

Abstract Abstract
Hypoxia stimulates pulmonary hypertension (PH), in part by increasing the proliferation of human pulmonary artery smooth muscle cells (HPASMCs) via sustained activation of mitogen-activated protein kinase, extracellular signal–regulated kinases 1 and 2 (ERK 1/2), and nuclear factor-kappa B (NF-κB); elevated expression of NADPH oxidase 4 (Nox4); and downregulation of peroxisome proliferator–activated receptor gamma (PPARγ) levels. However, the upstream mediators that control these responses remain largely unknown. We hypothesized that proline-rich tyrosine kinase 2 (Pyk2) plays a critical role in the mechanism of hypoxia-induced HPASMC proliferation. To test this hypothesis, HPASMCs were exposed to normoxia or hypoxia (1% O2) for 72 hours. Hypoxia activated Pyk2 (detected as Tyr402 phosphorylation), and inhibition of Pyk2 with small interfering RNA (siRNA) or tyrphostin A9 attenuated hypoxia-induced HPASMC proliferation. Pyk2 inhibition attenuated ERK 1/2 activation as early as 24 hours after the onset of hypoxia, suggesting a proximal role for Pyk2 in this response. Pyk2 inhibition also attenuated hypoxia-induced NF-κB activation, reduced HPASMC PPARγ messenger RNA levels and activity, and increased NF-κB-mediated Nox4 levels. The siRNA-mediated PPARγ knockdown enhanced Pyk2 activation, whereas PPARγ overexpression reduced Pyk2 activation in HPASMCs, confirming a reciprocal relationship between Pyk2 and PPARγ. Pyk2 depletion also attenuated hypoxia-induced NF-κB p65 activation and reduced PPARγ protein levels in human pulmonary artery endothelial cells. These in vitro findings suggest that Pyk2 plays a central role in the proliferative phenotype of pulmonary vascular wall cells under hypoxic conditions. Coupled with recent reports that hypoxia-induced PH is attenuated in Pyk2 knockout mice, these findings suggest that Pyk2 may represent a novel therapeutic target in PH.
Keywords: pulmonary hypertension, signaling, nuclear factor-kappa B, NADPH oxidase 4
Pulmonary hypertension (PH) is characterized by increases in pulmonary artery pressure and pulmonary vascular resistance that cause significant morbidity and mortality.1 Mounting evidence suggests that a nuclear hormone transcription factor, peroxisome proliferator–activated receptor gamma (PPARγ), participates in maintenance of normal pulmonary vascular function. For example, PPARγ expression is reduced in the lungs of rodents with hypoxia-induced PH,2,3 in the vascular lesions of patients with idiopathic pulmonary arterial hypertension (IPAH), in the lungs of rats with severe PH,4 and in pulmonary artery endothelial cells isolated from IPAH patients.5 Furthermore, targeted and constitutive genetic ablation of PPARγ from endothelial6 or vascular smooth muscle cells7 stimulates the development of spontaneous PH in mice under normoxic conditions. On the other hand, exogenous activation of PPARγ with synthetic thiazolidinedione ligands attenuates PH and pulmonary vascular remodeling in several experimental models of PH.2,3,8-10 Taken together, these reports suggest that reductions in PPARγ contribute to PH pathogenesis and that activation of PPARγ attenuates pulmonary vascular dysfunction and PH.
Hypoxia reduces both PPARγ expression and PPARγ activity through oxidative-stress signals and increases in NADPH oxidase 4 (Nox4) expression in the pulmonary vasculature.3,11 Nox4-derived H2O2 reduces PPARγ expression and activity in pulmonary artery smooth muscle cells (PASMCs)12 and similarly reduces PPARγ in endothelial cells in vitro.13 Hypoxia activates mitogen-activated protein kinases (MAP kinases) that regulate PPARγ transcriptional activity and the proinflammatory transcription factor NF-κB (nuclear factor-kappa B).14,15 Hypoxia increases Nox4 expression in human PASMCs (HPASMCs) by inducing NF-κB p65 binding to the Nox4 promoter.16 We recently demonstrated that hypoxia induces extracellular signal–regulated kinase (ERK)-mediated-NF-κB activation, Nox4 expression, H2O2 generation, and PPARγ downregulation in HPASMCs and that Nox4-derived H2O2 is in turn required for ERK 1/2 activation, suggesting the existence of cyclic signaling cascades underlying chronic hypoxia–induced derangements in pulmonary vascular wall cells.12 Although these studies clarify mechanisms involved in hypoxia-induced reductions in PPARγ expression, the early events upstream of ERK 1/2 activation attributable to PPARγ downregulation are not well defined.
Emerging evidence suggests that proline-rich tyrosine kinase 2 (Pyk2), a Ca2+-dependent, nonreceptor tyrosine kinase (and a member of the focal adhesion kinase [FAK] family), plays a crucial upstream role in vascular dysfunction associated with acute and chronic inflammatory diseases. For example, in models of sepsis and intravascular injury, Pyk2 promotes endothelial dysfunction by activating p38 MAP kinase and NF-κB.17,18 In systemic hypertension caused by angiotensin II, Pyk2 activates ERK 1/2 and upregulates cell cycle proteins to promote proliferation of vascular smooth muscle cells.19 Pyk2 phosphorylation promotes hypoxia-induced metalloproteinase 9 activation and human vascular smooth muscle cell migration.20 The role of Pyk2 in PH pathobiology has not been extensively examined. A recent study demonstrated that Pyk2 participates in hypoxia-induced HIF-1α (hypoxia-inducible factor 1α) expression and pulmonary vascular wall cell proliferation;21 however, the precise role of Pyk2 in PH pathogenesis remains to be defined.
Our study, therefore, examines the role of Pyk2 in hypoxic signaling cascades that contribute to pulmonary vascular cell proliferation and vascular remodeling. Our findings demonstrate that Pyk2 plays a proximal and critical upstream role in hypoxia-induced HPASMC proliferation by activating the ERK1/2–NF-κB–Nox4 signaling axis and downregulating PPARγ. Our findings also provide novel evidence that loss of PPARγ promotes Pyk2 activation in HPASMCs. Taken together with previous reports, these findings emphasize the importance of Pyk2 in pulmonary vascular cell biology and PH pathogenesis. Strategies that target suppression of these pathways may preserve PPARγ function in the pulmonary vascular wall and may provide a novel therapeutic strategy for PH.
Methods
Reagents
The Pyk2 inhibitor tyrphostin A9 (TA9) was purchased from Calbiochem (La Jolla, CA). Antibodies against phospho-(Tyr402)-Pyk2 and total Pyk2 were purchased from Abcam (Cambridge, MA). Antibodies against phospho-(Thr202/Tyr204)-ERK 1/2, total ERK 1/2, and phospho-(Ser536)-NF-κB were purchased from Cell Signaling Technology (Beverly, MA). Antibodies against PPARγ, total NF-κB, and Nox4 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and Abcam. GAPDH (glyceraldehyde 3-phosphate dehydrogenase) antibodies were purchased from Sigma-Aldrich (St. Louis, MO). All other materials were purchased from VWR Scientific (Gaithersburg, MD) or Fisher Scientific (Pittsburgh, PA).
Cell culture and small interfering RNA (siRNA) transfections
HPASMCs or human pulmonary artery endothelial cells (HPAECs) were purchased from Lonza (Basel, Switzerland). HPASMC monolayers (passages 3–4) were grown at 37°C in a 5% CO2 atmosphere in culture media (SmGM-2, Lonza) containing 2% fetal calf serum, growth factors, and antibiotics, as previously reported.12 In separate experiments, HPAEC monolayers were grown in EGM-2 media (Lonza). Upon reaching 50%–60% confluency, the cells were transfected with 50 nM nontargeting siRNA (control siRNA) or siRNA targeting human Pyk2 using Dharmafect transfection reagent (Dharmacon, Waltham, MA) for 12 hours. Cells were then washed with serum-free media and recovered for 12 hours in complete growth media before being exposed to normoxia (21% O2, 5% CO2) or hypoxia (1% O2, 5% CO2) for 72 hours. In separate experiments, HPASMCs were transfected with siRNA targeting human PPARγ for 72 hours under normoxic conditions, as described previously,22 and analyzed for Pyk2 activation.
Reporter gene assays
Confluent HPASMCs were transfected for 3 hours with PPREx3-TK-Luc plasmid containing 3 consensus PPARγ binding sites upstream of the firefly luciferase gene or with NF-κB-Luc plasmid containing 5 consensus NF-κB binding sites upstream of the firefly luciferase gene; lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) was used, as described previously.12 Transfection efficiencies were normalized by cotransfecting with renilla luciferase plasmid (0.1 μg/mL) in which the renilla luciferase gene is under the control of the constitutively active thymidine kinase promoter. After incubation with smooth muscle cell growth media for 12 hours, cells were exposed to hypoxia or normoxia for 72 hours and lysed with 300 μL of passive lysis buffer, and the lysates were analyzed for PPARγ or NF-κB luciferase activity. Transfected cells were treated with dimethyl sulfoxide (DMSO) or TA9 during the final 24 hours of the 72-hour normoxia or hypoxia exposure to assess the effect of Pyk2 inhibition on PPARγ or NF-κB luciferase activity.
Overexpression of PPARγ in HPASMCs
To ascertain the impact of PPARγ gain of function on selected signaling pathways, HPASMC monolayers were grown at 37°C in a 5% CO2 atmosphere in culture media (SmGM-2, Lonza) containing 5% fetal calf serum, growth factors, and antibiotics. Cells were incubated for 4 hours in 2% fetal bovine serum media with adenoviruses containing either human PPARγ (Ad-hPPARγ; multiplicity of infection [MOI] = 28) or green fluorescent protein (Ad-GFP; Vector Biolabs, Philadelphia, PA; MOI = 3). Media were then replaced with fresh SmGM-2 media, and HPASMCs were cultured for 72 hours.
Cell proliferation assays
HPASMC proliferation was determined with a quantitative colorimetric assay employing dimethylthiazol (MTT assay; ATCC), as described previously.12 Briefly, cells were incubated with MTT reagent for 4 hours. The mitochondrial reductase present in living cells reduces MTT to purple formazan, which is detected by spectrophotometry (optical density = 570 nm), and values from treated cells were normalized to values from corresponding control cells. Cell proliferation was also determined with a bromodeoxyuridine (BrdU) cell proliferation assay kit (EMD Millipore, Bellerica, MA) following the manufacturer’s instructions.
Quantitative real-time polymerase chain reaction (PCR)
Total RNA was isolated with an RNeasy Mini Kit (Qiagen, Valencia, CA), and RNA was quantified by Nanodrop spectrophotometry (Thermo Scientific, Wilmington, DE). The complementary DNA (cDNA) was prepared with the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Quantitative PCR was performed to assess the expression level of PPARγ1 RNA, using primers based on human PPARγ1 messenger RNA (mRNA) sequences: forward: 5′-GTGGC CGCAGATTTG AAAGA AG-3′; reverse: 5′-TGTCA ACCAT GGTCA TTTCG-3′. Real-time PCR was performed with SYBR Green real-time PCR mastermixes (Invitrogen) and a 7500 real-time PCR system (Applied Biosystems, Carlsbad, CA). Amplicon expression in each sample was normalized to 9s RNA levels. The relative abundance of target mRNA in each sample was calculated with ΔΔCT methods (Applied Biosystems).
Preparation of nuclear extracts
HPASMC nuclear extracts were prepared as described previously.18 Briefly, cells were washed twice with ice-cold phosphate-buffered saline and resuspended in 400 μL of buffer A (10 mM KCl, 0.1 mM EDTA [ethylene diamine tetraacetic acid], 10 mM HEPES [pH 7.9], 1 mM DTT [dithiothreitol], 0.1 mM EGTA [ethylene glycol tetraacetic acid], and 0.5 mM PMSF [phenylmethylsulfonyl fluoride]). After 20 minutes, Nonidet P-40 was added to a final concentration of 0.6%. Samples were centrifuged to collect the cytosolic extract. The pellets obtained were resuspended in 100 μL of buffer B (0.4 M NaCl, 1 mM EDTA, 20 mM HEPES [pH 7.9], 1 mM DTT, 1 mM EGTA, and 1 mM PMSF) and homogenized with a mortar-driven pestle. After 30 minutes of incubation on ice, lysates were centrifuged, and supernatants containing the nuclear proteins were transferred to new vials. The protein concentration of the nuclear extract was determined by the bicinchoninic acid protein assay method, and the nuclear extracts were analyzed by Western blot for NF-κB nuclear translocation.
Western blot analysis
HPASMC protein lysates (30–40 µg/sample) were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis; subjected to Western blot analysis for phospho-ERK 1/2, phospho-NF-κB/p65, and phospho-Pyk2; and normalized to their respective total forms (nonphosphorylated). Western blots for PPARγ or Nox4 were normalized to GAPDH. Relative levels of immunoreactive proteins were quantified with the Licor or Image J software system.
Statistical analysis
When more than two groups were compared, data were analyzed with analysis of variance (ANOVA). Post hoc analysis using the Student-Neuman-Keuls test was employed to detect differences between individual groups. In studies comparing only two experimental groups, data were analyzed with the Student t test to determine significance of treatment effects. The level of statistical significance was taken as P < 0.05.
Results
Activation of Pyk2 is required for hypoxia-induced HPASMC proliferation
To determine whether chronic hypoxia induces Pyk2 activation in HPASMCs, cells were exposed to normoxia (21% O2) or hypoxia (1% O2) for 72 hours, and lysates were analyzed by immunoblotting to determine Pyk2 activation. As illustrated in Figure 1A, hypoxia stimulated HPASMC Pyk2 activation, as determined by its phosphorylation at Tyr402 without altering overall Pyk2 protein levels. The effect of Pyk2 inhibition on hypoxia-induced HPASMC proliferation was next determined by treating cells with graded concentrations of the Pyk2 inhibitor TA917,18 during the final 24 hours of the 72-hour exposure to normoxia or hypoxia. TA9 caused dose-dependent inhibition of hypoxia-induced cell proliferation, as determined by MTT assay (Fig. 1B). Because treatment with 0.1-μM TA9 was sufficient to inhibit hypoxia-induced HPASMC cell proliferation (Fig. 1B), this lower concentration of TA9 was employed in all subsequent studies examining Pyk2 inhibition. The role of Pyk2 in hypoxic HPASMC proliferation was further confirmed by transfecting HPASMCs with siRNA targeting human Pyk2. Depletion of Pyk2 caused significant reductions in hypoxia-induced HPASMC proliferation, as detected by MTT assay (Fig. 1C). To ensure that Pyk2 depletion had no nonspecific effects on the expression of other kinases in the same family, Pyk2-depleted cell lysates were immunoblotted for FAK in a separate experiment, and Pyk2 siRNA had no effect on FAK protein levels (representative immunoblot in Fig. 1C). These findings suggest that reductions in cell proliferation were specific to Pyk2 depletion. The ability of Pyk2 depletion to attenuate hypoxia-induced HPASMC proliferation detected by MTT assay was also confirmed in parallel studies that examined HPASMC proliferation by BrdU assay (Fig. 1D).
Figure 1.
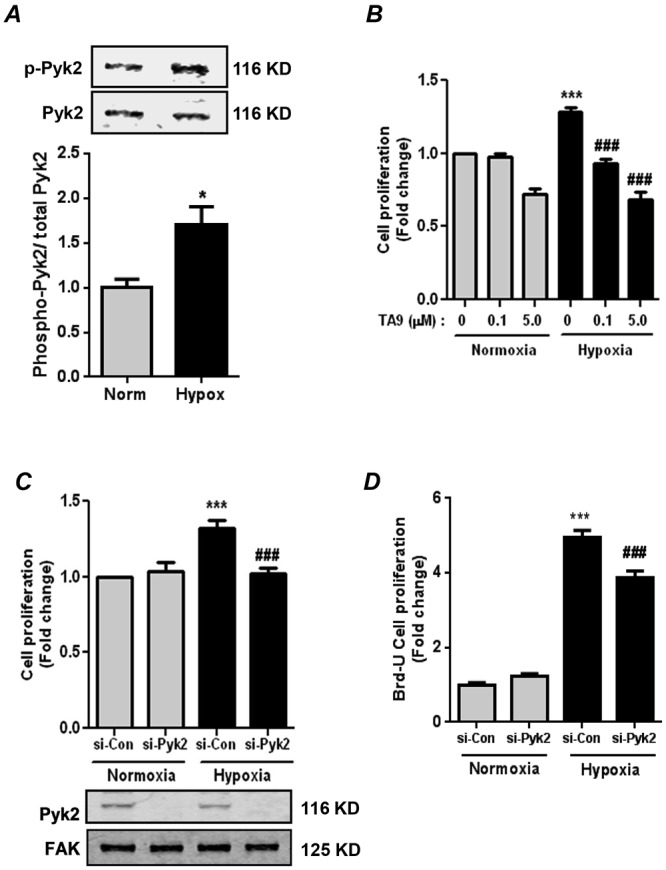
Proline-rich tyrosine kinase 2 (Pyk2) activation is required for hypoxia-induced human pulmonary artery smooth muscle cell (HPASMC) proliferation. A, HPASMCs were exposed to normoxia or hypoxia (1% O2) for 72 hours. Cell lysates were immunoblotted with anti-phospho-(Tyr402)-Pyk2 antibody to determine Pyk2 activation. Each bar represents mean ± SEM phospho-Pyk2 levels relative to total Pyk2 in the same sample, expressed as fold change versus control; n = 6. *P < 0.05. Representative immunoblots are shown above the bar graph. B, HPASMCs were exposed to normoxia or hypoxia for 72 hours. During the final 24 hours of this exposure, HPASMCs were treated with the Pyk2 inhibitor tyrphostin A9 (TA9) or dimethyl sulfoxide (DMSO; i.e., TA9 = 0) vehicle as indicated. Cell proliferation was determined by MTT (dimethylthiazol) assay. Each bar represents mean ± SEM cell proliferation, expressed as fold change versus control; n = 4. ***P < 0.001 versus normoxia + DMSO. ###P < 0.001 versus hypoxia + DMSO. C, D, HPASMCs were transfected with 50 nM control small interfering RNA (si-Con) or Pyk2 siRNA (si-Pyk2) and exposed to normoxia or hypoxia for 72 hours. Cell proliferation was determined by MTT assay (C) and confirmed by bromodeoxyuridine (BrdU) incorporation assay (D). Each bar represents mean ± SEM cell proliferation, expressed as fold change versus control; n = 4–6. ***P < 0.001 versus normoxia + si-Con. ###P < 0.001 versus hypoxia + si-Con. A representative immunoblot is presented below C, demonstrating the effectiveness and specificity of siRNA-mediated Pyk2 knockdown. FAK: focal adhesion kinase.
Pyk2 inhibition attenuates hypoxia-induced HPASMC ERK 1/2 activation and NF-κB nuclear translocation
We previously reported that hypoxia activates HPASMC ERK 1/2 upstream of NF-κB activation.12 To determine whether Pyk2 serves as an early upstream activator of ERK 1/2, HPASMCs were pretreated with the Pyk2 inhibitor TA9 or an equivalent volume of DMSO vehicle for 1 hour and then for the duration of exposure to normoxic or hypoxic conditions for 24 hours. Cell lysates were immunoblotted with an anti-phospho-(Thr202/Tyr204)-ERK 1/2 antibody to determine ERK 1/2 activation. Pyk2 inhibition significantly attenuated hypoxia-induced ERK 1/2 activation in HPASMCs (Fig. 2A). Because our previous studies demonstrated that NF-κB activation and nuclear translocation were downstream consequences of hypoxia-induced ERK 1/2 activation,12 we examined the impact of Pyk2 inhibition on HPASMC NF-κB activation. During the final 24 hours of 72-hour exposure to normoxia or hypoxia, cells were treated with TA9 or DMSO. Nuclear extracts were prepared and immunoblotted with an anti-NF-κB p65 antibody. The nuclear protein fibrillarin was used as a loading control. Pyk2 inhibition with TA9 attenuated hypoxia-induced NF-κB nuclear translocation, suggesting that Pyk2 activation is required for both hypoxia-induced ERK 1/2 and NF-κB activation in HPASMCs (Fig. 2B). Similar to nuclear extracts, cytoplasmic extracts also revealed elevated levels of NF-κBp65 under hypoxia (data not shown), which is consistent with hypoxia-induced increases in total p65 levels, as we previously demonstrated.12 However, we focused only on nuclear NF-κB levels, since this fraction is most relevant to transcriptional activation.
Figure 2.
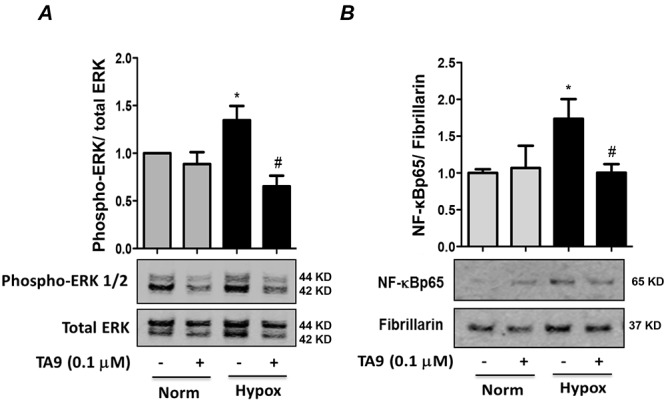
Hypoxia-induced proline-rich tyrosine kinase 2 (Pyk2) activation is required for extracellular signal–regulated kinase (ERK) 1/2 activation and nuclear factor-kappa B (NF-κB) nuclear translocation in human pulmonary artery smooth muscle cells (HPASMCs). A, HPASMCs were pretreated with tyrphostin A9 (TA9) or vehicle before exposure to normoxia (Norm) or hypoxia (Hypox) for 24 hours. Cell lysates were immunoblotted with an anti-phospho-(Thr202/Tyr204)-ERK 1/2 antibody to determine ERK 1/2 activation and normalized with total ERK 1/2. Each bar represents mean ± SEM phospho-ERK/total ERK, expressed as fold change versus control; n = 4. B, HPASMCs were exposed to hypoxia or normoxia for 72 hours and treated with TA9 during the final 24 hours of exposure. Nuclear extracts were immunoblotted for NF-κB p65 and normalized to the nuclear marker fibrillarin. Each bar represents mean ± SEM p65/fibrillarin expressed as fold change versus control; n = 4. In both panels, *P < 0.05 versus normoxia + TA9, and #P < 0.05 versus hypoxia + TA9.
To further confirm the effects of Pyk2 inhibition on NF-κB transcriptional activity, HPASMCs were transfected with NF-κB luciferase and renilla luciferase plasmids and exposed to normoxia or hypoxia for 72 hours. Cells were treated with TA9 or DMSO during the final 24 hours of normoxia or hypoxia exposure. Passive cell lysates were analyzed for NF-κB luciferase activity. These studies confirmed that Pyk2 inhibition with TA9 attenuated hypoxia-induced NF-κB transcriptional activity (Fig. 3A). Furthermore, inhibiting Pyk2 with siRNA attenuated hypoxia-induced expression of the NF-κB target gene, Nox4, in HPASMCs (Fig. 3B).
Figure 3.
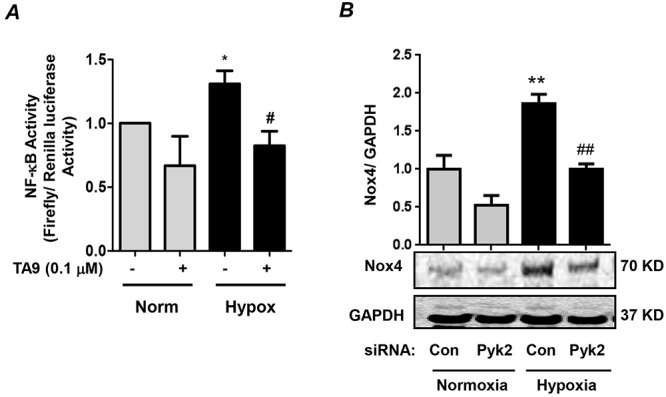
Hypoxia-induced proline-rich tyrosine kinase 2 (Pyk2) activation mediates nuclear factor-kappa B (NF-κB) transcriptional activity and NADPH oxidase 4 (Nox4) protein expression in human pulmonary artery smooth muscle cells (HPASMCs). A, HPASMCs were transfected with firefly luciferase vector containing 5 consensus NF-κB binding sites, along with a renilla luciferase vector to normalize for transfection efficiency. Cells were exposed to hypoxia or normoxia for 72 hours and treated with tyrphostin A9 (TA9) or vehicle during the final 24 hours of exposure. Cell lysates were prepared and analyzed for firefly luciferase activity. Each bar represents mean ± SEM firefly luciferase activity/renilla luciferase activity, expressed as fold change versus control; n = 4. *P < 0.05 versus normoxia + TA9. #P < 0.05 versus hypoxia + TA9. B, HPASMCs were transfected with 50 nM control siRNA (Con) or Pyk2 siRNA (Pyk2) and exposed to normoxia or hypoxia for 72 hours. Cell lysates were immunoblotted with an anti-Nox4 antibody. GAPDH (glyceraldehyde 3-phosphate dehydrogenase) levels were used for normalization. Each bar represents mean ± SEM Nox4/GAPDH levels, expressed as fold change versus control; n = 3. **P < 0.01 versus normoxia + Con. ##P < 0.01 versus hypoxia + Con. siRNA: small interfering RNA.
Inhibition of Pyk2 attenuates hypoxia-induced reductions in PPARγ expression levels and activity in HPASMCs
Because our previous report demonstrated that hypoxia-induced activation of the ERK 1/2–NF-κB–Nox4 pathway was required for hypoxic reductions in HPASMC PPARγ,12 the role of Pyk2 activation in hypoxia-induced reductions in PPARγ was determined. Selected HPASMCs were treated with TA9 during the final 24 hours of 72-hour exposure to normoxia or hypoxia, and RNA was isolated. Analysis of PPARγ mRNA levels demonstrated that Pyk2 inhibition attenuated hypoxia-induced reductions in PPARγ mRNA levels (Fig. 4A). To confirm these observations, PPARγ activity was measured in HPASMCs transfected with PPRE x3-TK-luciferase and renilla luciferase plasmids and exposed to normoxia or hypoxia for 72 hours with or without TA9 treatment during the final 24 hours of exposure. Hypoxia caused significant reductions in PPARγ transcriptional activity, which were attenuated by Pyk2 inhibition (Fig. 4B).
Figure 4.
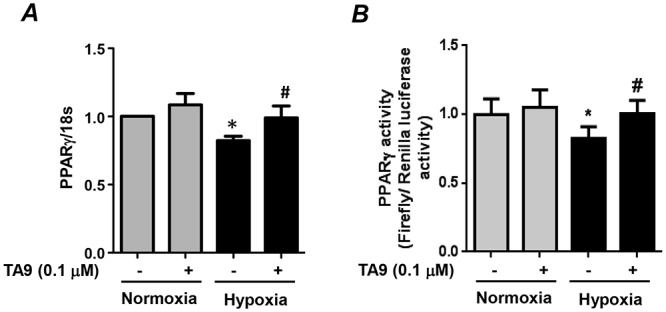
Hypoxia-induced proline-rich tyrosine kinase 2 (Pyk2) activation promotes reductions in peroxisome proliferator–activated receptor gamma (PPARγ) messenger RNA (mRNA) levels and transcriptional activity. A, Human pulmonary artery smooth muscle cells (HPASMCs) were exposed to hypoxia or normoxia for 72 hours and treated with tyrphostin A9 (TA9) during the final 24 hours of exposure. PPARγ mRNA levels were determined by quantitative real-time polymerase chain reaction. Levels of 18s ribosomal RNA in the same sample were used for normalization. Each bar represents mean ± SEM PPARγ/18s RNA, expressed as fold change versus control; n = 3. *P < 0.05 versus normoxia + vehicle. #P < 0.05 versus hypoxia + vehicle. B, HPASMCs were transfected with firefly luciferase vector containing 3 consensus PPARγ response elements, along with renilla luciferase vector to normalize for transfection efficiency. Cells were exposed to hypoxia or normoxia for 72 hours and treated with TA9 or vehicle during the final 24 hours of exposure. Passive cell lysates were analyzed for firefly luciferase activity. Each bar represents mean ± SEM firefly luciferase/renilla luciferase activity expressed as fold change versus control; n = 6. *P < 0.05 versus normoxia + TA9. #P < 0.05 versus hypoxia + TA9.
PPARγ negatively regulates Pyk2 in HPASMCs
To better define the relationship between PPARγ and Pyk2 activation, HPASMCs were transfected with control or PPARγ siRNA for 72 hours to reduce PPARγ levels to a degree comparable to that observed under hypoxic conditions.12 Alternatively, PPARγ was overexpressed in HPASMCs by means of an adenoviral construct. As illustrated in Figure 5, PPARγ knockdown stimulated Pyk2 activation (Fig. 5A), whereas PPARγ overexpression attenuated Pyk2 activation (Fig. 5B). These results provide novel evidence for a reciprocal relationship between Pyk2 and PPARγ activities.
Figure 5.
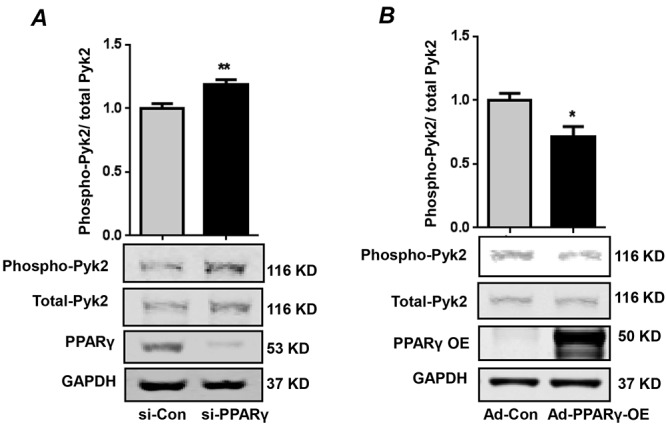
Peroxisome proliferator–activated receptor gamma (PPARγ) negatively regulates proline-rich tyrosine kinase 2 (Pyk2) in human pulmonary artery smooth muscle cells (HPASMCs). A, HPASMCs were transfected with control small interfering RNA (si-Con) or PPARγ small interfering RNA (si-PPARγ) for 72 hours. Cell lysates were analyzed for Pyk2 activation with an anti-phospho-(Tyr402)-Pyk2 antibody. Total Pyk2 levels were used for normalization. PPARγ levels were determined by Western blotting to verify depletion. n = 6. **P < 0.01 versus si-Con. B, HPASMCs were transduced with adenovirus containing control vector (Ad-Con) or adenovirus containing PPARγ to a multiplicity of infection of 28. Cell lysates were immunoblotted for phospho-Pyk2 and total Pyk2. PPARγ levels were determined to verify its overexpression (PPARγ-OE). A lower exposure was taken to highlight the sharpness of the overexpressed PPARγ protein. n = 3. *P < 0.05 versus Ad-Con. In both panels, each bar represents the ratio phospho-Pyk2/total Pyk2, expressed as fold change versus control. GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
Pyk2 promotes hypoxia-induced NF-κB activation and reduction in PPARγ protein levels in HPAECs
Given that hypoxia-induced NF-κB activation and PPARγ downregulation have also been described in HPAECs,23,24 we sought to further explore the role of Pyk2 in hypoxic signaling in HPAECs. HPAECs were transfected with control siRNA or Pyk2 siRNA and exposed to normoxia or hypoxia for 72 hours. Cell lysates were immunoblotted with an anti-phospho-(Ser536)-p65 antibody to determine NF-κB transcriptional capacity or with PPARγ antibody to determine total PPARγ levels. Similar to the effect in HPASMCs, Pyk2 knockdown significantly attenuated hypoxia-induced NF-κB activation and reductions in PPARγ protein levels in HPAECs (Fig. 6A, 6B).
Figure 6.
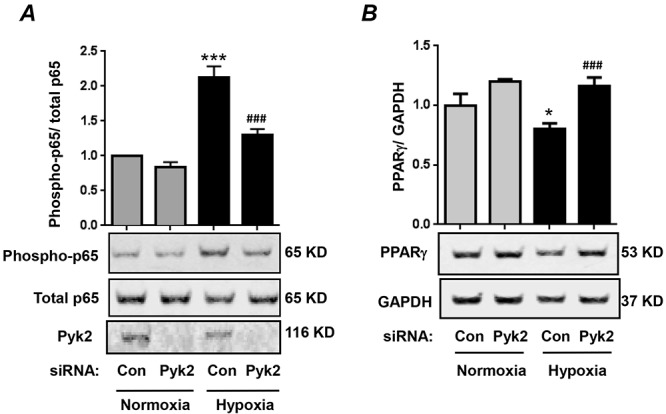
Proline-rich tyrosine kinase 2 (Pyk2) promotes hypoxia-induced nuclear factor-kappa B (NF-κB) activation and reductions in peroxisome proliferator–activated receptor gamma (PPARγ) levels in human pulmonary artery endothelial cells (HPAECs). HPAECs were transfected with control siRNA (Con) or Pyk2 siRNA (Pyk2) and exposed to normoxia or hypoxia for 72 hours. A, Cell lysates were immunoblotted with an anti-phospho-(Ser536)-p65 antibody to determine NF-κB activation. Total p65 levels were used for normalization. Total Pyk2 levels were determined to verify depletion. Each bar represents the ratio phospho-p65/total p65, expressed as fold change versus control; n = 3. ***P < 0.001 versus Con + normoxia. ###P < 0.001 versus Con + hypoxia. B, Cell lysates were immunoblotted for PPARγ and normalized to GAPDH (glyceraldehyde 3-phosphate dehydrogenase) levels in the same sample. Each bar represents the ratio PPARγ/GAPDH, expressed as fold change versus control; n = 3. *P < 0.05 versus Con + normoxia. ###P < 0.05 versus Con + hypoxia. siRNA: small interfering RNA.
Discussion
The pathogenesis of PH is complex and characterized in part by proliferation of pulmonary vascular wall cells that lead to pulmonary vascular remodeling and increases in pulmonary vascular resistance. In an attempt to better understand mechanisms of pulmonary vascular cell proliferation, our group has recently reported on two factors that play a critical role in pulmonary vascular cell proliferation and PH: (1) generation of reactive oxygen species (ROSs) by Nox4 and (2) downregulation of the transcription factor PPARγ. PPARγ plays an important role in the regulation of metabolism, inflammation, and cell proliferation, all basic cellular processes implicated in PH pathogenesis. While activation of PPARγ with synthetic thiazolidinedione ligands, such as rosiglitazone, attenuates experimental PH and pulmonary vascular cell dysfunction in rodent models of PH,2,3,8-10,23 their clinical application in the treatment of type 2 diabetes has been associated with significant deleterious side effects.25 Thus, a better definition of pathways involved in reductions in PPARγ in PH may identify novel therapeutic targets that inhibit or reverse reductions in PPARγ and proliferative pulmonary vascular wall cell phenotypes that mediate PH pathogenesis and PH. In this study, we identify the tyrosine kinase Pyk2 as an upstream signaling mediator of hypoxia-induced reductions in PPARγ expression.
We previously reported that 72 hours of hypoxia promotes HPASMC PPARγ downregulation and proliferation via activation of an ERK–NF-κB–Nox4 signaling axis. To further examine the role of Pyk2 in these established hypoxic HPASMC signaling pathways, we employed two strategies to inhibit Pyk2: treatment with the pharmacological inhibitor TA9 and siRNA-mediated Pyk2 knockdown. Hypoxia increases the intracellular Ca2+ concentrations26-28 that are required for Pyk2 activation, and hypoxic Pyk2 activation was recently reported in HPASMCs from 0.5 to 16 hours after the onset of hypoxia.21 As demonstrated in Figure 2A, Pyk2 inhibition during the first 24 hours of hypoxic exposure attenuated HPASMC ERK 1/2 activation. Because ERK 1/2 activation plays an upstream role in mediating (1) hypoxic increases in NF-κB activity and Nox4 expression and (2) hypoxic reductions in PPARγ levels and activity,12 we postulated that Pyk2 activation would play a critical role in these downstream signaling events. Our findings confirm that Pyk2 contributes to hypoxic HPASMC proliferation and that interventions to inhibit Pyk2, even if introduced after 48 hours of hypoxic exposure, can successfully attenuate HPASMC proliferation caused by hypoxic activation of a Pyk2–ERK 1/2–NF-κB–Nox4 signaling axis. Recent work by others also provided evidence that Pyk2 participates in stimulation of hypoxic PASMC migration.21
The NF-κB subunits p65/p50 bind to the Nox4 promoter to cause hypoxia-induced Nox4 protein expression and Nox4-derived H2O2 production.16 In the present study, Pyk2 depletion reduced the hypoxic induction of Nox4 protein levels, emphasizing the importance of Pyk2 in NF-κB regulation and ROS production. These findings are consistent with previous reports that Pyk2 regulates hypoxia-induced generation of ROSs in HPASMCs.21 Given that Nox4 is a major source of hypoxia-induced H2O2 in vascular wall cells16 and the recently reported role of Pyk2 in mediating mitochondrial ROS generation via activation of the mitochondrial Ca2+ uniporter,29 our observations reinforce the crucial role of Pyk2 in hypoxia-induced ROS generation in HPASMCs.
Our study demonstrates a novel role for Pyk2 in mediating hypoxic reductions in PPARγ levels and transcriptional activity. Our findings suggest that proximal Pyk2 activation stimulates the ERK 1/2–NF-κB–Nox4 signaling axis, which was previously shown to be sufficient to downregulate PPARγ.12 While our current and previous findings have demonstrated that hypoxia stimulates transcriptional suppression of PPARγ,12 posttranslational modifications of PPARγ may also contribute to alterations in PPARγ activity. Phosphorylation at Ser112 inhibits PPARγ transcriptional activity,30-32 and PPARγ phosphorylation by ERK 1/2 promotes its ubiquitination and proteosomal degradation.33,34 Pyk2 may therefore recruit ERK 1/2 or other MAP kinases to mediate reductions in PPARγ transcriptional activity via these posttranslational modifications.
An important aspect of hypoxia-induced pulmonary vascular remodeling is the existence of feed-forward signaling cascades mediated by Nox4-derived H2O2.12 Hypoxia-induced increases in Nox4-derived H2O2 were required for reductions in HPASMC PPARγ.12 Further, siRNA-induced reductions in HPASMC PPARγ levels under normoxic conditions were sufficient to activate the ERK 1/2–NF-κB–Nox4–H2O2 signaling axis and cause HPASMC proliferation.22 Our results here provide novel evidence that loss of PPARγ promotes Pyk2 activation, whereas PPARγ overexpression inhibits Pyk2 activation. Coupled with evidence that adenoviral PPARγ overexpression attenuates hypoxic HPASMC proliferation (K. M. Bijli, B.-Y. Kang, R. L. Sutliff, and C. M. Hart, unpublished observations), these findings support a mutually repressive relationship between Pyk2 and PPARγ and suggest that hypoxic reductions in PPARγ may contribute to sustained Pyk2 activation during chronic hypoxia exposure.
In models of acute inflammation, Pyk2 promotes endothelial dysfunction via NF-κB activation following inflammatory stimuli such as endotoxin or thrombin.17,18 Chronic hypoxia causes endothelial dysfunction via expression of the vasoproliferative mediator endothelin 1 (an NF-κB and HIF-1α target gene) and via reduction in PPARγ levels through upregulation of microRNA-27a.26 Similar to observations in HPASMCs, our study further demonstrates that Pyk2 serves as an important upstream regulator of hypoxia-induced NF-κB activation and PPARγ reductions in HPAECs (Fig. 6). Taken together, these findings emphasize that Pyk2 plays an important role in hypoxia-induced signaling events in pulmonary vascular wall cells.
In conclusion, our findings demonstrate that Pyk2 activation plays a central role in the proliferative responses of pulmonary vascular wall cells to hypoxia. They illustrate that Pyk2 regulates NF-κB and PPARγ in both HPASMCs and HPAECs to promote proliferative phenotypes in response to hypoxia. These findings are consistent with and extend recent evidence that hypoxia-induced PH was attenuated in Pyk2 knockout mice.21 Figure 7 summarizes the regulatory function of Pyk2 in promoting hypoxia-induced HPASMC proliferation. Taken together, the findings from this study, in concert with recent reports,21 demonstrate that Pyk2 activation plays a critical upstream role in the complex signaling mechanisms involved in hypoxia-induced pulmonary vascular cell proliferation and PH. These results further implicate Pyk2 as a novel therapeutic target in PH.
Figure 7.
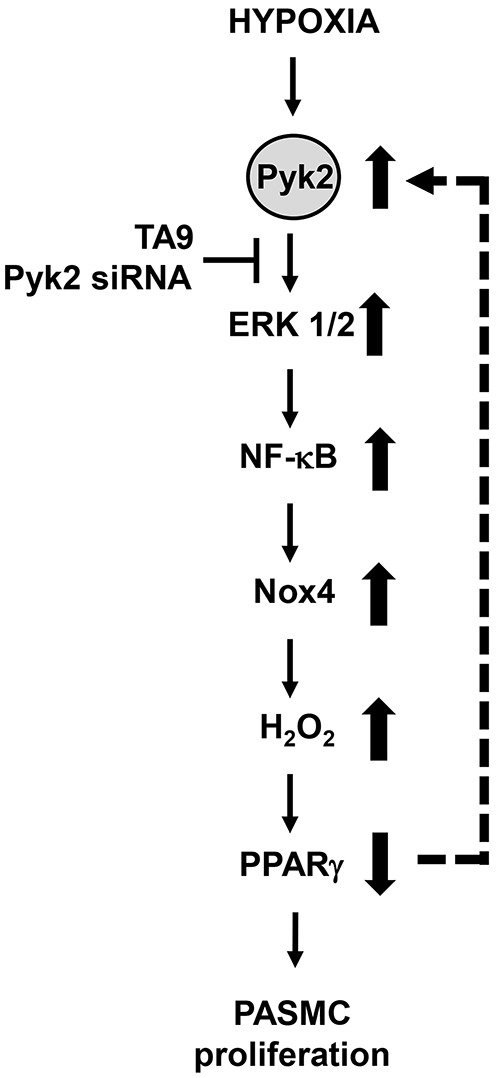
Schematic diagram summarizing the proximal upstream role of proline-rich tyrosine kinase 2 (Pyk2) in hypoxia-induced pulmonary artery smooth muscle cell (PASMC) proliferation. Hypoxia induces activation of Pyk2, which in turn activates extracellular signal–regulated kinase (ERK) 1/2, leading to nuclear factor-kappa B (NF-κB) activation, NADPH oxidase 4 (Nox4) expression, H2O2 generation, and downregulation of peroxisome proliferator–activated receptor gamma (PPARγ). Our previous reports demonstrated that the ERK 1/2–NF-κB–Nox4–H2O2 pathway was required for hypoxic human PASMC proliferation12 and that reductions in PPARγ were sufficient to activate this pathway and the proliferation of pulmonary vascular wall cells.22 Collectively, these findings indicate that hypoxia-induced Pyk2 activation triggers the ERK 1/2–NF-κB–Nox4–H2O2 cascade to reduce PPARγ levels. Reductions in PPARγ can promote feed-forward activation of Pyk2 (dashed arrow), thereby contributing to sustained activation of this proliferative signaling cascade. Depletion or inhibition of Pyk2 can attenuate these signaling mechanisms, restore PPARγ levels, attenuate proliferation of PASMCs, and thereby potentially reduce pulmonary hypertension.
Acknowledgments
We thank Jennifer Kleinhenz, Tamara Murphy, and Sherry Adesina for their technical help in the experiments. The contents reported here do not represent the views of the Department of Veterans Affairs or the United States Government.
Source of Support: This article is based on work that was supported in part by Merit Review funding from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development (1I01BX001910; CMH); by National Institutes of Health grant R01HL102167 (CMH and RLS); and by American Heart Association Scientist Development Grants (KMB and BYK).
Conflict of Interest: None declared.
References
- 1.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 2008;118(7):2372–2379. [DOI] [PMC free article] [PubMed]
- 2.Kim EK, Lee JH, Oh YM, Lee YS, Lee SD. Rosiglitazone attenuates hypoxia-induced pulmonary arterial hypertension in rats. Respirology 2010;15(4):659–668. [DOI] [PubMed]
- 3.Nisbet RE, Bland JM, Kleinhenz DJ, Mitchell PO, Walp ER, Sutliff RL, Hart CM. Rosiglitazone attenuates chronic hypoxia–induced pulmonary hypertension in a mouse model. Am J Respir Cell Mol Biol 2010;42(4):482–490. [DOI] [PMC free article] [PubMed]
- 4.Ameshima S, Golpon H, Cool CD, Chan D, Vandivier RW, Gardai SJ, Wick M, Nemenoff RA, Geraci MW, Voelkel NF. Peroxisome proliferator–activated receptor gamma (PPARγ) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ Res 2003;92(10):1162–1169. [DOI] [PubMed]
- 5.Green DE, Murphy TC, Kang BY, Kleinhenz JM, Szyndralewiez C, Page P, Sutliff RL, Hart CM. The NOX4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. Am J Respir Cell Mol Biol 2012;47(5):718–726. [DOI] [PMC free article] [PubMed]
- 6.Guignabert C, Alvira CM, Alastalo TP, Sawada H, Hansmann G, Zhao M, Wang L, El-Bizri N, Rabinovitch M. Tie2-mediated loss of peroxisome proliferator-activated receptor-γ in mice causes PDGF-receptor β-dependent pulmonary arterial muscularization. Am J Physiol Lung Cell Mol Physiol 2009;297(6):L1082–L1090. [DOI] [PMC free article] [PubMed]
- 7.Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, et al. An antiproliferative BMP-2/PPARγ/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest 2008;118(5):1846–1857. [DOI] [PMC free article] [PubMed]
- 8.Crossno JT Jr., Garat CV, Reusch JE, Morris KG, Dempsey EC, McMurtry IF, Stenmark KR, Klemm DJ. Rosiglitazone attenuates hypoxia-induced pulmonary arterial modeling. Am J Physiol Lung Cell Mol Physiol 2007;292(4):L885–L897. [DOI] [PubMed]
- 9.Hansmann G, Wagner RA, Schellong S, de Jesus Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator–activated receptor-γ activation. Circulation 2007;115(10):1275–1284. [DOI] [PubMed]
- 10.Matsuda Y, Hoshikawa Y, Ameshima S, Suzuki S, Okada Y, Tabata T, Sugawara T, Matsumura Y, Kondo T. Effects of peroxisome proliferator-activated receptor gamma ligands on monocrotaline-induced pulmonary hypertension in rats [in Japanese]. Nihon Kokyuki Gakkai Zasshi 2005;43(5):283–288. [PubMed]
- 11.Mittal M, Roth M, Konig P, Hofmann S, Dony E, Goyal P, Selbitz AC, et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res 2007;101(3):258–267. [DOI] [PubMed]
- 12.Lu X, Bijli KM, Ramirez A, Murphy TC, Kleinhenz J, Hart CM. Hypoxia downregulates PPARγ via an ERK 1/2–NF-κB–Nox4-dependent mechanism in human pulmonary artery smooth muscle cells. Free Radic Biol Med 2013;63:151–160. [DOI] [PMC free article] [PubMed]
- 13.Blanquicett C, Kang BY, Ritzenthaler JD, Jones DP, Hart CM. Oxidative stress modulates PPARγ in vascular endothelial cells. Free Radic Biol Med 2010;48(12):1618–1625. [DOI] [PMC free article] [PubMed]
- 14.Du J, Xu R, Hu Z, Tian Y, Zhu Y, Gu L, Zhou L. PI3K and ERK-induced Rac1 activation mediates hypoxia-induced HIF-1α expression in MCF-7 breast cancer cells. PLoS ONE 2011;6(9):e25213. doi:10.1371/journal.pone.0025213. [DOI] [PMC free article] [PubMed]
- 15.Hung HF, Wang BW, Chang H, Shyu KG. The molecular regulation of resistin expression in cultured vascular smooth muscle cells under hypoxia. J Hypertens 2008;26(12):2349–2360. [DOI] [PubMed]
- 16.Lu X, Murphy TC, Nanes MS, Hart CM. PPARγ regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-κB. Am J Physiol Lung Cell Mol Physiol 2010;299(4):L559–L566. [DOI] [PMC free article] [PubMed]
- 17.Anand AR, Cucchiarini M, Terwilliger EF, Ganju RK. The tyrosine kinase Pyk2 mediates lipopolysaccharide-induced IL-8 expression in human endothelial cells. J Immunol 2008;180(8):5636–5644. [DOI] [PMC free article] [PubMed]
- 18.Bijli KM, Fazal F, Rahman A. Regulation of Rela/p65 and endothelial inflammation by proline-rich tyrosine kinase 2. Am J Respir Cell Mol Biol 2012;47(5):660–668. [DOI] [PMC free article] [PubMed]
- 19.Perez J, Torres RA, Rocic P, Cismowski MJ, Weber DS, Darley-Usmar VM, Lucchesi PA. Pyk2 signaling is required for PDGF-dependent vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol 2011;301(1):C242–C251. [DOI] [PMC free article] [PubMed]
- 20.Revuelta-López E, Castellano J, Roura S, Gálvez-Montón G, Nasarre L, Benitez S, Bayes-Genis A, Badimon L, Llorente-Cortés V. Hypoxia induces metalloproteinase-9 activation and human vascular smooth muscle cell migration through low-density lipoprotein receptor–related protein 1–mediated Pyk2 phosphorylation. Arterioscler Thromb Vasc Biol 2013;33(12):2877–2887. [DOI] [PubMed]
- 21.Fukai K, Nakamura A, Hoshino A, Nakanishi N, Okawa Y, Ariyoshi M, Kaimoto S, et al. Pyk2 aggravates hypoxia-induced pulmonary hypertension by activating HIF-1α. Am J Physiol Heart Circ Physiol 2015;308(8):H951–H959. [DOI] [PubMed]
- 22.Bijli KM, Kleinhenz JM, Murphy TC, Kang BY, Adesina SE, Sutliff RL, Hart CM. Peroxisome proliferator-activated receptor gamma depletion stimulates Nox4 expression and human pulmonary artery smooth muscle cell proliferation. Free Radic Biol Med 2015;80:111–120. [DOI] [PMC free article] [PubMed]
- 23.Kang BY, Kleinhenz JM, Murphy TC, Hart CM. The PPARγ ligand rosiglitazone attenuates hypoxia-induced endothelin signaling in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol 2011;301(6):L881–L891. [DOI] [PMC free article] [PubMed]
- 24.Kang BY, Park KK, Green DE, Bijli KM, Searles CD, Sutliff RL, Hart CM. Hypoxia mediates mutual repression between microRNA-27a and PPARγ in the pulmonary vasculature. PLoS ONE 2013;8(11):e79503. doi:10.1371/journal.pone.0079503. [DOI] [PMC free article] [PubMed]
- 25.Rizos CV, Elisaf MS, Mikhailidis DP, Liberopoulos EN. How safe is the use of thiazolidinediones in clinical practice? Expert Opin Drug Saf 2009;8(1):15–32. [DOI] [PubMed]
- 26.Berna N, Arnould T, Remacle J, Michiels C. Hypoxia-induced increase in intracellular calcium concentration in endothelial cells: role of the Na+-glucose cotransporter. J Cell Biochem 2002;84(1):115–131. [DOI] [PubMed]
- 27.Ginnan R, Singer HA. CaM kinase II-dependent activation of tyrosine kinases and ERK1/2 in vascular smooth muscle. Am J Physiol Cell Physiol 2002;282(4):C754–C761. [DOI] [PubMed]
- 28.Fan C, Su Q, Li Y, Liang L, Angelini DJ, Guggino WB, Johns RA. Hypoxia-induced mitogenic factor/FIZZ1 induces intracellular calcium release through the PLC-IP3 pathway. Am J Physiol Lung Cell Mol Physiol 2009;297(2):L263–L270. [DOI] [PMC free article] [PubMed]
- 29.O-Uchi J, Jhun BS, Xu S, Hurst S, Raffaello A, Liu X, Yi B, et al. Adrenergic signaling regulates mitochondrial Ca2+ uptake through Pyk2-dependent tyrosine phosphorylation of the mitochondrial Ca uniporter. Antioxid Redox Signal 2014;21(6):863–879. [DOI] [PMC free article] [PubMed]
- 30.Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARγ. Science 1996;274(5295):2100–2103. [DOI] [PubMed]
- 31.Camp HS, Tafuri SR, Leff T. c-Jun N-terminal kinase phosphorylates peroxisome proliferator-activated receptor-γ1 and negatively regulates its transcriptional activity. Endocrinology 1999;140(1):392–397. [DOI] [PubMed]
- 32.Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. Transcriptional activation by peroxisome proliferator-activated receptor γ is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem 1997;272(8):5128–5132. [DOI] [PubMed]
- 33.Hauser S, Adelmant G, Sarraf P, Wright HM, Mueller E, Spiegelman BM. Degradation of the peroxisome proliferator-activated receptor γ is linked to ligand-dependent activation. J Biol Chem 2000;275(24):18527–18533. [DOI] [PubMed]
- 34.Burgermeister E, Chuderland D, Hanoch T, Meyer M, Liscovitch M, Seger R. Interaction with MEK causes nuclear export and downregulation of peroxisome proliferator-activated receptor γ. Mol Cell Biol 2017;27(3):803–817. [DOI] [PMC free article] [PubMed]