 6a (∗)) suggests a common origin; we therefore attribute this vibration to an overtone or combination vibration of the out-of-plane OH bending mode of PEG carboxylic acid.
The bands at 1730 and 1740 cm–1 (Figure 6 (+)) that were found in all samples except in the precipitation purified NPs and OA-NPs might originate from PEG-COOH or COOR groups from unreacted PEG or cross-reacted NDA-PEG (O-Ph). However, these byproducts were removed by precipitation/magnetic decantation.
An important observation is that the NPs purified by column show deviations from the IR absorption spectrum for iron oxide nanoparticles purified by other means. The column material Sephadex dextran shows strong similarities (bands at 1074–974 and 790–740 cm–1, Figure 5a (■), OH vibrations at 3600–3000 cm–1) with these additional peaks in the spectrum, indicating that NPs purified by column are additionally covered with small amounts of dextran.
A final important question is the presence of significant amounts of weakly or strongly bound oleic acid after ligand replacement and purification. In particular, free or weakly bound oleic acid can greatly influence the colloidal stability. The presence of free oleic acid can be determined from absorptions at 1702 cm–1 in the FTIR spectra (Figure 6 (△)) assigned to the C=O stretch of H-bonded (dimeric) alkylcarboxylic acids and from other bands characteristic for free acid groups such as the OH in-plane and out-of-plane bending at 1410 and 965 cm–1 which is overlaid (Figure 6 (ω)).9a,27 The absence of the bands at 1702 cm–1 in all samples (also the raw sample) demonstrates that free OA was successfully removed by the DMF–hexane extraction. Oleate can be strongly bound by complexation to the particle surface.9a,14 The presence of oleate can be evaluated at 1605, 1520, and 1410 cm–1 corresponding to two asymmetric and one symmetric stretching vibrations of the carboxylate headgroup.27,28 Whereas the peaks at 1605 and 1520 cm–1 are overlaid by bands from the PEG, a slight shoulder of the band at 1410 cm–1 can be found in all samples. This band, however, is more prominent in the raw and the dialyzed samples than in the precipitated samples. For the column purified NPs this peak is overlaid by a sugar band from the dextran column material.
Multistep TGA profiles observed below 400 °C have been attributed alternatively to vaporization of physisorbed oleic acid, to chemisorbed oleate species with different binding strengths,29 or to partial cleavage of the capping agent.28 Our data (Figure 6b) show that a significant second step in TGA only is observed when free or weakly bound impurities such as physisorbed oleate or dextran are present. A one-step profile is only observed for NPs purified by magnetic decantation.
We conclude from the IR measurements that the ligand replacement is not complete in any sample. This is also suggested by that the measured high surface coverage of ∼1 NDA-PEG(5 kDa)/nm2 is still lower than the theoretical maximum surface coverage of >2 NDA-dispersants/nm2.9a,20a However, for samples purified by precipitation and magnetic extraction only a minor amount of strongly complexed oleate remains on the particle surface shielded by a dense PEG brush. If oleate with a negligible desorption rate is distributed on the particle surface, its short extension combined with the high PEG-brush density still results in excellent colloidal stability as shown in the next section.
Long-Term Stability
The long-term stability of samples that were purified by dialysis and magnetic decantation was investigated with TEM, DLS, and SAXS. Long-term stability of dialyzed samples was only investigated for 4.9 nm core size as the dispersions of NPs with larger core diameters showed visible aggregation after a few days. Also, samples purified by membrane filtration were not subjected to long-term stability studies as the dispersions showed visible aggregates after 2 days.
For long-term studies of core–shell NPs purified by precipitation and magnetic decantation, 7.1 nm NPs were dispersed at a high concentration of 4 mg/mL that speeds up aggregation in both H2O and PBS. PBS tests the stability at physiological ionic strengths and is additionally known to cause aggregation and precipitation of poorly stabilized iron oxide NPs due to the strong interaction of phosphates with iron ions. TEM, DLS, and SAXS were measured after 1 week and 4 months. For NPs in water and PBS a RH of 16 and 23 nm (intensity weighted), respectively, was measured by DLS (Table 2), which is in good agreement with previous findings for individually stabilized core–shell NPs.7a,11,25,30 The RH is also in good agreement with theoretical calculations for a core–shell NP with NDA-PEG monolayer, considering the length of hydrated NDA-PEG(5 kDa) to be ∼4 nm23 and the core 7.1 nm, resulting in a RH(theoretical) of ∼15 nm. These values are significantly smaller than in other recent publications investigating direct ligand replacement on oleic acid-capped nanoparticles with PEG dispersants. The narrow size distributions strongly support the interpretation of individually stabilized and colloidally stable core–shell NPs.30b No precipitation and negligible change in the hydrodynamic radius were found for NPs in Milli-Q as well as PBS after storage of the dispersions for 4 months at 4 °C (Table 2).
Table 2. Hydrodynamic Radii RH for 7.1 nm NPs Purified by Magnetic Decantation and 4.9 nm NPs Purified by Dialysis, Dispersed in H2O and PBS at 4 mg/mL, As Prepared and after Storage in Solution for 4 Months at 4 °Ca.
| RH as prepared [nm] | RH after 4 months storage [nm] | |
|---|---|---|
| NPs in H2O/magn dec | 16.4 ± 0.0 | 15.0 ± 0.1 |
| NPs in PBS/magn dec | 23.2 ± 0.1 | 25.5 ± 0.4 |
| NPs in H2O/dialyzed | 16.5 ± 1.0 | 18.3 ± 0.1 |
Errors are standard deviations of three separate measurements.
The RH of dialyzed 4.9 nm cores was measured to be 16 nm and increased slightly after 4 months to 18 nm. TEM imaging of the nanoparticles stored for 4 months in Milli-Q and PBS showed no trace of core aggregation or aging of the particles (Figure S5). The stability of the dialyzed sample during storage, but not to further dialysis, indicates a degradation mechanism related to the dialysis membranes.
The stability of NPs purified by magnetic decantation was supported by SAXS. Scattering curves and fits for samples in Milli-Q and PBS fresh and after 4 months are shown in Figure 7b. The high monodispersity of the cores is evident from the scattering curves. A core size of 6.8 ± 0.4 nm could be fitted to the data.30b This is in excellent agreement with the core size determined by image analysis of the TEM data of the same particles (7.1 ± 0.4 nm). After storage for 4 months at 4 °C there is no significant deviation in the scattering attributable to aggregates, such as a structure factor peak and/or a change in the low q range.
Figure 7.

(a) DLS measurements of 7.1 nm NPs purified by magnetic decantation; 3 mg/mL in PBS with 10% v/v fetal calf serum added. Temperature cycle from 20 to 70 °C (circles) and back (triangles) with 5 min equilibration time at each temperature. (b) SAXS curves of 7.1 nm NPs in PBS and H2O, freshly prepared and after storage for 4 months at 4 °C. Inset: clear colloidal dispersion of NPs from measurement (a) stored in PBS/FCS 10% v/v for 2 months at 4 °C after temperature cycling.
The colloidal stability of the NPs was further challenged by heating the NPs (3 mg/mL) up to 70 °C in PBS with 10% v/v fetal calf serum (FCS). This test is applied to distinguish between particles that show stability in serum but interact with denatured protein aggregates and those for which interactions between core and denatured proteins also do not occur.7aFigure 7a shows the evolution of the hydrodynamic radius with temperature. No increase in RH or precipitation of nanoparticles during the temperature cycle was observed. The inset in Figure 7b shows a picture of the same NP dispersion subjected to temperature cycling after being stored for 2 months at 4 °C. Aggregation or precipitation also cannot be observed after storage; the dispersion remains clear and colloidally stable. TEM micrographs (Figure S6) show that the NPs in PBS/FCS remain well-dispersed after storage for 2 months in serum.
Discussion
First-generation, hydrophilic iron oxide NPs were mainly stabilized with dextran5a,5b or carbohydrate derivatives31 physically adsorbed without using a separately defined anchor such as nitrodopamine. A second generation of polymer-coated iron oxide NPs could be said to have polydisperse cores but defined polymer shells end-grafted with defined anchor groups.7a This defined core–shell architecture allows for exact characterization and functionalization, which is crucial to medical applications. A third generation with monodisperse iron oxide cores to perfectly tailor uniform properties, however, requires ligand replacement. Several mutually contradicting conditions during the ligand replacement reaction have to be fulfilled: (1) to dissolve the capping agent (oleic acid), (2) to solubilize the dispersant, (3) to keep the dispersant at low coil size, quantitatively described by e.g. RG, which determines the grafting footprint, and (4) to provide the right conditions (protonation) of the anchor group to irreversibly bind to the core.
We could optimize the ligand exchange procedure within the boundaries of the contradicting requirements and thus produce NPs with grafting densities consistently around 1 NDA-PEG(5 kDa)/nm2. The grafting densities of ∼1 NDA-PEG(5 kDa)/nm2 for monodisperse NPs were shown to be sufficient to render the NPs long-term stable in biologically relevant media. This is in contrast to what was reported earlier for polydisperse systems.7a,11,20a For polydisperse cores a grafting density of ∼2 NDA-PEG(5 kDa)/nm2 was found as the threshold for colloidal stability at temperatures above body temperature and in biological fluids. We hypothesize that the accurate exchange of ligands into a homogeneously coated shell is what enables the improved stability at a lower average grafting density for monodisperse, spherical NPs. On the other hand, we were unable to achieve grafting densities higher than ∼1 NDA-PEG(5 kDa)/nm2 by direct ligand replacement. That the grafting density was constant with respect to core size indicates that surface curvature is not the determinant of the ultimate grafting density by ligand replacement in the superparamagnetic size range. This is similar to previous observations for ligand replacement of hydrophobic dispersants9a and NDA-PEG grafting in the melt,20a for which within the experimental error a difference in grafting density with respect to curvature could not be demonstrated. In the present case the constant grafting density is lower and suggests that the size of the PEG-coil is the main determinant of the grafting density.
Recent work by e.g. Mefford and co-workers reported very high ligand densities from ligand replacement in their study of oleic acid displacement.14,21 In their study, one precipitation step followed by dialysis with low MWCO membranes was used to remove excess dispersant. Our results show that this is insufficient to remove excess dispersants so their unusually high dispersant density in comparison with literature can likely be traced to this fact. A higher polydispersity of the cores can also have contributed to uncertainty in the grafting density.
In analogous recent work by Lak et al.,13 large monodisperse cores were grafted with NDA-PEG and purified by repeated ultracentrifugation from aqueous solution, which is only feasible with very large cores. They report a grafting density of ∼1 NDA-PEG(5 kDa)/nm2, which is inconsistent with the presented TGA results that yield a grafting density within a large interval of 5–28 NDA-PEG(5 kDa)/nm2. This organic content, which corresponds to an order of magnitude higher grafting density than the theoretical maximum, demonstrates that also a single centrifugation step and dialysis are insufficient to purify also large core NPs. These samples even showed low colloidal stability, which is typically associated with a low grafting density. However, one should keep in mind that stabilizing ferrimagnetic nanoparticles with large cores using thin polymer brushes is more challenging than in the superparamagnetic range. A different ligand replacement protocol in this study might also have left a higher fraction of destabilizing or heterogeneously distributed oleate bound to the NP surface.
Finally, it was recently shown that grafting of polymer shells under solvent-free, polymer-melt conditions can lead to a near complete coverage of end-grafted polymer dispersants, with unique shell structure and colloidal properties as the result.20a,30b While this might seem superior we show here that optimized protocols for grafting-to can yield the required colloidal properties for standard medical applications. Our grafting-to by one-step ligand replacement has the benefits of (a) providing near-total yields (98%) under optimal conditions, while melt-grafting requires considerable excess dispersant and was reported with a maximum yield of ∼30% calculated on nanoparticle cores; (b) multifunctional particles can be constructed by molar mixing of differently functionalized dispersants without risking degradation of the functional group under the melt-grafting conditions; and (c) direct ligand replacement is easier to scale up for synthesis of large batches.
Purification of as-synthesized core–shell NPs is crucial for proper determination of NP properties, and we emphasize the thorough analysis that we have done of the efficiency and effect of different purification methods as a central result of the presented work. To our knowledge, there has been no previous study on the colloidal stability of core–shell nanoparticles for different dialysis times. We report a strong time-dependent degradation of the particles when dialyzed through high MWCO regenerated cellulose membranes. Cellulose ester should be inert and not the cause of the observed particle degradation, which from TEM clearly included the particle core. A similar trend was found for NPs purified by membrane filtration: NP degradation in terms of removal of surface-bound NDA-PEG was observed, and a colloidally stable product could not be obtained. Degradation of the core can occur by that a dispersant with a strongly binding anchor group leaves the core surface and removes the surface iron ion with it.11 Since the other purification methods do not lead to this degradation, i.e., the NDA anchor is stably bound to the core, we hypothesize that the degradation during dialysis and membrane filtration involves an osmotic/mechanical strain related to dispersant interaction with the pores of the membranes where the particles are also found to precipitate. Stretching of the PEG chain leads to a spring-like increase in energy; it could effectively weaken the bond to the surface and lead to polymer detachment and the observed surface dissolution.
Size-exclusion column chromatography with cross-linked dextran as column material is often used for purification of biotechnological samples in the size range of nanoparticles; it was previously advocated by us and others as a suitable way to purify core–shell nanoparticles after grafting-to and simultaneously testing their grafting density and colloidal stability. Dextran has high affinity to iron oxide and has been used in commercial contrast agent coatings of iron oxide nanoparticles. Incompletely grafted NPs therefore stick to a dextran column and are not collected.11,12,20a Our new results show that for highly but not extremely densely grafted NPs dextran can transfer from the column to the NPs. The cross-linked dextran employed in a size-exclusion column should not transfer to the NPs, but the IR results unambiguously show its presence in the eluted NP fractions even after passing the same fraction through fresh columns twice. Although dextran was not found by IR for the same type of NP cores with grafting densities >2 NDA-PEG(5 kDa)/nm2,20a a lower grafting density seems to result in transfer of column material to the free core area. The high PEG grafting density of ∼1 NDA-PEG(5 kDa)/nm2 obviously prevents further NP aggregation or sticking to the column. We interpret this result as that transferred dextran also can stabilize nanoparticles that are not extremely densely grafted. This additional physisorbed dextran coating can explain the high organic content in other particle fractions that passed the column. High-molecular-weight polymer physisorbed to NPs induce bridging interactions that cluster multiple cores; this leads to effective differences in size and column retention time, which spreads the NPs over several fractions and pushes down the yield of the main fraction. Thus, despite the stability of the NPs purified by column chromatography, the application of cross-linked dextran as column material cannot be generally recommended for purification that should yield particles with controlled shell properties. The choice of another column material with less strong interaction with iron oxide could remedy this drawback. However, the yield and throughput would based on our study remain low compared to magnetic decantation.
Among the many investigated purification methods, our study suggests a combination of precipitation and magnetic decantation repeated in multiple cycles as the preferred choice. This method reproducibly yields NPs with exceptional long-term colloidal stability, at high yield and without free dispersant. All excess ligand is removed in three decantation cycles at near total yield. This very high yield is far superior to the inherent losses of NPs when column purification or dialysis is used and can be attributed to the efficient magnetic extraction. Similar approaches have previously been applied in combination with centrifugation,20b,32 which is sensitive to the choice of solvent ratio and polymer shell molecular weight for their success and do not make use of the superparamagnetic core. The crucial step for successful separation by magnetic decantation was to discard the supernatant prior to sedimentation of precipitated excess PEG, since PEG and densely PEG-grafted NPs have similar solubility. Aggregation of the superparamagnetic cores enables rapid collection of the NPs within 2–5 min depending on their size; the spontaneous precipitation and sedimentation of the free PEG takes ∼10 min. The petrolether used in the magnetic decantation also aided removal of remaining OA from the nanoparticle sample that cannot be removed by dialysis or centrifugation, since OA was found in the removed supernatant.
A final major advantage of the magnetic decantation method is that in addition to its high yield and purity it can be scaled up and automated. Large volumes and samples can be handled in a short time (<1 h for >3 cycles necessary for free dispersant removal), while dialysis is inherently time-consuming (relying on diffusion), membrane centrifugation is limited by the volume of the filtration units, and columns are difficult to scale. We show magnetic decantation for synthesis of gram quantities and further scaling up is possible.
Conclusion
We conclude that direct ligand replacement of oleic acid-coated monodisperse iron oxide NPs can be optimized to consistently yield grafting densities of ∼1 NDA-PEG(5 kDa)/nm2 regardless of core size in the investigated superparamagnetic range (diameter 3–10 nm). The obtained grafting density is sufficient to keep the hydrophilic core–shell NPs colloidally stable over many months in biologically relevant media as well as withstanding demanding tests such as temperature cycling in serum. The choice of purification method is as important as the synthesis protocol for reproducible results and greatly affects yield, purity, speed, and amount that can be produced. The best purification method that we found was magnetic decantation, which compared to dialysis, centrifugation filtration, and Sephadex size-exclusion chromatography provides the highest quality product and a means to verify purity; it also provides the highest yield and sample volumes that can be extracted per time unit. The methods described in this work form the fundament for development of a whole set of diversely functionalized NPs that are suitable for biomedical applications such as targeted MRI, multifunctional superparamagnetic NPs, and drug delivery.
Acknowledgments
We thank Prof. Dieter Baurecht for access to FTIR. We acknowledge the VIBT Extremophile Center for access to TGA and Dr. Hofinger-Horvath for NMR measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.langmuir.6b00919.
Additional experimental details and spectral data (PDF)
Author Contributions
The study was planned by E.R., R.Z., and A.L. A.L. prepared all samples and performed most measurements. O.B. contributed to the IR analysis. T.G. and H.L. performed the SAXS measurements and analysis. The manuscript was written by A.L. and E.R. All authors have given approval to the final version of the manuscript.
The research leading to these results received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP/2007–2013)/ERC Grant Agreement no. 310034 and BMWFW IGS BioNanoTech.
The authors declare no competing financial interest.
Supplementary Material
References
- a Lewin M.; Carlesso N.; Tung C.-H.; Tang X.-W.; Cory D.; Scadden D. T.; Weissleder R. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat. Biotechnol. 2000, 18, 410–414. 10.1038/74464. [DOI] [PubMed] [Google Scholar]; b Pittet M. J.; Swirski F. K.; Reynolds F.; Josephson L.; Weissleder R. Labeling of immune cells for in vivo imaging using magnetofluorescent nanoparticles. Nat. Protoc. 2006, 1, 73–79. 10.1038/nprot.2006.11. [DOI] [PubMed] [Google Scholar]
- a Pankhurst Q. A.; Thanh N. K. T.; Jones S. K.; Dobson J. Progress in applications of magnetic nanoparticles in biomedicine. J. Phys. D: Appl. Phys. 2009, 42, 224001/1–224001/15. 10.1088/0022-3727/42/22/224001. [DOI] [Google Scholar]; b Halbreich A.; Roger J.; Pons J. N.; Geldwerth D.; Da Silva M. F.; Roudier M.; Bacri J. C. Biomedical applications of maghemite ferrofluid. Biochimie 1998, 80, 379–390. 10.1016/S0300-9084(00)80006-1. [DOI] [PubMed] [Google Scholar]
- Namdeo M.; Saxena S.; Tankhiwale R.; Bajpai M.; Mohan Y. M.; Bajpai S. K. Magnetic nanoparticles for drug delivery applications. J. Nanosci. Nanotechnol. 2008, 8, 3247–3271. 10.1166/jnn.2008.399. [DOI] [PubMed] [Google Scholar]
- a Weissleder R.; Elizondo G.; Wittenberg J.; Rabito C. A.; Bengele H. H.; Josephson L. Ultrasmall superparamagnetic iron oxide: characterization of a new class of contrast agents for MR imaging. Radiology (Oak Brook, IL, U. S.) 1990, 175, 489–93. 10.1148/radiology.175.2.2326474. [DOI] [PubMed] [Google Scholar]; b Weissleder R.; Hahn P. F.; Stark D. D.; Rummeny E.; Saini S.; Wittenberg J.; Ferrucci J. T. MR imaging of splenic metastases: ferrite-enhanced detection in rats. AJR, Am. J. Roentgenol. 1987, 149, 723–6. 10.2214/ajr.149.4.723. [DOI] [PubMed] [Google Scholar]; c McCarthy J. R.; Weissleder R. Multifunctional magnetic nanoparticles for targeted imaging and therapy. Adv. Drug Delivery Rev. 2008, 60, 1241–1251. 10.1016/j.addr.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hong R. Y.; Feng B.; Chen L. L.; Liu G. H.; Li H. Z.; Zheng Y.; Wei D. G. Synthesis, characterization and MRI application of dextran-coated Fe3O4 magnetic nanoparticles. Biochem. Eng. J. 2008, 42 (3), 290–300. 10.1016/j.bej.2008.07.009. [DOI] [Google Scholar]; b Lacava L. M.; Lacava Z. G. M.; Da Silva M. F.; Silva O.; Chaves S. B.; Azevedo R. B.; Pelegrini F.; Gansau C.; Buske N.; Sabolovic D.; Morais P. C. Magnetic resonance of a dextran-coated magnetic fluid intravenously administered in mice. Biophys. J. 2001, 80 (5), 2483–2486. 10.1016/S0006-3495(01)76217-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tassa C.; Shaw S. Y.; Weissleder R. Dextran-Coated Iron Oxide Nanoparticles: A Versatile Platform for Targeted Molecular Imaging, Molecular Diagnostics, and Therapy. Acc. Chem. Res. 2011, 44, 842–852. 10.1021/ar200084x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni A.; Bardi G.; Bertero A.; Cappello V.; Emdin M.; Flori A.; Gemmi M.; Innocenti C.; Menichetti L.; Sangregorio C.; Villa S.; Piazza V. Design and optimization of lipid-modified poly(amidoamine) dendrimer coated iron oxide nanoparticles as probes for biomedical applications. Nanoscale 2015, 7, 7307–7317. 10.1039/C5NR01148E. [DOI] [PubMed] [Google Scholar]
- a Amstad E.; Textor M.; Reimhult E. Stabilization and functionalization of iron oxide nanoparticles for biomedical applications. Nanoscale 2011, 3 (7), 2819–2843. 10.1039/c1nr10173k. [DOI] [PubMed] [Google Scholar]; b Frey N. A.; Peng S.; Cheng K.; Sun S. Magnetic nanoparticles: synthesis, functionalization, and applications in bioimaging and magnetic energy storage. Chem. Soc. Rev. 2009, 38 (9), 2532–2542. 10.1039/b815548h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.; Joo J.; Kwon S. G.; Jang Y.; Hyeon T. Synthesis of monodisperse spherical nanocrystals. Angew. Chem., Int. Ed. 2007, 46, 4630–4660. 10.1002/anie.200603148. [DOI] [PubMed] [Google Scholar]
- a Bixner O.; Lassenberger A.; Baurecht D.; Reimhult E. Complete Exchange of the Hydrophobic Dispersant Shell on Monodisperse Superparamagnetic Iron Oxide Nanoparticles. Langmuir 2015, 31 (33), 9198–9204. 10.1021/acs.langmuir.5b01833. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ling D.; Hyeon T. Iron Oxide Nanoparticles: Chemical Design of Biocompatible Iron Oxide Nanoparticles for Medical Applications (Small 9–10/2013). Small 2013, 9, 1449. 10.1002/smll.201370057. [DOI] [PubMed] [Google Scholar]
- a Lu A. H.; Salabas E. L.; Schueth F. Magnetic nanoparticles: synthesis, protection, functionalization, and application. Angew. Chem., Int. Ed. 2007, 46, 1222–1244. 10.1002/anie.200602866. [DOI] [PubMed] [Google Scholar]; b Jun Y.-w.; Lee J.-H.; Cheon J. Chemical design of nanoparticle probes for high-performance magnetic resonance imaging. Angew. Chem., Int. Ed. 2008, 47, 5122–5135. 10.1002/anie.200701674. [DOI] [PubMed] [Google Scholar]
- Amstad E.; Gillich T.; Bilecka I.; Textor M.; Reimhult E. Ultrastable Iron Oxide Nanoparticle Colloidal Suspensions Using Dispersants with Catechol-Derived Anchor Groups. Nano Lett. 2009, 9 (12), 4042–4048. 10.1021/nl902212q. [DOI] [PubMed] [Google Scholar]
- Gillich T.; Acikgoez C.; Isa L.; Schluter A. D.; Spencer N. D.; Textor M. PEG-Stabilized Core-Shell Nanoparticles: Impact of Linear versus Dendritic Polymer Shell Architecture on Colloidal Properties and the Reversibility of Temperature-Induced Aggregation. ACS Nano 2013, 7 (1), 316–329. 10.1021/nn304045q. [DOI] [PubMed] [Google Scholar]
- Lak A.; Dieckhoff J.; Ludwig F.; Scholtyssek J. M.; Goldmann O.; Luensdorf H.; Eberbeck D.; Kornowski A.; Kraken M.; Litterst F. J.; Fiege K.; Mischnick P.; Schilling M. Highly stable monodisperse PEGylated iron oxide nanoparticle aqueous suspensions: a nontoxic tracer for homogeneous magnetic bioassays. Nanoscale 2013, 5 (23), 11447–11455. 10.1039/c3nr02197a. [DOI] [PubMed] [Google Scholar]
- Davis K.; Qi B.; Witmer M.; Kitchens C. L.; Powell B. A.; Mefford O. T. Quantitative Measurement of Ligand Exchange on Iron Oxides via Radiolabeled Oleic Acid. Langmuir 2014, 30, 10918–10925. 10.1021/la502204g. [DOI] [PubMed] [Google Scholar]
- Mondini S.; Ferretti A. M.; Puglisi A.; Ponti A. Pebbles and PebbleJuggler: software for accurate, unbiased, and fast measurement and analysis of nanoparticle morphology from transmission electron microscopy (TEM) micrographs. Nanoscale 2012, 4 (17), 5356–5372. 10.1039/c2nr31276j. [DOI] [PubMed] [Google Scholar]
- Hyeon T.; Lee S. S.; Park J.; Chung Y.; Na H. B. Synthesis of Highly Crystalline and Monodisperse Maghemite Nanocrystallites without a Size-Selection Process. J. Am. Chem. Soc. 2001, 123 (51), 12798–12801. 10.1021/ja016812s. [DOI] [PubMed] [Google Scholar]
- Napolitano A.; D’Ischia M.; Costantini C.; Prota G. A new oxidation pathway of the neurotoxin 6-aminodopamine. Isolation and characterization of a dimer with a tetrahydro[3,4a]iminoethanophenoxazine ring system. Tetrahedron 1992, 48 (39), 8515–22. 10.1016/S0040-4020(01)86599-6. [DOI] [Google Scholar]
- El-Faham A.; Funosas R. S.; Prohens R.; Albericio F. COMU: A Safer and More Effective Replacement for Benzotriazole-Based Uronium Coupling Reagents. Chem. - Eur. J. 2009, 15, 9404–9416. 10.1002/chem.200900615. [DOI] [PubMed] [Google Scholar]
- Finger L. W.; Hazen R. M.; Hofmeister A. M. High-pressure crystal chemistry of spinel (MgAl2O4) and magnetite (Fe3O4): comparisons with silicate spinels. Phys. Chem. Miner. 1986, 13 (4), 215–20. 10.1007/BF00308271. [DOI] [Google Scholar]
- a Zirbs R.; Lassenberger A.; Vonderhaid I.; Kurzhals S.; Reimhult E. Melt-Grafting for the Synthesis of Core-Shell Nanoparticles with Ultra-High Dispersant Density. Nanoscale 2015, 7, 11216. 10.1039/C5NR02313K. [DOI] [PubMed] [Google Scholar]; b Stone R. C.; Qi B.; Trebatoski D.; Jetti R.; Bandera Y. P.; Foulger S. H.; Mefford O. T. A versatile stable platform for multifunctional applications: synthesis of a nitroDOPA-PEO-alkyne scaffold for iron oxide nanoparticles. J. Mater. Chem. B 2014, 2, 4789–4793. 10.1039/C4TB00198B. [DOI] [PubMed] [Google Scholar]
- Saville S. L.; Stone R. C.; Qi B.; Mefford O. T. Investigation of the stability of magnetite nanoparticles functionalized with catechol based ligands in biological media. J. Mater. Chem. 2012, 22 (47), 24909–24917. 10.1039/c2jm34902g. [DOI] [Google Scholar]
- Jokerst J. V.; Lobovkina T.; Zare R. N.; Gambhir S. S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine (London, U. K.) 2011, 6, 715–728. 10.2217/nnm.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Linegar K. L.; Adeniran A. E.; Kostko A. F.; Anisimov M. A. Hydrodynamic radius of polyethylene glycol in solution obtained by dynamic light scattering. Colloid J. 2010, 72, 279–281. 10.1134/S1061933X10020195. [DOI] [Google Scholar]; b Ziebacz N.; Wieczorek S. A.; Kalwarczyk T.; Fialkowski M.; Holyst R. Crossover regime for the diffusion of nanoparticles in polyethylene glycol solutions: influence of the depletion layer. Soft Matter 2011, 7 (16), 7181–7186. 10.1039/c0sm01357a. [DOI] [Google Scholar]; c Lee H.; Venable R. M.; MacKerell A. D. Jr.; Pastor R. W. Molecular dynamics studies of polyethylene oxide and polyethylene glycol: hydrodynamic radius and shape anisotropy. Biophys. J. 2008, 95 (4), 1590–1599. 10.1529/biophysj.108.133025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Burda C.; Chen X.; Narayanan R.; El-Sayed M. A. Chemistry and properties of nanocrystals of different shapes. Chem. Rev. 2005, 105 (4), 1025–1102. 10.1021/cr030063a. [DOI] [PubMed] [Google Scholar]; b Kang S. M.; Choi I. S.; Lee K.-B.; Kim Y. Bioconjugation of poly(poly(ethylene glycol) methacrylate)-coated iron oxide magnetic nanoparticles for magnetic capture of target proteins. Macromol. Res. 2009, 17 (4), 259–264. 10.1007/BF03218689. [DOI] [Google Scholar]
- Liu D.-F.; Wu W.; Ling J.-J.; Wen S.; Gu N.; Zhang X.-Z. Effective PEGylation of iron oxide nanoparticles for high performance in vivo cancer imaging. Adv. Funct. Mater. 2011, 21 (8), 1498–1504. 10.1002/adfm.201001658. [DOI] [Google Scholar]
- Cornard J.-P.; Rasmiwetti; Muerlin J.-C. Molecular structure and spectroscopic properties of 4-nitrocatechol at different pH: UV-visible, Raman, DFT and TD-DFT calculations. Chem. Phys. 2005, 309 (2–3), 239–249. 10.1016/j.chemphys.2004.09.020. [DOI] [Google Scholar]
- Bronstein L. M.; Huang X.; Retrum J.; Schmucker A.; Pink M.; Stein B. D.; Dragnea B. Influence of iron oleate complex structure on iron oxide nanoparticle formation. Chem. Mater. 2007, 19 (15), 3624–3632. 10.1021/cm062948j. [DOI] [Google Scholar]
- Roonasi P.; Holmgren A. A Fourier transform infrared (FTIR) and thermogravimetric analysis (TGA) study of oleate adsorbed on magnetite nano-particle surface. Appl. Surf. Sci. 2009, 255, 5891–5895. 10.1016/j.apsusc.2009.01.031. [DOI] [Google Scholar]
- Sahoo Y.; Pizem H.; Fried T.; Golodnitsky D.; Burstein L.; Sukenik C. N.; Markovich G. Alkyl Phosphonate/Phosphate Coating on Magnetite Nanoparticles: A Comparison with Fatty Acids. Langmuir 2001, 17 (25), 7907–7911. 10.1021/la010703+. [DOI] [Google Scholar]
- a Xie J.; Xu C.; Kohler N.; Hou Y.; Sun S. Controlled PEGylation of monodisperse Fe3O4 nanoparticles for reduced non-specific uptake by macrophage cells. Adv. Mater. (Weinheim, Ger.) 2007, 19, 3163–3166. 10.1002/adma.200701975. [DOI] [Google Scholar]; b Gruenewald T. A.; Lassenberger A.; van Oostrum P. D. J.; Rennhofer H.; Zirbs R.; Capone B.; Vonderhaid I.; Amenitsch H.; Lichtenegger H. C.; Reimhult E. Core-Shell Structure of Monodisperse Poly(ethylene glycol)-Grafted Iron Oxide Nanoparticles Studied by Small-Angle X-ray Scattering. Chem. Mater. 2015, 27 (13), 4763–4771. 10.1021/acs.chemmater.5b01488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lattuada M.; Hatton T. A. Functionalization of Monodisperse Magnetic Nanoparticles. Langmuir 2007, 23, 2158–2168. 10.1021/la062092x. [DOI] [PubMed] [Google Scholar]; b Boyer C. The design and utility of polymer-stabilized iron-oxide nanoparticles for nanomedicine applications. NPG Asia Mater. 2010, 2, 23–30. 10.1038/asiamat.2010.6. [DOI] [Google Scholar]
- Na H. B.; Palui G.; Rosenberg J. T.; Ji X.; Grant S. C.; Mattoussi H. Multidentate Catechol-Based Polyethylene Glycol Oligomers Provide Enhanced Stability and Biocompatibility to Iron Oxide Nanoparticles. ACS Nano 2012, 6, 389–399. 10.1021/nn203735b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.