

Abstract Abstract
Right ventricular (RV) failure is the primary cause of death in pulmonary arterial hypertension (PAH) and is a significant cause of morbidity and mortality in other forms of pulmonary hypertension. There are no approved therapies directed at preserving RV function. F-series and E-series isoprostanes are increased in heart failure and PAH, correlate to the severity of disease, and can signal through the thromboxane-prostanoid (TP) receptor, with effects from vasoconstriction to fibrosis. The goal of these studies was to determine whether blockade of the TP receptor with the antagonist CPI211 was beneficial therapeutically in PAH-induced RV dysfunction. Mice with RV dysfunction due to pressure overload by pulmonary artery banding (PAB) were given vehicle or CPI211. Two weeks after PAB, CPI211-treated mice were protected from fibrosis with pressure overload. Gene expression arrays and immunoblotting, quantitative histology and morphometry, and flow cytometric analysis were used to determine the mechanism of CPI211 protection. TP receptor inhibition caused a near normalization of fibrotic area, prevented cellular hypertrophy while allowing increased RV mass, increased expression of antifibrotic thrombospondin-4, and blocked induction of the profibrotic transforming growth factor β (TGF-β) pathway. A thromboxane synthase inhibitor or low-dose aspirin failed to replicate these results, which suggests that a ligand other than thromboxane mediates fibrosis through the TP receptor after pressure overload. This study suggests that TP receptor antagonism may improve RV adaptation in situations of pressure overload by decreasing fibrosis and TGF-β signaling.
Keywords: pulmonary arterial hypertension, thromboxane-prostanoid receptor, adaptation, isoprostane, cardiomyocyte, right ventricle
Introduction
Right ventricular (RV) failure is the primary cause of death in pulmonary arterial hypertension (PAH) and is a source of significant morbidity and mortality in other forms of pulmonary hypertension. Loss of RV function can progress despite treatments decreasing pulmonary arterial pressure.1 The RV ability to adapt to chronic pressure overload often determines clinical outcome; however, there are no specific treatments to aid RV adaptation.2 Adaptive ventricular hypertrophy with increased protein synthesis sustains function, whereas fibrosis and cardiomyocyte hypertrophy can cause arrhythmias and contractile dysfunction. This dysfunction can progress to RV dilatation and failure.3 Consequently, treatment strategies promoting adaptation in the face of chronic load stress could preserve cardiac function and improve outcomes.4
The development of cellular hypertrophy and myocardial fibrosis that occurs with chronic pressure overload is also associated with increased oxidative stress and lipid peroxidation.5 The 15-F2t isoprostane (8-isoPGF2α, or 8-isoF) is a common biomarker for oxidative stress.6 Its levels increase with ventricular dilatation and correlate with the severity of heart failure.7,8 In addition to being a biomarker, it is suggested that 8-isoF and other isoprostanes can play a direct role in cardiomyopathy. F-series and E-series isoprostanes are known to signal through the G protein-coupled thromboxane-prostanoid (TP) receptor, with effects ranging from vasoconstriction to fibrosis.9-11 The cyclooxygenase products of cyclic endoperoxide and thromboxane A2 (TxA2) are also ligands of the TP receptor, and TP receptor activation contributes to cardiac hypertrophy in models of chronic hypertension12,13 and decreases cardiac function in Gh-overexpressing mice.14 The TP receptor is found not only in platelets and vessels but also in mouse and rat ventricular cardiomyocytes15 and the human RV, where receptor density is increased in patients with PAH.16 This G protein-coupled receptor can signal through Gαq/11 or Gα12/13 subunits, and so receptor activation can result in increased intracellular calcium; in cardiomyocytes, this may contribute to arrhythmia.17 Given the predominantly deleterious consequences of TP receptor activation in conditions of cardiac stress and the production of isoprostanes associated with cardiomyopathy, we examined the effects of TP receptor antagonism in a pulmonary artery banding (PAB) model of RV pressure overload.
Material and Methods
Animal protocols
This study was performed in accordance with the National Institutes of Health’s Public Health Service Policy of Humane Care and Use of Laboratory Animals and the Animal Welfare Act. All protocols were approved by the Vanderbilt University Institutional Animal Care and Use Committee (IACUC protocol M/13/030). Animals were housed with a 12L∶12D cycle and with ad libitum access to food and water. Experiments were performed using appropriate anesthetics and analgesics, and every effort was made to minimize animal pain and distress.
PAB model
Equal numbers of male and female age-matched (approximately 4 months old) FVB/NJ mice, obtained by in-house breeding, were used for PAB. Except for gene expression analysis, all data presented consist of a sex-mixed group. PAB was performed as described elsewhere.18 Briefly, mice were anesthetized with 3% isoflurane and orotracheally intubated. A sternal incision was made, the pericardium was removed, and a tantalum clip (Weck, Research Triangle Park, NC) was placed around the pulmonary artery for mechanical vasoconstriction. Sham-operated animals underwent the same procedure without placement of a banding clip. Animals were then sutured closed and allowed to recover from anesthesia. Mice received 7.5 mg/kg of carprofen subcutaneously as postoperative analgesia.
The day after surgery, surviving mice were randomized to receive either 25 mg/kg/day CPI211 (ifetroban; Cumberland Pharmaceuticals, Nashville, TN) in drinking water or normal drinking water (vehicle) for 2 weeks before hemodynamic evaluation. In a follow-up experiment, 10 mg/kg/day of veterinary aspirin (QC Supply, Schuyler, NE) with 70% ethanol vehicle was given to PAB or sham-operated mice, or 50 mg/kg/day ozagrel HCl hydrate (Combi-Blocks, San Diego, CA) was given to PAB mice, and several mice were alternatively treated with ethanol vehicle or CPI211 as negative or positive controls. Final concentration of ethanol in drinking water was approximately 0.04%. All drugs were pretested for palatability to ensure normal consumption of drinking water. Mice were weighed and water was changed once a week.
To visualize monocytes, LysM-GFP mice were obtained by cross-breeding LysM-Cre × mTomato/GFP reporter mice B6.129P2-Lyz2tm1(cre)Ifo/J (a gift from Tim Blackwell) × Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo (JAX stock #007676). Age- and sex-matched C57Bl/6 mice, obtained by in-house breeding, were used to match the LysM-GFP background.
Echocardiography
One day before sacrifice, 2-dimensional echocardiography (Vivo 770 High-Resolution Image System, VisualSonics, Toronto, Canada) was performed as described elsewhere,19 while mice were anesthetized with 3% isoflurane. The following RV-specific parameters were reliably attained in mice from this setup: area of pulmonary artery, pulmonary artery velocity time integral and acceleration time, and stroke volume with resulting cardiac output and index.
Hemodynamics
Open-chested catheterization of the pulmonary artery was performed in PAB mice as described elsewhere.19 Briefly, mice were anesthetized with 5% isoflurane and orotracheally intubated. During catheterization, animals were mechanically ventilated under 2% isoflurane. A 1.4F Mikro-tip catheter (Millar Instruments, Houston, TX) was introduced into the surgically exposed heart of an anesthetized mouse, and hemodynamic data were acquired at 1,000 Hz and continuously recorded with a Millar MPVS-300 unit coupled to a Powerlab 8-SP analog-to-digital converter (AD Instruments, Colorado Springs, CO).
Histology
Human RV tissue was obtained under consent via the Pulmonary Hypertension Breakthrough Initiative (PHBI) or autopsy patients from Vanderbilt University, according to the Second International Helsinki Declaration. Collection and storage of paraffin-mounted samples were approved by the Vanderbilt University Institutional Review Board (protocol 9401). Human RV was stained with antibody raised against the C-terminal of the human TP receptor alpha (Cayman Chemical #10004452). For mice, whole hearts or RVs were flash-frozen alone or in embedding media before sectioning. Samples were typically sliced at the midpoint before sectioning; thus, RV slices used for histology tended to be from the mid-RV. Slices (5 µm) were mounted on glass slides for direct visualization of fluorescence or stained with Masson’s trichrome. All scale bars shown are 25 microns. Fibrosis and myocyte diameter were assessed by blind reading as described elsewhere19 using Nikon NIS Elements software. Myocyte diameters were determined from 4–6 fields per sample, 25 cells per field at ×20. All PAB mice with RV systolic pressure (RVSP) > 30 mmHg were analyzed by histology for fibrosis when possible; any differences in n between hemodynamic data and histological data reflect tissue availability or quality (for cross-sectional analysis only). Fluorescently labeled tissue was visualized using confocal imaging on a Nikon Eclipse Ti outfitted with a Nikon DS-Fi1 camera.
Gene expression arrays
Gene expression array experiments were designed using a design: (sham/PAB) × (vehicle/CPI211) × (male/female), for a total of 8 arrays. Each array consisted of RNA from the RV of a pool of 3 mice, and only mice with RVSP ≥ 30 were used for the PAB groups. After RNA quality assurance, expression analysis was performed using Mouse Genome 2.0 Affymetrix (Foster City, CA) expression arrays, and data were analyzed as described elsewhere.20 Raw data were normalized using the Affymetrix RMA protocol. Of the original 41,345 probe sets, 12,179 remained after removing sets with universal low expression (<7.0) and low variability (log2 variation between maximum and minimum across all samples <0.2). A total of 9,529 probe sets remained after further limiting data to probe sets with accession data.
Principal components analysis was performed on this set of 9,529 probe sets using JMP 11 (SAS Institute, Cary, NC). Gene expression differences between groups were ranked by starting from the log-based group differences and subtracting twice the standard deviation in probe set expression within groups, summed by root mean square. Ranking was thus a hybrid of fold change and statistical significance, effectively the 95% confidence level minimum fold change. Heat maps were generated within JMP 11. Gene ontology analyses were performed using Webgestalt (Vanderbilt University).
Immunoblotting
Western blotting was performed as described elsewhere.21 PVDF membranes were used for transfer, and antibodies used include anti-phosphoSMAD2/3 (1∶1,000; Cell Signaling, Danvers, MA), anti-TSP-4 (1 µg/mL; R&D Systems, Minneapolis, MN), anti-HSP-70 (1∶1,000; Enzo Life Sciences, Farmingdale, NY), anti-TP receptor (Cayman Chemical, Ann Arbor, MI), anti-α- and β-MHC (1∶4,000 and 1∶2,000; Proteintech, Chicago, IL), and anti-PAI-1 (1∶1,000; Abcam, Cambridge, MA). All antibodies to phosphorylated proteins were diluted in 5% BSA/TBST, and the rest were diluted in 2% milk/TBST.
Flow cytometry
C57Bl/6 mice or mice expressing eGFP under the LysozymeM promoter, derived as described above, were banded and treated as above for 2 weeks. Right hearts were dissected and prepared for flow cytometry as described in Chow et al.,22 using a BD Fortessa for analysis (BD Biosciences, San Jose, California). Cells from C57Bl/6 mice were stained with antibodies to detect CD45+ cells as described elsewhere.23 Fluorescence minus one (FMO) controls were used to set gates. GFP+ and CD45+ cells from LysozymeM-eGFP mice were analyzed by flow cytometry using GFP-positive LV or hearts from genotyped GFP-negative littermates as FMO controls. The compensation controls were established as cells only, cells plus DAPI, cells plus APC-CD45 antibody, and cells plus PerCP or FITC or PE antibody. Alternatively, comp beads were used. The gating strategy included FSC/SSC, DAPI+Ter119 to exclude dead and red blood cells, followed by gating on the CD45+ population. For LysM-eGFP mice, the GFP+ population was gated to the FMO generated by wild-type RV. Antibodies used were CD45-APC-780 (emits 750), F4/80-PE-Cy7, CD11b-APC-660, CD14-APC, CD86-FITC, CD206-PE, and also Ter119–450 in the PAC Blue dump gate. All were from eBioscience (San Diego, CA), with the exception of CD206-PE (Biolegend, San Diego, CA). Gating strategies are shown in Supplemental Figure 2 (3MB, pdf) .
Statistics
Values are expressed as mean ± SEM, and sample size is given for each figure. Two-way analysis of variance (ANOVA) or 1-way ANOVA followed by the Sidak post test was performed on Prism (GraphPad, San Diego, CA) to determine statistical significance.
Results
Differential expression of TP receptor in patients with PAH
As rationale for a role for TP receptor signaling in pulmonary hypertension, we examined RV taken from human patients with PAH and healthy control subjects for the TP receptor by immunohistochemistry, because earlier studies in PAH heart were confined to radioligand binding.16 Stained with an antibody raised against the C-terminus of the TPα isoform, cardiomyocytes of patients with PAH robustly expressed the TP receptor throughout the cell surface, whereas expression in normal RV was primarily perinuclear (Fig. 1A–1D). This intracellular pool probably indicates unglycosylated immature receptor.24 Interestingly, tissue from a former patient with PAH who had undergone lung transplantation 13 years earlier contained both an intracellular pool and light cell surface expression (Fig. 1E); thus, receptor expression may revert after RV load stress is reduced.
Figure 1.

Distribution of thromboxane-prostanoid (TP) receptor in the right ventricles (RVs) of healthy patients and patients with pulmonary arterial hypertension (PAH). By immunohistochemistry, RV taken from patients with idiopathic (A) or hereditary (B) PAH expressed the TPα receptor (in brown) at the cell membrane, whereas internalized pools of receptor were seen in healthy control subjects (C, D; arrowheads). E, RV from a patient with PAH who had undergone curative lung transplant 13 years earlier containing expression features associated with both PAH and healthy control subjects. Scale bars in all photographs are 25 microns.
Increased ventricular efficiency in PAB mice receiving CPI211
Mice underwent PAB or sham surgery and were treated for 2 weeks with vehicle or CPI211 (competitive antagonist of TPα/TPβ25) in the drinking water. The dose of CPI211 was chosen on the basis of doses previously used in rodent models.26,27 RVSP was elevated in PAB mice (Fig. 2A), indicating successful mechanical vasoconstriction. An RVSP of 30 mmHg was used to define successful PAB surgery, and only samples from mice at or above this threshold were used for additional analysis of PAB groups. At 2 weeks after banding, there was no significant change in cardiac output, and total RV mass similarly increased in vehicle- and CPI211-treated PAB groups (Fig. 2B, 2C). However, by echocardiography, the E/A wave ratio was increased in mice given TP receptor antagonist (Fig. 2D). Although this measurement can be confounded by loading conditions, it suggests increased filling efficiency. A low 1/10 dose of CPI211, 3 mg/kg, did not have any significant effect on E/A wave ratio in PAB mice ( for PAB-3 mg/kg vs. for sham-vehicle and for PAB-25 mg/kg). A full table of all echocardiography parameters is available in the data supplement (3MB, pdf) .
Figure 2.
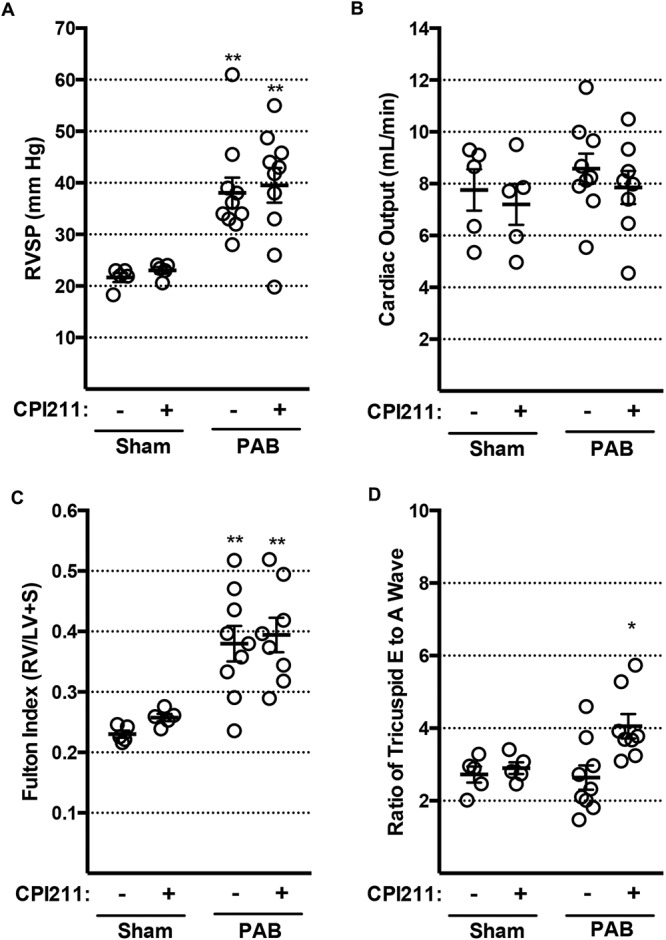
Hemodynamic characteristics in pulmonary artery banding (PAB) mice. In each figure, each circle represents data from one mouse; dark bars represent mean values with standard error of the mean. A, PAB induced an increase in right ventricular systolic pressure (RVSP) from ∼22 to ∼40 mmHg (2 asterisks: from sham by 2-way ANOVA followed by Sidek post test). B, Cardiac output by echocardiography was not reduced in PAB mice after 2 weeks. C, Ratio of right ventricular weight to left ventricle and septum weight increases in PAB mice (2 asterisks: from sham by 2-way ANOVA followed by Sidek post test); this increase is not blocked by CPI211. D, Tricuspid E/A wave ratio, a measure of diastolic function, is improved by CPI211 (asterisk: from PAB vehicle by 2-way ANOVA followed by Sidek post test) but only in PAB mice. Sham and PAB (A); after removing mice where PAB was not successful (RVSP < 30 mmHg), PAB vehicle and PAB CPI211 (B–D).
Decreased cardiac fibrosis with TP receptor antagonism
PAB mice developed significant RV fibrosis by 2 weeks, which was almost completely abolished with TP receptor antagonism (Fig. 3A, 3B). The 3 mg/kg dose of CPI211 was ineffective in preventing fibrosis with a mean (±SEM) fibrotic area of compared with PAB vehicle control (). Thus, from this point forward, all experiments were confined to the 25 mg/kg/day dose of CPI211. A “low dose”28 of aspirin (10 mg/kg/day), chosen to avoid anti-inflammatory or salicylate-mediated effects, or a 50 mg/kg/day dose of ozagrel, a thromboxane A2 synthase inhibitor, failed to prevent fibrosis in this model (Fig. 3A, 3B), suggesting that thromboxane A2 is not the ligand mediating fibrosis. Neither ozagrel nor aspirin changed the E/A wave ratio in PAB mice (data not shown).
Figure 3.

CPI211 prevents cardiac fibrosis following pressure overload. A, Two weeks after pulmonary artery banding (PAB), fibrosis is evident in histologic sections of frozen right ventricle (RV) stained with Masson trichrome. This effect is blocked by CPI211 but not by aspirin (ASA) or the thromboxane synthetase inhibitor ozagrel (oza). B, Quantification of fibrotic area. Each symbol indicates one animal, with bars for the mean. Two asterisks: from sham or from vehicle PAB where indicated by 1-way ANOVA followed by Sidek post test. CPI211-treated PAB RV was not significantly different from either CPI211-treated or vehicle-treated control. Data are compiled from 3 separate experiments. Sham ( for ASA sham); PAB for ASA and oza, PAB for vehicle (veh) and CPI211. Groups consist of an even mix of male and female animals; no sex-specific difference in fibrosis was seen at 2 weeks ( by unpaired t test). Scale bars are 25 microns.
Changes in RV gene expression in CPI211-treated PAB mice
To delineate the mechanism through which antagonism of the TP receptor reduced fibrosis and protected RV contractility, we performed gene expression arrays on sets of pooled RV from mice with sham or PAB surgery, with and without drug treatment. By principal components analysis, we found a strong effect of banding (X-axis in Fig. 4A) and an effect of CPI211 only in mice that had undergone PAB. CPI211 did not significantly change gene expression in mice with only sham surgeries (Y-axis in Fig. 4A). In PAB mice, CPI211 treatment enhanced transcription of some genes, whereas the effect of banding on other genes was blocked with TP receptor antagonism (Fig. 4B). In particular, CPI211 treatment changed gene expression associated with adhesion, collagen organization, extracellular structure, and developmental processes (Fig. 4C). We explore functional correlates of these findings through the rest of this study.
Figure 4.

Gene expression arrays from pooled RNA from right hearts of mice with and without CPI211, with pulmonary arterial banding (PAB) or sham surgery (A). Principal components analysis found PAB to account for the most variation in genes (principal component 1). Treatment with CPI211 (principal component 3) caused substantial changes in gene expression only in mice with PAB. Blue indicates CPI211, orange indicates vehicle, heavy outline indicates PAB, and no outline indicates sham. In sham-operated mice, expression is primarily separated by sex (circles: female pool; square: male pool). B, Heat map of the top 200 genes affected by banding shows CPI211-enhanced banding effect on some genes (top group), blocked banding effect on other genes (middle group), and did not affect banding effect in still others (bottom group). C, Gene ontology group analysis of the top 200 genes impacted by CPI211 treatment in banding. Angular width corresponds to number of genes altered with CPI211 treatment compared with vehicle-treated PAB mice; radius corresponds to −log10 (statistical significance, so longer is more significant).
Decreased cellular hypertrophy in CPI211-treated PAB mice
PAB mice receiving TP receptor antagonist increased expression of genes associated with myogenic differentiation29,30 compared with banded controls (Fig. 5A). Although RV weight increased similarly in vehicle-treated and CPI211-treated PAB mice (Fig. 1C), the increase in cardiomyocyte diameter normally associated with PAB was blocked with CPI211 treatment (Fig. 5B, 5C). Aspirin- or ozagrel-treated mice had cardiomyocyte diameters similar to those of vehicle-treated mice (Fig. 5B, 5C). Interestingly, CPI211 treatment of PAB mice caused an induction of β-myosin heavy chain (β-MHC) protein with no change in α-MHC expression (Fig. 5D). In an aortic banding model of pressure overload, this induction is associated with decreased individual hypertrophy in β-MHC–expressing cells.31,32 The induction of β-MHC after TP receptor antagonism was not due to increased animal age, as the mean (±SEM) ages of vehicle-treated and CPI211-treated PAB mice were and days, respectively.
Figure 5.

Adaptive remodeling (A) gene expression arrays showed CPI211-induced genes associated with adaptive remodeling but only in the context of pressure overload. Each point represents the mean of the male and female pools of RNA from 3 right ventricles (RVs) each, normalized to sham-operated control; error bars show standard error of the mean. Meox1: mesenchyme homeobox 1; Myh7: myosin heavy chain-7; Nkd2: naked cuticle 2; Nmrk2: nicotinamide riboside kinase 2; Tnc: tenascin C. B, Cardiomyocyte size increases in mice with pulmonary arterial banding (PAB). This effect is blocked by CPI211 but not by aspirin (ASA) or ozagrel (oza). Each symbol indicates the mean diameter of 25 cells/field, 4 separate fields/mouse, taken from one mouse by a blinded observer. Sham ; PAB as shown; data are compiled from 3 separate experiments. Two asterisks represent and 3 asterisks represent from sham, and 2 asterisks represent from vehicle (veh) PAB where indicated by 1-way ANOVA followed by Sidek post test. Despite an effort to obtain RV cross-sections, cardiomyocytes were sometimes in different orientations; however, no significant difference in diameter was observed between cross-sectioned and longitudinal cardiomyocytes. C, Representative figures at ×40, demonstrating increase in cardiomyocyte size following PAB. D, Western blot demonstrating α myosin heavy chain (α-MHC) and β-MHC expression in RV. α-MHC is variable but unchanged, whereas β-MHC is strongly induced in PAB mice given the thromboxane-prostanoid receptor antagonist. Each band represents RV from one mouse.
Decreased TGF-β signaling and enhanced TSP-4
We examined the effect of thromboxane receptor antagonism on known fibrotic and antifibrotic signaling pathways. Mice receiving the TP receptor antagonist had a −2.4-fold decrease in phospho-SMAD2/3 associated with PAB, compared with vehicle-treated mice (Fig. 6A). Accordingly, we saw, by microarray, a reduced expression of genes associated with fibrosis, with increased expression of antifibrotic genes (Fig. 6B). CPI211-treated H9C2 cardiomyocytes given exogenous TGF-β in culture did not change PAI-1 expression or promoter activity compared with vehicle-treated cells (see Supplemental Fig. 3 (3MB, pdf) ), indicating no direct blockade of TGF-β signaling from its receptor and suggesting that the in vivo block of TGF-β occurs upstream of ligand binding. Opposing the profibrotic TGF-β, pressure overload induces antifibrotic thrombospondin-4 (Thbs4; TSP-4) messenger RNA (mRNA).33 CPI211 treatment enhances Thbs4 mRNA expression in PAB RV only (Fig. 6B), which translates to TSP-4 protein increased 18-fold in CPI211-treated banded mice (Fig. 6C). While localization of TSP-4 was confined to fibrotic patches in vehicle-treated RV, PAB mice receiving CPI211 demonstrated increased cardiomyocyte expression of TSP-4 (Fig. 6D).
Figure 6.

Mechanisms leading to decreased fibrosis. A, CPI211 blocks phosphorylation of transforming growth factor β signaling molecule Smad2/3 in vivo. Each band represents right ventricle (RV) from one pulmonary artery banding (PAB) mouse. B, Gene expression arrays from RV showed CPI211 reduced profibrotic genes and induced antifibrotic genes but only in the context of pressure overload. Expression is shown normalized to sham-operated control. Each point represents the mean of the male and female pools of RNA (3 RV/pool), normalized to sham-operated control; error bars represent standard error of the mean. Some error bars are smaller than the symbols and so may be hidden. Aspn: asporin 1; Fbxo32: F box protein 32; Ltbp2: latent transforming growth factor β binding protein 2; Pdgfd: platelet-derived growth factor D; Thbs4: thrombospondin 4 (Tsp4). C, D, PAB induces Tsp4 protein expression in RV, which is strongly enhanced with CPI211 treatment. Each band represents a different mouse. RV was immunostained for Tsp4 (shown in dark brown; D) and counterstained with methyl green before being photographed at ×40. Scale bars are 25 microns. veh: vehicle.
Cardiac monocytes in PAB mice
CPI211 treatment after PAB decreased RV expression of Cd14, a marker for mature macrophages, as well as Tlr8 and other proinflammatory genes by expression array (Fig. 7A). TP receptors are also expressed on monocytes,34 and blockade of these receptors may have immune-specific effects. To determine whether the decrease in fibrosis seen with TP receptor inhibition was due to decreased macrophage infiltration or proliferation in the RV, we performed PAB on LysM-Cre mice expressing eGFP under the LysozymeM promoter, where all cells of monocyte lineage are labeled with GFP, even if they have later differentiated. While the majority of monocytes located to the left heart or septum (Stadtfield et al.35; see Supplemental Fig. 1 (3MB, pdf) ), PAB appeared to increase the number of RV monocytes in vehicle-treated and aspirin-treated mice (Fig. 7B). Flow cytometry of RV taken from wild-type mice revealed no significant changes with banding in any macrophage population examined, with a nonsignificant trend toward an increased percentage of CD45+ cells in vehicle-treated, but not CPI211-treated, PAB mice compared with sham-operated controls (sham-vehicle: ; sham-CPI211: ; PAB-vehicle: ; and PAB-CPI211: ; see Supplemental Table 2 (229.6KB, pdf) for full table). This mismatch to the LysM-Cre mice suggests that the cells may lose their circulating differentiation markers upon lodging in the heart. To further examine this, we analyzed GFP+ and CD45+ RV cells from PAB or sham-operated LysM-Cre mice by flow cytometry. Similar to our previous experiments, the CD45+ population was not quite significantly increased after PAB ( for surgery; Fig. 7C), and the increase in GFP+ cells in vehicle-treated PAB mice was also nonsignificant ( for interaction; Fig. 7C). There was no change in the percentage of CD45+ cells that were of monocyte lineage. However, mice that underwent PAB lost CD45 expression in a population of GFP+ monocyte lineage cells, indicating differentiation of these cells; this differentiation was blocked with CPI211 treatment (; Fig. 7C, 7D). This modest effect on monocyte number and differentiation suggests that the anti-inflammatory effects seen by expression array may be more likely caused by a change in cell signaling within the RV following TP receptor antagonism than by a direct effect on monocytes.
Figure 7.

Inflammatory cell recruitment. A, Gene expression arrays showed that CPI211 reduced expression of genes associated with inflammatory cell recruitment but only in the context of pressure overload. Expression is shown normalized to sham-operated control, and error bars reflect SEM (3 right ventricles [RVs]/pool of RNA; male and female pools used for ). Cd14: cluster of differentiation 14; Cd74: cluster of differentiation 74; Ly6c1: lymphocyte antigen 6 complex C1; Tap1: transporter 1 adenosine triphosphate (ATP)–binding cassette; Tlr8: toll-like receptor 8. B, transgenic LysM-Cre × flox-mTomato-flox eGFP mice were used to visualize cells of monocyte derivation in the RV. These mice express a fluorescent red, except cells derived from monocyte lineage, which express green fluorescent protein (GFP) even if they have later differentiated. By microscopy, PAB increases numbers of green cells in RV of vehicle (veh) and ASA mice, but not in CPI211-treated mice (representative images from 2–3 transgenic hearts shown at ×20). Scale bars are 25 microns. C, Results of flow sorting for CD45+ and GFP+ cells in the viable RV cell population of LysM-eGFP mice after PAB; the means and SEM are shown (, where each mouse). Any changes in total CD45 or GFP expression did not reach statistical significance (C, top; for PAB and for interaction, respectively, by 2-way ANOVA). PAB mice lost CD45 marker expression in GFP+ cells, which is blocked by treatment with CPI211 (C, lower right; for interaction by 2-way ANOVA (D). Representative scatterplots of viable cells from PAB-veh (left) and PAB-CPI211 (right) RV are shown. Fuchsia cells are CD45+, whereas green cells are GFP+; gating strategy and additional data are shown in the data supplements.
Discussion
The effect of TP receptor antagonism was studied in mice with mechanical constriction of the pulmonary artery, a pressure overload model of PAH-associated RV hypertrophy. Treatment with the TP receptor antagonist CPI211 reduced RV fibrosis and cardiomyocyte hypertrophy in PAB mice and increased E/A ratio, which is an indicator of cardiac efficiency. This was associated with augmented RV expression of antifibrotic and muscularization genes as well as with decreased expression of genes associated with inflammation and a decrease in RV phospho-SMAD2/3. Few differences were found in sham-operated mice receiving CPI211, indicating that CPI211-mediated gene expression changes are specific to PAB. This requirement of PAB surgery for the effects of CPI211 creates difficulties in modeling cellular events in vitro.
The PAB model is usually insufficient to induce RV failure36 but is instead a model of RV adaptation. Although it may not model the cardiac metabolic defects seen in hereditary PAH,37 PAB does mimic the immediate pressure overload of pulmonary embolism and chronic thromboembolytic pulmonary hypertension. Although the PAB model allows us to examine the direct effects of increased afterload on the RV and does not rely on direct cardiotoxicity or an extensive initial inflammatory response, like the monocrotaline or sugen/hypoxia models, the sudden increase in pressure differs from the more gradual onset of most PAH. Thus, the role of the TP receptor may differ in animal models that allow for gradual RV adaptation.
The cardioprotective effects seen with TP receptor antagonism were not duplicated with low-dose aspirin treatment, the dose chosen to avoid confounding anti-inflammatory or salicylate-mediated effects, or with the thromboxane synthase inhibitor ozagrel. This suggests that platelet-generated or locally generated TxA2 does not mediate the fibrosis and cellular hypertrophy associated with PAB. Similarly, aspirin did not duplicate the effects of CPI211 in reducing myocardial infarct size in a coronary occlusion/reperfusion model.38 Locally available isoprostanes, such as 8-iso-PGF2α, are also known to signal through the TP receptor39,40 and can cause increased collagen production and fibrogenic effects.41 Indeed, reducing oxidative stress and isoprostane generation through enhancement of superoxide dismutase or introduction of free radical scavengers can have antifibrotic, cardioprotective effects in pressure overload.42-44 Alternatively, the effects of CPI211 could be mediated by another endogenous ligand for the TP receptor, such as prostaglandin H2 or 20-HETE,45 or a combination of ligands.
By immunostaining, the TP receptor localized primarily to cardiomyocytes in the RV, similar to the gene and protein expression demonstrated by others.15,17 We also demonstrate here an upregulated cell surface expression of the TP receptor in RV from patients with PAH, a finding in accord with the radioligand binding work of Katugampola and Davenport.16 Human TPα receptor expression can be augmented posttranslationally by reactive oxygen species in a feed-forward mechanism,24 which would correspond with both the pattern of expression seen in patients with PAH as well as availability of an isoprostane ligand. While TP receptor protein and mRNA are also robustly expressed in murine RV, at 2 weeks after PAB, we do not see an upregulation in RV protein extracts when normalized to HSP70 expression (see Supplemental Fig. 4 (3MB, pdf) ). This may reflect differential regulation of the murine TP receptor; alternatively, it may reflect a decrease in cardiomyocyte contribution to the total RV protein in PAB mice, because the percentage by volume occupied by fibrotic tissue is greater.
In this study, we also examined the effects of TP receptor inhibition on RV macrophage number and cell marker expression in PAB mice. Analysis of cell surface markers did not find any significant differences in monocyte population, not only with CPI211 treatment compared with vehicle, but also between PAB and sham-operated mice. When we analyzed mice containing a lineage tracer for monocytes, we found that TP receptor antagonism modestly blocked the differentiation of monocytes into nonmonocytic cells in PAB mice. Future experiments will be necessary to determine the identity of the CD45−/GFP+ population, but because a subpopulation of cardiac fibroblasts are monocyte derived,46 a monocyte-to-fibroblast transition is an intriguing possibility. The absence of overt effects of CPI211 on macrophage number suggests that CPI211 does not directly affect either proliferation of existing macrophages or vascular permeability to increase monocyte extravasation. This also implies cardiomyocyte-mediated TP receptor activity after RV pressure overload.
At 2 weeks after PAB, CPI211 treatment modestly increased the E/A wave ratio in PAB mice, indicating possible increased contractile efficiency. This is similar to a previous study, which demonstrated that TP receptor antagonism preserves RV efficiency after endotoxic shock.47 It is unknown whether this is due to the decrease in cardiac fibrosis, the decrease in cardiomyocyte hypertrophy associated with PAB, or perhaps a combination of these 2 mechanisms. The decreased cellular hypertrophy we find with CPI211 is consistent with the known hypertrophic effects of TP receptor activation in mice14,48 but is likely a concerted in vivo effect and not due to direct TP receptor inhibition on cardiomyocytes.49 Here, with the prevention of individual cellular hypertrophy, we also noted concomitant increased RV expression of β-MHC in TP antagonist–treated PAB mice, with no change in α-MHC expression. This corresponds with an earlier study by Lopez et al.31 in which, by size-sorting individual cardiomyocytes after pressure overload, they found that β-MHC expression only occurred in nonhypertrophic cardiomyocytes, whereas all cells had similar α-MHC expression.
Ventricular hypertrophy is useful as a compensatory mechanism in functional adaptation to increased load stress; however, this occurs on a continuum, the other extreme of which is maladaptive remodeling and pathological hypertrophy.50 Interestingly, nonadaptive hypertrophy can result from activation of other Gq/11-coupled, G protein–coupled receptors, such as the angiotensin II receptor or endothelin receptor, in association with protein kinase C or phosphoinositide-3-kinase activity.50,51 Although individual cells did not increase in diameter, PAB mice given CPI211 had increases in RV weight similar to vehicle-treated mice, as evidenced by increased Fulton index. Among the genes activated by CPI211 in conjunction with banding were a number of genes associated with cell growth and reprogramming. We theorize that TP receptor inhibition induces muscularization in response to pressure overload, whether by decreasing cardiomyocyte loss or increasing total number or contractility of cardiomyocytes, while also decreasing the fibrotic response. One group has found that stimulation of the TP receptor dose-dependently causes DNA fragmentation and cell death in cultured primary cardiomyocytes.15 These effects were reversed with a TP receptor antagonist, suggesting that CPI211 treatment could prevent cell death in the RV during pressure overload; however, additional studies will be needed to ascertain this possibility. Reduced TGFβ activity is probably partially responsible for the diminished fibrotic response, although the CPI211-initiated event behind the decrease in active TGFβ is yet unknown.
TSP-4, which is increased by PAB and further upregulated with CPI211 treatment, is not known to regulate TGFβ activation but itself can decrease fibrosis and hypertrophy and increase contractility and cardiac adaptation in pressure overload.33,52,53 Exogenous TSP-4 also decreases cardiac fibroblast activation and collagen production when added in cell culture, whereas coculture with cardiomyocytes from TSP-4 knockout mice increases fibroblast expression of collagen and activation markers in response to angiotensin II (Ang II).53 Thus, cardiomyocyte TSP-4 acts in vitro and in vivo as a signaling protein to cardiac fibroblasts to decrease activation and fibrosis, and it likely mediates some antifibrotic effects of TP receptor antagonism.
Expression of TSP-4 is normally repressed by Kruppel-like transcription factor 6 (KLF6), and the removal of this repression decreases fibrosis as well as increases the E/A wave ratio in hearts of mice infused with Ang II.53 We see both of these effects in the PAB mice treated with TP receptor antagonist. By immunostaining, we found that TSP-4 protein localized to cardiomyocytes in PAB CPI211-treated mice, as opposed to the interstitial space in vehicle-treated mice, which also corresponds with its localization in hearts of KLF6-haploinsufficient versus wild-type mice after Ang II infusion.53 This cardiomyocyte localization would also support intracellular signaling, and within cardiomyocytes, TSP-4 is thought to partially act protectively by inducing protective endoplasmic reticulum (ER) stress signaling via (Atf6α), an ER stress response transcription factor.54 In our model, there was no difference in Atf6α mRNA levels either after PAB or TP receptor antagonism (data not shown). It is entirely possible that our 2-week time point missed any increase in mRNA or that the RV stress response differs from the LV model where the Atf6α response to TSP-4 was delineated.
In summary, these studies demonstrate that CPI211 is cardioprotective against pressure overload by moving the right heart toward adaptation rather than toward a maladaptive fibrosis, inflammation, and cellular hypertrophy. TP receptor antagonists have been studied clinically in humans for years and are orally bioavailable25 but could not compete with aspirin in the primary end point of platelet aggregation.55 The TPr antagonist seratrodast is currently approved in India, China, and Japan as asthma treatment and is well tolerated. Because there are no existing approved therapies to support right heart function under stress, CPI211 and other TP receptor antagonists are excellent candidates for additional translational research in this area.
Source of Support: This work was supported by Cumberland Pharmaceuticals and the National Heart, Lung, and Blood Institute of the National Institutes of Health (P01-HL108800, R01-HL095797).
Conflict of Interest: Cumberland Pharmaceuticals, which funded this project, owns CPI211 through Cumberland Emerging Technologies, a joint initiative between Cumberland Pharmaceuticals, Vanderbilt University, and Tennessee Technology Development. JDW, BMV, LP, and EJC have filed US patent application no. 14/715,143, “Compositions and Methods of Treating Cardiac Fibrosis with Ifetroban.”
Supplements
Data Supplement (3MB, pdf)
Supplemental Tables (229.6KB, pdf)
References
- 1.van de Veerdonk MC, Kind T, Marcus JT, et al. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol 2011;58:2511–2519. [DOI] [PubMed]
- 2.Pokreisz P, Marsboom G, Janssens S. Pressure overload-induced right ventricular dysfunction and remodelling in experimental pulmonary hypertension: the right heart revisited. Eur Heart J Suppl 2007;9:H75–H84.
- 3.Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 2009;135:794–804. [DOI] [PubMed]
- 4.Vonk Noordegraaf A, Galie N. The role of the right ventricle in pulmonary arterial hypertension. Eur Respir Rev 2011;20:243–253. [DOI] [PMC free article] [PubMed]
- 5.Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007;49:241–248. [DOI] [PubMed]
- 6.Montuschi P, Barnes PJ, Roberts LJ 2nd. Isoprostanes: markers and mediators of oxidative stress. FASEB J 2004;18:1791–1800. [DOI] [PubMed]
- 7.Mallat Z, Philip I, Lebret M, Chatel D, Maclouf J, Tedgui A. Elevated levels of 8-iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure: a potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation 1998;97:1536–1539. [DOI] [PubMed]
- 8.Nonaka-Sarukawa M, Yamamoto K, Aoki H, et al. Increased urinary 15-F2t-isoprostane concentrations in patients with non-ischaemic congestive heart failure: a marker of oxidative stress. Heart 2003;89:871–874. [DOI] [PMC free article] [PubMed]
- 9.Acquaviva A, Vecchio D, Arezzini B, Comporti M, Gardi C. Signaling pathways involved in isoprostane-mediated fibrogenic effects in rat hepatic stellate cells. Free Radic Biol Med 2013;65:201–207. [DOI] [PubMed]
- 10.Crankshaw D. Effects of the isoprostane, 8-epi-prostaglandin F2 alpha, on the contractility of the human myometrium in vitro. Eur J Pharmacol 1995;285:151–158. [DOI] [PubMed]
- 11.Janssen LJ, Tazzeo T. Involvement of TP and EP3 receptors in vasoconstrictor responses to isoprostanes in pulmonary vasculature. J Pharmacol Exp Ther 2002;301:1060–1066. [DOI] [PubMed]
- 12.Francois H, Athirakul K, Mao L, Rockman H, Coffman TM. Role for thromboxane receptors in angiotensin-II-induced hypertension. Hypertension 2004;43:364–369. [DOI] [PubMed]
- 13.Francois H, Makhanova N, Ruiz P, et al. A role for the thromboxane receptor in L-NAME hypertension. Am J Physiol Renal Physiol 2008;295:F1096–F1102. [DOI] [PMC free article] [PubMed]
- 14.Zhang Z, Vezza R, Plappert T, et al. COX-2-dependent cardiac failure in Gh/tTG transgenic mice. Circ Res 2003;92:1153–1161. [DOI] [PubMed]
- 15.Touchberry CD, Silswal N, Tchikrizov V, et al. Cardiac thromboxane A2 receptor activation does not directly induce cardiomyocyte hypertrophy but does cause cell death that is prevented with gentamicin and 2-APB. BMC Pharmacol Toxicol 2014;15:73. [DOI] [PMC free article] [PubMed]
- 16.Katugampola SD, Davenport AP. Thromboxane receptor density is increased in human cardiovascular disease with evidence for inhibition at therapeutic concentrations by the AT1 receptor antagonist losartan. Br J Pharmacol 2001;134:1385–1392. [DOI] [PMC free article] [PubMed]
- 17.Wacker MJ, Kosloski LM, Gilbert WJ, et al. Inhibition of thromboxane A2-induced arrhythmias and intracellular calcium changes in cardiac myocytes by blockade of the inositol trisphosphate pathway. J Pharmacol Exp Ther 2009;331:917–924. [DOI] [PMC free article] [PubMed]
- 18.Johnson JA, West J, Maynard KB, Hemnes AR. ACE2 improves right ventricular function in a pressure overload model. PLoS One 2011;6:e20828. [DOI] [PMC free article] [PubMed]
- 19.Hemnes AR, Maynard KB, Champion HC, et al. Testosterone negatively regulates right ventricular load stress responses in mice. Pulm Circ 2012;2:352–358. [DOI] [PMC free article] [PubMed]
- 20.Lane KL, Talati M, Austin E, et al. Oxidative injury is a common consequence of BMPR2 mutations. Pulm Circ 2011;1:72–83. [DOI] [PMC free article] [PubMed]
- 21.Johnson JA, Hemnes AR, Perrien DS, et al. Cytoskeletal defects in Bmpr2-associated pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2012;302:L474–L484. [DOI] [PMC free article] [PubMed]
- 22.Chow KS, Jun D, Helm KM, Wagner DH Jr, Majka S. Isolation & characterization of Hoechst(low) CD45(negative) mouse lung mesenchymal stem cells. J Vis Exp 2011;56:e3159. [DOI] [PMC free article] [PubMed]
- 23.Marriott S, Baskir RS, Gaskill C, et al. ABCG2pos lung mesenchymal stem cells are a novel pericyte subpopulation that contributes to fibrotic remodeling. Am J Physiol Cell Physiol 2014;307:C684–C698. [DOI] [PMC free article] [PubMed]
- 24.Wilson SJ, Cavanagh CC, Lesher AM, Frey AJ, Russell SE, Smyth EM. Activation-dependent stabilization of the human thromboxane receptor: role of reactive oxygen species. J Lipid Res 2009;50:1047–1056. [DOI] [PMC free article] [PubMed]
- 25.Rosenfeld L, Grover GJ, Stier CT Jr. Ifetroban sodium: an effective TxA2/PGH2 receptor antagonist. Cardiovasc Drug Rev 2001;19:97–115. [DOI] [PubMed]
- 26.Cathcart MC, Tamosiuniene R, Chen G, et al. Cyclooxygenase-2-linked attenuation of hypoxia-induced pulmonary hypertension and intravascular thrombosis. J Pharmacol Exp Ther 2008;326:51–58. [DOI] [PubMed]
- 27.Tesfamariam B, Ogletree ML. Dissociation of endothelial-cell dysfunction and blood-pressure in SHR. Am J Physiol-Heart C 1995;269:H189–H194. [DOI] [PubMed]
- 28.Carlson L-M, Rasmuson A, Idborg H, et al. Low-dose aspirin delays an inflammatory tumor progression in vivo in a transgenic mouse model of neuroblastoma. Carcinogenesis 2013;34:1081–1088. [DOI] [PubMed]
- 29.Li J, Mayne R, Wu C. A novel muscle-specific beta 1 integrin binding protein (MIBP) that modulates myogenic differentiation. J Cell Biol 1999;147:1391–1398. [DOI] [PMC free article] [PubMed]
- 30.Mankoo BS, Skuntz S, Harrigan I, et al. The concerted action of Meox homeobox genes is required upstream of genetic pathways essential for the formation, patterning and differentiation of somites. Development 2003;130:4655–4664. [DOI] [PubMed]
- 31.Lopez JE, Myagmar B, Swigart PM, et al. β-Myosin heavy chain is induced by pressure overload in a minor subpopulation of smaller mouse cardiac myocytes. Circ Res 2011;109:629–638. [DOI] [PMC free article] [PubMed]
- 32.Pandya K, Smithies O. β-MyHC and cardiac hypertrophy: size does matter. Circ Res 2011;109:609–610. [DOI] [PubMed]
- 33.Frolova EG, Sopko N, Blech L, et al. Thrombospondin-4 regulates fibrosis and remodeling of the myocardium in response to pressure overload. FASEB J 2012;26:2363–2373. [DOI] [PMC free article] [PubMed]
- 34.Allan CJ, Halushka PV. Characterization of human peripheral blood monocyte thromboxane A2 receptors. J Pharmacol Exp Ther 1994;270:446–452. [PubMed]
- 35.Stadtfield M, Ye M, Graf T. Identification of interventricular septum precursor cells in the mouse embryo. Dev Biol 2007;302:195–207. [DOI] [PubMed]
- 36.Bogaard HJ, Natarajan R, Henderson SC, et al. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009;120:1951–1960. [DOI] [PubMed]
- 37.West J, Niswender KD, Johnson JA, et al. A potential role for insulin resistance in experimental pulmonary hypertension. Eur Respir J 2013;41:861–871. [DOI] [PMC free article] [PubMed]
- 38.Gomoll AW, Ogletree ML. Failure of aspirin to interfere with the cardioprotective effects of ifetroban. Eur J Pharmacol 1994;271:471–479. [DOI] [PubMed]
- 39.Audoly LP, Rocca B, Fabre J-E, et al. Cardiovascular responses to the isoprostanes iPF2alpha-III and iPE2-III are mediated via the thromboxane A2 receptor in vivo. Circulation 2000;101:2833–2840. [DOI] [PubMed]
- 40.Kinsella BT, O’Mahony DJ, Fitzgerald GA. The human thromboxane A2 receptor alpha isoform (TP alpha) functionally couples to the G proteins Gq and G11 in vivo and is activated by the isoprostane 8-epi prostaglandin F2 alpha. J Pharmacol Exp Ther 1997;281:957–964. [PubMed]
- 41.Comporti M, Arezzini B, Signorini C, Vecchio D, Gardi C. Oxidative stress, isoprostanes and hepatic fibrosis. Histol Histopathol 2009;24:893–900. [DOI] [PubMed]
- 42.Gupta PK, DiPette DJ, Supowit SC. Protective effect of resveratrol against pressure overload-induced heart failure. Food Sci Nutr 2014;2:218–229. [DOI] [PMC free article] [PubMed]
- 43.Iglarz M, Touyz RM, Viel EC, Amiri F, Schiffrin EL. Involvement of oxidative stress in the profibrotic action of aldosterone. Interaction with the renin-angiotension system. Am J Hypertens 2004;17:597–603. [PubMed]
- 44.Lu Z, Xu X, Hu X, et al. Oxidative stress regulates left ventricular PDE5 expression in the failing heart. Circulation 2010;121:1474–1483. [DOI] [PMC free article] [PubMed]
- 45.Toth P, Rozsa B, Springo Z, Doczi T, Koller A. Isolated human and rat cerebral arteries constrict to increases in flow: role of 20-HETE and TP receptors. J Cereb Blood Flow Metab 2011;31:2096–2105. [DOI] [PMC free article] [PubMed]
- 46.Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol 2010;225:631–637. [DOI] [PMC free article] [PubMed]
- 47.Lambermont B, Kolh P, Ghuysen A, et al. Effect of a novel thromboxane A2 inhibitor on right ventricular-arterial coupling in endotoxic shock. Shock 2004;21:45–51. [DOI] [PubMed]
- 48.Iismaa SE, Graham RM. Dissecting cardiac hypertrophy and signaling pathways: evidence for an interaction between multifunctional g proteins and prostanoids. Circ Res 2003;92:1059–1061. [DOI] [PubMed]
- 49.Kunapuli P, Lawson JA, Rokach JA, Meinkoth JL, FitzGerald GA. Prostaglandin F2alpha (PGF2alpha) and the isoprostane, 8, 12-iso-isoprostane F2alpha-III, induce cardiomyocyte hypertrophy: differential activation of downstream signaling pathways. J Biol Chem 1998;273:22442–22452. [DOI] [PubMed]
- 50.Drazner MH. The progression of hypertensive heart disease. Circulation 2011;123:327–334. [DOI] [PubMed]
- 51.Carreño JE, Apablaza F, Ocaranza MP, Jalil JE. Cardiac hypertrophy: molecular and cellular events. Rev Esp Cardiol 2006;59:473–486. [PubMed]
- 52.Cingolani OH, Kirk JA, Seo K, et al. Thrombospondin-4 is required for stretch-mediated contractility augmentation in cardiac muscle. Circ Res 2011;109:1410–1414. [DOI] [PMC free article] [PubMed]
- 53.Sawaki D, Hou L, Tomida S, et al. Modulation of cardiac fibrosis by Kruppel-like factor 6 through transcriptional control of thrombospondin 4 in cardiomyocytes. Cardiovasc Res 2015;107:420–430. [DOI] [PMC free article] [PubMed]
- 54.Lynch JM, Maillet M, Vanhoutte D, et al. A thrombospondin-dependent pathway for a protective ER stress response. Cell 2012;149:1257–1268. [DOI] [PMC free article] [PubMed]
- 55.Davì G, Santilli F, Vazzana N. Thromboxane receptors antagonists and/or synthase inhibitors. Handb Exp Pharmacol 2012:261–286. [DOI] [PubMed]