

Abstract
Foxp3+ regulatory T cells (Treg) playing a crucial role in the maintenance of immune tolerance and prevention of autoimmune diseases consist of thymus-derived naturally-occurring CD4+Foxp3+ Treg cells (nTreg) and those that can be induced ex vivo with TGF-β (iTreg). Although both Treg subsets share similar phenotypes and functional characteristics, they also have potential biologic differences on their biology. The role of iTreg in regulating B cells remains unclear so far. The suppression assays of Treg subsets on activation, proliferation and antibodies production of B cells were measured using a Treg and B cell co-culture system in vitro. Transwell and antibody blockade experiments were performed to assess the roles of cell contact and soluble cytokines. Treg were adoptively transferred to lupus mice to assess in vivo effects on B cells. Like nTreg, iTreg subset also directly suppressed activation and proliferation of B cells. nTreg subset suppressed B cell responses through cytotoxic manner related to expression of Granzyme A, Granzyme B and Perforin; while the role of iTreg subset on B cells did not involve in cytotoxic action, but depending on TGF-β signaling. Furthermore, iTreg subset can significantly suppress antibody produced by lupus B cells in vitro. Comparison experiments using autoantibodies microarrays demonstrated that adoptive transfer of iTreg had a superior effect than nTreg subset on suppressing lupus B cell responses in vivo. Our data implicate a role and advantage of iTreg subset in treating B cell-mediated autoimmune diseases, boosting the translational potential of these findings.
Keywords: regulatory T cells, TGF-β, Foxp3, B cells, lupus
Introduction
Substantial evidence has revealed that the thymus-derived naturally-occurring CD4+Foxp3+ regulatory T cells (nTreg) are essential for immune homeostasis (1). Lack or dysfunction of nTreg in mice and human was associated with many autoimmune diseases (1–3). In addition, adoptive transfer of nTreg prevents or alters the course of autoimmune diseases in several animal models (4, 5).
It is notable that the therapeutic effect of nTreg on established autoimmune diseases is fairly unsatisfactory (5, 6). Several reasons may account for this inability. First, the low frequency makes sufficient numbers for cell immunotherapy a significant challenge. Although expansion in vitro can overcome this problem, it has been observed that repeated stimulation leads to diminished Foxp3 expression and suppressive activity (7). In addition, T effector cells contaminating in the nTreg may preferentially be expanded following this protocol and therefore this possibly limits their use in autoimmune disease treatment. Third, it is very likely that when antigen-specific effector cells are induced, nTreg cannot counteract the effector T cell responses and function. Additionally, when inflammation is established, a large number of proinflammatory cytokines, such as IL-6, IL-1 and TNF-α are released. It has been reported that increased levels of IL-6 released by activated DCs abolished the suppressive ability of nTreg (8). TNF-α possibly has a similar ability to affect the suppressive activity of nTreg (9). Furthermore, T cells from autoimmune conditions may be resistant to nTreg even though the properties of the nTreg are normal (10). And a final possible reason could be dynamic alteration and plasticity of nTreg in the inflammatory milieu.
Considerable studies have also documented that iTreg induced ex vivo with IL-2 and TGF-β share similar phenotypic and functional characteristics with nTreg (11). iTreg can suppress T cell activation, proliferation, cytokine production and autoimmunity diseases in animal models (12). Moreover, iTreg but not nTreg are resistant to IL-6-deriven Th17 cell conversion (13). While previous studies by others have addressed the possibility of direct suppression on B cell by nTreg subset (14–16), it is less clear whether the iTreg subset has the similar suppressive capacity on B cells. We herein demonstrate that both nTreg and iTreg directly suppress B cells, but unlike nTreg, the suppression of B cells by iTreg does not involve cytotoxicity. We further show that TGF-β signaling is important for the effects of iTreg on B cells. Moreover, iTreg directly suppress autoreactive B cells in vitro and in vivo in lupus mice and display a superior effect on lupus B cell responses compared to nTreg subset.
Materials and methods
Mice
Female C57BL/6 (B6), female Granzyme B deficient, female Perforin deficient mice, female NZB/NZW F1 (NZBWF1/J, BWF1), B6 Rag1−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME). C57BL/6 Foxp3-GFP knock-in mice were generously provided by Dr. Talil Chatilla (University of California Los Angeles). Female New Zealand mixed (NZM) 2328 mice were gifted by Chaim O. Jacob (University of Southern California, Los Angeles). NZM2328 Foxp3-GFP mice were generated by breeding NZM2328 and C57BL/6 Foxp3-GFP together for 12 generations on NZM2328 background. Lymphocyte-specific TGF-β type II receptor (TβRII) conditional knockout (CKO) mice on a C57BL/6 background were generously provided by Dr. Wei Shi (University of Southern California, Los Angeles). All animals were treated according to National Institutes of Health guidelines for the use of experimental animal with the approval of the Penn State University and Sun Yat-sen University Committee for the Use and Care of Animals.
Flow cytometry
The following fluorescence conjugated mouse antibodies were used for flow cytometric analysis from Biolegend (San Diego, CA): FITC-CD69 (H1.2F3), FITC-CD86 (PO3), APC-CD80 (16-10A1), APC-CD138(281-2), PE-B220 (RA3-6B2), Alexa Fluor 647-Granzyme B (GB11), AlexaFlur 488-Foxp3 (150D), PE-IL-10 (JES5-16E3), PE-TNF-α (MP6-XT22), PerCP/cy5.5-CD5 (53-7.3), PE-MHCII-I-Ab (AF6-120.1), APC-CD24 (30-F1), PE-CD23 (B3B4), PE-Ki-67 (16A8); From ebioscience (San Diego, CA): FITC-CD21/CD35 (eBio4E3 (4E3)), PE-Perforin (eBioOMAK-D), PE-CD357(GITR) (DTA-1); From Santa Cruz Biotechnology (Santa Cruz, CA): PE-Granzyme A (3G8.5). Cell subset was stained with mAbs and isotype control indicated above, and analyzed on a FACS Calibur flow cytometer using Cell Quest Software (Becton Dickinson). For intracellular staining, such as Foxp3, Granzyme A, Granzyme B, and Perforin, cells were first stained with surface marker CD4, and further fixed, permeabilized for intracellular staining. Plots figures were prepared using FlowJo Software (Treestar Inc. Ashland, OR).
The generation of CD4+ induced regulatory T cells (iTreg)
Naïve CD4+CD62L+CD25−CD44low T cells were isolated from spleen cells of C57BL/6, TβRII CKO, Granzyme B KO, Perforin KO, BWF1 or NZM2328 mice using naïve CD4+ T cell isolation kit (MiltenyiBiotec, Auburn, CA). Cells were cultured in 48-well plates and stimulated with anti-CD3/CD28 microbeads (1 bead per 5 cells, Invitrogen) in the presence of IL-2 (50 U/ml; R&D) with (iTreg) or without (CD4med) TGF-β (2ng/ml; R&D) for 3 days. RPMI 1640 medium supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin, and 10mM HEPES (Invitrogen) and 10% heat-inactivated FCS (HyClone Laboratories) was used for all cultures. Foxp3 expression was determined by flow cytometry.
Natural regulatory T cell (nTreg) expansion
CD4+CD25+ cells sorted from the thymus in C57BL/6 mice were expanded with anti-CD3/CD28 coated beads (1 bead per 3 cells, Invitrogen) and IL-2 (300 U/ml) for 3 days. 300 U/ml IL-2 was renewed at day 2. In one experiment (Figure 6), CD4+CD25+ cells were isolated from spleen cells. After cultures, cells were harvested and beads were removed. The percentage of Foxp3+ cells was examined by flow cytometry before and after 3 days expansion.
FIGURE 6.
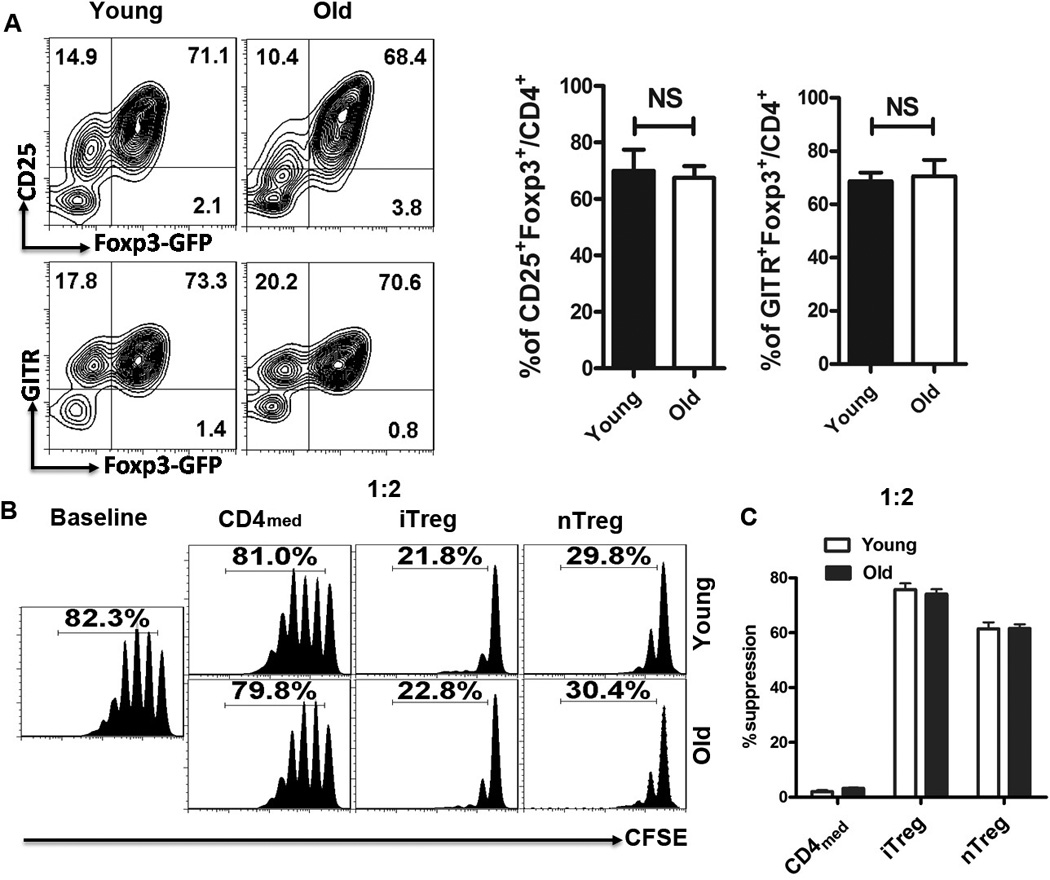
Similar phenotypes and function of iTreg derived from young and old lupus mice. Splenic naive CD4+T cells isolated from either young or old NZM2328 mice were stimulated with TCR signal ± TGF-β for 3 days to generate iTreg and CD4med control cells. Splenic CD4+Foxp3(GFP)+ cells (nTreg) were sorted from the NZM2328 Foxp3-GFP reporter mice and expanded with TCR signal and IL-2 for 3 days. (A) The expression of Foxp3-GFP, CD25, CD4 and GITR of iTreg subsets generated from young and old NZM2328 mice was determined by flow cytometry. (B) Fresh T cells labeled with carboxyfluorescein succinimidyl ester (CFSE) were cultured with or without nTreg, iTreg or CD4med cells (The ratio of T cells: B cell was 1:2 ) in the presence of anti-CD3 (0.025µg/mL) and irradiated APCs (30 Gy, 1:1 ratio) for 3 days. T-cell proliferation was determined by the CFSE dilution rates after 3 days of culture by flow cytometry. All data indicated Mean ± SEM of three separate experiments.
In vitro suppression assays
To examine the suppressive activity of Treg on B cell in vitro, B cells from C57BL/6, TβRII CKO, or BWF1 mice were isolated by B220 positive sorting. Then B cells were stimulated with or without Lipopolysaccharides (LPS) (2µg/mL, Escherichia coli serotype 0111: B4, Sigma-Aldrich) in the presence or absence of nTreg, iTreg or CD4med cells. The graded concentration of Treg to B cell ratios was 1:2–1:16. Cells were cultured for 3 days, and 1 µCi/well of 3H-thymidine was added for the last 16 hours of culture. The proliferation was analyzed by the value of thymidine incorporation. Fresh B cells labeled with carboxyfluorescein succinimidyl ester (CFSE) were cultured with nTreg, iTreg or CD4med cells (The ratio of T cells: B cell was 1:2 to 1:8) in the presence of LPS (2µg/mL), B-cell proliferation was determined by the CFSE dilution rates after 2 days of culture. In addition, CFSE-labeled B cells were pre-incubated with LPS (2µg/mL) for 24 hours and then cells were extensively washed to remove LPS. These B cells were re-cultured with Treg or control cells for additional two days. The proliferative levels of B cells were judged by the rates and intensity of CFSE dilution with flow cytometry.
To determine the mechanism of Treg inhibition, different antibodies, including anti-TGF-β (10 µgml−1; R&D Systems), TGF-β receptor I (ALK5) inhibitor (10 µgml−1; Sigma-Aldrich), anti-IL-10 (10 µgml−1; R&D Systems), anti-IL-10R (10 µgml−1; Biolegend), anti-PD-1 (10 µgml−1; Biolegend), anti PD-L1 (10 µgml−1; Biolegend), anti-CTLA4 (10 µgml−1; Biolegend), anti-GITR (10 µgml−1; Biolegend), Adenosine 5'-(α,β-methylene) diphosphate (APCP, 100 µM; Sigma-Aldrich), Sodium polyoxotungstate (POM1, 100 µM; Tocris Bioscience) and their correlated isotype controls, were added to the culture system described as above.
Apoptosis assays
iTreg, nTreg, or CD4med cells were co-cultured with B cells in the presence or absence of LPS (2µg/mL) for 16 hours. The cells were then collected and stained with Annexin V and 7-amino-actinomycin D (7-AAD) using an Annexin V apoptosis detection kit (BD Biosciences) following the manufacturer’s instructions. Both Annexin-V and 7-AAD expression was measured by FACSCalibur flow cytometer (BD Biosciences) gated on B220+ cells.
Autoantibody detection
To compare the IgG antibody production from B cells in vitro, the supernatants were collected from different cultural systems after 3 days culture. For the in vivo autoantibody detection, mice were bled at the indicated time points, and sera were collected. IgG, IgM and anti-dsDNA autoantibodies were measured by an ELISA as previously described (17). All samples tested for anti-DNA were performed at the same time. Sera were diluted 1/400 or 1/800 for anti-DNA and 1/40,000 for measuring IgG.
Quantitative RT-PCR
Total RNA was extracted from cells by using TRIzol reagent and used to determine the expression and relative level of the TGF-β receptor I (TβRI) in Treg subsets. The mRNA levels of targeted genes were measured by quantitative RT-PCR (Applied Biosystems) by using the SYBR Green RT-PCR reagents Kit (Life technologies). The relative expression of TβRI on T cells before and after LPS stimulation was determined by normalizing expression of each target to hypoxanthine guanine phosphoribosyl transferase (HPRT). TβRI forward primer: 5'-TGT GCA CCA TCT TCA AAA ACA-3' and reverse primer: 5'-ACC AAG GCC AGC TGA CTG-3'; HPRT forward primer: 5'-TGA AGA GCTACT GTA ATG ATC AGT CAA C-3' and reverse primer: 5'-AGC AAG CTT GCA ACC TTA ACC A-3'.
Assessment of lupus-prone mice and adoptive transfer
BWF1 mice were monitored regularly. The sera IgG and anti-DNA autoantibodies were measured by ELISA. Proteinuria was assessed using Albustix reagent strips (Bayer, Elkart, IN). When the proteinuria developed, BWF1 mice were received with a single dose of 300µg anti-mouse CD3 antibody (17A2, BioLegend) to deplete the endogenous CD3+ T cells (18), or the isotype control. 7 days later, CD4med or iTreg were adoptively transferred to the mice with antibody treatment. The percentages of plasma were detected and sera were collected 0, 1, 2 weeks after cell transfer for IgG measurement. While homogeneous iTreg (derived from 10–12 weeks age NZM2328) and nTreg (sorted from the thymus in 10–12 weeks age NZM2328) were adoptively transferred to another lupus-prone mouse, NZM2328 (more than 4 months age), the sera were also collected. Proteinuria and anti-DNA autoantibodies were measured. Sera IgG and IgM autoantibodies cluster was measured by Autoantigen Microarrays.
In vivo suppression assays
Fresh B cells were obtained from NZM2328 (16 weeks age) and stimulated with LPS (2µg/mL) for 24 hours, then washed to remove LPS. These B cells were co-transferred with nTreg, iTreg or CD4med cells (The ratio of Treg cells: B cell was 1:2, B cells were 8 × 106 for each mouse) into B6 Rag1−/− mice. In some groups, TGF-β receptor I (ALK5) inhibitor (1mg/kg), anti-IL-10R (1mg/kg), or IgG (control for anti-IL-10R) or DMSO (control for ALK5i) were given to mice by IP injection at day 0 and day 2. The mice were sacrificed and splenic B cells were harvested and prepared to test Ki-67 expression by flow cytometry.
Autoantibody profiling using Autoantigen Microarrays
Autoantibody activities against a penal of 123 autoantigen specificities were measured using an Autoantigen Microarray platform developed by University of Texas Southwestern Medical Center (19, 20). Briefly, sera samples were pretreated with DNAse-I and then diluted 1:50 in PBST buffer for autoantibody profiling. The autoantigen array bearing autoantigens and control proteins were printed in duplicates onto Nitrocellulose film slides (Grace Bio-Labs). The diluted sera samples were incubated with the autoantigen arrays and autoantibodies were detected with cy3-labeled anti-mouse IgG and cy5-labeled anti-mouse IgM using a Genepix 4200A scanner (Molecular Device) with laser wavelength of 532 nm and 635 nm. The resulting images were analyzed using Genepix Pro 6.0 software (Molecular Devices). The median of the signal intensity for each spot was calculated and subtracted the local background, and data obtained from duplicate spots were averaged. Finally, the net fluorescence intensity (NFI) of the antibody against each antigen was used to generate heatmaps using Cluster and Treeview software (http://rana.bl.gov/EisenSoftware.htm). Each row in the heatmap represents an autoantibody and each column represents a sample. Red color represents the signal intensity higher than the mean value of the raw and green color means signal intensity is lower than the mean value of the raw. Black color represents the signal closing or equal to the mean value of the raw, and gray color indicates value is 0 or missing. Microarray data has been deposited in the ArrayExpress public database (https://www.ebi.ac.uk/arrayexpress/browse.ht ml, accession number, E-MTAB-4441, for reviewer access, please login with username: Reviewer_E-MTAB-4441, password: 4yTK1190).
Statistical analysis
Data were expressed as Mean ± SEM unless otherwise indicated. Data were analyzed using the unpaired t-tests (Mann-Whitney) or paired t-tests for comparison between two groups or ANOVA for comparison among multiple groups as appropriate. Differences were considered statistically significant when p < 0.05.
Results
Both iTreg and nTreg subsets directly suppress B cell responses
To determine whether iTreg can suppress B cell responses, the effects of both nTreg and iTreg cells on the activation, proliferation and autoantibody production of stimulated B cells were compared. We detected the expression of CD69, CD80, CD86 and CD138 on B cells in the Treg-B cell co-culture system. Unlike the control CD4med (CD4+ T cells stimulated with TCR and IL-2 without TGF-β), both iTreg and nTreg similarly suppressed the expression of CD69, CD80, CD86 and CD138 on B cells (Fig. 1A), indicating that iTreg also suppress B cell activation and differentiation. It was noted that CD4med also slightly suppressed B cell differentiation (Fig. 1A), possible explanations include 1) CD4med may contain minor Foxp3+ cells; 2) CD4med express high levels of CD25 that consume IL-2 and then indirectly reduce the B cell differentiation.
FIGURE 1.
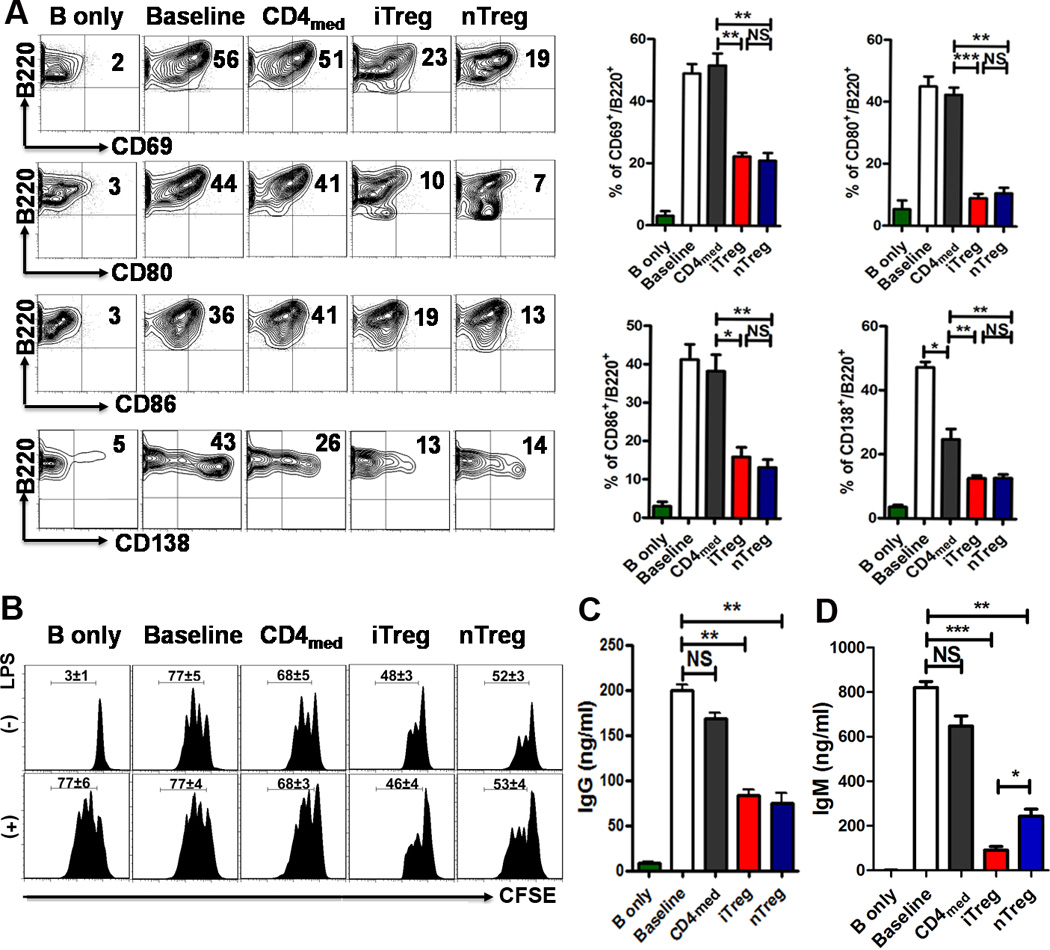
Both iTreg and nTreg directly suppress B cells. B cells were isolated from C57BL/6, stimulated with (Baseline) or without LPS (2 µg/ml) (B only) in the presence or absence of nTreg, iTreg and CD4med (ratio of T to B cells was 1:2 to 1:4). (A) The expression of B cell activation (CD69, CD80, and CD86) and differentiation (CD138) markers was detected after 20 and 48 hours of culture by flow cytometry. Typical FACS plots (left) and summary data (right) were shown. (B) Fresh B cells labeled with carboxyfluorescein succinimidyl ester (CFSE) were cultured with nTreg, iTreg or CD4med cells (The ratio of T cells: B cell was 1:2 to 1:8) in the presence of LPS (2µg/mL), B-cell proliferation was determined by the CFSE dilution rates after 2 days of culture. In lower panel, CFSE-labeled B cells were pre-incubated with LPS (2µg/mL) for 24 hours and then cells were extensively washed out to remove LPS. Treg or CD4med were co-cultured with LPS pretreat-B cells for another 2 days, the proliferation was analyzed by the flow cytometry. (C and D) The supernatants were collected from above cultural systems after 3 days culture, and the IgG, IgM secretion was detected by ELISA. The data indicate the Mean ± SEM of 3 separated experiments (*p<0.05, **p<0.01, ***p<0.001; baseline or CD4med vs. Treg group).
Using CFSE labeling, it is clearly observed that both Treg subsets significantly suppressed B cell proliferation (1:2 ratio; CD4med vs. Treg subsets, p<0.001) (Fig. 1B). Interestingly, these Treg subsets even inhibited the proliferation of B cells that had been pre-treated with LPS (Fig.1B low panel). 3H incorporation assay further validated that both Treg subsets suppressed B cell proliferation (data not shown). Moreover, both nTreg and iTreg subsets also significantly suppressed TNF-α production by B cells (Supplemental Fig. 1A), but only iTreg cells upregulated IL-10 production by B cells (Supplemental Fig. 1A). However, both Treg subsets did not up- or down-regulate MHC-II, CD21/35, CD23 and CD24 related to antigen presenting ability and Breg phenotypes (Supplemental Fig. 1B). Further work is needed to determine whether iTreg can induce Breg cell development.
The in vitro suppression by the iTreg subset on B cell proliferation was equivalent to the nTreg subset at the ratios of 1:2 to 1:8 (Fig. 1B), and exhibited a dose-dependent effect (data not shown). To determine if the iTreg subset suppresses B cell function, supernatants from a Treg-B cell co-culture system were tested, and IgG production by activated B cells was markedly reduced in cultures containing either nTreg or iTreg cells than that in cultures containing CD4med or no T cells (Fig. 1C). The suppressive effect of both nTreg and iTreg on IgG levels produced by B cells was similar (Fig.1C). Furthermore, both nTreg and iTreg subsets, but not CD4med, markedly suppressed IgM production by B cells (Fig. 1D). The IgM levels were significantly lower in cultures containing iTreg than those containing nTreg cells (Fig. 1D), implicating the iTreg subset as having a stronger effect on B cell-mediated IgM production.
iTreg subset suppresses B cell response in a cell-contact dependent manner involving TGF-β but not cytoxicity
Given previous studies attribute the suppressive ability of nTreg subset on B cell responses to cytotoxic mechanisms (14, 18), we next tested if the iTreg subset also has a similar target cell killing effect on B cells. When B cell and CD4med were co-cultured, the ongoing apoptosis levels of B cells were substantially elevated, and co-culturing with nTreg subset further increased the B cell apoptosis, confirming the previous reports that nTreg suppress B cells through their killing (14). Nonetheless, the addition of iTreg subset did not change the levels of B cell apoptosis (Fig. 2A, 2B). To learn whether this functional difference is related to the expression levels of cytotoxic molecules on different Treg subsets, we detected the expression of Granzyme A, Granzyme B and Perforin since they are expressed with some extents on nTreg subsets (14). While the nTreg subset expressed Granzyme A and Granzyme B (8–12%), as well as Perforin (2%), the expression of these cytotoxic molecules in iTreg subset was almost undetectable (Fig. 2C, 2D). To exclude the possibility that the cell culture may change the expression of these molecules on Treg subsets, we also analyzed the expression of Granzyme A, Granzyme B and Perforin before and after co-cultures. These data revealed that Granzyme A and Granzyme B (Supplemental Fig. 2) and Perforin (data not shown) were only expressed on the nTreg subset and cell culture did not change their levels, while the iTreg subset did not express these molecules before or after the co-cultures (Supplemental Fig. 2).
FIGURE 2.
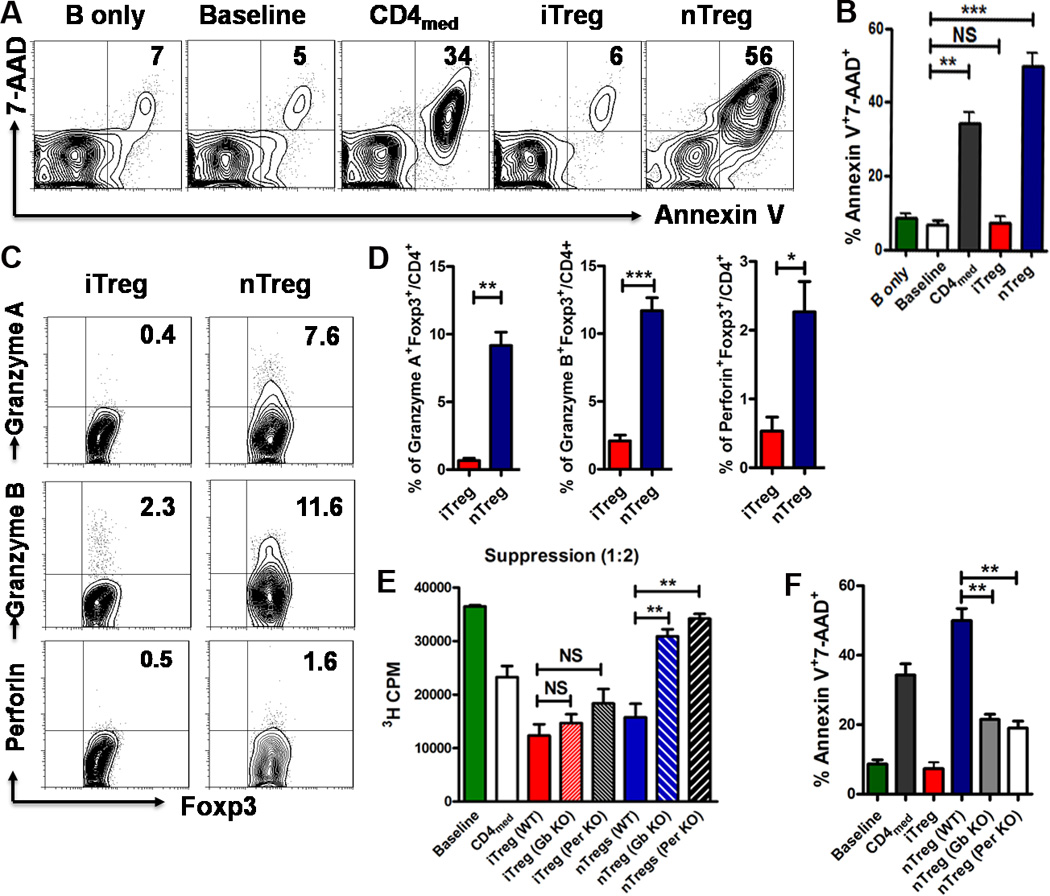
iTreg and nTreg have a different mechanism of action on suppressing B cell responses regarding to cell killing and TGF-β signals. (A and B) iTreg, nTreg, or CD4med cells were co-cultured with B cells in the presence or absence of LPS as Fig. 1A. After 16 hours of culture, the expression of cell apoptosis marker (Annexin-V, 7-AAD) was detected by Flow Cytometry. Representative FACS plots gated on B220+ cells (left) and the summarized data (right) of three separated experiments were shown. (C and D) The expression of Granzyme A, Granzyme B and Perforin gated on Foxp3+ iTreg or nTreg was determined by FACS with intracellular staining (n=3). (E and F) CD4med, nTreg or iTreg isolated or induced from WT, Granzyme B-deficient (Gb KO) or Perforin-deficient mice (Per KO) were co-cultured with WT B cells (+/− LPS) for 3 days, 3H-thymidine was added for the last 16 hours of culture. The proliferation was analyzed by 3H-thymidine incorporation. The expresson of Annexin-V and 7-AAD on B220+ cells was measured by flow cytometry. The data indicated Mean ± SEM of three separate experiments.
We then studied the functional link of these molecules between Treg subset and B cell. While the nTreg subset isolated from WT mice suppressed B cell proliferation and induced B cell apoptosis, these cells failed to suppress B cell proliferation and apoptosis when isolated from Granzyme B or Perforin knockout mice (Fig. 2E, 2F). Conversely, the suppression of iTreg subsets generated from WT, Granzyme B or Perforin knockout mice on B cell responses were similar and sustained (Fig. 2E). Thus, the TGF-β-induced iTreg subset suppresses B immune responses independent of the cytotoxicity mechanism that characterizes the nTreg subset.
Several studies have demonstrated that cell contact is required for suppression by both nTreg and iTreg subsets. To determine the importance of cell contact in Treg-mediated suppression of B cells, Treg subsets were separated from target B cells by Transwell inserts. Under these conditions, little suppression was observed in either nTreg or iTreg co-culture system (Fig. 3A), suggesting that cell contact is needed for suppressive function of both Treg subsets on B cells. To further explore the mechanisms responsible for iTreg-mediated suppression on B cells, we analyzed several potential candidates, including anti-IL-10, anti-IL-10R, anti-TGF-β, anti-PD-1, anti PD-L1, anti-CTLA4, anti-GITR, APCP (CD73 inhibitor) or POM1 (CD39 inhibitor), and no effect was observed on Treg-mediated B cell suppression (data not shown). However, using the TGF-β receptor I (TβRI) antagonist-ALK5 inhibitor (Fig. 3B) or B cells isolated from TβRII knockout mice (Fig. 3C), we found the suppressive effect of iTreg subset on B cells disappeared. This result is not surprising, since we previously have reported that the iTreg subset expressed surface TGF-β and secreted activated TGF-β (21). We further demonstrated that iTreg also express increased levels of TβRI, but LPS stimulation did not change the TβRI expression on Treg subsets (Fig. 3D). That is reasonable since LPS mainly stimulates B cells but not T cells. This finding demonstrates that iTreg suppression is different from that of the nTreg, whose suppression is independent upon TGF-β signal (22).
FIGURE 3.

iTreg suppress B cells in a cell-contact dependent manner involving TGF-β signals. (A and B) iTreg, nTreg, and CD4med cells were prepared and co-cultured with B cells with or without LPS for 3 days as described in Figure 1A. In some experiments, transwell was used for separating the Treg from B cells. 3H-thymidine was added for the last 16 hours of culture. The proliferation was analyzed by the value of thymidine incorporation in triplicate wells. Data was presented as the mean ± SEM of three separate experiments. **p<0.01 means no transwell group versus transwell group. (B) ALK5i and DMSO were added to the culture system as described above. 3H-thymidine was added for the last 16 hours of culture. Results (Mean ± SEM) were the summary of three independent experiments. ***p<0.001 means ALK5i group vs. DMSO group. (C) iTreg, nTreg, and CD4med cells were co-cultured with B cells from WT or TβRII lymphocyte conditional knockout (CKO) mice with or without LPS for 3 days. The suppressive activity of Treg or control cells from WT and TβRII CKO mice was analyzed. The data indicated Mean ± SEM of three separate experiments. **p<0.01 means WT group versus TβRII CKO group. (D) Total RNA was extracted from iTreg, nTreg, and CD4med cells (+/− LPS) by using TRIzol reagents, the mRNA levels of targeted genes were measured by quantitative RT-PCR (Applied Biosystems) by using the SYBR Green RT-PCR reagents Kit (Life technologies). The relative expression of TβRI was determined by normalizing expression of each target to hypoxanthine guanine phosphoribosyl transferase (HPRT). Data were presented as Mean ± SEM of triplicate samples by one experiment and similar results were repeated twice.
An in vivo experiment further validates that the iTreg suppression does need TGF-β signal. B cell pretreated with LPS were co-transferred with CD4med, iTreg or nTreg (2:1 ratio) into Rag1−/− mice, in some groups, ALK5i, anti-IL-10R antibody, control IgG or DMSO (for Alk5i control) were i.p. injected (Fig. 4A). Ki-67 expression by LPS pretreated B cells was dramatically increased 3 days after cell transfer; compared to co-transfer of CD4med, co-transfer of iTreg or nTreg significantly prevented the upregulation of Ki-67 on B cells in vivo (Fig. 4B, 4C). iTreg effect was also superior to nTreg against B cell proliferation in vivo (Fig. 4B, 4C). iTreg suppression was mostly dependent upon TGF-β signal since blockade of TβRI almost completely abolished the suppression of iTreg on B cell proliferation in vivo. However, IL-10 seems to also play a partial role in iTreg-mediated B cell suppression in vivo (Fig. 4B, 4D).
FIGURE 4.
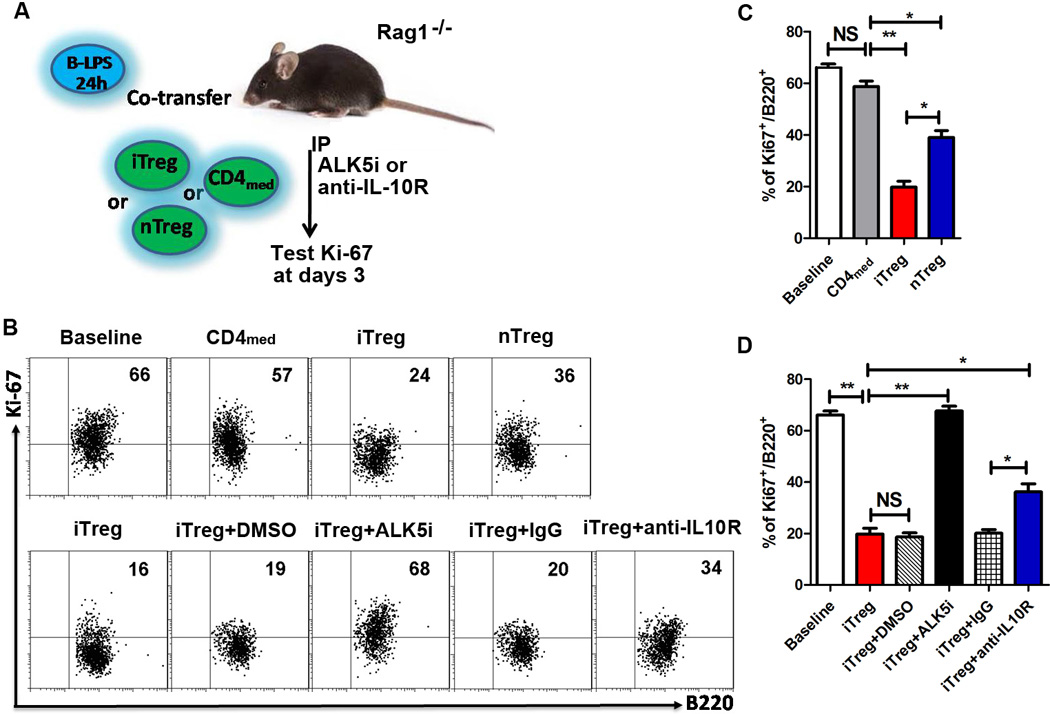
Inhibitor role of iTreg on B cells proliferation is mostly dependent upon TGF-β signal in vivo, and partially on IL-10 signal. (A–D) NZM2328 fresh B cells were stimulated with LPS (2µg/mL) for 24 hours, then co-transferred with nTreg, iTreg or CD4med (2:1 ratio) into Rag1−/− mice, in some groups, ALK5i (1mg/kg), anti-IL-10R antibody (1mg/kg), DMSO (for Alk5i control) or control IgG were i.p. injected at day 0 and day 2. The mice were sacrificed and spleen B cells were harvested and prepared to test Ki-67 expression on B cells by flow cytometry at days 3. There were five mice in each group. The data indicated Mean ± SEM of two separate experiments. *p<0.05, **p<0.01 means control cell group vs. iTreg group after cell transfer.
Although some groups have a concern that iTreg are unstable (23, 24), we and other have demonstrated that iTreg are stable under the right protocol to generate iTreg (13, 25, 26). New data here demonstrated that iTreg were fairly stable in vivo 3 weeks after cell transfer (Supplemental Fig. 3). iTreg were completely resistant to Th17 cell conversion although few of them had converted into Th1 cells (Supplemental Fig. 3). Previous study has revealed that iTreg subset maintains their functional feature even they began to express IFN-γ (27).
iTreg subset directly suppresses autoreactive B cells in vitro and in vivo in lupus mice
Previous study has documented that nTreg have a mildly suppressive effect on lupus (5). To determine whether the iTreg subset displays suppressive effects on lupus B cells, we first approached this using in vitro co-culture experiments as described above, and showed that iTreg but not CD4med control cells generated from young or old BWF1 mice significantly suppressed IgG production by lupus B cells (Fig. 5A).
FIGURE 5.
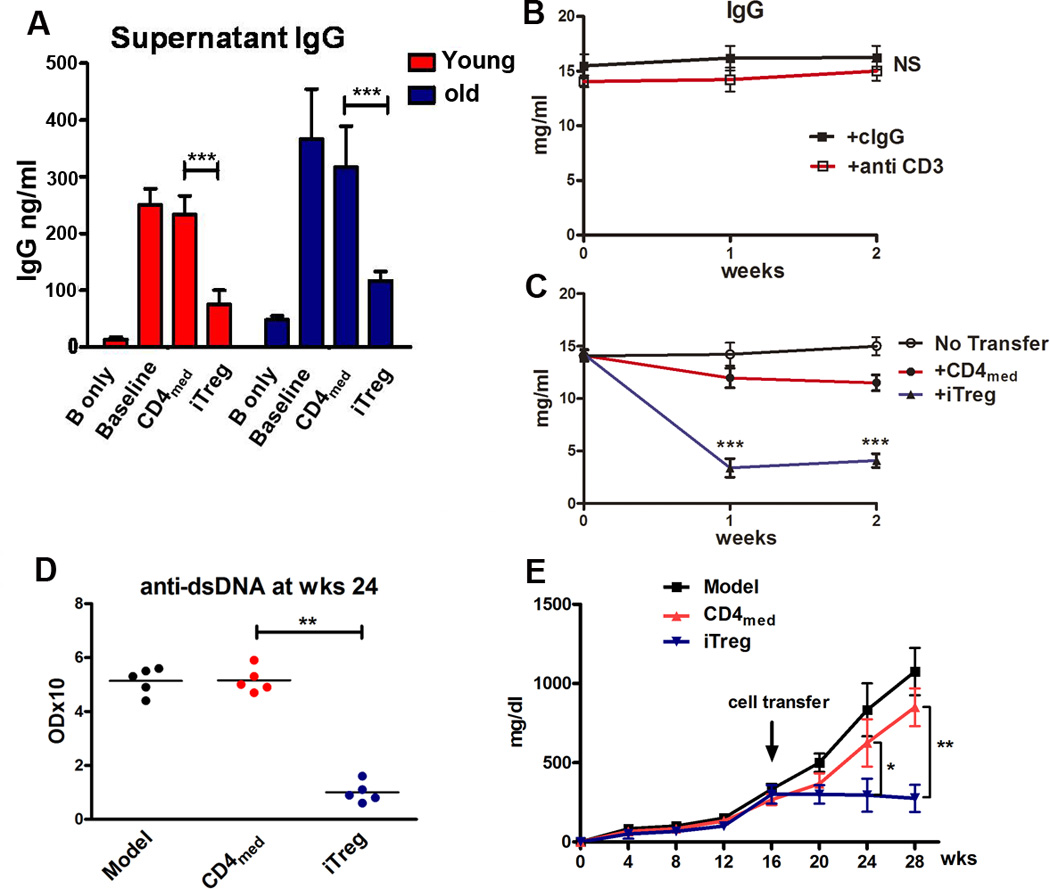
iTreg directly suppress autoreactive B cells in vitro and in vivo in lupus-prone mice. (A) iTreg and CD4med cells induced from young or old BWF1 mice were co-cultured with B cells in the presence or absence of LPS for 3 days, the supernatants were harvested for measurement of IgG production by ELISA. Data were presented as the Mean ± SEM. ***p<0.001 means control CD4med cell group versus iTreg group. (B and C) iTreg or control CD4med cells induced from BWF1 mice that had developed high titers of anti-DNA and proteinuria were adoptively transferred to old lupus mice with evident lupus syndromes. These lupus mice have been previously depleted of endogenous T cells (300ug anti-mouse CD3 antibody, iv) 1 day before the cell transfer. The IgG antibody production was measured in different lupus mice groups at the 0, 1, 2 week time points after cell transfer. Data were presented as the Mean ± SEM. ***p<0.001 means CD4med cell group versus iTreg group. (D and E) iTreg or CD4med cells induced from NZM2328 mice with evident lupus syndromes were adoptively transferred to established lupus mice (16 weeks age), the anti-dsDNA IgG and proteinurina level were measured. *p<0.05, **p<0.01 means CD4med control cell group vs. iTreg group at weeks 24 and 28.
To further determine whether iTreg directly suppress B cells in lupus mice in vivo, iTreg or CD4med control cells were adoptively transferred to old lupus mice that had been previously depleted of endogenous T cells. Treatment with iTreg but not CD4med cells significantly decreased IgG antibody and anti-dsDNA titers in lupus mice (Fig. 5B, 5C, 5D). Moreover, adoptive transfer of iTreg to 16-week old lupus mice that had developed some levels of proteinuria almost completely prevented the increase in proteinuria after 12 weeks of cell transfer, while CD4med did not inhibit the development of proteinuria in lupus mice (Fig. 5E). We also found that the percentages of blood CD138+ plasma cells were significantly reduced in iTreg treatment group compared to CD4med cell treatment group (Supplemental Fig. 4A, 4B). Although iTreg also slightly reduced the frequency of CD138+ plasma cells in spleen and bone marrow, the effect was not significantly different from CD4med treatment (Supplemental Fig. 4C–E).
We demonstrated that TGF-β similarly induced CD4+CD25+Foxp3+GITR+ cells from either young (10 weeks) or old (16 weeks) NZM2328 mice (Fig. 6A). Moreover, the suppressive activity exerted by iTreg induced from either young or old mice was comparable, as well as similar to splenic nTreg subsets from same aged mice (Fig. 6B). We choose mice with age of 16 weeks due to the fact that NZM2328 mice at 12–16 weeks have developed renal pathology (28, 29). This result suggests one may induce iTreg from the established autoimmune diseases, having an important clinical implication.
We directly compared the effects of nTreg and iTreg subsets on lupus B cells in NZM2328 lupus mice. While the titers of almost all of antibodies (both IgG and IgM) as indicated by Net fluorescence intensity (NFI) are elevated in lupus mice from days 0 to days 32, infusion of either nTreg or iTreg not only prevented the continuous elevation of autoantibodies, but reduced these antibodies, particularly at 32 days after cell transfer (Fig. 7). The mircoassays have included almost all of autoantibodies related to lupus. Except for anti-KU(P70/P80), -PCNA, and -TTG IgG, the levels of all auto-antibodies (IgG) were significantly decreased in lupus mice received iTreg than in lupus mice received nTreg subset (Fig. 7A). Regarding autoantibodies (IgM), iTreg subset treatment displayed a superior suppressive effect on production of auto-antibodies compared to nTreg subset treatment (Fig. 7B).
FIGURE 7.
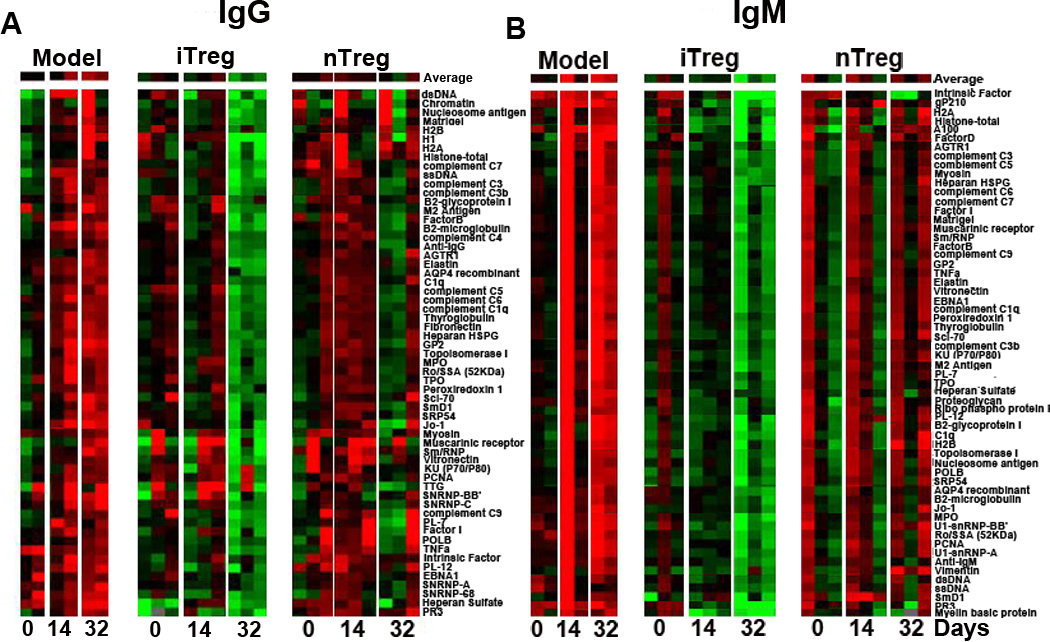
Superior effect of iTreg on B cells in vivo in lupus mice. Each row in the heatmap represents an autoantibody and each column represented a sample. Red color represents the signal intensity higher than the mean value of the raw and green color means signal intensity is lower than the mean value of the raw. Black color represents the signal close to or equal to the mean value of the raw, and gray color indicates value is 0 or missing. (A and B) iTreg induced from NZM2328 lupus-prone mice (10–12 weeks old) and nTreg sorted from the thymus of 10–12 weeks old NZM2328 lupus-prone mice were adoptively transferred into old NZM2328 mice (>4 months age) with established lupus. The serum IgG and IgM autoantibody clusters were detected by microarrays at days 0, 14, 32 after cell transfer. Green, black, and red represent Net Fluorescent Intensity (NFIs) below, close to, and above the mean. Data are the representative of serum autoantibody heatmap from NZM2328 mice (n=2), NZM2328 mice received iTreg (n=3), and NZM2328 received nTreg (n=3) at days 0, 14, 32 after cell transfer. Experiments were repeated twice with the similar results.
We also measured the dynamic changes of total IgG and IgM auto-antibodies. As shown in Figure 8A and 8B, infusion of either nTreg or iTreg reduced IgG levels on 2 weeks after cells transfer. This effect was further enhanced on 4 weeks after cell transfer. Nonetheless, iTreg infusion resulted in greater suppression against autoantibodies production, particularly on 4 weeks after cell transfer. iTreg but not nTreg subset controlled IgM auto-antibodies production on 2 weeks after cell transfer, although nTreg restored their suppression on week 4, the effect of iTreg subset on IgM autoantibodies production suppression was significantly greater than nTreg subset at that time point (Fig. 8A, 8B).
FIGURE 8.

Immunoglobulin (Ig)G and IgM reactivity profiles to selected antigens in lupus mice following cell therapy. (A and B) the mean of Net Fluorescent Intensity of total serum IgG and IgM was shown in NZM2328 model (n=4), NZM2328 received iTreg (n=6) and NZM2328 received nTreg (n=6) at days 0, 14, 32 after cell transfer. (C–L) the IgG and IgM seroreactivities to dsDNA, Matrigel, Thyroglobulin, Histone and Myosin determined by autoantigen arrays in NZM2328 lupus mice (n=4), or NZM2328 received iTreg (n=6) or nTreg (n=6) in indicated days. P-values indicate t-test results between different groups. *p<0.05; **p<0.01.
We simultaneously observed the changes of several important auto-antibodies related to lupus development such as subsets of auto-antibodies of IgG and IgM: anti-dsDNA, anti-Matrigel, anti-Thyroglobulin, anti-Histone, and anti-Myosin. nTreg subset infusion significantly suppressed IgG anti-Thyroglobulin, IgM anti-Thyroglobulin, IgM anti-Matrigel and IgG anti-Histone at 4 weeks after cell transfer, however, they failed to suppress IgG anti-dsDNA, IgM anti-dsDNA, IgG anti-Matrigel, IgM anti-Histone, IgG anti-Mysin and IgM anti-Myosin at 4 weeks after cell transfer. Conversely, iTreg subset infusion significantly suppressed all of IgG and IgM antibodies mentioned above at 4 weeks after cell transfer (Fig 8C–L). These results strongly imply that iTreg subset may have more potent suppressive effects on antibody production by autoreactive B cells in lupus mice.
Discussion
Treg cells modulate immune tolerance through immune regulation mechanisms. Treg cells suppress T and other cells through complicated but as yet poorly understood mechanisms. Although T cells are considered to be the main target of Treg cells, recent studies have also demonstrated that Treg cells act on other immune cells as well as non-immune cells (30–34), implicating Treg cells in regulating tolerance balance through a wide range of influence.
Although pathogenic T cells are a key culprit, B cells are also required for the pathogenesis of autoimmune diseases. B cells are clearly needed for development of collagen-induced arthritis in mice and mice develop an arthritis that can be driven almost entirely by immunoglobulins (35–37). B cells can contribute to autoimmune diseases by the secretion of autoantibodies, presentation of autoantigen, secretion of inflammatory cytokines, modulation of antigen processing and presentation, and generation of ectopic germinal center (37).
Thus, the suppression of B cell function provides a new approach to combat autoimmune and inflammatory disease. B-cell depletion using Rituximab has been used for the treatment of a number of autoimmune and chronic inflammatory diseases (38, 39). Rituximab treatment results in nearly undetectable circulating B-cell levels one month after therapy and B cell counts remain low for 6–12 months (40). However, its role is limited by the fact that long-lived plasma cells and bone marrow stem cells are not directly depleted (37, 41), and B cells located in the peritoneal cavity are also strongly resistant to depletion therapy (42).
Treg cell therapy may provide an innovative strategy for controlling B cell-mediated diseases. It has been reported that nTreg suppress B cell responses as T cells (14, 18), nonetheless, the mechanisms underlying are completely different. While iTreg cells suppress T cell by cell contact or suppressive cytokines, the role played by nTreg cells on B cells is related to direct B cell killing (14). This raises a concern as to whether nTreg cells are real suppressor cells and whether nTreg cell therapy is appropriate to treat B cell-mediated diseases.
iTreg are another Treg subset that has displayed similar phenotypes and suppressive activity in some autoimmune diseases (43–45). We herein demonstrated that iTreg also directly suppress B cells but their mechanism of action is completely different from nTreg and is almost completely independent of cell killing. This difference could be explained by different expression of Perforin and Granzyme A/B on both Treg subsets since nTreg express much higher levels of these molecules compared to iTreg subset. This observation was further confirmed by the effect of Treg subsets using Perforin and Granzyme knockout mice.
We further revealed that the iTreg subset suppresses B cell responses mainly through the TGF-β receptor signaling pathway. This was shown by either adding TGF-β receptor I antagonist, or by using B response cells derived from TβRII knockout mice. It is consistent with previous studies showing that iTreg subset expresses surface TGF-β and secreted active TGF-β (21), and suppress T and dendritic cells through TGF-β signaling (46).
The current study demonstrates that iTreg subsets also suppressed B cell differentiation to plasma cell in circulating blood, this is likely that iTreg cells eventually reduce autoantibodies production in lupus. Although the effect of iTreg on plasma cell development in spleen and bone marrow was minimal, given most short and long-lived plasma cells locate on spleen and bone marrow, it is unclearly whether the long-term effect of iTreg subsets on autoantibodies in lupus will be compromised.
We also studied the phenotype and cytokine profiles of B cells. iTreg significantly suppressed production of inflammatory cytokines but increased IL-10 by B cells. Although the B cells do not display the phenotypic characteristic of B regulatory (Breg) cell after iTreg treatment, the increase of IL-10 production by B cells is likely significant since IL-10+ B cells have been considered as B regulatory (Breg) cells that also play an important role in controlling inflammatory diseases. As IL-10 is an intracellular cytokine, we are currently unable to sort IL-10+ B cells to analyze their functional activity. We are developing IL-10 reporter and Foxp3/IL-10 double reporter mice that will enable us to determine whether iTreg can induce Breg cells and IL-10-producing Tr1 cells eventually. Although all of these Treg subsets have a capacity to regulate immune responses, their effects are different. For example, Foxp3+ cells are important for initial immune suppression, while Tr1 cells are more important for long-term immune tolerance in a transplantation model (47). We previously have demonstrated that iTreg cells can induce the formation of tolerogenic DC or of new generation of Treg cells that builds the foundation of “infectious tolerance” and contributes to the long-term therapeutic role in lupus and other autoimmune diseases (30, 48). It has been noted that iTreg subset did not suppress MHC-II expression on B cells, it warrants a further study to determine if the antigen-presenting ability of B cells keeps normal after iTreg treatment.
One of the interesting observations is that the iTreg subset has an advantage in controlling B cell-mediated immune response. While nTreg cell subset mildly suppressed IgG and IgM autoantibodies, iTreg cell subset markedly suppressed both IgG and IgM autoantibodies in lupus mice. Using antibody microassays, we have analyzed most autoantibodies related to lupus. These observations have strongly suggested that iTreg subset has a superior effect on the production of IgG, particularly of IgM antibodies compared to nTreg cells in lupus mice. It is possible that iTreg cells are stable in the inflammatory condition (13, 27). Although others reported that iTreg subset is unstable (24), using the comparison study, we recently demonstrated iTreg are stable and functional as long as the appropriate protocol for the differentiation of iTreg is used (49). Thus, the current findings may provide a new strategy of using iTreg cell for treating lupus and other B cells-mediated autoimmune and inflammation diseases without side effect of B cell killing/depletion. The clinical trial on iTreg therapy will determine this feasibility. Nonetheless, this observation may at least have translational applications.
Supplementary Material
Acknowledgments
The study was in part supported by NIH AR059103, AI084359, Natural Science Foundation of Guangdong Province (Grant No. 2014A030308005) and National Natural Science Foundation of China (Grant No. 81400739).
S.G.Z and AP. X conceived the study. Y.L, WQ.C, JL.W, J.L, M.Y, YQ.X, F.H, LM.R, DH.L and QZ.L performed experiments. Y.L, WQ.C, B.L, JX.S, and S.G.Z analyzed the data. Y.L, WQ.C and S.G.Z wrote the manuscript. N.J.O critically revised the manuscript.
Abbreviations used in this article
- Treg
Foxp3+ regulatory T cells
- B6
C57BL/6
- BWF1
NZB/NZW F1
- NZM
New Zealand mixed
- TβRI
TGF-β type I receptor
- TβRII
TGF-β type II receptor
- CKO
conditional knockout
- LPS
Lipopolysaccharides
- CFSE
carboxyfluorescein succinimidyl ester
- ALK5i
TGF-β receptor I inhibitor
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 2.Yan B, Ye S, Chen G, Kuang M, Shen N, Chen S. Dysfunctional CD4+,CD25+ regulatory T cells in untreated active systemic lupus erythematosus secondary to interferon-alpha-producing antigen-presenting cells. Arthritis Rheum. 2008;58:801–812. doi: 10.1002/art.23268. [DOI] [PubMed] [Google Scholar]
- 3.Morgan ME, Sutmuller RP, Witteveen HJ, van Duivenvoorde LM, Zanelli E, Melief CJ, Snijders A, Offringa R, de Vries RR, Toes RE. CD25+ cell depletion hastens the onset of severe disease in collagen-induced arthritis. Arthritis Rheum. 2003;48:1452–1460. doi: 10.1002/art.11063. [DOI] [PubMed] [Google Scholar]
- 4.DiPaolo RJ, Glass DD, Bijwaard KE, Shevach EM. CD4+CD25+ T cells prevent the development of organ-specific autoimmune disease by inhibiting the differentiation of autoreactive effector T cells. J Immunol. 2005;175:7135–7142. doi: 10.4049/jimmunol.175.11.7135. [DOI] [PubMed] [Google Scholar]
- 5.Scalapino KJ, Tang Q, Bluestone JA, Bonyhadi ML, Daikh DI. Suppression of disease in New Zealand Black/New Zealand White lupus-prone mice by adoptive transfer of ex vivo expanded regulatory T cells. Journal of immunology (Baltimore, Md. : 1950) 2006;177:1451–1459. doi: 10.4049/jimmunol.177.3.1451. [DOI] [PubMed] [Google Scholar]
- 6.Bardos T, Czipri M, Vermes C, Finnegan A, Mikecz K, Zhang J. CD4+CD25+ immunoregulatory T cells may not be involved in controlling autoimmune arthritis. Arthritis Res Ther. 2003;5:R106–R113. doi: 10.1186/ar624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chai JG, Tsang JY, Lechler R, Simpson E, Dyson J, Scott D. CD4+CD25+ T cells as immunoregulatory T cells in vitro. Eur J Immunol. 2002;32:2365–2375. doi: 10.1002/1521-4141(200208)32:8<2365::AID-IMMU2365>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 8.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 9.Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. 2006;108:253–261. doi: 10.1182/blood-2005-11-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu L, Zhang L, Yi Y, Kang HK, Datta SK. Human lupus T cells resist inactivation and escape death by upregulating COX-2. Nat Med. 2004;10:411–415. doi: 10.1038/nm1005. [DOI] [PubMed] [Google Scholar]
- 11.Zhou X, Kong N, Wang J, Fan H, Zou H, Horwitz D, Brand D, Liu Z, Zheng SG. Cutting edge: all-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. Journal of immunology (Baltimore, Md.: 1950) 2010;185:2675–2679. doi: 10.4049/jimmunol.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lan Q, Fan H, Quesniaux V, Ryffel B, Liu Z, Zheng SG. Induced Foxp3(+) regulatory T cells: a potential new weapon to treat autoimmune and inflammatory diseases? J Mol Cell Biol. 2012;4:22–28. doi: 10.1093/jmcb/mjr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng SG, Wang J, Horwitz DA. Cutting edge: Foxp3+CD4+CD25+ regulatory T cells induced by IL-2 and TGF-beta are resistant to Th17 conversion by IL-6. Journal of immunology (Baltimore, Md.: 1950) 2008;180:7112–7116. doi: 10.4049/jimmunol.180.11.7112. [DOI] [PubMed] [Google Scholar]
- 14.Zhao DM, Thornton AM, DiPaolo RJ, Shevach EM. Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood. 2006;107:3925–3932. doi: 10.1182/blood-2005-11-4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim HW, Hillsamer P, Banham AH, Kim CH. Cutting edge: direct suppression of B cells by CD4+ CD25+ regulatory T cells. Journal of immunology (Baltimore, Md.: 1950) 2005;175:4180–4183. doi: 10.4049/jimmunol.175.7.4180. [DOI] [PubMed] [Google Scholar]
- 16.Gotot J, Gottschalk C, Leopold S, Knolle PA, Yagita H, Kurts C, Ludwig-Portugall I. Regulatory T cells use programmed death 1 ligands to directly suppress autoreactive B cells in vivo. Proc Natl Acad Sci U S A. 2012;109:10468–10473. doi: 10.1073/pnas.1201131109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lan Q, Zhou X, Fan H, Chen M, Wang J, Ryffel B, Brand D, Ramalingam R, Kiela PR, Horwitz DA, Liu Z, Zheng SG. Polyclonal CD4+Foxp3+ Treg cells induce TGFbeta-dependent tolerogenic dendritic cells that suppress the murine lupus-like syndrome. Journal of molecular cell biology. 2012;4:409–419. doi: 10.1093/jmcb/mjs040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iikuni N, Lourenco EV, Hahn BH, La Cava A. Cutting edge: Regulatory T cells directly suppress B cells in systemic lupus erythematosus. Journal of immunology (Baltimore, Md.: 1950) 2009;183:1518–1522. doi: 10.4049/jimmunol.0901163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li QZ, Xie C, Wu T, Mackay M, Aranow C, Putterman C, Mohan C. Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. The Journal of clinical investigation. 2005;115:3428–3439. doi: 10.1172/JCI23587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li QZ, Zhou J, Wandstrat AE, Carr-Johnson F, Branch V, Karp DR, Mohan C, Wakeland EK, Olsen NJ. Protein array autoantibody profiles for insights into systemic lupus erythematosus and incomplete lupus syndromes. Clinical and experimental immunology. 2007;147:60–70. doi: 10.1111/j.1365-2249.2006.03251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu L, Zhou X, Wang J, Zheng SG, Horwitz DA. Characterization of protective human CD4CD25 FOXP3 regulatory T cells generated with IL-2, TGF-beta and retinoic acid. PloS one. 2010;5:e15150. doi: 10.1371/journal.pone.0015150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piccirillo CA, Letterio JJ, Thornton AM, McHugh RS, Mamura M, Mizuhara H, Shevach EM. CD4(+)CD25(+) regulatory T cells can mediate suppressor function in the absence of transforming growth factor beta1 production and responsiveness. The Journal of experimental medicine. 2002;196:237–246. doi: 10.1084/jem.20020590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, Klein-Hessling S, Serfling E, Hamann A, Huehn J. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS biology. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koenecke C, Czeloth N, Bubke A, Schmitz S, Kissenpfennig A, Malissen B, Huehn J, Ganser A, Forster R, Prinz I. Alloantigen-specific de novo-induced Foxp3+ Treg revert in vivo and do not protect from experimental GVHD. European journal of immunology. 2009;39:3091–3096. doi: 10.1002/eji.200939432. [DOI] [PubMed] [Google Scholar]
- 25.Gu J, Lu L, Chen M, Xu L, Lan Q, Li Q, Liu Z, Chen G, Wang P, Wang X, Brand D, Olsen N, Zheng SG. TGF-beta-induced CD4+Foxp3+ T cells attenuate acute graft-versus-host disease by suppressing expansion and killing of effector CD8+ cells. Journal of immunology (Baltimore, Md.: 1950) 2014;193:3388–3397. doi: 10.4049/jimmunol.1400207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, O'Shea JJ, Shevach EM. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. The Journal of experimental medicine. 2008;205:1975–1981. doi: 10.1084/jem.20080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Connor RA, Floess S, Huehn J, Jones SA, Anderton SM. Foxp3+ Treg cells in the inflamed CNS are insensitive to IL-6-driven IL-17 production. Eur J Immunol. 2012;42:1174–1179. doi: 10.1002/eji.201142216. [DOI] [PubMed] [Google Scholar]
- 28.Perry D, Sang A, Yin Y, Zheng YY, Morel L. Murine models of systemic lupus erythematosus. Journal of biomedicine & biotechnology. 2011;2011:271694. doi: 10.1155/2011/271694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waters ST, Fu SM, Gaskin F, Deshmukh US, Sung SS, Kannapell CC, Tung KS, McEwen SB, McDuffie M. NZM2328: a new mouse model of systemic lupus erythematosus with unique genetic susceptibility loci. Clinical immunology (Orlando, Fla.) 2001;100:372–383. doi: 10.1006/clim.2001.5079. [DOI] [PubMed] [Google Scholar]
- 30.Lan Q, Zhou X, Fan H, Chen M, Wang J, Ryffel B, Brand D, Ramalingam R, Kiela PR, Horwitz DA, Liu Z, Zheng SG. Polyclonal CD4+Foxp3+ Treg cells induce TGFβ-dependent tolerogenic dendritic cells that suppress the murine lupus-like syndrome. J Mol Cell Biol. 2012;4:409–419. doi: 10.1093/jmcb/mjs040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaiss MM, Axmann R, Zwerina J, Polzer K, Guckel E, Skapenko A, Schulze-Koops H, Horwood N, Cope A, Schett G. Treg cells suppress osteoclast formation: a new link between the immune system and bone. Arthritis Rheum. 2007;56:4104–4112. doi: 10.1002/art.23138. [DOI] [PubMed] [Google Scholar]
- 32.Kong N, Lan Q, Chen M, Zheng T, Su W, Wang J, Yang Z, Park R, Dagliyan G, Conti PS, Brand D, Liu Z, Zou H, Stohl W, Zheng SG. Induced T regulatory cells suppress osteoclastogenesis and bone erosion in collagen-induced arthritis better than natural T regulatory cells. Ann Rheum Dis. 2012;71:1567–1572. doi: 10.1136/annrheumdis-2011-201052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galani IE, Wendel M, Stojanovic A, Jesiak M, Müller MM, Schellack C, Suri-Payer E, Cerwenka A. Regulatory T cells control macrophage accumulation and activation in lymphoma. Int J Cancer. 2010;127:1131–1140. doi: 10.1002/ijc.25132. [DOI] [PubMed] [Google Scholar]
- 34.Ralainirina N, Poli A, Michel T, Poos L, Andrès E, Hentges F, Zimmer J. Control of NK cell functions by CD4+CD25+ regulatory T cells. J Leukoc Biol. 2007;81:144–153. doi: 10.1189/jlb.0606409. [DOI] [PubMed] [Google Scholar]
- 35.Mori L, de Libero G. Genetic control of susceptibility to collagen-induced arthritis in T cell receptor beta-chain transgenic mice. Arthritis Rheum. 1998;41:256–262. doi: 10.1002/1529-0131(199802)41:2<256::AID-ART9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 36.Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999;286:1732–1735. doi: 10.1126/science.286.5445.1732. [DOI] [PubMed] [Google Scholar]
- 37.Hampe CS. B Cell in Autoimmune Diseases. Scientifica (Cairo) 2012;2012 doi: 10.6064/2012/215308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, Gottlieb PA, Marks JB, McGee PF, Moran AM, Raskin P, Rodriguez H, Schatz DA, Wherrett D, Wilson DM, Lachin JM, Skyler JS T. D. T. A.-C. S. Group. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dörner T, Burmester GR. New approaches of B-cell-directed therapy: beyond rituximab. Curr Opin Rheumatol. 2008;20:263–268. doi: 10.1097/BOR.0b013e3282f5e08d. [DOI] [PubMed] [Google Scholar]
- 40.Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, Langer-Gould A, Smith CH H. T. Group. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 41.Leandro MJ, Cooper N, Cambridge G, Ehrenstein MR, Edwards JC. Bone marrow B-lineage cells in patients with rheumatoid arthritis following rituximab therapy. Rheumatology (Oxford) 2007;46:29–36. doi: 10.1093/rheumatology/kel148. [DOI] [PubMed] [Google Scholar]
- 42.Hamaguchi Y, Uchida J, Cain DW, Venturi GM, Poe JC, Haas KM, Tedder TF. The peritoneal cavity provides a protective niche for B1 and conventional B lymphocytes during anti-CD20 immunotherapy in mice. J Immunol. 2005;174:4389–4399. doi: 10.4049/jimmunol.174.7.4389. [DOI] [PubMed] [Google Scholar]
- 43.Xu W, Lan Q, Chen M, Chen H, Zhu N, Zhou X, Wang J, Fan H, Yan CS, Kuang JL, Warburton D, Togbe D, Ryffel B, Zheng SG, Shi W. Adoptive transfer of induced-Treg cells effectively attenuates murine airway allergic inflammation. PloS one. 2012;7:e40314. doi: 10.1371/journal.pone.0040314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kong N, Lan Q, Chen M, Wang J, Shi W, Horwitz DA, Quesniaux V, Ryffel B, Liu Z, Brand D, Zou H, Zheng SG. Antigen-specific transforming growth factor beta-induced Treg cells, but not natural Treg cells, ameliorate autoimmune arthritis in mice by shifting the Th17/Treg cell balance from Th17 predominance to Treg cell predominance. Arthritis and rheumatism. 2012;64:2548–2558. doi: 10.1002/art.34513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Su H, Ye DQ, Wang BL, Fang XH, Chen J, Wang Q, Li WX, Zhang N. Transforming growth factor-beta1-induced CD4+CD25+ regulatory T cells in vitro reverse and prevent a murine lupus-like syndrome of chronic graft-versus-host disease. The British journal of dermatology. 2008;158:1197–1209. doi: 10.1111/j.1365-2133.2008.08555.x. [DOI] [PubMed] [Google Scholar]
- 46.Zheng SG, Gray JD, Ohtsuka K, Yamagiwa S, Horwitz DA. Generation ex vivo of TGF-beta-producing regulatory T cells from CD4+CD25− precursors. Journal of immunology (Baltimore, Md.: 1950) 2002;169:4183–4189. doi: 10.4049/jimmunol.169.8.4183. [DOI] [PubMed] [Google Scholar]
- 47.Gagliani N, Jofra T, Valle A, Stabilini A, Morsiani C, Gregori S, Deng S, Rothstein DM, Atkinson M, Kamanaka M, Flavell RA, Roncarolo MG, Battaglia M. Transplant tolerance to pancreatic islets is initiated in the graft and sustained in the spleen. Am J Transplant. 2013;13:1963–1975. doi: 10.1111/ajt.12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng SG, Wang JH, Gray JD, Soucier H, Horwitz DA. Natural and induced CD4+CD25+ cells educate CD4+CD25− cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J Immunol. 2004;172:5213–5221. doi: 10.4049/jimmunol.172.9.5213. [DOI] [PubMed] [Google Scholar]
- 49.Gu J, Lu L, Chen M, Xu L, Lan Q, Li Q, Liu Z, Chen G, Wang P, Wang X, Brand D, Olsen N, Zheng SG. TGF-β-induced CD4+Foxp3+ T cells attenuate acute graft-versus-host disease by suppressing expansion and killing of effector CD8+ cells. J Immunol. 2014;193:3388–3397. doi: 10.4049/jimmunol.1400207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.