

Abstract
IGFs are critical for normal intrauterine and childhood growth and sustaining health throughout life. We showed previously that the production of IGF-1 and IGF-2 requires interaction with the chaperone glucose-regulated protein 94 (GRP94) and that the amount of secreted IGFs is proportional to the GRP94 activity. Therefore, we tested the hypothesis that functional polymorphisms of human GRP94 affect IGF production and thereby human health. We describe a hypomorphic variant of human GRP94, P300L, whose heterozygous carriers have 9% lower circulating IGF-1 concentration. P300L was found first in a child with primary IGF deficiency and was later shown to be a noncommon single-nucleotide polymorphism with frequencies of 1%–4% in various populations. When tested in the grp94−/− cell-based complementation assay, P300L supported only approximately 58% of IGF secretion relative to wild-type GRP94. Furthermore, recombinant P300L showed impaired nucleotide binding activity. These in vitro data strongly support a causal relationship between the GRP94 variant and the decreased concentration of circulating IGF-1, as observed in human carriers of P300L. Thus, mutations in GRP94 that affect its IGF chaperone activity represent a novel causal genetic mechanism that limits IGF biosynthesis, quite a distinct mechanism from the known genes in the GH/IGF signaling network.
Glucose-regulated protein 94 (GRP94) is a ubiquitously expressed endoplasmic reticulum (ER)-resident chaperone that is essential for growth and development of multicellular organisms (1–4). It is a unique chaperone in being selective toward a small subset of membrane and secreted proteins. GRP94 client proteins include IGs (5); β1-integrins (6); glycoprotein Ib-IX-V complex (7); Toll-like receptors 2, 4, 5, 7, and 9 (8); and, most importantly for this study, IGF-1 and IGF-2 (1, 9). The presence of GRP94 and its intact ATPase activity are prerequisites for proper IGF-1/2 folding and cannot be substituted by other chaperones (2, 9). Consequently, variability in GRP94 activity impacts IGF-1/2 production.
The ATPase activity of GRP94 requires not only the nucleotide binding pocket in the GRP94 N-terminal domain but also the second, acidic domain (residues 265–344) that follows (10), as well as residues in the middle domain of the protein (11). Site-directed mutations in these domains affect the ATPase and chaperone activities of GRP94 (9), as we have previously shown by biochemical and cell-based assays (12). Therefore, sequence variability in several domains of GRP94 affects its function and may have phenotypic consequences.
Because grp94+/− mice have no detectable phenotype, half of GRP94 expression level or activity is sufficient for normal development. However, reducing the total GRP94 activity below 50% leads to multiple defects (1). Interestingly, inhibition or knockdown of GRP94 does not cause an unfolded protein response as would be the case for other ER chaperones but rather leads to a limited up-regulation of specific genes such as Ig binding protein (BiP) and protein disulfide isomerase A6 (PDIA6) (13). These observations support the substrate selectivity of GRP94.
Of all the known substrates, IGFs depend most stringently on GRP94, as shown in cell culture and in animal models (4, 9). The interaction of GRP94 with IGF-1/2 is direct, transient, and specific; distinct pro-IGF intermediates bind to GRP94 after synthesis, whereas mature IGFs dissociate from GRP94 (9). We created a muscle-specific GRP94 knockout (KO) that resulted in dramatic reduction of locally produced IGF-1, 30% reduction in circulating IGF-1 concentration, small muscle mass, and a whole body growth phenotype (4). These studies demonstrate the biological importance of the GRP94-IGF interaction.
The mammalian IGF system consists of two homologous hormones (IGF-1 and IGF-2) that bind to type 1 IGF receptors (IGF1R), insulin receptors, or hybrid insulin/IGF1R receptors (14). Receptor activation initiates signaling pathways that regulate growth and survival at the cellular and organismal levels (15–18). Expression of IGF-1 itself is linked to GH signaling through a feedback loop (19–21). Many monogenic disorders that lead to perturbations in growth have been identified (22), including mutations that affect members of the GH/IGF axis and modulate activity of the system. Among the known ones are mutations in the genes that code for GHRH and its receptor (23–25), GH itself (26), GH receptor (27, 28), signal transducer and activator of transcription (STAT)-5b (29), IGF-1 (30–32), IGF1R (33, 34), and the acid-labile subunit protein (IGFALS gene), which forms a circulating complex with IGF-1 (35).
So far, human GRP94 variants have not been reported to play a role in GH/IGF-1 axis modulation or in the context of any other growth phenotype. Nonetheless, the experimental evidence from animal models and cell culture studies (1, 2, 4, 9) compelled us to postulate that natural activity variants of human GRP94 exist, and we set out to uncover and analyze nonsynonymous variants of the HSP90B1 gene that encodes GRP94 in different human populations. We initially selected a population of children diagnosed with primary IGF deficiency (PIGFD) who presented with growth failure and unexplained extremely low levels of circulating IGF-1 (GH deficiency and mutations in currently common candidate genes have been excluded) and in whom we suspected GRP94 variants to play a critical role. In this report we describe a rare human variant of GRP94, first discovered in a child with PIGFD, in whom proline 300 of the mature protein is substituted with leucine (P300L). In vitro, this variant shows 42% diminished activity in support of IGF-2 production. Purified recombinant P300L variant showed impaired nucleotide binding and subtle changes in the protein structure. The P300L variant affects circulating IGF-1 levels and height of even heterozygous adult carriers. Therefore, we have identified the first hypomorphic variant of GRP94 and relate it to a specific IGF-1 phenotype in humans.
Materials and Methods
GRP94 sequence analysis
Genomic DNA was collected for sequencing of the HSP90B1 gene (encoding GRP94) from nine subjects enrolled in the PIGFD Consortium in whom mutations in candidate genes for IGF-1, IGF1R, GH receptor, or STAT5B had already been excluded (36). The PIGFD Consortium is an international registry of patients with growth failure (defined as height < −2.0 SD) and unexplained PIGFD (circulating IGF-1 levels < −3.0 SD) who consented to provide DNA for genetic studies. Additionally, we obtained DNA of seven subjects from the Leiden University Medical Center cohort where patients presented with apparent GH insensitivity based on variably low height and IGF-1 yet normal GH secretion (37). Eighteen exons of the gene were PCR amplified from genomic DNA (1–10 ng per run) using eight pairs of primers and GoTaq Hot Start green mix (Promega) according to the manufacturer's protocol. The resulting mixture was purified and sequenced. For probe design and sequences, see the Supplemental Data file.
Exon sequence variants resulting in altered protein structure were investigated further by in vitro functional studies as described below. Sequencing of the HSP90B1 gene from the parents of identified probands was also conducted to determine whether the variant had been inherited or represented a de novo mutation. Of note, the amino acid numbering used in the work corresponds to the mature GRP94 protein sequence; thus, 21 should be added to any residue numbered based on the gene sequence.
The prevalence of identified mutations was determined using public domain databases, including the National Center for Biotechnology Information db single-nucleotide polymorphism (dbSNP) and National Heart, Lung, and Blood Institute Exome Sequencing Project Exome Variant Server and the Exome Aggregation Consortium (Cambridge, MA).
Plasmids and site-directed mutagenesis
The cDNA for canine GRP94 was cloned into pCDNA3.1 with eF1α promoter and fused after codon 777 to monomeric green or cherry fluorescent proteins, ending with a KDEL C-terminal sequence as reported elsewhere (12). Site-directed mutagenesis (Quikchange; Stratagene) was used to generate all point mutants described. The primers used in mutagenesis are detailed in the Supplemental Methods. Canine and human GRP94 have 98% sequence identity, and the canine GRP94 expression complements several features of human or murine GRP94-deficient cells (2, 12, 13)
Cell culture
Grp94−/− mouse embryonic fibroblast (MEF) lines were described previously (1). They were maintained on fibronectin-coated plates in endotoxin-free DMEM (Gibco/Life Technologies), supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL), L-glutamine (2.92 mg/mL), sodium pyruvate, nonessential amino acids, ESGRO (Chemicon International), and 10% fetal bovine serum (HyClone, Thermo Scientific). Human embryonic kidney-293T cells were maintained in DMEM supplemented with antibiotics and 10% fetal bovine serum (Atlanta Biologicals).
IGF-dependent cell survival assay
This cell survival assay uses grp94−/− MEFs, which in the absence of active GRP94 cannot produce endogenous IGF-2 and therefore cannot survive growth in low serum. The assay and its validation were performed as described elsewhere (9, 12). Briefly, 2 × 105 grp94−/− MEFs 18 hours after plating were transfected with plasmids coding for wild-type (WT) or variants of canine GRP94-green fluorescent protein (GFP), using Lipofectamine 2000 (Life Technologies). After subsequent growth for 30 hours, the cells were washed twice in serum-free medium and incubated in the absence of serum for up to 39 hours. Where specified, 300 or 900 pg/mL of murine IGF-2 (R%D Systems) was added to the media. At the indicated times, cells were washed and photographed (15 pictures per sample). GRP94-positive cells were counted, and the changes in their numbers as a function of time were calculated. The survival index of cells expressing WT or GRP94 variants (12) was calculated at the time point within each experiment in which survival of the cells expressing GRP94 was one-tenth the survival of the cells expressing WT GRP94. More experimental details are provided in the Supplemental Methods.
Recombinant N34–355 GRP94
The truncated, His-tagged mouse GRP94 containing the N-terminal half of the protein (residues 34–355) was expressed in Escherichia coli and purified by affinity chromatography as described elsewhere (38). This recombinant protein retains the peptide binding and calcium binding activities of the apolipoprotein as well as the epitope for the conformation-sensitive antibody 9G10 (39).
Determination of ATP binding
His-tagged N34–355 GRP94 WT, E82A, or P300L recombinant proteins were incubated with γ-phosphate-linked ATP resin (gift of Dr Timothy Haystead, Duke University, Durham, NC) as described previously (40). In separate experiments, preformed complexes of GRP94 and the ATP resin were incubated with 0–40 μM concentrations of ATP (Sigma-Aldrich) for an additional 3 hours at 4°C to allow for dissociation. Samples were then subjected to SDS-PAGE and immunoblotting as described above, and the fraction of GRP94 bound to the ATP resin was quantified.
Assessment of GRP94 conformation by antibody 9G10
The monoclonal antibody 9G10 recognizes an epitope within the second domain of GRP94, which changes its conformation when the protein binds nucleotides via its first domain (39). Therefore, reactivity with 9G10 provides a convenient assay for GRP94 conformation. A solid-phase indirect binding assay was used (see Supplemental Methods) and of 9G10 bound to GRP94 epitope quantified with an Odyssey scanner at 680 nm. Specificity was ensured by inhibition with the GRP94 inhibitor radicicol.
Intracellular localization of GRP94 variants
Grp94−/− MEFs were transiently transfected with plasmids coding for ER-targeted GFP, cherry-tagged WT GRP94, and GFP-tagged P300L GRP94. Cells were fixed 24 hours after the transfection and processed for imaging, as described in the Supplemental Methods.
Human populations used
Leiden cohort of short-statured children
Subjects were selected from a previously described cohort with apparent GH insensitivity based on variably low height and low IGF-1 SDs (37). All had normal GH secretion.
Health In Men Study (HIMS; Australia)
This study enrolled only older Caucasian males, aged 70–89 years, from Perth, Australia, and thus includes a relatively homogenous population. A detailed description of this cohort has been published previously (41, 42).
HSP90B1 was genotyped from 3862 subjects using custom-designed probes for TaqMan SNP genotyping assay from Life Technologies (see Supplemental Materials). For the measurement of total IGF-1, samples were pretreated with acid to displace IGF-1 from its binding proteins, followed by neutralization and addition of binding inhibitors prior to measurement using the DSL-10-2800 kit (Diagnostic Systems Laboratories, Inc). Blood concentrations of IGF-1 and IGF binding protein (IGFBP)-3 were measured using DSL-10-280 and DSL-10-6600 kits, respectively, per the manufacturer's protocols (both from Diagnostic Systems Laboratories, Inc).
IGF-1 was measured in 98 carriers of P300L, 3568 controls, and the homozygous P300L subject. The measurements used one standardized technique (ELISA as described above) in the same clinical laboratory and were made during the span of only 3 months. These factors helped limit the variability in reported IGF-1 levels.
Longevity Genes Project (New York)
The Longevity Genes Project (LGP) is a large prospective cohort study based at Albert Einstein College of Medicine (New York, New York) (43). The project consists of Ashkenazi Jewish probands with exceptional longevity (mean age 98.2 y), their offspring (mean age 68.3 y), and a control group age matched to the offspring. HSP90B1 sequencing was performed on 459 cases (probands and their offspring) and 1807 controls. IGF-1 concentration was measured using a DSL-10-2800 kit as above.
Danish Cohort Study
This cohort included 15 663 subjects from Denmark from five combined studies, described previously (44). In brief, the cohort is based on the general Danish population in which approximately half of the subjects were diagnosed with type 2 diabetes. Every subject was genotyped for 16 192 SNPs, whereas whole-exome sequencing was performed on a subset of 2000 subjects. Height data were available in this population, but IGF-1 was not measured.
Health, Aging, and Body Composition study
We sequenced 100 cases from the National Institute of Aging Health, Aging, and Body Composition cohort (http://www.grc.nia.nih.gov/branches/leps/healthabc/) with IGF-1 data available. The study population consisted of 3075 persons aged 70–79 years at baseline, with about equal numbers of men and women. We selected three sample groups with circulating IGF-1 levels below 57 ng/mL, between 66 and 100 ng/mL, and greater than 200 ng/mL.
Statistics
For normally distributed continuous variables, means (±SD) were compared between P300L carriers and controls using independent-samples t tests. In contrast, nonnormally distributed data were summarized as median (interquartile range) and compared using the nonparametric (Mann-Whitney U) test. Extremely high IGF-1 values in an elderly population may represent pathology (ie, acromegaly) or laboratory errors (eg, assay artifact from IGFBP interference) (45). For this reason, all IGF-1 measurements above +3 SD from the mean (above 315.5 ng/mL) were excluded as biologically implausible from the HIMS population (26 WT, two P300L carriers). Data were analyzed using SPSS version 22.0 (IBM).
Other methods
Immunoblotting, antibodies, GRP94, and murine IGF-2 ELISA, immunofluorescence techniques, circular dichroism measurements, and software used for the alignment of sequences and prediction of structure are described in the Supplemental Methods.
Results
GRP94 analysis in patients with primary IGF deficiency
We sequenced the HSP90B1 (grp94) gene from children diagnosed with PIGFD, nine from the cohort of the PIGFD Consortium and seven from the Leiden University Medical Center cohort (37). One subject, a male with in utero growth retardation, postnatal growth failure, and nondetectable circulating IGF-1 at age 4 months (Figure 1A), was found to be heterozygous at chromosome 12 position 103938446 (GRCh38), with a C-to-T substitution in one allele and a WT sequence in the other (Figure 1B). This substitution changed the amino acid coding sequence from proline 300 to leucine (note that our numbering system assigned 1 to the N terminal asparagine created after cleavage of the signal sequence to align with the mature protein sequence, rather than to the initiating methionine, as is the convention in genomics; therefore, P300 in this paper is identical with P321 in genomic databases). The patient's mother also carried this mutation and had a height in the lower half of the reference population (height −0.9 SD) (Figure 1B). Her circulating IGF-1 concentration was not available.
Figure 1.
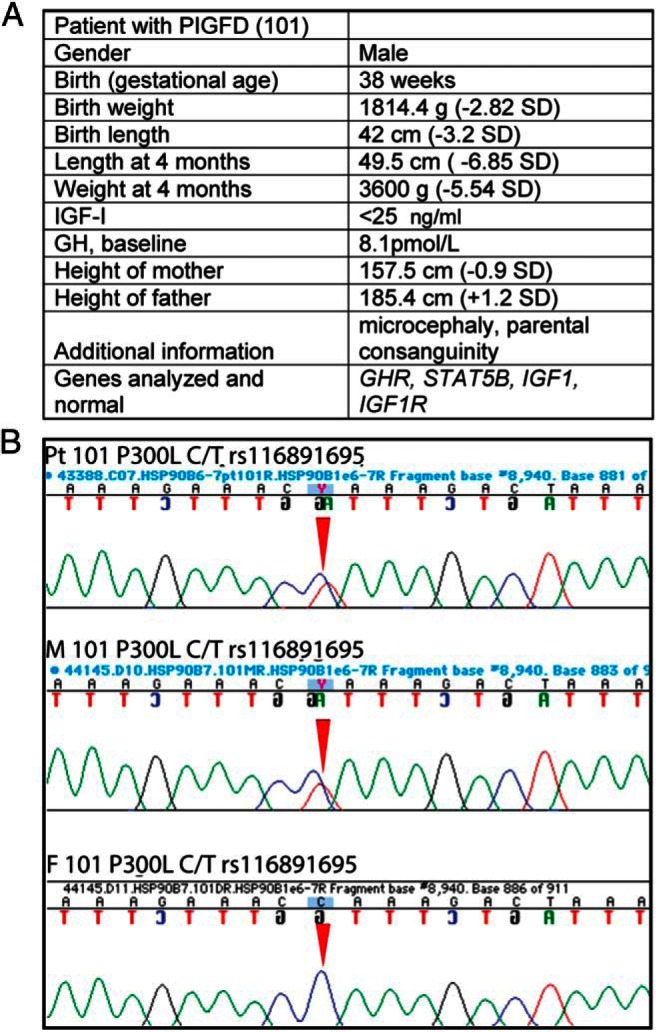
Phenotypic and genotypic data on a patient with PIGFD and P300L substitution of GRP94. A, Table with the patient's clinical and sequencing data. B, Fluorescent peak trace chromatogram depicting the sequences of the HSP90B1 gene encoding GRP94 from the patient and his parents. Note heterozygous peaks at base pair position 962 with a C-to-T substitution (red arrow) in one allele from the patient with PIGFD (top panel) and his mother (middle panel). The father's sequence (bottom panel) presents a homozygous peak with both alleles consisting of C. The C-to-T substitution changes proline 300 to leucine (amino acid position has been derived from the mature GRP94 N terminus).
To determine whether P300L was a unique mutation, we interrogated several public-domain databases with GRP94 sequences. We found that P300L was a minor allele frequency variant (rs116891695) that occurs in several human cohorts with frequencies ranging from 0.8% (African population, ESP-AA, Table 1) to 4% (Danish population, D15K, Table 1). In every case that P300 was substituted (n = 2319), it was changed into leucine and not to another amino acid, even though a single base change can convert proline to five amino acids other than leucine.
Table 1.
Genotype Frequency of P300L Variant in the Respective Human Cohorts
| Cohort | D15K | LGP Probands | LGP Controls | HIMS | ESP-EA | ESP-AA | ExAC |
|---|---|---|---|---|---|---|---|
| Heterozygotes | 619/15 663 | 9/459 | 25/1897 | 112/3862 | 140/4158 | 18/2185 | 1629/57 207 |
| Percentage | 3.95 | 1.96 | 1.38 | 2.9 | 3.36 | 0.82 | 2.84 |
| Homozygotes | 4 (11) | 0 | 0 | 1 (3) | 2 | 0 | 20 (24) |
P300L is a minor frequency allele (<5%). Population frequency of P300L variant within the indicated cohorts include the following: NCBI dbSNP, National Center for Biotechnology Information Database of Single Nucleotide Polymorphisms; D15K, Danish general population; LGP, Longevity Genes Project protein centenarians and controls; ESP-EA and ESP-AA, Exome Sequencing Project European-American and African-American cohorts, respectively; ExAC, Exome Aggregation Consortium. Note that homozygotes were found, albeit at reduced numbers compared with the expected frequency from the Hardy-Weinberg equation (in parentheses).
In vitro analysis of the effects of the P300L mutation on GRP94 function
P300L is hypomorphic in supporting IGF-2 production
Because the P300L variant was initially found in an PIGFD patient, we sought to determine its functionality in supporting IGF-1 production. The best in vitro system available is the expression of the variant in grp94−/− MEFs (9). These cells produce IGF-2, but our previous work showed that IGF-1 is regulated by GRP94 variants in the same manner as IGF-2 (2, 4). Expression of GFP-tagged P300L GRP94 in knockout MEFs showed reticular localization that overlapped with that of cherry-tagged WT GRP94 (Figure 2A), as expected from an ER resident protein. When grp94−/− MEFs, transfected with various forms of GRP94, were analyzed by Western blots, the expression levels of P300L were similar to WT and the E82A mutant (impaired in ATP hydrolysis [2]) and higher than that of the triple-point mutant E82A/D128N/G132A (impaired in both ATP binding and hydrolysis; Figure 2B). Therefore, P300L exhibits normal cellular localization and is stably expressed with levels similar to WT GRP94.
Figure 2.

P300L GRP94 is properly expressed but less effective than WT GRP94 in supporting the production of IGF-2. A, Fluorescence microscopy analysis of expression and intracellular localization of GRP94 in grp94−/− cells. WT GRP94 mCherry-tagged was coexpressed with P300L GRP94 mGFP-tagged. Cells were fixed after 24 hours and imaged with a ×60 objective (n = 2). Bar, 5 μm. B, Comparative expression of GFP-tagged WT GRP94 or P300L, E82A, or E82A/D128N/G132A mutants of GRP94. Human embryonic kidney-293T cells were harvested 24 hours after transfection with plasmids coding for empty vector (EV) or the indicated GRP94 constructs, and protein extracts were analyzed by immunoblotting with the anti-GRP94 antibody. The lanes were run on the same gel but were noncontiguous. Quantitation of expression levels of exogenous GRP94 as compared with endogenous GRP94 is provided below lanes (n = 2). C–E, Differential ability of GRP94 mutants to support IGF-2-mediated cell survival. Grp94−/− MEFs were transiently transfected with a plasmid expressing GFP-tagged WT, P300L, E82A, or E82A/D128N/G132A variants of GRP94. Twenty-eight hours later, cell cultures were serum starved and scored at the indicated intervals by microscopy. C, Pictures of GRP94-expressing cells acquired at the indicated time points from a representative experiment. D, The number of GFP-positive cells was counted at given time points from 15 separate fields. The cell counts were plotted as a percentage of the initial time point (representative experiment shown). E, Activity of GRP94 was calculated as the survival index of cells expressing GRP94 mutants. The index was calculated at the time point within each experiment in which survival of the cells transfected with E82A/D128N/G132A GRP94 mutant was one-tenth the value of the WT-transfected cells (WT, E82A, E82A/D128N/G132A, n = 8, P300L, n = 5). Means and SDs are graphed. F, ELISA measurements of IGF-2 in supernatants of grp94−/− cells 16 hours after serum withdrawal. Grp94−/− MEFs were prepared as in panel C and secreted the indicated levels of IGF-2 in the absence of serum. The detection limit of this assay is approximately 60 pg/mL. Means and SDs are shown for two independent experiments.
To test the functionality of P300L variant, we used the previously described cell-based assay (9), in which the survival of serum-starved grp94−/− MEFs relies on their ability to produce and secrete IGF-2. Grp94−/− MEFs were transiently transfected with WT, P300L, E82A or E82A/D128N/G132A, GFP-tagged GRP94. Twenty-eight hours later, cells were serum starved and at the indicated time points, images of GFP-positive cells were captured (Figure 2C) and scored (Figure 2D). Cells expressing WT GRP94 survived longer (more GFP positive cells at different time points) than cells expressing the completely inactive GRP94, E82A/D128N/G132A variant, all of which died within 25 hours. The point mutant E82A had some residual activity (Figure 2, D and E), consistent with our previous results (12), and P300L was clearly more active than E82A but consistently less so than WT GRP94. The calculated survival index of GFP-positive cells, representing the functionality of the tested GRP94 variants (Figure 2E), was 58% of WT for the P300L variant (P < .01). Coexpression of the P300L variant with WT GRP94 in the same cells did not diminish cell survival, indicating that P300L does not act in a dominant-negative fashion. Thus, we conclude that P300L is a hypomorphic mutant of GRP94.
Survival of serum-deprived grp94−/− MEFs depends greatly on their ability to secrete mouse IGF-2 (mIGF-2). We first estimated the levels of recombinant mIGF-2 necessary to rescue grp94−/− MEFs from apoptosis. The cells were transfected with WT GRP94 or nonactive Kpn deletion mutant (12) alone or Kpn-transfected cells were supplemented with 300 or 900 pg/mL of mouse IGF-2. Addition of the lower dose of mIGF-2 rescued cell survival partially, whereas increasing it just 3-fold was sufficient to provide a rescue similar to that achieved by WT GRP94 and to alter the cells' fate during the starvation period (Supplemental Figure 1A).
Therefore, using the same experimental approach, in which grp94−/− MEFs were transiently transfected with variants of GRP94 and subsequently stressed by serum withdrawal, we quantified the concentration of mIGF-2 secreted over the span of the 16 hours of starvation. grp94−/− MEFs that were not transfected, or were transfected with inactive variants of GRP94, produced similar mIGF-2 concentrations of 85 pg/mL. Cells transfected with WT and P300L variants secreted 141 and 118 pg/mL of mIGF-2, respectively (Figure 2F). Assuming the signal detected from nontransfected cells represents leakage of intracellular, inactive mIGF-2 from dying cells, which cannot support growth of IGF-dependent cells, the P300L variant supported approximately 58% of IGF-2 secretion relative to WT GRP94. The difference in the amount of secreted mIGF-2 seems to be sufficient to dictate survival or death of the cells in the cell-based assay (see also reference 12).
The P300L mutation is predicted to impact the structure of GRP94
Pro300 is an evolutionarily conserved residue (Figure 3A) located in the second, charged linker domain of GRP94. This domain is absent from the previously determined crystal structures of GRP94 (46, 47), making it difficult to predict the structural effect of the specific substitution. We therefore used two computational predictions (Phyre2 and PSIPRED; Figure 3B) and inferred that Pro300 is flanked by two α-helices, and its substitution with leucine or any other amino acid may cause fusion of the two helices into one 25-residue-long α-helix (Figure 3B). Previous studies showed that deletion of the linker domain (amino acids 280–345) impacted the nucleotide-binding pocket of GRP94 (10), and the generation of a long α-helix by P300L may similarly affect the nucleotide binding of GRP94.
Figure 3.

P300L substitution affects tertiary but not secondary structure of GRP94 and is partially refractory to radicicol induced changes in secondary structure. A, Conservation of proline 300 of human GRP94 and other species. Amino acid sequence alignment between positions 282 and 319 of human GRP94, in the linker domain, and the corresponding sequences of indicated organisms. B, Secondary structure of amino acids 285–314 from WT, P300L, P300A, and P300K substitutions predicted using the algorithm PSIPRED (SS, secondary structure; ?, prediction of a region most likely structurally disordered). C and D, Far UV CD spectra of native N34-355 GRP94 WT and P300L variant. CD of purified GRP94 at the concentration of 1.9 μM was measured in 0.1 cm path length cuvette at 20°C. The spectra were buffer corrected. The graph represents the average of nine independent experiments. D, GRP94 WT and P300L variant were incubated for 4 hours at 4°C with 5 μM of radicicol and CD of the proteins were measured. Decrease of the spectra intensity after radicicol treatment was observed for WT GRP94 (left panel) and P300L variant (right panel). The spectra were corrected with the buffer containing radicicol. The graphs represent four independent experiments. E, Table with α-helix and turns fractions from panels D and E calculated by DichroWeb using the Selcon3 program. F, Near UV CD spectra of native N34-355 WT and P300L GRP94. Purified GRP94 was desalted into 10 mM ammonium acetate buffer to a final concentration of 7 μM. WT GRP94 was additionally incubated in 95°C or pH 2 for the period of 1 hour. CD of the proteins was measured in 1 cm path length cuvette at 20°C. The spectra were buffer corrected. The graph represents the average of four independent experiments. G, Table provides wavelength specific molecular ellipticity values for five independent experiments.
When functional aspects of P300L substitution were analyzed by Sorting Intolerant From Tolerant and Polymorphism Phenotyping, both algorithms predicted that Pro300 substitution to most amino acids (with exceptions of P300E and P300K) has 0.995 probability of being damaging to protein function. Interestingly, a P300K substitution, predicted to be tolerable (functionally neutral) by Sorting Intolerant From Tolerant would generate (like P300L) one 25-residue-long helix (Figure 3B), raising the possibility that P300L is actually on the spectrum of tolerable rather than fully damaging substitutions, a characteristic one would expect from a rare population variant.
P300L reduces the structural response of GRP94 to radicicol binding and changes the tertiary structure of GRP94
Secondary structure of native N34-355 WT and P300L GRP94 was measured by far-UV circular dichroism (CD; Figure 3C). The average content of the α-helix was 23% and the β-sheet 7% (as analyzed by SELOCON 3) and did not differ between WT and P300L (Figure 3C). However, when GRP94 was incubated for 4 hours in 4°C with 5 μM of radicicol, a small molecule GRP94 inhibitor that binds at its ATP pocket, a bigger decrease in α-helix content was observed in WT (2.8%, P = .01) than in P300L variant (1.8%, P = .74; Figure 3, D and E). It is important to note that the result may not represent changes in the percentage of α-helices but rather reflect their different packing against other secondary structure elements in the core of GRP94. Such a difference in the magnitude of structural changes on inhibitor binding may reflect reduced flexibility of the linker domain of P300L and/or lower affinity for radicicol as compared with WT GRP94.
The near-UV CD measurements of native N34-355 P300L showed that this protein folded to a defined structure resembling that of WT GRP94. Compared with WT, there was a decreased signal at the wavelengths corresponding to the aromatic residues tyrosine and tryptophan. However, this was not due to a global loss of tertiary structure as shown by the spectra of heat-treated or acid-treated samples (Figure 3, F and G). Rather, this change suggests subtle tertiary structure changes in P300L (values for WT/P300L changes were P = .75 for 260 nm, P = .03 for 280 nm, and P = .04 for 290 nm).
The P300L variant affects the nucleotide binding function of GRP94
To test the predicted effects of structural changes in P300L, we used in vitro assays for recombinant GRP94 function. It was previously shown that a truncated WT GRP94 containing amino acids 34–355 had retained the nucleotide binding activities and could be inhibited by nucleotide mimetics (48). First, we established that 10 μg of His-tagged, N34-355 GRP94 is sufficient to saturate 15 μL of γ-linked ATP resin beads (Dr T. Haystead, Duke University; Supplemental Figure 1B) (40), and then we measured the relative ATP-binding capacities of point mutants of recombinant GRP94. As shown in Figure 4, A and B, P300L was able to bind γ-linked ATP beads but only 75% the amount of WT GRP94, and its diminished binding was similar to that of the mutant E82A (82%). In addition to the on-rate of binding, we estimated the off-rate by charging the ATP beads with each GRP94 variant and then eluting the bound proteins with increasing concentrations of free ATP. WT GRP94 was eluted by free ATP more readily than P300L, whereas the nonhydrolyzing mutant E82A showed only minimal elution (Figure 4, C and D). In summary, these data show that P300L has a negative impact on the ability of the ATP pocket to bind and release ATP, a biochemical activity that is essential for the chaperone function of GRP94 (12).
Figure 4.

The P300L mutation affects the nucleotide-binding pocket of GRP94. A–D, His-tagged N34-355 portion of GRP94 was allowed to bind to γ-phosphate-linked ATP resin for 3 hours. Subsequently, aliquots have been analyzed by Western blotting with anti-His antibody to determine the ability of WT and the indicated mutants to bind ATP. Quantitation of eight independent binding experiments with means and SDs are graphed in panel A and one representative experiment shown in panel B. Additionally, the aliquots were incubated with increasing concentrations of free ATP for 3 hours at 4°C. The amount of the remaining ATP-bound GRP94 was analyzed by immunoblotting with anti-His antibody. Trend lines graphs of four independent competition experiments are shown in panel C with one representative experiment shown in panel D. E and F, His-tagged N34-355 GRP94 was bound to Ni-covered plates and preincubated for 4 hours at room temperature with the indicated concentrations of radicicol. Note that most protein was bound to the walls of the wells (circumference). Subsequently, plates were washed and incubated with the anti-GRP94 antibody 9G10 with the same radicicol concentrations for 1 hour. 9G10 binding was detected with an IRDye 800CW-conjugated secondary antibody and scanned on a Licor Odyssey. Trend lines graphs of four independent competition experiments are shown in panel E with one representative experiment shown in panel F.
Ligand-induced conformational change is diminished in P300L variant
As reported previously (48), GRP94 undergoes structural changes in response to ligand binding. When the inhibitor radicicol binds to the nucleotide-binding pocket of GRP94, the epitope recognized by the monoclonal antibody 9G10, approximately 200 amino acids away from the nucleotide-binding pocket, is altered, precluding the binding of 9G10 (48). We adapted this procedure and covered Ni-plates with equal amounts of His-tagged GRP94 variants. The variants were then incubated with increasing concentrations of radicicol, and after 4 hours were probed with the 9G10 antibody. As shown in Figure 4, E and F, WT, P300L, and E82A GRP94 were recognized by 9G10 with the same efficiency. However, the loss of antibody binding in WT GRP94 was observed with concentrations of radicicol as low as 0.1 μM, whereas a 100-fold higher concentration of radicicol was needed for the P300L or E82A proteins. Thus, P300L variant is partially refractory to radicicol binding and to the conformational change that is reported by the loss of the 9G10 epitope.
Assessing the impact of P300L in humans
To assess the impact of P300L on human physiology, we analyzed four human populations. First, we sequenced 200 GRP94 alleles from the National Institute of Aging Health, Aging, and Body Composition cohort, but none contained the P300L substitution. This is possibly due to the high prevalence of African American subjects, in whom the minor allele frequency is lower (Table 1).
Second, we genotyped the HIMS cohort (49) to assess whether carriers of P300L have lower circulating levels of IGF-1. This cohort had a narrow age distribution and a sufficient number of circulating IGF-1 measurements to allow meaningful statistical analyses. Among 3862 genotyped cases, 112 subjects (2.9%) were heterozygous for P300L, one was homozygous for this mutation (0.03%), and 3619 (97.07%) were homozygous for WT GRP94. IGF-1 was measured in 98 carriers of P300L, 3568 controls, and the homozygous subject. IGF-1 concentrations were not normally distributed within these groups. The median (interquartile range) IGF-1 concentration in heterozygous P300L carriers was 120 ng/mL (91–165) and in the noncarriers, 134 ng/mL (101–172) (Table 2; P = .037, Mann-Whitney U test). Unlike circulating IGF-1 concentration, there was no significant difference between P300L carriers and subjects with WT GRP94 in the following: circulating IGFBP-3 concentration, height, body mass index, or age (Table 2). Except for IGF-1, all other measurements in this cohort were distributed normally. Therefore, consistent with our hypothesis, heterozygous carriers of P300L (n = 98) had a decrease of approximately 10% in serum IGF-1 levels, as compared with WT subjects (n = 3568). Given the large variability in IGF-1 measurements (50) and the multitude of factors that influence the circulating IGF-1 levels in vivo, we were impressed that a decrease of 10% could be found with a large-enough sample size.
Table 2.
Phenotypic Data for HIMS Cohort
| HIMS Cohort | Wild Type (n = 3568) | P300L (n = 98) | P Value |
|---|---|---|---|
| IGF-1, ng/mL | 134.0 (101–172)a | 120.0 (91–165)a | .037a |
| IGFBP-3, mg/L | 3.8 ± 0.9 | 3.7 ± 1.0 | .25 |
| Height, cm | 171.9 ± 6.7 | 173.0 ± 6.4 | .13 |
| BMI, kg/m2 | 26.6 ± 3.7 | 26.7 ± 3.8 | .73 |
| Age, y | 77.1 ± 3.6 | 76.9 ± 3.5 | .57 |
Abbreviation: BMI, body mass index. In the HIMS cohort, carriers of P300L have lower circulating IGF-1 levels. The table shows the values for individuals with heterozygous P300L or WT GRP94. IGF-1 values were distributed in non-Gaussian fashion, so the interquartile ranges are given and comparisons were made using the nonparametric Mann-Whitney U test. IGFBP-3, body mass index, and height were distributed normally, and thus, these comparisons were made using an independent-samples Student's t test.
Comparisons were made using the nonparametric Mann-Whitney U test.
The relation between P300L and circulating IGF-1 was also analyzed in the LGP (Albert Einstein College of Medicine, New York, New York). Thirty-four P300L carriers were found, but IGF-1 measurements were available for only 10 of them. This small sample did not enable assessment of the impact of P300L on IGF-1 production. However, height data were available in this cohort, and height is often correlated with circulating IGF-1 (51). Indeed, when P300L carriers were compared with noncarriers, their mean height was lower, consistent with the hypothesis (Table 3). The lower height was statistically significant in females and was just below the significance level in males, although the trend was also consistent with the hypothesis.
Table 3.
Phenotypic Data for LGP Cohort
| LGP Cohort | Number of Heterozygotes | Number of Homozygotes | Total | Height in Carriers, in. | Height in Noncarriers | P Value |
|---|---|---|---|---|---|---|
| rs116891695 (P300L) | 28 | 1341 | 1369 | |||
| Females | 14 (age 82.3 ± 13.2 y) | 713 (age 71.5 ± 8.7 y) | 727 | 61.0 ± 1.8 | 62.8 ± 2.9 | .007a |
| Males | 14 (age 78.1 ± 13.0 y) | 628 (age 72.6 ± 8.5 y) | 642 | 67.2 ± 2.7 | 68.5 ± 3.4 | .082a |
Mann-Whitney U test.
The fourth population analyzed was the Danish 15K cohort. Among the 7681 male subjects, 299 were carriers of P300L and four were homozygous, but no IGF-1/-2 levels had been measured in this cohort. There was also no difference in heights between the P300L carriers and noncarriers in this cohort, in either males (177.7 ± 7.1 cm, n = 299 vs 177.7 ± 6.9 cm, n = 7687) or females (165.8 ± 6.7 cm, n = 320 vs 165.0 ± 6.5 cm, n = 7357).
Discussion
This work shows that hypomorphic variants of the chaperone GRP94 exist in humans and that such variants affect circulating levels of IGF-1. Specifically, we used a cell-based complementation assay (12) to show that the human variant P300L supports IGF production less efficiently than WT GRP94. P300L GRP94 is expressed properly in the ER and at levels comparable with WT GRP94 but has lower chaperone activity toward IGF-2. This lower activity is explained by three molecular differences: 1) altered ATP binding and dissociation of P300L GRP94, implying reduced ATPase activity; 2) a smaller conformational change induced by ligand binding to GRP94, and 3) an altered global tertiary structure of the variant protein compared with WT GRP94.
The P300L substitution occurs in the second domain of GRP94, which is an acidic domain that links the N-terminal domain with the middle domain. This acidic linker domain is conformationally sensitive: it does not crystalize with the rest of the protein (47), it contains the epitope for a conformation-sensitive monoclonal antibody 9G10 (39), and it also contains one to two calcium binding sites whose occupancy affects the binding activity of the protein (38). We presume that the helix-breaking nature of proline 300 is required for one or more of these conformational aspects and that a leucine substitution disrupts the conformation, as we see in the CD and nucleotide binding experiments. This interpretation is consistent with the fact that proline 300 is substituted only with leucine and not another amino acid. Presumably, even though leucine 300 is disruptive, it is not as deleterious as other substitutions.
The P300L variant was reported recently by both the 1000 Genome Project and the Exome Aggregation Consortium (http://exac.broadinstitute.org; accessed May 2015). P300L is a minor allele frequency (rs116891695) variant found in 0.8% of Africans and up to 4% of Caucasians. Interestingly, all but two of the approximately 200 nonsynonymous variants in the human HSP90B1 gene are found only in single cases. The two exceptions are P300L and DelE770 (rs5800607; an in-frame, single amino acid deletion present in up to 26% of human population). So far, P300L is the only GRP94 variant with associated phenotypic data.
Pro300 is evolutionarily conserved in all tested organisms with the exception of Caenorhabditis elegans and is located within the second domain of GRP94, the charged linker domain. This domain is essential for the activity of GRP94 because it mediates a conformational change that is required for the ATPase activity of GRP94 (10, 48, 52). The structure of this domain cannot be resolved by crystallography (47), so structural inferences must rely on indirect methods. One method is binding of 9G10, an anti-GRP94 monoclonal antibody, which binds to the linker domain when the nucleotide-binding N-terminal domain is available (10, 48). As shown in this work, leucine 300 does not impact the binding of this conformation-sensitive antibody. Another indirect method is binding of calcium: the E/D-rich sequence within the linker domain contains one to two calcium binding sites, which induce the conformational change that impacts the N-terminal domain (38). P300L is in the basic sequence C terminal to the calcium-binding site, and it would be informative to measure the calcium-binding capacity of P300L because it may account for the partial defect in the ATPase activity shown here.
Our previous mouse work showed that GRP94 is an essential gene whose complete ablation is embryonic lethal, starting at the stage when IGF-2 is first produced, whereas mice hemizygous for HSP90B1 are phenotypically normal (1). Thus, it is likely that only hypomorphic variants with activity greater than 50% of WT will be found in asymptomatic adult humans. Like other HSP90 family members, GRP94 is a homodimeric chaperone with an ATPase cycle that induces folding of its client proteins, including IGFs (reviewed in reference 53). Measurements of nucleotide binding in vitro show saturation at 1 mol per GRP94 dimer, suggesting that perhaps each monomer acts independently of the other (54). If this is the case, then even heterodimers of P300L-WT GRP94, as found in carriers of the variant, should have lower binding capacity and therefore lower biological activity than WT, but there is evidently strong evolutionary selection against more severe mutants.
The existence of a minimal activity threshold is supported by data from mouse KO studies and cell culture gene-silencing experiments. Conditional deletion of GRP94 in mouse skeletal muscle is very informative because Cre-mediated deletion in this syncytial tissue is often incomplete, a graded series of expression is created, and muscles with 30% less GRP94 than WT have been analyzed (4). These muscles have a significant reduction in their mass and decreased IGF protein content (Figures 2 and 6 in Ref. 4), without altered expression of other GH-IGF axis members (4). Therefore, reduced activity of GRP94 leads directly to lower IGF production that in turn diminishes muscle growth in this animal model.
Echoing the animal models, in cell culture, many cell types are known to depend on autocrine or paracrine production of IGF-1 and IGF-2 to grow and/or avoid apoptosis. Progressive silencing of GRP94 expression using short hairpin RNA, or inhibition of its activity using small molecules, also shows that reducing the expression below 50% of WT level gives rise to progressively lower growth rates and higher susceptibility to stress conditions (9, 55–57). Importantly, Eletto et al (13) showed by complementation analysis that cells sense the level of GRP94 activity, not merely its level of expression. Thus, there is solid experimental knowledge that not only predicted the existence of hypomorphic human GRP94 variants, as shown in this work, but also explains the biomedical context of these variants.
The effects of the P300L mutation on GRP94 structure and function are obvious in cell-free and cell culture assays yet are subtler in the clinical context. Based on our data, we would have predicted that a role for GRP94 would have been detected already in the previously published genome-wide association studies of human growth or aging, yet it has not. In the context of PIGFD, the patient in whom we initially found P300L was heterozygous for the mutation, his mother also carried this mutation and although she was shorter than the population mean (157.5 cm; −0.9 SDs), her short stature was less severe than her child's. This relationship reflects the study of Walenkamp et al (58) in which heterozygous carriers of IGF-1 mutation exhibited only 1 SD loss of height, whereas height of homozygotes was −8 SD. Therefore, we surmise that P300L was a contributing factor, rather than sole cause, of the patient's short stature. Age may also play a role; although IGF-1 is important for fetal and postnatal growth, IGF-2 has been described so far mainly as a prenatal growth factor (59), and production of both factors requires GRP94 activity. Recently Begemann et al (60) postulated that IGF-2 is involved in growth through direct contribution and/or fetal programming. However, lower IGF-2 levels delay bone maturation (60) so that adult height may be less decreased than growth in childhood. Thus, the phenotype may be more severe in neonates and infants (the patient was 18 mo old), with some catch-up growth afterward, resulting in improved height by adulthood. Some catch-up growth may also occur if the loss of IGF-1-negative feedback from the GRP94 hypomorph stimulates GH or another factor that can then compensate to some degree for the IGF loss. Similarly, in cohorts such as LGP, in which longevity was already shown to be related to IGF-1 signaling (61), there was no robust relationship between individuals with P300L GRP94 and lower IGF levels, arguably because of a small sample size, heterogeneity of the P300L carriers (males vs females, younger vs older) or different mechanisms that block the deleterious effect of the mutation (45). In contrast, the homogenous HIMS cohort (70–89 y old Caucasian males) displayed lower IGF-1 levels among P300L carriers, consistent with the hypothesis.
There are several potential reasons for the lack of obvious clinical impact of P300L in humans despite the clear effects of P300L in the in vitro experiments. For one, the cell culture and in vitro assays measured the activity of the GRP94 variant in the absence of any other GRP94, whereas human subjects are mostly heterozygous. It will be interesting to use genome editing to create a heterozygous GRP94 cell line. The second reason for the in vivo vs in vitro discrepancy is the nature of the measurements: because GRP94 is a chaperone, it affects a number of proteins expressed in different body systems, so a hypomorphic variant may have multiple effects that do not necessarily augment each other. It should be pointed out in this context that there is no evidence that any of the IGF-1-nteracting proteins, relevant receptors, or transcription factors are affected by GRP94, which is a highly selective chaperone protein. Yet a third reason is that in most genome-wide association studies, the variable assessed may be very much downstream from the primary, biochemical action of the variant GRP94. Even when one measures a more proximal parameter such as IGF-1 levels, the measurement is known to be variable, partly because of biological feedback mechanisms that may compensate for the lower IGF-1 production. Such mechanisms that counteract the effect of low IGF-1 was already suggested by Milman et al (45), and will be important to explore further.
It is likely that when GRP94 activity is reduced, two types of compensatory mechanisms are activated to restore its folding potential (and thus IGF production). One relates directly to the number of functional GRP94 molecules in the ER; because cells can monitor the total activity, rather than expression of GRP94 (13), there may be compensatory up-regulation of P300L expression, which we have not yet detected in cell culture. The second possible compensatory mechanism may be due to the hormonal feedback network between IGF-1 and GH. Under normal physiological conditions when IGF-1 levels are low, such as in starvation, loss of negative feedback results in a compensatory rise in GH levels (62–64). However, because GRP94 is a limiting factor in IGF-1 production, compensatory GH-dependent increases in IGF-1 production would be possible, mostly through higher levels of GRP94 expression in response to GH and more chaperone molecules available for IGF-1 folding. This could be determined in future studies. We note that the analysis of the GRP94 promoter indicates binding sites of STAT5, a major transcription factor activated through GH binding to its receptor (18, 65), so increased GH/STAT5 signaling could potentially lead to the up-regulation of GRP94. Another consequence of higher GH levels in P300L carriers would be its IGF-1ndependent contributions to linear growth. Evidence shows that GH acts directly on epiphyseal cartilage (66–71) and mediates 20% of linear growth independently of IGF-1, which could be another regulatory network operating in humans but not recapitulated in cell culture models.
P300L is not the only human hypomorphic variant of GRP94. We found a second nonsynonymous SNP (rs34482425) in a European female, which changes lysine 513 to asparagine, and this variant also had reduced chaperoning activity toward IGF in the same assay system (Argon, Y., D. Eletto, and M. Marzec, unpublished observations). Thus, variants of GRP94 that affect its expression and/or activity exist in human populations and should be evaluated for their biomedical relevance. It is likely that, given the specificity of GRP94 for client proteins like Toll-like receptors (8), Igs (5), or low-density lipoprotein receptors (Braakman, I., personal communication), hypomorphic GRP94 variants may be discovered also in some patients with immune or cardiovascular dysfunctions.
This work shows that when seeking explanations for defects in human growth or drug targets that impact growth, the search should not be limited to the candidate genes within the GH/IGF axis described to date. GRP94 is not a direct participant in GH/IGF signaling cascades, nor is it a transcription factor for expression of hormones or their receptors. Rather, it is a chaperone required for hormone protein folding and therefore its secretion. It is remarkable that a housekeeping protein has such a highly selective mode of action toward IGFs (1, 7–9). This selectivity, in turn, suggests that the function of the chaperone is important in limiting the bioavailability of IGFs and that this mode of growth regulation should be explored further.
Study approval
For the PIGFD Consortium, institutional review board protocol approved by the Institutional Review Board (IRB) of Oregon Health and Science University reference number FWA00000161, IRB00000148.
For the PIGFD Leiden University Medical Center, patients were studied under an IRB of the Leiden University Medical Center (reference number P07.021).
For the HIMS, IRB protocol was approved by the Human Research Ethics Committee of the University of Western Australia (reference number RA/4/1/5765).
For the LGP, the protocol was approved by the IRB of Albert Einstein College of Medicine of Yeshiva University Montefiore Medical Center (reference number 1998-125).
Acknowledgments
We are grateful to Drs J. Burkhardt, D. Dersh, T. Gidalevitz, T. Mandrup-Poulsen, and B. Eriksen for their useful advice and comments throughout this work. We also thank the Exome Aggregation Consortium and the groups that provided the exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about.
Current address for V.H.: Division of Pediatric Endocrinology, Cincinnati Children's Hospital Medical Center, Cincinnati, Ohio.
Current address for M.M.: Department of Biomedical Sciences, Section of Immuno-Endocrinology, University of Copenhagen, Copenhagen, Denmark.
Author contributions included the following: M.M. conceived the experimental design, performed the experiments, analyzed the data, and wrote the manuscript. C.P.H. generated the population data and performed the statistical analysis. D.E. and S.B. performed the experiments. R.R., V.H., J.M.W., H.A.v.D., W.O., M.L., O.B.P., C.T.H., B.B.Y., L.F., G.A., and N.B. acquired and provided the population samples and helped analyze the data. A.G. conceived the experimental design and analyzed the data. C.P.H., V.H., J.M.W., B.B.Y., G.A., and A.G. helped in the manuscript preparation. Y.A. designed the project, performed the experiments, analyzed the data and wrote the manuscript. All coauthors reviewed the manuscript.
This work was supported by Grants AG18001 and GM-077480 (to Y.A.) as well as Award KO1 AG-040153 and the Junior Investigator Pilot Grant Program Grant UL1 RR024134 (to M.M.). The IGF-1 and IGF binding protein-1 and BP3 assays in the Health In Men Study were supported by Project Grant 513823 from the National Health and Medical Research Council of Australia. The Health, Aging, and Body Composition Study Project was supported by National Institute on Aging Contracts N 01-AG-6-2101; N01-AG-6-2103; N01-AG-6-2106; National Institute on Aging Grant AG-028050, and National Institute of Nursing Research Grant NR-012459.
Disclosure Summary: A.G. and Y.A. declare that they have an interest in the intellectual property in growth control, held by the Children's Hospital of Philadelphia, related to the subject of this work. V.H. received lecture fees from EMD Serono and also received sponsored travel from Pfizer, Inc. R.G.R. received payments for consulting or lectures from Ipsen. This potential conflict has been reviewed and managed by Oregon Health and Science University. J.M.W. received payments for consulting or lectures from Pfizer, Sandoz, Versartis, Merck-Serono, OPKO, Teva, and Biopartners. W.O. received unrestricted grant support from Novo Nordisk and Ferring for other studies. The other authors have nothing to disclose.
Appendix (see Table 4)
Table 4.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised (Monoclonal or Polyclonal) | Dilution Used |
|---|---|---|---|---|---|
| GRP94 | Native chicken Grp94 | 9G10 | SPA-850, Stressgen Biotechnologies | Rat monoclonal | 1:4000 |
| Anti-His | Not known | H-3 | Santa Cruz Biotechnologies | Mouse monoclonal | 1:1000 |
Footnotes
- CD
- circular dichroism
- dbSNP
- The Single Nucleotide Polymorphism database
- ER
- endoplasmic reticulum
- GFP
- green fluorescent protein
- GRP94
- glucose-regulated protein 94
- HIMS
- Health In Men Study
- IGFBP
- IGF binding protein
- IGF1R
- IGF-1 receptor
- IRB
- institutional review board
- KO
- knockout
- LGP
- Longevity Genes Project
- MEF
- mouse embryonic fibroblast
- mIGF-2
- mouse IGF-2
- PIGFD
- primary IGF deficiency
- P300L
- proline 300 of the mature protein substituted with leucine
- SNP
- single-nucleotide polymorphism
- STAT
- signal transducer and activator of transcription
- WT
- wild type.
References
- 1. Wanderling S, Simen BB, Ostrovsky O, et al. GRP94 is essential for mesoderm induction and muscle development because it regulates insulin-like growth factor secretion. Mol Biol Cell. 2007;18:3764–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ostrovsky O, Eletto D, Makarewich C, Barton ER, Argon Y. Glucose regulated protein 94 is required for muscle differentiation through its control of the autocrine production of insulin-like growth factors. Biochim Biophys Acta. 2010;1803:333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chang SC, Erwin AE, Lee AS. Glucose-regulated protein (GRP94 and GRP78) genes share common regulatory domains and are coordinately regulated by common trans-acting factors. Mol Cell Biol. 1989;9:2153–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barton ER, Park S, James JK, et al. Deletion of muscle GRP94 impairs both muscle and body growth by inhibiting local IGF production. FASEB J. 2012;26:3691–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Melnick J, Dul JL, Argon Y. Sequential interaction of the chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature. 1994;370:373–375. [DOI] [PubMed] [Google Scholar]
- 6. Reddy RK, Lu J, Lee AS. The endoplasmic reticulum chaperone glycoprotein GRP94 with Ca(2+)-binding and antiapoptotic properties is a novel proteolytic target of calpain during etoposide-induced apoptosis. J Biol Chem. 1999;274:28476–28483. [DOI] [PubMed] [Google Scholar]
- 7. Staron M, Wu S, Hong F, et al. Heat-shock protein gp96/grp94 is an essential chaperone for the platelet glycoprotein Ib-IX-V complex. Blood. 2011;117:7136–7144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang Y, Liu B, Dai J, et al. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity. 2007;26:215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ostrovsky O, Ahmed NT, Argon Y. The chaperone activity of GRP94 toward insulin-like growth factor II is necessary for the stress response to serum deprivation. Mol Biol Cell. 2009;20:1855–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schulte TW, Akinaga S, Murakata T, et al. Interaction of radicicol with members of the heat shock protein 90 family of molecular chaperones. Mol Endocrinol. 1999;13:1435–1448. [DOI] [PubMed] [Google Scholar]
- 11. Ali MM, Roe SM, Vaughan CK, et al. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ostrovsky O, Makarewich CA, Snapp EL, Argon Y. An essential role for ATP binding and hydrolysis in the chaperone activity of GRP94 in cells. Proc Natl Acad Sci USA. 2009;106:11600–11605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eletto D, Maganty A, Eletto D, et al. Limitation of individual folding resources in the ER leads to outcomes distinct from the unfolded protein response. J Cell Sci. 2012;125:4865–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009;30:586–623. [DOI] [PubMed] [Google Scholar]
- 15. Yakar S, Kim H, Zhao H, et al. The growth hormone-insulin like growth factor axis revisited: lessons from IGF-1 and IGF-1 receptor gene targeting. Pediatr Nephrol. 2005;20:251–254. [DOI] [PubMed] [Google Scholar]
- 16. Montarras D, Aurade F, Johnson TJ, II, Gros F, Pinset C. Autonomous differentiation in the mouse myogenic cell line, C2, involves a mutual positive control between insulin-like growth factor II and MyoD, operating as early as at the myoblast stage. J Cell Sci 1996;109(Pt 3):551–560. [DOI] [PubMed] [Google Scholar]
- 17. Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- 18. Teglund S, McKay C, Schuetz E, et al. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93:841–850. [DOI] [PubMed] [Google Scholar]
- 19. Romero CJ, Pine-Twaddell E, Sima DI, et al. Insulin-like growth factor 1 mediates negative feedback to somatotroph GH expression via POU1F1/CREB binding protein interactions. Mol Cell Biol. 2012;32:4258–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berelowitz M, Szabo M, Frohman LA, Firestone S, Chu L, Hintz RL. Somatomedin-C mediates growth hormone negative feedback by effects on both the hypothalamus and the pituitary. Science. 1981;212:1279–1281. [DOI] [PubMed] [Google Scholar]
- 21. Kaplan SA, Cohen P. The somatomedin hypothesis 2007: 50 years later. J Clin Endocrinol Metab. 2007;92:4529–4535. [DOI] [PubMed] [Google Scholar]
- 22. Dauber A, Rosenfeld RG, Hirschhorn JN. Genetic evaluation of short stature. J Clin Endocrinol Metab. 2014;99:3080–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krzisnik C, Grguric S, Cvijovic K, Laron Z. Longevity of the hypopituitary patients from the island Krk: a follow-up study. Pediatr Endocrinol Rev. 2010;7:357–362. [PubMed] [Google Scholar]
- 24. Salvatori R, Hayashida CY, Aguiar-Oliveira MH, et al. Familial dwarfism due to a novel mutation of the growth hormone-releasing hormone receptor gene. J Clin Endocrinol Metab. 1999;84:917–923. [DOI] [PubMed] [Google Scholar]
- 25. Baumann G, Maheshwari H. The Dwarfs of Sindh: severe growth hormone (GH) deficiency caused by a mutation in the GH-releasing hormone receptor gene. Acta Paediatr Suppl. 1997;423:33–38. [DOI] [PubMed] [Google Scholar]
- 26. Besson A, Salemi S, Gallati S, et al. Reduced longevity in untreated patients with isolated growth hormone deficiency. J Clin Endocrinol Metab. 2003;88:3664–3667. [DOI] [PubMed] [Google Scholar]
- 27. Laron Z. Laron syndrome (primary growth hormone resistance or insensitivity): the personal experience 1958–2003. J Clin Endocrinol Metab. 2004;89:1031–1044. [DOI] [PubMed] [Google Scholar]
- 28. Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011;3:70ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kofoed EM, Hwa V, Little B, et al. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med. 2003;349:1139–1147. [DOI] [PubMed] [Google Scholar]
- 30. Fuqua JS, Derr M, Rosenfeld RG, Hwa V. Identification of a novel heterozygous IGF1 splicing mutation in a large kindred with familial short stature. Horm Res Paediatr. 2012;78:59–66. [DOI] [PubMed] [Google Scholar]
- 31. Woods KA, Camacho-Hubner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med. 1996;335:1363–1367. [DOI] [PubMed] [Google Scholar]
- 32. Bonapace G, Concolino D, Formicola S, Strisciuglio P. A novel mutation in a patient with insulin-like growth factor 1 (IGF1) deficiency. J Med Genet. 2003;40:913–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tazearslan C, Huang J, Barzilai N, Suh Y. Impaired IGF1R signaling in cells expressing longevity-associated human IGF1R alleles. Aging Cell. 2011;10:551–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burkhardt S, Gesing J, Kapellen TM, et al. Novel heterozygous IGF1R mutation in two brothers with developing impaired glucose tolerance. J Pediatr Endocrinol Metab. 2015;28:217–225. [DOI] [PubMed] [Google Scholar]
- 35. Domene HM, Bengolea SV, Martinez AS, et al. Deficiency of the circulating insulin-like growth factor system associated with inactivation of the acid-labile subunit gene. N Engl J Med. 2004;350:570–577. [DOI] [PubMed] [Google Scholar]
- 36. Rosenfeld RG, von Stein T. A database and website for molecular defects of the GH-IGF axis (http://www.growthgenetics.com) Horm Res Paediatr. 2013;80:443–448. [DOI] [PubMed] [Google Scholar]
- 37. Wit JM, van Duyvenvoorde HA, et al. Genetic analysis of short children with apparent growth hormone insensitivity. Horm Res Paediatr. 2012;77:320–333. [DOI] [PubMed] [Google Scholar]
- 38. Biswas C, Ostrovsky O, Makarewich CA, Wanderling S, Gidalevitz T, Argon Y. The peptide-binding activity of GRP94 is regulated by calcium. Biochem J. 2007;405:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gidalevitz T, Biswas C, Ding H, et al. Identification of the N-terminal peptide binding site of glucose-regulated protein 94. J Biol Chem. 2004;279:16543–16552. [DOI] [PubMed] [Google Scholar]
- 40. Hughes PF, Barrott JJ, Carlson DA, et al. A highly selective Hsp90 affinity chromatography resin with a cleavable linker. Bioorg Med Chem. 2012;20:3298–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yeap BB, Chubb SA, Ho KK, et al. IGF1 and its binding proteins 3 and 1 are differentially associated with metabolic syndrome in older men. Eur J Endocrinol. 2010;162:249–257. [DOI] [PubMed] [Google Scholar]
- 42. Yeap BB, Chubb SA, McCaul KA, et al. Associations of IGF1 and IGFBPs 1 and 3 with all-cause and cardiovascular mortality in older men: the Health In Men Study. Eur J Endocrinol. 2011;164:715–723. [DOI] [PubMed] [Google Scholar]
- 43. Barzilai N, Atzmon G, Schechter C, et al. Unique lipoprotein phenotype and genotype associated with exceptional longevity. JAMA. 2003;290:2030–2040. [DOI] [PubMed] [Google Scholar]
- 44. Albrechtsen A, Grarup N, Li Y, et al. Exome sequencing-driven discovery of coding polymorphisms associated with common metabolic phenotypes. Diabetologia. 2013;56:298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Milman S, Atzmon G, Huffman DM, et al. Low insulin-like growth factor-1 level predicts survival in humans with exceptional longevity. Aging Cell. 2014;13:769–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Soldano KL, Jivan A, Nicchitta CV, Gewirth DT. Structure of the N-terminal domain of GRP94. Basis for ligand specificity and regulation. J Biol Chem. 2003;278:48330–48338. [DOI] [PubMed] [Google Scholar]
- 47. Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol Cell. 2007;28:41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vogen S, Gidalevitz T, Biswas C, et al. Radicicol-sensitive peptide binding to the N-terminal portion of GRP94. J Biol Chem. 2002;277:40742–40750. [DOI] [PubMed] [Google Scholar]
- 49. Norman PE, Flicker L, Almeida OP, Hankey GJ, Hyde Z, Jamrozik K. Cohort profile: The Health In Men Study (HIMS). Int J Epidemiol. 2009;38:48–52. [DOI] [PubMed] [Google Scholar]
- 50. Ketha H, Singh RJ. Clinical assays for quantitation of insulin-like-growth-factor-1 (IGF1). Methods. 2015;81:93–98. [DOI] [PubMed] [Google Scholar]
- 51. Yakar S, Rosen CJ, Beamer WG, et al. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest. 2002;110:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wearsch PA, Voglino L, Nicchitta CV. Structural transitions accompanying the activation of peptide binding to the endoplasmic reticulum Hsp90 chaperone GRP94. Biochemistry. 1998;37:5709–5719. [DOI] [PubMed] [Google Scholar]
- 53. Marzec M, Eletto D, Argon Y. GRP94: an HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim Biophys Acta. 2012;1823:774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rosser MF, Nicchitta CV. Ligand interactions in the adenosine nucleotide-binding domain of the Hsp90 chaperone, GRP94. I. Evidence for allosteric regulation of ligand binding. J Biol Chem. 2000;275:22798–22805. [DOI] [PubMed] [Google Scholar]
- 55. Duerfeldt AS, Peterson LB, Maynard JC, et al. Development of a Grp94 inhibitor. J Am Chem Soc. 2012;134:9796–9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Patel HJ, Patel PD, Ochiana SO, et al. Structure-activity relationship in a purine-scaffold compound series with selectivity for the endoplasmic reticulum hsp90 paralog grp94. J Med Chem. 2015;58:3922–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Patel PD, Yan P, Seidler PM, et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat Chem Biol. 2013;9:677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Walenkamp MJ, Karperien M, Pereira AM, et al. Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J Clin Endocrinol Metab. 2005;90:2855–2864. [DOI] [PubMed] [Google Scholar]
- 59. Gicquel C, Le Bouc Y. Hormonal regulation of fetal growth. Horm Res. 2006;65(suppl 3):28–33. [DOI] [PubMed] [Google Scholar]
- 60. Begemann M, Zirn B, Santen G, et al. Paternally inherited IGF2 mutation and growth restriction. N Engl J Med. 2015;373:349–356. [DOI] [PubMed] [Google Scholar]
- 61. Suh Y, Atzmon G, Cho MO, et al. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci USA. 2008;105:3438–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fichter MM, Pirke KM, Holsboer F. Weight loss causes neuroendocrine disturbances: experimental study in healthy starving subjects. Psychiatry Res. 1986;17:61–72. [DOI] [PubMed] [Google Scholar]
- 63. Gianotti L, Lanfranco F, Ramunni J, Destefanis S, Ghigo E, Arvat E. GH/IGF-1 axis in anorexia nervosa. Eat Weight Disord. 2002;7:94–105. [DOI] [PubMed] [Google Scholar]
- 64. Brooks AJ, Waters MJ. The growth hormone receptor: mechanism of activation and clinical implications. Nat Rev Endocrinol. 2010;6:515–525. [DOI] [PubMed] [Google Scholar]
- 65. Udy GB, Towers RP, Snell RG, et al. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci USA. 1997;94:7239–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Isaksson OG, Eden S, Jansson JO. Mode of action of pituitary growth hormone on target cells. Annu Rev Physiol. 1985;47:483–499. [DOI] [PubMed] [Google Scholar]
- 67. Russell SM, Spencer EM. Local injections of human or rat growth hormone or of purified human somatomedin-C stimulate unilateral tibial epiphyseal growth in hypophysectomized rats. Endocrinology. 1985;116:2563–2567. [DOI] [PubMed] [Google Scholar]
- 68. Schlechter NL, Russell SM, Spencer EM, Nicoll CS. Evidence suggesting that the direct growth-promoting effect of growth hormone on cartilage in vivo is mediated by local production of somatomedin. Proc Natl Acad Sci USA. 1986;83:7932–7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ohlsson C, Nilsson A, Isaksson O, Lindahl A. Growth hormone induces multiplication of the slowly cycling germinal cells of the rat tibial growth plate. Proc Natl Acad Sci USA. 1992;89:9826–9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Isaksson OG, Jansson JO, Gause IA. Growth hormone stimulates longitudinal bone growth directly. Science. 1982;216:1237–1239. [DOI] [PubMed] [Google Scholar]
- 71. Isgaard J, Nilsson A, Lindahl A, Jansson JO, Isaksson OG. Effects of local administration of GH and IGF-1 on longitudinal bone growth in rats. Am J Physiol. 1986;250:E367–E372. [DOI] [PubMed] [Google Scholar]