

Abstract
Hydrogen sulfide (H2S) is both a lethal gas and an emerging gasotransmitter in humans, suggesting that cellular H2S level must be tightly regulated. CstB is encoded by the cst operon of the major human pathogen Staphylococcus aureus (S. aureus) and is under the transcriptional control of the persulfide sensor CstR and H2S. Here we show that CstB is a multifunctional Fe(II)-containing persulfide dioxygenase (PDO), analogous to the vertebrate protein ETHE1 (Ethylmalonic Encephalopathy Protein 1). Chromosomal deletion of ethe1 is fatal in vertebrates. In the presence of molecular oxygen (O2), hETHE1 oxidizes glutathione persulfide (GSSH) to generate sulfite and reduced glutathione. In contrast, CstB oxidizes major cellular low molecular weight (LMW) persulfide substrates from S. aureus, coenzyme A persulfide (CoASSH) and bacillithiol persulfide (BSSH), directly to generate thiosulfate (TS) and reduced thiols, thereby avoiding the cellular toxicity of sulfite. Both Cys201 in the N-terminal PDO domain (CstBPDO) and Cys408 in the C-terminal rhodanese domain (CstBRhod) strongly enhance the TS generating activity of CstB. CstB also possesses persulfide transferase (PT; reverse rhodanese) activity which generates TS when provided with LMW persulfides and sulfite, as well as conventional thiosulfate transferase (TST; rhodanese) activity; both activities require Cys408. CstB protects S. aureus against H2S toxicity with C201S and C408S cstB genes unable to rescue a NaHS-induced ΔcstB growth phenotype. Induction of the cst operon by NaHS reveals that functional CstB impacts the cellular TS concentrations. These data collectively suggest that CstB may have evolved to facilitate the clearance of LMW persulfides that occur upon the elevation of the level of cellular H2S and hence may have an impact on bacterial viability under H2S stress, in concert with the other enzymes encoded by the cst operon.
Keywords: ETHE1, hydrogen sulfide, persulfide dioxygenase, sulfurtransferase, non-heme iron, reactive sulfure species
INTRODUCTION
Hydrogen sulfide (H2S) is a colorless and flammable gas that has a strong rotten egg smell and can freely pass through biological membranes. In the cell it primarily exists as hydrosulfide anion (HS−).1 H2S inhibits cellular respiration by poisoning the terminal electron acceptor cytochrome c oxidase at sub-micromolar concentrations.2-4 H2S may also exhibit toxicity when HS− reacts directly with the oxidized low molecular weight (LMW) thiols that collectively control the cellular redox potential, e.g., glutathione disulfide (GSSG) or bacillithiol disulfide (BSSB), to form reactive sulfur species (RSS),5 glutathione or bacillithiol persulfides (GSSH, BSSH).6 Recent studies reveal that H2S can also play positive biological roles. In mammalian systems, H2S has been characterized as a gasotransmitter or signaling molecule that influences a number of processes, including vasorelaxation, cardioprotection and neurotransmission.7-9 In many bacteria, endogenous production of H2S by three major H2S-generating enzymes, cystathionine β-synthase (CBS), cystathionine Δ-lyase (CSE), and 3-mercaptopyruvate sulfur transferase (3-MST), has been shown to enhance resistance to oxidative stress induced by antibiotic treatment via an unknown mechanism.10
This combination of toxicity and utility of H2S suggests a physiological need to regulate intracellular H2S concentrations. One example of such regulation in eukaryotic systems is the mitochondrial persulfide dioxygenase (PDO), ethylmalonic encephalopathy protein 1 (ETHE1). Chromosomal deletion of ethe1 causes ethylmalonic encephalopathy and is ultimately fatal, due to H2S toxicity.11 Although there is a report of a HS− efflux transporter from the human pathogen Clostridium difficile,12 major H2S detoxification mechanisms in bacteria remain poorly understood.
Staphylococcus aureus is a Gram-positive opportunistic pathogen that causes a wide variety of hospital- and community-acquired infections, ranging from minor skin infections to life-threatening diseases13 With no gene encoding 3-MST, S. aureus relies on CBS and CSE for the synthesis of endogenous H2S.10 We recently described the cst (copper-sensing operon repressor-like sulfur transferase) operon14 which appears to be crucial for sulfide detoxification in S. aureus (Fig. 1). Transcription of the cst operon is regulated by a per- and polysulfide-sensing repressor CstR and is induced upon addition of exogenous NaHS or polysulfides to cells grown aerobically in liquid culture.15 Three of the genes encoded by the cst operon, including cstA, cstB and sqr, the latter encoding a sulfide:quinone oxidoreductase, are necessary to mitigate the effects of cellular sulfide toxicity.15 The core cst-encoded genes, tauE, cstR, cstA and cstB, are often duplicated in methicillin-resistant strains of S. aureus (MRSA), and a synteny analysis reveals that this copy is flanked by methicillin-resistance determinants from the major staphylcoccal cassette chromosome mec element types (SCCmec) (Fig. S1).
Figure 1.

Genomic region of S. aureus subspecies aureus strains Newman (top) and COL (middle, bottom) encompassing the cst operon region. Locus tag identifiers are indicated as NWMN_0029 to 0026 (top) encoding SQR (shaded green), CstB (orange), CstA (blue), CstR (yellow) and TauE (red), SACOL0065 to SACOL0061 (middle) and duplicated in the strain COL as SACOL0045 to SACOL0050 (bottom), shaded as indicated for the Newman strain. The duplicated core cst operon region (SACOL0046 to SACOL0049; cstB2, cstA2, cstR2, tauE2) is upstream of a methicillin resistance determinant (SACOL0050, pls or methicillin resistance surface protein) and downstream of the mecA, mecR1, and mecI genes (not shown), encoding the penicillin binding protein 2A, a β-lactam sensor/signal transducer membrane protein that binds methicillin and the methicillin-resistance repressor, respectively (Fig. S1). CstB encoded by NWMN_0028 is the subject of the studies reported here.
CstA has recently been characterized as a multidomain sulfurtransferase that reacts with a persulfide formed on the cysteine desulfurase SufS, as well as with inorganic polysulfides such as sodium tetrasulfide.16 SQR and CstB, on the other hand, are predicted to comprise a system analogous to the first two steps in human mitochondrial H2S oxidation. Human SQR (hSQR) initially oxidizes H2S to glutathione persulfide (GSSH); subsequently, ETHE1 oxidizes GSSH to sulfite in the presence of molecular oxygen, while regenerating reduced glutathione (GSH).17 In the mitochondrion, toxic sulfite18 can be further oxidized to thiosulfate (TS), via a persulfide transferase (PT; also known as a reverse rhodanese) activity, or to sulfate via the molybdopterin-requiring enzyme sulfite oxidase. Since S. aureus encodes no known sulfite oxidase, we surmise that sulfite is either assimilated by one of four identified cellular rhodaneses including those found in CstA and CstB,19 or is effluxed from the cell via an unknown mechanism.20
CstB is predicted to be a three-domain enzyme containing an N-terminal metallo-β-lactamase-like (MBL), non-heme Fe(II)-containing PDO domain, followed by a pseudo-rhodanese homology (RHD) domain, and a conventional C-terminal rhodanese (Rhod) domain (Fig. 2A). The N-terminal PDO domain of CstB is homologous to full-length human ETHE1, with 21% identity (Fig. S2). The conserved iron-binding residues in the active site (Fig. S2) and highlighted in the crystal structure of hETHE1 shown in the left panel of Fig. 2B.21 Although this enzyme forms the basis for our hypotheses regarding the enzymatic activity of CstB, such relatively low sequence identity leads us to anticipate important structural differences that impact enzyme function.
Figure 2.

Structural overview of CstB and related enzymes. (A) Domain organization of S. aureus CstB, with N-terminal persulfide dioxygenase (PDO) domain, middle rhodanese homology domain (RHD), and C-terminal rhodanese domain (Rhod). (B) Structures of CstB homologs previously published or from structural genomics efforts: hETHE1 (PDB 4CHL, left panel), S. aureus COL CstB2PDO-RHD (PDB 3R2U, middle panel), and A. acidocaldarius homolog of unknown function (PDB 3TP9, right panel). PDO domains are shaded green, RHD in light blue, and rhodanese domains in dark blue. Conserved metal-binding side chain ligands are highlighted in orange and conserved cysteines in yellow. The extended loop containing C201 that is not present in hETHE1 is highlighted in purple in the other two structures. (C) Cartoon model of S. aureus CstB highlighting the three catalytic activities documented here: coupled persulfide dioxygenase-persulfide transferase (cPDO-PT) activity, reaction 1; persulfide transferase (PT) activity, reaction 2; rhodanese or thiosulfate transferase (TST) activity, reaction 3. Approximate locations of conserved cysteines and Fe center are indicated by the red and brown circles, respectively. In all panels, the PDO domain is shaded green, RHD light blue, and Rhod domain dark blue.
A number of structural genomics initiatives have provided several crystal structures that collectively provide additional perspectives on CstB function. These structures include CstB2 from S. aureus strain COL which shows the N-terminal PDO and middle RHD domains, but lacks the C-terminal Rhod domain (Fig. 2B, middle panel). This protein is 77% identical to CstB from S. aureus strain Newman, and is representative of CstBs contained within a duplicated core cst operon in the COL (SACOL0046; Fig. 1) and other methicillin-resistant S. aureus strains (Fig. S1); SACOL0064-encoding CstB1 is 100% identical to the Newman-strain CstB studied here (Fig. 1). The structure of the CstB2 PDO domain is similar to that of hETHE1 and that of an ETHE1-like protein from Arabidopsis thaliana,22 except for the presence a long, cysteine-containing loop near the iron active site, highlighted in Fig 2B. Like ETHE1, the CstB2 structure also shows Fe(II) bound in a semi-facial triad composed of the side chains of H56, H119 and D145 (Fig. 2B, inset). Another related structure comes from an Alicyclobacillus acidocaldarius protein with a very similar domain organization to full-length CstB (3TP9PDO-RHD-Rhod) (Fig. 2B, right panel). This protein is 52% identical to CstB and contains all three cysteine residues that are conserved in S. aureus CstB (equivalent to C20, C201, and C408, highlighted in Figs. 2B and S1). The N-terminal and middle domains are oriented similarly to the CstB2 structure, suggesting that the three domains of CstB might pack together in a similar orientation in order to function collaboratively.
In this work, we show that CstB catalyzes the oxidation of major LMW persulfides from S. aureus, coenzyme A persulfide (CoASSH) and bacillithiol persulfide (BSSH), and to a lesser extent cysteine persulfide (CSSH) and glutathione persulfide (GSSH), to TS and reduced thiols, in contrast to the sulfite-generating activity of ETHE1.17 CstB is also shown to be a multifunctional enzyme that harbors persulfide transferase (PT) and conventional thiosulfate transferase (TST) activities, both catalyzed by the CstBRhod domain. All three cysteine residues of CstB, C20, C201 and C408, are crucial for cellular viability under NaHS stress, despite the fact that C20 and C201 are not conserved in eukaryotic PDOs. This work represents the first characterization of a multidomain bacterial PDO-RHD-Rhod fold protein from a human pathogen, and the potential significance of these findings on S. aureus physiology is discussed, in concert with the other known activities of cst-encoded gene products.15,16
MATERIALS AND METHODS
Cloning and purification of recombinant CstB proteins
Full-length CstBs (wild-type, C20S, C201S, C408S and C201S/C408S) and CstBPDO-RHD were cloned into a pET15b expression plasmid using the Nco1 and BamH1 restriction sites. CstBRhod was cloned into pHis parallel expression plasmid using Nco1 and Sal1 restriction sites. CstB mutants (C20S, C201S and C408S) were generated by site-directed mutagenesis. All CstB expression plasmids were transformed into the E. coli Rosetta strain, cultured in LB medium at 37 ºC until the OD600 reached 0.6-0.8, induced with 1 mM IPTG, and expressed at 16 ºC for 16 h. Cells were harvested by centrifugation and stored at −80 ºC.
Full-length CstBs (wild-type, C201S, C408S and C201S/C408S) and CstBPDO-RHD were all purified using similar protocols. The cell pellet was resuspended in 25 mM HEPES, 150 mM NaCl, 5 mM DTT, 2 mM EDTA, pH 7.0 and lysed by sonication. The cell lysate was clarified by centrifugation and 0.015% (w/v) polyethyleneimine (PEI) was added to the supernatant to precipitate nucleic acids and nucleic acid binding proteins. Full-length CstBs co-precipitated with nucleic acids with the addition of 0.015% (w/v) PEI and were re-dissolved by stirring the pellet in 25 mM Tris-HCl, 1 M NaCl, 5 mM DTT, 2 mM EDTA, pH 8.0 for 2 h at 4 ºC. Ammonium sulfate (AS) precipitation was next performed to salt out full-length CstBs between 40% and 60% (w/v) AS. CstBPDO-RHD remained in solution with the addition of 0.015% (w/v) PEI and was salted out between 30% and 50% (w/v) AS directly. Ammonium sulfate pellets were then resuspended in 25 mM Tris-HCl, 50 mM NaCl, 5 mM DTT, 2 mM EDTA, pH 8.0, and CstB proteins were purified by anion exchange chromatography (Q-sepharose) followed by size exclusion chromatography (G200 16/60). Fractions containing target proteins with a purity of >95% were pooled and stored at −80 ºC until use with 20% (v/v) final concentration of glycerol added.
CstBRhod was purified by Ni-NTA affinity chromatography followed by removal of the N-terminal His6-tag. The cell pellet was resuspended in 25 mM Tris-HCl, 500 mM NaCl, 20 mM imidazole, 2 mM TCEP, pH 8.0 and lysed by sonication. The cell lysate was clarified by centrifugation and the supernatant was loaded on a Ni-NTA column, which was pre-equilibrated with the lysis buffer. The column was washed with lysis buffer containing 50mM imidazole, followed by elution with 500 mM imidazole. His6-CstBRhod was exchanged into lysis buffer and incubated with Tobacco Etch Virus (TEV) protease at 4 ºC overnight. The cleaved His6 tag was removed by reapplication of the TEV-incubated protein to the Ni-NTA column and the cleaved CstBRhod was collected in the flow-through and in the 50 mM imidazole wash fractions, followed by further purification by size exclusion chromatography (G75 16/60). Fractions containing CstBRhod with a purity of >95% were pooled and stored at −80 ºC until use with 20% (v/v) final concentration of glycerol added.
Metal analysis
Wild-type CstB was analyzed by Inductively Coupled Plasma Mass Spectrometry (ICP-MS) to confirm that iron (Fe) was the only significant metal found, with undetectable zinc (Zn), cobalt (Co), manganese (Mn) and nickel (Ni). The Fe content for all CstBs was then quantified by Atomic Absorption Spectroscopy (AAS), utilizing proteins prepared at 20 μM in 25 mM Tris-HBr, 100 mM NaBr, pH 8.0 as follows: all CstBs were exchanged into degassed and chelexed, metal-free 25 mM Tris-HBr, 100 mM NaBr, pH 8.0 in an anaerobic chamber to remove reducing reagents and EDTA, then loaded with Fe by exchanging into 25 mM Tris-HBr, 100 mM NaBr, 10 mM FeSO4, 10 mM Na2S2O4, pH 8.0, and finally into 25 mM Tris-HBr, 100 mM NaBr, pH 8.0 to remove free Fe.
EPR spectroscopy
X-band EPR spectroscopy was carried out in the NMR Facility, Department of Chemistry, and was used to estimate the total Fe(III) content in wild-type CstB following anaerobic loading of Fe to a stoichiometry of 1:1 and subsequent reduction with sodium dithionite.17 Fe(III) was quantified by comparison to a series of standard solutions acquired under the same condition. 200 μM Fe(II)-loaded CstB was found to contain 18 μM Fe(III), revealing ≈90% Fe(II) in this preparation. Multiple preparations of CstB contained similar specific activities. All other mutant CstB preparations were assumed to harbor largely reduced Fe(II) as a result of this anaerobic reconstitution procedure.
Gel filtration chromatography
All CstBs containing stoichiometric Fe were chromatographed on an analytical G200 GL column by loading 100 μL at 10 μM protomer concentration protein and eluting at 0.5 mL/min at 4 ºC. CstBRhod was chromatographed on an analytical G75 column by loading 100 μL of 20 μM protomer concentration protein and eluting at 0.5 mL/min at 4 ºC.
Preparation of LMW persulfide substrates
Bacillithiol disulfide (BSSB) was synthesized by the Vanderbilt Chemical Synthesis Core and reduced to BSH according to published procedures.23 All other reduced thiols and disulfides were obtained from commercial sources and used without further purification. LMW persulfides were freshly prepared by mixing a five-fold molar excess of sodium sulfide (Na2S) with oxidized LMW disulfide and incubating anaerobically at 30 ºC for 30 min in degassed 300 mM phosphate, pH 7.4. The concentration of generated LMW persulfides was determined using a cold cyanolysis assay and used without further purification in enzymatic assays at the indicated final concentration.17
Coupled persulfide dioxygenase-persulfide transferase (cPDO-PT) activity assays
The cPDO-PT activity was measured using a fluorescence-based HPLC assay to detect the production of TS as outlined previously.15 Typical 100 μL reactions contained 40 nM CstBs and a specific LMW persulfide substrate ranging from 40 μM to 2.56 mM, buffered by air-saturated 25 mM MES, 100 mM NaBr, pH 6.0 at 25 ºC. These reactions were initiated with the addition of enzyme or BSA (as a control), incubated for 2 min and terminated by addition of 0.5 μL 5 M methanesulfonic acid (MA). Proteins were removed by ultrafiltration and 25 μL of the filtered solution was labeled in the dark by 75 μL monobromobimane (mBBr) labeling solution containing 25 mM Tris-HBr, 2 M mBBr, pH 8.0, for 30 min at room temperature. The labeling reaction was terminated by addition of 100 μL 16.4% methanol, 0.25% acetic acid, pH 3.9. 20 μL samples were injected in duplicate onto a Kinetex C18 reversed-phase column (Phenomenex, P/No. 00F-4601-E0, 4.6 mm × 150 mm, 5 μm, 100 Å) outfitted with a Zorbax Eclipse Plus C18 guard column, followed by chromatographic analysis on a Waters 600 high-performance liquid chromatography system equipped with a Waters 717 plus auto-sampler, a Waters 474 scanning fluorescence detector (λex=384 nm and λem=478 nm) and Empower chromatography software installed on a standard PC running Windows XP. A methanol-based gradient system was employed to analyze the samples at 25 ºC (Solvent A: 16.8% methanol, 0.25% acetic acid, pH 3.9; Solvent B: 90% methanol, 0.25% acetic acid, pH 3.9) with a flow rate of 1.2 mL/min and an elution protocol as follows: 0-10 min, 0% B isocratic; 10–12 min, 0-100% B, linear gradient; 12-17 min, 100% B isocratic followed by re-equilibration to 0% B. Quantitation of TS was enabled by running a series of authentic TS standards in separate chromatographic runs with an identical protocol. For time course studies, 200 μL reactions containing 10 μM CstB and 200 μM persulfide substrate in 25 mM MES, 100 mM NaBr, pH 6.0 at 25 ºC were used. Here, the reactions were initiated as above, and terminated at various times, from 0 min to 32 min, by adding 1 μL 5 M methanesulfonic acid (MA). 50 μL of the ultrafiltered solution was labeled in the dark by 50 μL mBBr labeling solution containing 25 mM Tris-HBr, 2 M mBBr, pH 8.0 for 30 min at room temperature. The labeling reaction was terminated by addition of 100 μL 16.8% methanol, 0.25% acetic acid, pH 3.9 and 40 μL samples were subjected to chromatographic analysis as described above.
Persulfide transferase (PT) activity assays
The PT activity was measured using the fluorescence-based HPLC assay described above for TS production. 500 μL reactions contained 200 nM C201S CstB, CstB2CS or BSA (as a control) and a specific persulfide substrate ranging from 8 μM to 384 μM and 200 μM sulfite buffered in degassed 25 mM MES, 100 mM NaBr, pH 6.0 at 25 ºC. The reaction was initiated in the anaerobic chamber by sulfite, incubated for 2 min and terminated by addition of 2.5 μL 5 M MA. 50 μL of the ultrafiltered solution was labeled in the dark with 50 μL mBBr labeling solution containing 25 mM Tris-HBr, 2 M mBBr, pH 8.0, for 30 min at room temperature. These reactions were analyzed as described above for the cPDO-PT activity assays.
Thiosulfate transferase (TST) activity assays
Purified CstBs were pre-incubated with 1000-fold molar excess of potassium cyanide (KCN) (prepared as 2 M KCN stock in 100 mM Tris-HCl, pH 8.0) at room temperature for 1 h to strip any persulfide sulfur on the cysteine residues that may have occurred during purification. KCN-treated CstBs were then extensively exchanged into fully degassed 25 mM Tris-HBr, 100 mM NaBr, pH 8.0 in anaerobic chamber to remove KCN. The TST activity was measured using a cold cyanolysis assay for the thiocyanate production essentially as described previously for CstA proteins.16 1.0 mL reactions contained 400 nM CstBs, 50 mM KCN, and TS ranging from 100 μM to 6.4 mM, buffered by 100 mM MES, pH 6.0 at 37 ºC, with the reactions initiated with the addition of enzyme, incubated for 4 min and terminated by addition of 2 mL Fe/formaldehyde solution [50 g/L Fe(NO3)3 in 65% HNO3:37% formaldehyde = 1:3 (v/v)]. The potassium thiocyanate (KSCN) production was measured using a 96-well plate reader for absorption at 460 nm for the generated thiocyanato iron complex. Quantitation of KSCN was enabled by running a series of authentic KSCN standards in separate runs with the identical protocol.
Cell growth
S. aureus Newman strains were inoculated from glycerol stocks and grown in 5 mL Tryptic Soy Broth (TSB) medium with 10 μg/ml chloramphenicol overnight, as all the strains carry either the empty pOS1 vector (wild-type strain and ΔcstB strain) or the indicated cstB alleles constructed into pOS1 vector.15 The overnight cultures were pelleted by centrifugation, resuspended in equal volume of Hussain-Hastings-White modified (HHWm) medium and diluted into 15 mL HHWm supplemented with 0.5 mM TS, in the presence and absence of 0.2 mM NaHS. All cultures (≈25 mL) were grown aerobically at 37 ºC in duplicate with shaking at 200 rpm in loosely capped 50 mL Falcon tubes. Cell density was recorded hourly for 10 h by removal of 0.5 mL and measurement of optical density at 600 nm. The starting OD600 of each culture was ≈0.008.
Measurement of cellular TS concentration
All the S. aureus wild-type, ΔcstB and complemented strains were grown in 10 mL TSB medium with 10 μg/mL chloramphenicol overnight. Cells were pelleted, washed with phosphate buffered saline (PBS) and cultures were initiated at OD600 of ≈ 0.02 in HHWm medium with 10 μg/mL chloramphenicol and 0.5 mM TS as sole sulfur source. NaHS was added to the cultures at a final concentration of 0.2 mM when the OD600 reached ≈0.2. All cultures were grown in loosely capped 50 mL Falcon tubes at 37 °C with shaking at 200 rpm. Aliquots were removed at 0, 10, 30, 60 and 120 min following addition of NaHS (equivalent to 15 mL of OD600 = 0.2 cells) and harvested by centrifugation at 3000 rpm for 10 min with culture medium supernatant discarded. Cell pellets were then washed with PBS, pelleted again by centrifugation at 13,200 rpm for 5 min and stored frozen at −80 °C until analyzed. Cell pellets were then thawed, resuspended in 100 μL mBBr labeling solution containing 20 mM Tris-HBr, pH 8.0, 50% acetonitrile, 1 mM mBBr, and 2 μM N-acetyl-L-cysteine (NAC) for use as an internal standard, and incubated at 60 °C for 1 h in the dark in screw capped Eppendorf tubes to avoid liquid loss by evaporation. Cellular debris and proteins were then pelleted by centrifugation at 13,200 rpm for 5 min and the supernatant was transferred to a fresh Eppendorf tube containing 300 μL 10 mM MA to terminate the labeling reaction. These samples were centrifuged at 13,200 rpm for 5 min through 0.2 μm centrifugal filter unit to remove particulates and 40 μL samples were injected onto a Kinetex C18 reversed-phase column exactly as described above. Duplicate samples were typically analyzed using the a methanol-based gradient system (Solvent A: 10% methanol, 0.25% acetic acid, pH 3.9; Solvent B: 90% methanol, 0.25% acetic acid, pH 3.9) with the elution protocol at 25°C and a flow rate of 1.2 mL/min as follows: 0-10 min, 0% B isocratic; 10-22 min, 0-24% B, linear gradient; 24-32 min, 24% B isocratic; 32-45 min, 24-45% B, linear gradient; 45-50 min, 45-82% B, linear gradient; 50-52 min, 82-100% B, linear gradient, followed by re-equilibration to 0% B. Quantitation of TS was enabled by running a series of authentic TS standards in separate chromatographic runs with the identical protocol.
RESULTS
Purification and biochemical characterization of CstB, CstB mutants, CstBPDO-RHD and CstBRhod
Intact wild-type and CstB mutants (C201S and C408S) were purified to >95% purity and confirmed by ESI mass spectrometry with cysteine residues fully reduced (Table 1). Because crystal structures show that both hETHE1 and the S. aureus COL CstB2 bind Fe(II) in a semi-facial triad of protein-derived ligands (Fig. 2B), we predicted that the side chains of H56, H119 and D145 coordinate Fe(II) in CstB. Indeed, purified CstBs contain ~0.5 mol equivalent of iron per protomer as isolated and each is capable of binding iron up to a ratio of 1:1 upon anaerobic addition of excess exogenous Fe(II), followed by separation of bound from free Fe(II) (Table 1). EPR studies of purified CstB show that the oxidation state is ≥90% Fe(II) following anaerobic reduction with dithionite.
Table 1.
Molecular weights and iron binding stoichiometries of wild-type and mutant CstBs.
| Protein | Molecular mass (Da)a |
Molecular mass (Da)b (calculated) |
Molecular mass (Da) gel filtrationc |
Fe content as purified (mol Fe/mol protomer) |
Fe content post loading (mol Fe/mol protomer) |
|---|---|---|---|---|---|
| CstB | 49,275.8 | 49,277.4 | 220,000 | 0.5 ± 0.1 | 1.0 ± 0.1 |
| C201S CstB | 49,262.5 | 49,261.3 | 221,000 | 0.5 ± 0.1 | 1.0 ± 0.1 |
| C408S CstB | 49,262.3 | 49,261.3 | 298,000 | 0.5 ± 0.1 | 1.0 ± 0.1 |
| CstB2CS | 49,243.9 | 49,245.2 | 293,000 | 0.5 ± 0.1 | 1.0 ± 0.1 |
| CstBPDO-RHD | 38,333.2 | 38,334.3 | 147,000 | 0.3 ± 0.2 | 1.0 ± 0.1 |
| CstBRhod | 11,132.9 | 11,133.3 | 34,000 | N/A | N/A |
Protomer molecular mass measured by ESI-MS.
Calculated from the amino acid sequence.
Native chromatography on Sepharose G200 GL, 10-20 μM protomer, pH 8.0.
Analytical gel filtration experiments show that Fe(II)-loaded wild-type CstB is tetrameric (226 kDa), as is C201S CstB and the truncated CstBPDO-RHD (Table 1; Fig. S4A-B). These findings are in contrast to hETHE1, which exists in a monomer-dimer equilibrium in solution,21 but are consistent with the D2-symmetric assembly state observed in the crystal structure of CstB2 (Fig. S3). This structure shows the middle RHD domain mediates most of the intersubunit contacts in the solid state; CstB from S. aureus strain Newman likely adopts a similar architecture in solution. In contrast, chromatography of C408S CstB results in a smaller retention time and larger apparent molecular weight, ≈298 kDa (Table 1; Fig. S4C). The same apparent molecular weight is obtained for the double C201S/C408S CstB, designated CstB2CS (Table 1; Fig. S4D). This suggests that C408S CstB and CstB2CS each exist as hexamers or alternatively, more poorly packed and hydrodynamically larger tetramers, perhaps harboring a destabilized C-terminal Rhod domain. A homonuclear 1H-1H NOESY spectrum of the isolated CstBRhod (residues 347-444) shows mostly broad lines and a few very sharp resonances found in the “random-coil” region of the amide spectrum (Fig. S5); this isolated domain also results in an oligomeric, mostly trimeric assembly state (Table 1; Fig. S4F). These data suggest that C-terminal Rhod domain may be poorly folded in the absence of the rest of CstB, consistent with the considerable interdomain packing shown in the structure of the A. acidocaldarius CstB-like protein (Fig. 2B, right panel). Likewise, substitution of the presumed Rhod active-site C408 also appears to destabilize the native fold. Efforts to purify a core ETHE1-like CstBPDO domain (residues 1-256) were also unsuccessful due to insolubility in bacterial lysates, rationalized by the appreciable packing of the N-terminal PDO and middle RHD domains (Fig. 2B, middle panel). This is validated by the analytical gel filtration result on CstBPDO-RHD, indicating the tetrameric status of purified CstBPDO-RHD (Table 1; Fig. S4E). These findings collectively highlight the substantial differences between the vertebrate core PDOs and bacterial PDO-RHD-Rhod fusion proteins in overall structural organization.
Coupled persulfide dioxygenase-persulfide transferase (cPDO-PT) activity of CstB
We employed a fluorescence-based assay to quantify reactant-product profiles obtained with a variety of LMW persulfides as substrates using Fe(II)-loaded wild-type and mutant CstBs to measure the cPDO-PT activity (see Fig. 2C, reaction 1). Briefly, reactions were carried out at pH 6.0 for some time t, with the products derivatized with monobromobimane (mBBr), quenched with excess thiol blocker, and acidified, filtered and subjected to C18 reversed phase HPLC using a methanol-based gradient system at pH 3.9 (Fig. 3A). The results of a representative assay carried out at high wild-type CstB concentration (400 nM protomer) using bacillithiol persulfide (BSSH) and molecular O2 to quantify the consumption of substrate and the yield of products are shown (Fig. 3A). Two points are apparent from these experiments. First, approximately two mols of BSSH are consumed to yield a single mol equivalent of TS, similar to the finding on CoASSH (Fig. 3B). No free sulfite can be detected as a product in these experiments under these conditions (Fig. 3A), consistent with tight coupling of the PDO activity and presumed persulfide transferase activity (see Fig. 2C, reaction 2; vide infra). The latter activity likely employs PDO-catalyzed formation of sulfite or a LMW S-sulfonate to generate TS and a second mol equiv of RSH, since in stand-alone persulfide dioxygenases, sulfite is the major product.17
Figure 3.

Coupled persulfide dioxygenase-persulfide transferase (cPDO-PT) activity of CstB with various persulfide substrates. (A) Representative LC-based assays used to measure reactant-product profiles of cPDO-PT activity with BSSH at t=0 min (top trace) and t=2 min (bottom trace) as substrates at 400 nM CstB. The chromatographic position of sulfite is indicated by the arrow.15 Note that B1 and B2 peaks are from labeling buffer; labeled BSH displays as two major peaks (BSH P1&P2) and one minor peak; BSSH is highly unstable in aerobic buffer used here to lyse the cells and thus is underestimated as two small peaks (BSSH P1 & P2). The concentration was alternatively measured using a cold cyanolysis assay.17 (B) Stoichiometry of persulfide substrates consumed (CoASSH and BSSH) versus TS generated for cPDO-PT activity of CstB. The molar ratio of CoASSH consumed to TS generated is ≈1.9 and BSSH consumed to TS generated is ≈2.1 in in multiple experiments carried out at 400 nM CstB. (C) Initial TS generation rates as a function of concentrations of CoASSH (open circles) vs. CoA (open squares), CoAox (open diamonds) and Na2S (crosses). The dashed, continuous line through the open circles is a fit to the Michaelis-Menten equation, with parameters summarized in Table 2. (D) Initial TS generation rates as a function of the concentrations of BSSH (open circles) vs. BSH (open squares), BSSB (open diamonds) and Na2S (crosses). The dashed, continuous line through the open circles is a fit to the Michaelis-Menten equation, with parameters summarized in Table 2.
Utilizing this assay, we next measured the initial rate of TS production as a function of LMW persulfide concentration and saturating O2 at 40 nM enzyme, and obtained Michaelis-Menten kinetic constants (Km, Vmax) for wild-type and mutant CstBs. We find that wild-type CstB preferentially catalyzes the oxidation of CoASSH with Km=1.1 ± 0.3 mM, Vmax=23.3 ± 2.5 μmol·min−1·mg−1 (Table 2; Fig. 3C) and BSSH, the latter with a Km=1.8 ± 0.4 mM, Vmax= 24.1 ± 3.0 μmol·min−1·mg−1 (Table 2; Fig. 3D). In parallel control experiments, CstB does not catalyze the formation of TS from reduced LMW thiol, oxidized LMW disulfide or Na2S, all used to generate LMW persulfide substrates in situ which were used without purification 17 (Figs. 3C-D). Cysteine persulfide (CSSH) and glutathione persulfide (GSSH) also serve as persulfide substrates for wild-type CstB but are poorer substrates, with Km ≥20 mM for CSSH and Km=7.8 ± 5.0 mM for GSSH (Table 2; Fig. S6A-B), the latter of which was reported as major persulfide substrate for human ETHE1. kcat/Km for these two substrates are only 10-15% that of preferred substrates CoASSH and BSSH (Table 2).
Table 2.
Kinetic characterization of the coupled persulfide dioxygenase-persulfide transferase (cPDO-PT) activity of wild-type CstB with various substrates.a
| Substrate |
Km
(x10−3 M) |
Vmax
(μmol TS · min− 1 · mg−1) |
kcat
(s−1) |
kcat/Km
(x10−3 M−1 · s−1) |
Specific activity ([substrate]=1 mM) (μmol TS · min−1 · mg− 1) |
|---|---|---|---|---|---|
| CoASSH | 1.1 ± 0.3 | 23.3 ± 2.5 | 19.3 ± 2.2 | 17.5 ± 5.1 | 11.2 ± 1.2 |
| BSSH | 1.8 ± 0.4 | 24.1 ± 3.0 | 19.7 ± 2.5 | 10.9 ± 2.8 | 8.6 ± 1.1 |
| CSSHb | 20 ± 21 | 34 ± 32 | 28 ± 27 | 1.4 ± 2.0 | 1.5 ± 0.1 |
| GSSHb | 7.8 ± 5.0 | 7.9 ± 4.0 | 6.5 ± 3.3 | 0.8 ± 0.7 | 0.9 ± 0.1 |
Conditions: 0.04 μM CstB (protomer), 25 mM MES, 100 mM NaBr, pH 6.0, 25 °C.
Very low activity (see Fig. S6) making it difficult to measure Km and Vmax accurately.
Further investigation of the requirements for cPDO-PT activity reveals that neither isolated CstBPDO-RHD or CstBRhod domain is capable of generating TS using CoASSH and BSSH as substrates since no detectable product is observed (Fig. 4A-B). Nor is TS observed when the two domains are mixed together and tested in trans, a result perhaps attributed to the poor folding of the isolated Rhod domain (vide supra). This finding was validated in time course experiments with CSSH and GSSH serving as persulfide substrates using higher enzyme concentrations due to the low specific activities with CSSH and GSSH (Fig. S7A-B). We also tested the activity of C201S and C408S mutant CstBs. C201S displays a specific activity at 1 mM persulfide substrate (≈Km) that is ≈25% that of wild-type CstB (Fig. 4A-B), with the same Km of 1.1 ± 0.1 mM, but reduced Vmax of 6.0 ± 0.3 μmol·min−1·mg−1 for CoASSH. The C408S enzyme is even less active, with a Km again comparable to that of wild-type and C201S CstBs, 1.2 ± 0.9 mM, but a substantially reduced Vmax of 1.0 ± 0.3 μmol·min−1·mg−1 using CoASSH as substrate (Fig. 4A). Similar findings characterize the reaction for C201S and C408S mutant CstBs with BSSH as substrate (Fig. 4B). Further investigation also validates in the time course experiments for C201S and C408S mutant CstBs with CSSH and GSSH serving as persulfide substrates using higher enzyme concentrations (Fig. S7C-D). There is no detectable sulfite generated in these assays with mutant CstBs, again consistent with a strong coupling of persulfide dioxygenase and persulfide transferase activities. These data taken collectively reveal that both conserved Cys play important roles in cPDO-PT catalytic efficiency with C408 clearly more important for formation of TS product (see Discussion).
Figure 4.

Specific coupled persulfide dioxygenase-persulfide transferase (cPDO-PT) activities of various CstBs determined at 1 mM LMW persulfide substrate. (A) CoASSH; (B) BSSH.
Growth phenotypes of ΔcstB strains under hydrogen sulfide stress
We showed in previous work that CstB is required for Staphylococcus aureus to recover from hydrogen sulfide stress and that the growth defect of the ΔcstB strain can be complemented by ectopic expression of the wild-type allele from an extrachromosomal plasmid (Fig. 5A).15 Since both C201 and C408 play catalytically important roles in the PDO activity of CstB (see Fig. 4), we tested the effect of complementation with mutant allelic cstBs under NaHS stress (0.2 mM). We find that the growth defect of ΔcstB strain under NaHS stress cannot be efficiently complemented by ectopic expression of either the C201S allele or C408S allele (Fig. 5B). The same is true for a strain expressing a C20S CstB allele (Fig. 5B). Since we were unable to express recombinant C20S CstB in E. coli, C20 may play a structural role in the assembly of active CstB; note that this residue is 100% buried in the structure of CstBPDO-RHD (Fig. 2A). Thus, all three Cys substitution mutants fail to efficiently rescue a ΔcstB strain stressed with exogenous hydrogen sulfide.
Figure 5.

Representative growth curves for wild-type (WT), ΔcstB, and plasmid-complemented ΔcstB allelic S. aureus Newman strains on HHWm media in the presence of 0 mM and 0.2 mM NaHS added at t=0 h. (A) WT (black square), ΔcstB (blue circle) and ΔcstB:pCstB (green diamond) strains in the presence of 0 mM NaHS; WT (red square), ΔcstB (magenta circle) and ΔcstB:pCstB (wine diamond) strains in the presence of 0.2 mM NaHS. Note that both the WT and ΔcstB:pCstB strains exhibit a growth lag (≈7 h) in the presence of 0.2 mM NaHS; ΔcstB exhibits a more significant growth lag (≈9 h) in the presence of 0.2 mM NaHS. Also note that all the strains grow normally in the presence of 0 mM NaHS. (B) ΔcstB:pCstBC20S (black square), ΔcstB:pCstBC201S (blue circle) and ΔcstB: pCstBC408S (green diamond) strains in the presence of 0 mM NaHS; ΔcstB:pCstBC20S (red square), ΔcstB:pCstBC201S (magenta circle) and ΔcstB: pCstBC408S (wine diamond) strains in the presence of 0.2 mM NaHS. Note that all the three strains exhibit a more significant growth lag (≈9 h) in the presence of 0.2 mM NaHS. Also note that all the strains grow normally in the presence of 0 mM NaHS.
Persulfide transferase (PT) activity of CstB
A key aspect of the reactant-product profile obtained as a result of PDO turnover is the consumption of two mol equivalents of LMW persulfide and lack of sulfite as an intermediate product (Fig. 3). This leads to the prediction that the C-terminal Rhod domain possesses intrinsic PT activity when provided with sulfite exogenously, independent of the Fe(II)-dependent PDO activity (Fig. 2C, reactions 1 and 2). It is also known that LMW thiols react noncatalytically to form TS (Fig. S8A-B). In order to test this, we measured PT activity in an anaerobic chamber with degassed buffers, using C201S CstB which has reduced O2-dependent PDO activity (Fig. 4), and relatively low concentrations of each of four LMW persulfide substrates (Fig. 6A-B; Fig. 9A-B). With correction of the background, non-enzyme-catalyzed rate of TS formation, we find that all three major LMW thiol persulfides from S. aureus, including CoASSH, BSSH and CSSH, can function as the persulfide substrates in this assay with similar catalytic efficiencies, and Km values are ≈20-fold lower than found for the preferred substrates for the cPDO-PT activity, CoASSH and BSSH (Table 3). GSSH is also a persulfide substrate for this reaction but is characterized by relatively lower substrate affinity and catalytic efficiency (Table 3). As verified above for the cPDO-PT activity, control experiments reveal that C201S CstB does not catalyze any reaction of sulfite with a reduced LMW thiol, oxidized LMW disulfide or Na2S to generate TS (Fig. 6A-B; Fig. S9A-B). In a second control experiment, we show that the double Cys substitution mutant CstB2CS has no PT activity above the background, non-catalyzed rates of TS formation (Fig. S8A-B) with any persulfide substrate (Fig. S9C-F). These findings collectively suggest that the C-terminal Rhod domain is capable of catalyzing PT activity, and this activity may function in concert with the initial Fe-oxidation step in the PDO activity to generate TS.
Figure 6.

C201S CstB displays persulfide transferase (PT) activity with LMW persulfide substrates. (A) Initial TS generation rates as a function of concentrations of CoASSH (open circles), CoA (open squares), CoAox (open diamonds) and Na2S (crosses). The dashed, continuous line through the open circles is a fit to the Michaelis-Menten equation, with parameters summarized in Table 3. (B) Initial TS generation rates as a function of concentrations of BSSH (open circles), BSH (open squares), BSSB (open diamonds) and Na2S (crosses). The dashed, continuous line through the open circles is a fit to the Michaelis-Menten equation, with parameters summarized in Table 3.
Figure 9.

Current model for cellular H2S detoxification in Staphylococcus aureus highlighting the known or proposed enzymatic activities encoded by the cst operon. (this work). Extracellular H2S freely penetrates the cell membrane and exists as HS− once in the cytoplasm. SQR then carries out the initial two-electron oxidation of HS− to form SQR-bound persulfide and the SQR-bound persulfide is transferred to reduced cellular LMW thiols (LMW-SH) to form LMW persulfides (LMW-SSH), which can also be generated from HS− directly via reaction with oxidized LMW thiols (LMW-S-S-LMW). In the presence of molecular oxygen (O2), CstBPDO further oxidizes LMW persulfides to possibly form a LMW thiol-S-sulfonate or CstB-S-sulfonate (denoted R-S-SO3−) intermediate, which requires further investigation. Free thiosulfate (TS) is then immediately generated either through the persulfide transferase activity of CstBRhod or other cellular rhodanese proteins (other Rhod), or directly generated via reaction of a LMW persulfide with LMW thiol-S-sulfonate or CstB-S-sulfonate. Finally, accumulating thiosulfate is processed by the thiosulfate sulfurtransferase activities of CstBRhod, CstARhod or other cellular rhodanese proteins. Sulfite (SO32−) is effluxed from the cell (perhaps by TauE) and sulfane sulfur (S0) proposed to be transferred to as yet unknown downstream cellular sulfur acceptor(s) for assimilation.
Table 3.
Comparison of the kinetic properties of the enzyme-catalyzed persulfide transferase (PT) activity of C201S CstB with various substrates.a
| Substrate | Km (x10−6 M) | Vmax (μmol TS · min−1 · mg−1) | kcat (s−1) | kcat/Km (x10−3 M−1·s−1) |
|---|---|---|---|---|
| CoASSH | 49 ± 13 | 3.3 ± 0.3 | 2.7 ± 0.2 | 55.1 ± 15.2 |
| BSSH | 40.1 ± 5.2 | 3.1 ± 0.1 | 2.5 ± 0.1 | 62.3 ± 8.4 |
| CSSH | 37.0 ± 7.5 | 2.9 ± 0.2 | 2.4 ± 0.1 | 64.8 ± 13.4 |
| GSSH | 110 ± 27 | 4.0 ± 0.4 | 3.3 ± 0.3 | 30.0 ± 7.9 |
| KSCN | NAb | NA | NA | NA |
Condition: 0.2 μM C201S CstB (protomer), 25 mM MES, 100 mM NaBr, pH 6.0, 25 °C, anaerobic condition.
NA, no observable activity.
Thiosulfate transferase (TST) activity of CstB
Free-standing rhodaneses as well as the N-terminal Rhod domain of CstA encoded by the cst operon catalyze TS breakdown to generate a protein persulfide and sulfite.16,19 When provided with an exogenous nucleophile, e.g., CN−, the Rhod Cys-derived persulfide turns over to generate thiocyanate anion (SCN−) and reduced active-site sulfhydryl (Fig. 2C, reaction 3). We therefore tested if the wild-type and various mutant forms of CstB possess TST activity and if so, obtained kinetic parameters under standard assay conditions. Wild-type CstB is characterized by Km=1.7 ± 0.2 mM and Vmax=10.4 ± 0.6 μmol·min−1·mg−1 towards TS in the typical TST assay (Table 4; Fig. 7). Consistent with the prediction of the functional roles of individual domains in CstB, both CstBPDO-RHD domain and C408S CstB are completely inactive in this assay. In contrast, even though C201S CstB is deficient in cPDO-PT activity, it displays nearly identical kinetic constants as wild-type CstB in this assay, with Km=2.2 ± 0.4 mM and Vmax=11.2 ± 0.9 μmol·min−1·mg−1 (Table 4; Fig. 7). As expected, this reveals that the TST activity is contained entirely in the C-terminal Rhod domain of CstB. Interestingly, the isolated CstBRhod domain is inactive in this TST activity, and mixing stoichiometric CstBRhod with CstBPDO-RHD also does not recover activity (Table 4; Fig. 7). One explanation for these findings is that the structure of the isolated CstBRhod is non-native (Fig. S5) and, as a result, the independently purified domains fail to interact productively (Table 4; Fig. 7).
Table 4.
Comparison of the thiosulfate transferase (TST) activities of various CstBs.a
| Enzyme |
Km
(x10−3 M) |
Vmax
(μmol SCN min−1 mg−1) |
kcat (s−1) |
kcat/Km
(x10−3 M− ·s−1) |
|---|---|---|---|---|
| CstB | 1.7 ± 0.2 | 10.4 ± 0.6 | 8.5 ± 0.5 | 5.0 ± 0.7 |
| C201S CstB | 2.2 ± 0.4 | 11.2 ± 0.9 | 9.2 ± 0.7 | 4.2 ± 0.8 |
| C408S CstB | NAb | NA | NA | NA |
| CstBPDO-RHD | NA | NA | NA | NA |
| CstBRhod | NA | NA | NA | NA |
| CstBPDO-RHD + CstBRhod | NA | NA | NA | NA |
Condition: 0.4 μM CstB (protomer), 50 mM KCN, 100 mM MES, pH 6.0, 25 °C.
NA, no activity observed.
Figure 7.
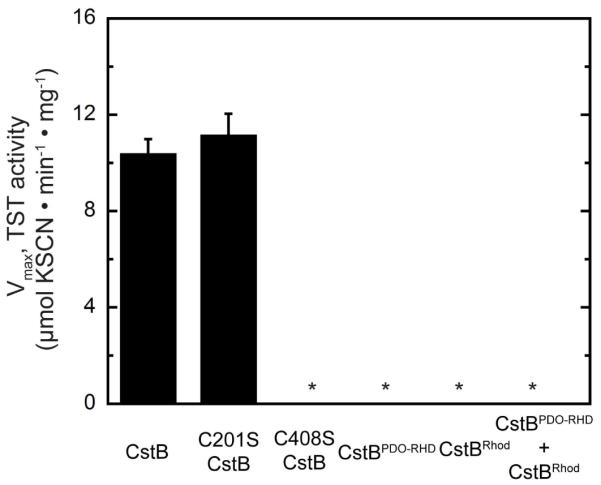
Maximal velocities (Vmax) for thiosulfate transferase (TST) activities of various CstBs using cyanide anion (CN−) as sulfane sulfur acceptor. *, none detected, ≤0.1 μmol min−1 mg−1 protein. Kinetic constants are summarized in Table 4.
Cellular thiosulfate concentrations in ΔcstB and complemented strains
The experiments described above reveal that CstB catalyzes three distinct reactions in vitro, two of which synthesize TS via reduction of LMW persulfides (Fig. 2C, reactions 1 and 2) and one of which consumes TS (Figure 2C, reaction 3). We therefore determined the impact of deletion of cstB and complementation with wild-type and mutant cstB genes on cellular TS concentrations in aerobic mid-log S. aureus cells, as a function of time following induction of the cst operon by 0.2 mM NaHS using TS as the sole sulfur source. Previous qRT-PCR experiments reveal that the cst operon is maximally induced 10 min following addition of NaHS under these conditions, with mRNA levels returning to near baseline at 30 min.15
As found previously, TS is below the limit of detection in cells prior to NaHS addition revealing that TS is efficiently utilized as a sole sulfur source once imported into the cell and does not efficiently accumulate.15 In the wild-type strain, the cellular TS level reaches 15 nmol per mg protein 10 min following NaHS addition and decreases ≈2-fold over the course of 2 h or approximately two culture doublings (Fig. 8). In the ΔcstB strain, the cellular TS level is significantly lower at t=10 min, ≈ 9 nmol per mg protein, decreasing to ≈6 nmol per mg protein by 2 h. This overall decrease in the cellular TS level in the ΔcstB strain can be complemented by extrachromosomal expression of the wild-type cstB allele, but not by the C20S or C201S cstB alleles (Fig. 8). This suggests that the PDO activity of CstB makes a significant contribution to the cellular TS level under NaHS stress immediately following induction.
Figure 8.

Cellular thiosulfate concentrations in mid-log (OD600 ≈0.2) wild-type (WT), ΔcstB, and plasmid-complemented ΔcstB allelic S. aureus Newman strains as a function of time following induction of the cst operon at t=0 by 0.2 mM NaHS.15 *, no TS detected (≤0.2 nmol/mg protein). The color of the columns is assigned as follows: cyan for WT strain, blue for ΔcstB strain, red for ΔcstB:pCstB strain, purple for ΔcstB:pCstBC20S strain, orange for ΔcstB:pCstBC201S strain and grey for ΔcstB:pCstBC408S strain.
In contrast, complementation with the C408S cstB allele “over-complements” the cstB deletion, with a wild-type-like cellular TS level 10 min post induction, at ≈15 nmol per mg protein, remaining elevated throughout the 2 h time course (Fig. 8). Since C408 plays roles in all three CstB activities, but gives rise to a TS level accumulation phenotype opposite to that observed with the C201S mutant, this suggests that the TST activity of the CstBRhod domain plays a more significant role in governing the cellular TS level following NaHS induction, particularly at longer time points post-induction.
DISCUSSION
In this work, we show that S. aureus CstB as a non-heme Fe(II)-containing multidomain persulfide dioxygenase-sulfurtransferase. We describe three separable enzymatic activities of CstB, including an Fe(II)-dependent cPDO-PT activity, the first half of which was previously described for human mitochondrial ETHE1, a PT activity and a TST activity, the latter two activities catalyzed by the C-terminal Rhod domain (see Fig. 2C). Thus, CstB possesses two activities that we hypotheisize to function collaboratively to clear LMW organic persulfides that accumulate in cells when exposed to H2S stress or as a result of misregulation of H2S homeostasis. The major observable products of the cPDO-PT reaction are TS and reduced LMW thiols, with no detectable sulfite formed as an intermediate (Fig. 3A). This finding is in contrast to previous studies of hETHE1, where sulfite is found as the major product, and addition of a classical Rhod domain in trans would be expected to lead to the formation of TS.17 We show here that the CstBPDO-RHD fragment when mixed with the CstBRhod does not generate sulfite as product, as could be expected if the two enzyme activities were uncoupled as a result of reconstitution in trans. Clearly, C201S CstB has significant PT activity using sulfite as an exogenous nucleophile, thus suggesting that it may not be likely that sulfite is channeled in some way to the C408 active site directly from the Fe active site. How the Fe and C408 active sites communicate with one another is therefore unknown. We do show that the cPDO-PT activity is highest with persulfide substrates derived from major cellular thiols in S. aureus including bacillithiol and coenzyme A, and is less efficient when glutathione and cysteine persulfides are used as substrates (Table 2).
CstB is a homotetramer in solution, and is characterized by a core PDO domain that is structurally superimposable on Arabidopsis ETHE1-like protein and hETHE1 fused to a middle RHD domain that is packed against the core PDO domain but lacks an active site cysteine (Fig. 2B). This RHD may well provide the majority of the interprotomer contacts that stabilize the tetramer in solution as well (Fig. S3). In CstB, this PDO-RHD is connected to a C-terminal classical Rhod domain that harbors an active site C408. This latter domain was not resolved in the crystal structure of CstB, but is found in a CstB-like protein from A. acidocaldarius (Fig. 2B). The biochemical activity of this A. acidocaldarius CstB-like protein to our knowledge is not yet known, so these structural comparisons must be taken with a degree of caution. Nonetheless, it is striking that all three Cys in S. aureus CstB are conserved in this CstB-like protein, including the equivalent of C201 which makes close approach to an active site metal ion, which itself is coordinated in exactly the same way as in CstB, hETHE1 and the Arabidopsis ETHE1-like protein. If this A. acidocaldarius CstB-like protein is a good structural model of CstB, then this reveals that C408 and the open coordination site of the Fe atom are indeed oriented toward one another within the same subunit, but they may be too far apart in this conformation to drive a concerted oxidation of the terminal persulfide sulfur atom at the Fe active site to TS without release of sulfite. Other plausible scenarios include a Rhod domain of one subunit making closer approach to the PDO domain Fe(II) atom of an adjacent subunit, or that the flexible loop containing C201 facilitates intramolecular transfer from the PDO active site to C408.
Interestingly, wild-type and C201S CstBs have identical TST activities and in fact exhibit kinetic parameters that are very similar to those measured for the N-terminal Rhod domain of S. aureus CstA (Table 4).16 This suggests that the CstBRhod domain has unfettered access to TS substrate even in the context of intact CstB, thus providing another argument against a close structural approach of the Fe(II) and C408 active sites. In fact, it is this TST activity that may dominate cellular TS metabolism following acute-phase NaHS stress (Fig. 8). CstA is also under the transcriptional control of the persulfide sensor CstR,15 and genes encoding CstA and CstB may comprise a minimal LMW persulfide clearance system that is duplicated in many methicillin-resistant strains (Fig. S1), either via reduction of RSSH to reduced RSH concomitant with the generation of TS via CstB, or as in the case of CstA, by reacting directly with LMW persulfides and inorganic polysulfides, to generate CstA-bound persulfides, as a means to assimilate sulfide-derived sulfur.16 In preliminary experiments, we have tested if a CstA-bound persulfide can function as a substrate (at 200 μM) for the cPDO-PT activity of CstB, and could measure no activity (J. Shen and H. Peng, unpublished results). Since the multidomain sulfurtransferase CstA is capable of shuttling persulfides between the N-terminal Rhod and middle TusA domain,16 the lifetime of a persulfide on the Rhod domain may be short; furthermore, we have not yet tested if the C-terminal TusD domain is part of this CstA persulfide shuttle, or is capable of functioning as a direct persulfide donor to CstB. Alternatively, the TS generated by the cPDO-PT activity of CstB is shuttled directly to CstA to generate mobilizable sulfur in the form of protein-bound persulfides (Fig. 9). Finally, it also seems possible by analogy with the mitochondrial sulfide oxidation system that an SQR-bound persulfide might function as a persulfide donor for CstB.24 Preliminary experiments to distinguish among these possibilities are in progress.
The chemical mechanism of the PDO activity is unknown, as is the functional role of the conserved C201. Reduced Fe(II) is required for activity, and by analogy to cysteine dioxygenases (CDO), may involve direct coordination of the persulfide substrate via one or both sulfur atoms, to the side of the Fe(II) that is free of protein-derived ligands (Fig. 2B)25 By further analogy to CDO, a high-valent Fe-oxo species is the likely oxidant, which leads to the formation of a Fe-bound organic LMW-S-sulfonate, RSSO2, followed by addition of H2O to generate RSSO3. Then, in the second phase of the reaction, the S-S bond is cleaved by an attacking LMW persulfide or a Rhod-bound persulfide formed as a result of the PT activity of CstB to generate TS and a second equivalent of LMW thiol. C201 could function in this second step by forming an enzyme-bound S-sulfocysteine intermediate which is then attacked by the C408 persulfide. The low, but detectable level of cPDO-PT activity of C408S CstB (Fig. 4) may derive from LMW persulfides reacting with LMW thiol-S-sulfonate or CstB-S-sulfonate, potentially generated by the PDO activity of C408S CstB. Additional experiments are required to substantiate key aspects of this mechanistic model, including efforts to trap an enzyme-bound covalent intermediate in this process. It is unknown, for example, if there are two physically distinct RSSH binding sites, one for the initial oxidation and another for the persulfide transfer step.
We note that candidate CstR-regulated genes near a clearly identifiable, consensus cst operator sequence14 are also found in two other Gram-positive human pathogens, Enterococcus facaelis (E. facaelis) and Bacillus anthracis (B. anthracis). In both instances, downstream genes include two stand-alone candidate Rhod homology domains that flank a gene that is annotated as a CoA disulfide reductase-rhodanese homology domain protein (CoADR-RHD). In both organisms, an authentic CoADR is encoded elsewhere in the genome,26 suggesting an alternate function for the CoADR-RHD. Interestingly, the crystal structure of B. anthracis CoADR-RHD has been solved, and biochemical experiments carried out prior to the knowledge of CstR-regulated cellular persulfide chemistry15 reveal that the substrate binding channel for CoA is unlikely to accommodate the CoA disulfide. Further, the two cysteines, one each in the core CoADR and rhodanese domains on a complementary subunit, although separated by ≈25 Å, are proposed to communicate with one another via the long swinging pantethiene arm.27 We speculate that both B. anthracis and E. facaelis CoADR-RHDs have evolved to catalyze the reduction of CoASSH formed under conditions of sulfide of RSS stress to the free thiol concomitant with the generation of a RHD enzyme-bound persulfide, which may then be assimilated by cellular components in some way via a subsequent persulfide transfer. Thus, CstB and CoADR-RHD may provide distinct solutions to a common problem, i.e., to clear CoASSH (CoA is a major cellular thiol in both S. aureus 26 and B. anthracis) that may accumulate under conditions of H2S toxicity. It will be of interest, for example, to determine if CoADR-RHD can functionally complement a ΔcstB S. aureus Newman strain. Finally, the function of the cst operon in general microbial physiology is not yet firmly established, but at least one transcriptomic experiment in S. aureus strain N315 ties strong induction of the cst operon-encoded genes, including cstB, to nitric oxide-mediated dispersal of staphylococcal biofilms.28 Efforts are underway to link this process with the need to regulate endogenous hydrogen sulfide levels in this human pathogen.
Supplementary Material
Acknowledgments
Funding
The authors gratefully acknowledge support by the NIH (R01 GM097225 to D. P. G., R01 GM030910 to R. N. A., and T32 ES007028 to M. E. K.). The NMR instrumentation at Indiana University was generously supported by the Indiana METACyt Initiative, funded in part through a major grant from the Lilly Endowment, Inc.
ABBREVIATIONS
- Cst
CsoR-like sulfurtransferase
- Rhod
rhodanese
- TS
thiosulfate
- TST
thiosulfate sulfurtransferase
Footnotes
SUPPORTING INFORMATION
This manuscript contains Supporting Information including Supplementary Methods and Supplementary Figures S1-S9 which can accessed free of charge via the Internet at http://pubs.acs.org.
Author Contributions
J. S. and D. P. G. designed the research; J. S. performed the research; M. E. K. and R. N. A. provided reduced and oxidized bacillithiol; K. A. H. helped J. S. perform EPR experiments with the data analyzed by K. A. H.; K. A. E. performed the bioinformatics and structural analysis of CstB and cst operon synteny; and J. S. and D. P. G. wrote the manuscript. This work will be submitted by J. S. to the Graduate School of Indiana University in partial fulfillment of the requirements for the Ph.D. in Biochemistry. The authors wish to thank Mr. Jay Levy for help in setting up the HPLC used for sulfur metabolite analyses. We also thank Drs. Hongwei Wu, Joey Braymer and Justin Luebke for many helpful discussions on this project.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Mathai JC, Missner A, Kugler P, Saparov SM, Zeidel ML, Lee JK, Pohl P. No facilitator required for membrane transport of hydrogen sulfide. Proc Natll Acad Sci U S A. 2009;106:16633–16638. doi: 10.1073/pnas.0902952106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Cooper CE, Brown GC. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr. 2008;40:533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- (3).Dorman DC, Moulin FJ, McManus BE, Mahle KC, James RA, Struve MF. Cytochrome oxidase inhibition induced by acute hydrogen sulfide inhalation: correlation with tissue sulfide concentrations in the rat brain, liver, lung, and nasal epithelium. Toxicol Sci. 2002;65:18–25. doi: 10.1093/toxsci/65.1.18. [DOI] [PubMed] [Google Scholar]
- (4).Truong DH, Eghbal MA, Hindmarsh W, Roth SH, O'Brien PJ. Molecular mechanisms of hydrogen sulfide toxicity. Drug Metabol Rev. 2006;38:733–744. doi: 10.1080/03602530600959607. [DOI] [PubMed] [Google Scholar]
- (5).Lin VS, Chen W, Xian M, Chang CJ. Chemical probes for molecular imaging and detection of hydrogen sulfide and reactive sulfur species in biological systems. Chem Soc Rev. 2014 doi: 10.1039/c4cs00298a. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, Suematsu M, Motohashi H, Fujii S, Matsunaga T, Yamamoto M, Ono K, Devarie-Baez NO, Xian M, Fukuto JM, Akaike T. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc Natl Acad Sci U S A. 2014;111:7606–7611. doi: 10.1073/pnas.1321232111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kabil O, Banerjee R. Redox biochemistry of hydrogen sulfide. J Biol Chem. 2010;285:21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kolluru GK, Shen X, Bir SC, Kevil CG. Hydrogen sulfide chemical biology: pathophysiological roles and detection. Nitric Oxide. 2013;35:5–20. doi: 10.1016/j.niox.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Paul BD, Snyder SH. H(2)S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol. 2012;13:499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- (10).Shatalin K, Shatalina E, Mironov A, Nudler E. H2S: a universal defense against antibiotics in bacteria. Science. 2011;334:986–990. doi: 10.1126/science.1209855. [DOI] [PubMed] [Google Scholar]
- (11).Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, Tiveron C, Levitt MD, Prelle A, Fagiolari G, Rimoldi M, Zeviani M. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med. 2009;15:200–205. doi: 10.1038/nm.1907. [DOI] [PubMed] [Google Scholar]
- (12).Czyzewski BK, Wang DN. Identification and characterization of a bacterial hydrosulphide ion channel. Nature. 2012;483:494–497. doi: 10.1038/nature10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kluytmans J, van Belkum A, Verbrugh H. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev. 1997;10:505–520. doi: 10.1128/cmr.10.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Grossoehme N, Kehl-Fie TE, Ma Z, Adams KW, Cowart DM, Scott RA, Skaar EP, Giedroc DP. Control of copper resistance and inorganic sulfur metabolism by paralogous regulators in Staphylococcus aureus. J Biol Chem. 2011;286:13522–13531. doi: 10.1074/jbc.M111.220012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Luebke JL, Shen J, Bruce KE, Kehl-Fie TE, Peng H, Skaar EP, Giedroc DP. The CsoR-like sulfurtransferase repressor (CstR) is a persulfide sensor in Staphylococcus aureus. Mol Microbiol. 2014;94:1343–1360. doi: 10.1111/mmi.12835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Higgins KA, Peng H, Luebke JL, Chang FM, Giedroc DP. Conformational analysis and chemical reactivity of the multidomain sulfurtransferase, Staphylococcus aureus CstA. Biochemistry. 2015;54:2385–2398. doi: 10.1021/acs.biochem.5b00056. [DOI] [PubMed] [Google Scholar]
- (17).Kabil O, Banerjee R. Characterization of patient mutations in human persulfide dioxygenase (ETHE1) involved in H2S catabolism. J Biol Chem. 2012;287:44561–44567. doi: 10.1074/jbc.M112.407411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gunnison AF. Sulphite toxicity: a critical review of in vitro and in vivo data. Food Cosmet Toxicol. 1981;19:667–682. doi: 10.1016/0015-6264(81)90519-8. [DOI] [PubMed] [Google Scholar]
- (19).Cipollone R, Ascenzi P, Visca P. Common themes and variations in the rhodanese superfamily. IUBMB Life. 2007;59:51–59. doi: 10.1080/15216540701206859. [DOI] [PubMed] [Google Scholar]
- (20).Weinitschke S, Denger K, Cook AM, Smits TH. The DUF81 protein TauE in Cupriavidus necator H16, a sulfite exporter in the metabolism of C2 sulfonates. Microbiology. 2007;153:3055–3060. doi: 10.1099/mic.0.2007/009845-0. [DOI] [PubMed] [Google Scholar]
- (21).Pettinati I, Brem J, McDonough MA, Schofield CJ. Crystal structure of human persulfide dioxygenase: structural basis of ethylmalonic encephalopathy. Hum Mol Gen. 2015;24:2458–2469. doi: 10.1093/hmg/ddv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).McCoy JG, Bingman CA, Bitto E, Holdorf MM, Makaroff CA, Phillips GN., Jr. Structure of an ETHE1-like protein from Arabidopsis thaliana. Acta Crystallogr D Biol Crystallogr. 2006;62:964–970. doi: 10.1107/S0907444906020592. [DOI] [PubMed] [Google Scholar]
- (23).Lamers AP, Keithly ME, Kim K, Cook PD, Stec DF, Hines KM, Sulikowski GA, Armstrong RN. Synthesis of bacillithiol and the catalytic selectivity of FosB-type fosfomycin resistance proteins. Org Lett. 2012;14:5207–5209. doi: 10.1021/ol302327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kabil O, Motl N, Banerjee R. HS and its role in redox signaling. Biochim Biophys Acta. 2014;1844:1355–1366. doi: 10.1016/j.bbapap.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Joseph CA, Maroney MJ. Cysteine dioxygenase: structure and mechanism. Chem Comm. 2007:3338–3349. doi: 10.1039/b702158e. [DOI] [PubMed] [Google Scholar]
- (26).delCardayre SB, Stock KP, Newton GL, Fahey RC, Davies JE. Coenzyme A disulfide reductase, the primary low molecular weight disulfide reductase from Staphylococcus aureus. Purification and characterization of the native enzyme. J Biol Chem. 1998;273:5744–5751. doi: 10.1074/jbc.273.10.5744. [DOI] [PubMed] [Google Scholar]
- (27).Wallen JR, Mallett TC, Boles W, Parsonage D, Furdui CM, Karplus PA, Claiborne A. Crystal structure and catalytic properties of Bacillus anthracis CoADR-RHD: implications for flavin-linked sulfur trafficking. Biochemistry. 2009;48:9650–9667. doi: 10.1021/bi900887k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Schlag S, Nerz C, Birkenstock TA, Altenberend F, Gotz F. Inhibition of staphylococcal biofilm formation by nitrite. J Bacteriol. 2007;189:7911–7919. doi: 10.1128/JB.00598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.