

Abstract
Alpha helices form a critical part of the binding interface for many protein-protein interactions, and chemically stabilized synthetic helical peptides can be effective inhibitors of such helix-mediated complexes. In particular, hydrocarbon stapling of peptides to generate constrained helices can improve binding affinity and other peptide properties, but determining the best stapled peptide variant often requires laborious trial and error. Here we describe the rapid discovery and optimization of a stapled-helix peptide that binds to Mcl-1, an anti-apoptotic protein that is overexpressed in many chemoresistant cancers. To accelerate discovery, we developed a peptide library synthesis and screening scheme capable of identifying subtle affinity differences among Mcl-1-binding stapled peptides. We used our method to sample combinations of non-natural amino-acid substitutions that we introduced into Mcl-1 inhibitors in the context of a fixed helix-stabilizing hydrocarbon staple that increased peptide helical content and reduced proteolysis. Peptides discovered in our screen contained surprising substitutions at sites that are conserved in natural binding partners. Library-identified peptide M3d is the most potent molecule yet tested for selectively triggering mitochondrial permeabilization in Mcl-1 dependent cell lines. Our library approach for optimizing helical peptide inhibitors can be readily applied to the study of other biomedically important targets.
Graphical Abstract
An important goal in modern chemical biology is to develop molecules for inhibiting, modifying or otherwise regulating protein-protein interactions (PPIs). PPIs control myriad biological phenomena in healthy and diseased cells, and differences in interactions in different cell states provide therapeutic opportunities. An ability to rapidly identify and optimize tight-binding and selective inhibitors of PPIs could change the landscape of pharmaceutical discovery and also benefit basic research. Antibodies and other large, engineered proteins can be used to block or alter PPIs outside the cell, and human or humanized monoclonal antibodies are powerful agents in the laboratory and the clinic.1 But developing smaller molecules is also important. Antibodies cannot currently be delivered into the cell cytoplasm or nucleus where many important therapeutic targets function. The large size of antibodies also limits their access to certain tissues and types of binding sites. From this perspective, small molecule or peptide inhibitors of PPIs are attractive, and peptides are good candidates for development because they can readily mimic natural interaction motifs and achieve a high degree of binding affinity and specificity even at interaction sites that lack deep binding pockets.2,3
Peptides composed of natural amino acids can be engineered using library screening coupled with DNA sequencing for hit decoding. For example, phage display and cell-surface display readily provide access to diverse libraries with 107–1014 members that can be screened for target binding.4–8 However, the natural L-amino acids that can be easily encoded using genetics sample only a small part of the potentially interesting peptide chemical space. Furthermore, many studies have demonstrated that chemical modifications such as cyclization and helix crosslinking can dramatically improve peptide binding.9 Although it is possible to chemically modify peptides on phage,10,11 synthesis provides the most versatile method of generating chemically diverse modified peptides for optimization. A few recent studies have demonstrated the chemical synthesis of libraries of cyclic peptides in a one-bead-one-compound format.12–16 Alpha helical peptides that include i, i+4 or i, i+7 crosslinks that stabilize helical conformations are another exciting class of peptide-based inhibitors that have shown activity against a range of targets.9,17–19 These peptides also have the potential to be optimized using combinatorial methods.
Anti-apoptotic Bcl-2 family proteins are an exciting class of targets for which inhibiting PPIs is a promising strategy for developing new cancer therapies.20–22 Human anti-apoptotic Bcl-2 family proteins, which include Bcl-2, Bcl-xL, Bcl-w, Mcl-1 and Bfl-1, counteract cell death signaling using a mechanism that involves binding to and inhibiting pro-apoptotic proteins. The anti-apoptotic proteins bind to a helical Bcl-2 homology region (BH3) in the pro-apoptotic partners.23 Thus, using BH3 mimetics to neutralize the function of Bcl-2 family proteins is an attractive strategy to restore apoptotic signaling in cancer cells and enhance the response to currently approved chemotherapeutics. Consistent with this, small molecule PPI inhibitors of Bcl-2 and Bcl-xL have shown dramatic effects shrinking tumors in mice and are now being tested in several clinical trials with exciting results.24–27 Amplification of Bcl-2 family protein Myeloid cell leukemia 1 (Mcl-1) is one of the most common genetic aberrations that drives the development and maintenance of a variety of human cancers, including adult acute myelogenous leukemias, acute lymphoblastic leukemias and triple negative breast cancers.28–30 Moreover, Mcl-1 contributes to the resistance of cancer cells to chemotherapies. Unfortunately, cancer cells that rely on anti-apoptotic protein Mcl-1 for survival are not susceptible to inhibition by compounds hat target Bcl-2 or Bcl-xL, and despite significant research efforts, small molecule Mcl-1 inhibitors have not advanced to the clinic.31
Several studies have led to synthetic BH3 motif peptides that bind selectively to Mcl-1 and show activity in assays using permeabilized cancer cells.7,8,32,33 BH3 mimics made entirely of natural amino acids are intrinsically disordered in the unbound state and undergo α-helical folding of the BH3 domain upon binding to anti-apoptotic partners. Thus, a number of studies have focused on making improved inhibitors by inserting an all-hydrocarbon staple into peptides derived from the BH3 motifs of Bcl-2 family proteins. Such an approach led to a 20-fold improvement in binding of a Mcl-1 targeting BH3 peptide.33 In recent years, peptides with improved binding to Mcl-1 have been developed,8 and these are also likely to benefit from stabilization using a stapling strategy. When optimizing staple-modified peptides, synthetic iteration is required to vary sequence composition, length and charge to achieve optimal solubility, structural stability and binding activity.34–37 Importantly, modification of the peptide may be required to compensate for changes introduced by stapling.
Here we describe a rapid design approach that applies hydrocarbon stapling to a potent lead peptide and then uses a specialized one-bead-one-compound library to perform further optimization. Working with the MS1 peptide, which displays high affinity and specificity for Mcl-1, we first identified favorable stapling positions along the BH3 sequence.8 Next, by applying a new library synthesis protocol that allowed us to evaluate a diverse library of peptides that include a chosen staple, we identified Mcl-1 selective binders using a screen for binding affinity and specificity. Our synthetic library of stabilized peptides included non-natural amino acids that promoted the discovery of protease resistant inhibitors and allowed us to access binding crevices not accessible to natural amino-acid side chains. The effectiveness of our approach is supported by our identification of molecules that achieve exceptionally tight and specific Mcl-1 targeting and show the greatest activity yet observed for inducing mitochondrial permeabilization in BH3 profiling assays of Mcl-1 dependent cancer cells.
Results and Discussion
Peptide MS1 was selected from a Bim BH3-based library using yeast-surface display, as reported by Foight et al.; this peptide binds to Mcl-1 with high affinity and selectivity over other anti-apoptotic proteins (Table 1 and Table S1).8 Hydrocarbon stapling, which involves installing a covalent, hydrophobic, olefin-bearing linker to connect sequential turns of an alpha helix, has been shown to increase the stability and cellular permeability of peptides that bind to protein targets17,38; stapling also increases binding affinity in some cases.39 To improve the binding and helicity of MS1 while minimizing its molecular weight, we made a truncated sequence and generated a panel of stapled variants to identify staple locations that maintained or enhanced binding activity (Figure S1). Three variants with staple positions that replaced key hydrophobic or charged residues at the binding interface showed impaired binding, but four stapled MS1 variants including M1d resulted in improved binding. The staple location in peptide M1d, which replaces Asn at 4b and Ala at 4f with the non-natural amino acids required for crosslinking, corresponds to a position previously shown to promote binding to Mcl-1 in a peptide called Mcl-1-SAHBD (see Figure S1 and Table 1 for sequences and position notation). Interestingly, the crystal structure of Mcl-1 bound to Mcl-1-SAHBD shows hydrophobic contacts made between the staple atoms and the edge of the Mcl-1 binding groove that likely contribute to the improved affinity (PDB ID: 3MK8).33 The secondary structure of M1d was measured by circular dichroism, showing that M1d is ~3-fold more helical in solution than unstapled MS1 (Figure 1a). The hydrocarbon stapling strategy also improved Mcl-1-binding affinity and increased peptide resistance to proteolysis, in comparison to unstapled MS1 (Figure 1). Having determined that M1d has attractive properties as a lead stapled peptide inhibitor, we considered strategies to optimize it further.
Table 1.
Alignment of parent peptides MS1 and M1d with library LB and selected Mcl-1-binding peptides from LB.

|
S5= α-4-pentenyl alanine, J= Aminoisobutyric acid (Aib), Z= Cyclohexylalanine (Cha), B= Norleucine (substituted for methionine to optimize activity of the ruthenium catalyst), X1 = Thr, Val, Aib, X2 = Phe, 5F-Phe, Cha, Leu, X3 = Arg, Asp, Gln, Trp, Tyr, Aib, D-Phenylglycine, α-methyl-L-leucine, α-methyl-L-phenylalanine.
Figure 1.

a) Circular dichroism analysis of unmodified and stapled variants. b) Competition of stapled peptides with fluorescently labeled 21mer Bim-BH3 (25 nM) for binding to Mcl-1. Error bars show standard errors for three experiments. c) Competition of M3d with fluorescently labeled 21mer Bim-BH3 (25 nM) for binding to Mcl-1, Bfl-1, Bcl-w, Bcl-xL or Bcl-2. Error bars show the standard error of four replicates. d) Half-lives of unstapled and stapled peptides exposed to chymotrypsin. e) Overlay of the leucine side chain at position 3a in Mcl-1 SAHBD (green side chain, PDB 3MK8) with the cyclohexylalanine side chain in M3d (yellow side chain, model based on 3MK8) illustrates how these ligands occupy the P2 pocket of Mcl-1. f) Kinetic analysis of M1d, M2d and M3d binding to Mcl-1, using biolayer interferometry.
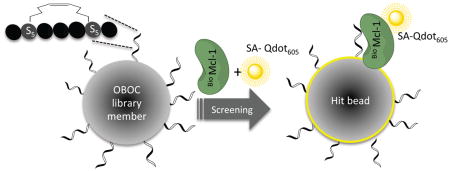
Although M1d is able to compete effectively for binding with a peptide corresponding to the BH3 region of native Bim, structural modeling suggested that M1d may not be maximally exploiting the available binding opportunities at three helix positions: 2e, 3a and 3b. However, the virtually unlimited hydrocarbon-stapled sequence variations that can be studied present a challenge. To date, hydrocarbon stapled peptides have typically been rationally optimized by iterative mutagenesis. Obtaining tight-binding and biologically active molecules often requires many rounds of laborious synthesis and stapling of different candidates. To accelerate the discovery of therapeutic peptides, we extended the application of one-bead-one-compound (OBOC) libraries40 by applying ring–closing metathesis reaction (RCM)41 to make libraries of BH3-like stapled peptides that can be sequenced using standard mass spectroscopy methods (Scheme 1).
Scheme 1.

Overview of a stapled-peptide library screen. Linear peptides synthesized on resin are cyclized using 1st generation Grubbs catalyst, and excess catalyst is removed by washing with tris(hydroxymethyl)phosphine. X1, X2 and X3 comprise the variable region. The one-bead-one-compound library of stapled peptides is incubated with a mixture of biotinylated target protein and non-biotinylated competitor to identify selective binders. Cyclized peptides that bind the target are visualized using a fluorescence microscope and then decoded by MALDI-TOF mass spectrometry.
To establish conditions that would produce a high-quality stapled peptide library suitable for analysis without purification (Figure S2), and to develop efficient on-bead competition screening procedures, we synthesized a first-generation 128-member stapled library based on a stapled Bim BH3 peptide that contains an i, i+4 all-hydrocarbon crosslink on the nonbinding surface of the α-helix (library LA) (Supporting Information). Library LA incorporated a number of mutations that have been studied previously to serve as controls, and it was designed so that all members had unique masses.7,8,32 Installation of a staple into peptides requires incorporation of two appropriately spaced α-4-pentenyl alanine residues, with defined stereochemical configuration, followed by RuIV-catalyzed RCM on the TentaGel resin.42 Ruthenium mediated olefin metathesis is fully compatible with solid-phase peptide synthesis and can be used in a synthetic library. However, excess ruthenium catalyst interferes with fluorescence-based on-bead screening and thus must be removed from beads prior to the screen. Here, this was accomplished using tris(hydroxymethyl)phosphine.
Library LA was screened for binding to Mcl-1 and Bcl-xL. Bcl-xL is a paralog of Mcl-1 and an undesired interaction target for candidate Mcl-1 inhibitors. Mcl-1 and Bcl-xL have the lowest sequence identity among five Bcl-2 family pro-survival proteins, but many BH3 peptide ligands bind indiscriminately to both of these proteins.7,43 When screening for Mcl-1 binding, we performed counter screening against Bcl-xL (and vice versa) to incorporate specificity criteria directly in our screens. The Mcl-1 screen led to isolation of Mcl-1 specific ligands that did not bind to Bcl-xL at micromolar concentration. The library synthesis was optimized to determine an appropriate ligand density (0.25 mmol/g) on the bead surface that allowed identification of the most active ligands in a narrow range of low-nanomolar dissociation constants. In addition, exhaustive testing of buffers and additives indicated that a blocking buffer composed of 1000-fold excess cell lysate greatly reduced the amount of nonspecific binding.44 Our screen entailed bead blocking, incubation with biotinylated Mcl-1 (BioMcl-1) and unlabeled Bcl-xL, washing, incubation with streptavidin-coated quantum dots (Qdots-SA605), and visualization using a fluorescence microscope. 173 hit peptides, which were selected manually under the microscope, were cleaved from the beads and identified using mass spectrometry. We isolated 96 BH3 peptides specific for binding Mcl-1 in preference to Bcl-xL and 77 with the opposite specificity (Figure 2). Both the stapling chemistry and subsequent tests for binding were carried out with peptides bound to resin, with one unique peptide per bead.
Figure 2.

Sequence logos for populations of peptides from library LA screening. 96 Mcl-1 specific stapled peptides were identified from competition screens using a mixture of BioMcl-1:mycBcl-xL (1:50). 77 Bcl-xL-specific peptides were identified in competition screening using BioBcl-xL:mycMcl-1 (1:50). The side chains at the 3c and 3g positions, marked X, were replaced with cross-linked α-4-pentenylalanine groups. Mutations were made at positions 2d, 2e, 2g, 3b, 3e, 4a and 4e. Figures were made using WebLogo.
The effects of residue substitutions in stapled peptides isolated from library LA were consistent with results from previously reported mutational analyses of non-cyclized peptides. When screening for selective binding to Mcl-1 vs. Bcl-xL (or Bcl-xL vs. Mcl-1) we observed enrichment of residues previously implicated as important for binding specificity (Figure 2 and Supporting Information). For example, at position 2e, Mcl-1 accommodates a range of amino acids, including Val, Pro and Thr. In contrast, Bcl-xL has a strong preference for Gly and Ala.8,33 We mutated this position to Ala or Thr and found, as expected, that Thr was selected more frequently than Ala when screening for selective Mcl-1 binding; the opposite preference was observed when screening for Bcl-xL selective binding. Mutation of Tyr to Lys at 4e is known to disfavor Mcl-1 binding and confer a preference for interaction with Bcl-xL.32 We also observed this effect in stapled peptides; Lys was more prevalent at 4e in the library screened for binding to Bcl-xL than in the library screened for binding to Mcl-1. More generally, position 3e is typically occupied by small amino acids in native BH3 sequences and does not tolerate substitution with residues larger than Gly, Ala or Ser for either Mcl-1 or Bcl-xL binding. In parallel to this result for non-stapled peptides, an absolute selection of Gly over Glu was observed for stapled peptides at position 3e. Other examples of good agreement between the library screening results and prior studies are listed in the Supporting Information. Selected hit peptides were also confirmed to bind to Mcl-1 in solution when not attached to beads (Supporting Information). These observations provided evidence that our stapled peptide library screen can efficiently isolate peptides with desired binding properties.
To apply library-based optimization to M1d, we prepared 108 stapled peptides designed to be enriched in Mcl-1 binders (library LB). Positions 2e, 3a and 3b were diversified with 3, 4 and 9 amino acids, respectively, varying the residue size, hydrophobicity and/or charge (Table 1). Many of the side chains were chosen to be hydrophobic to favor hydrophobic contacts with Mcl-1. We also introduced known helix-inducing side chains to improve the helical content of stapled peptides, which is expected to be poor at the peptide N-terminus because of two Gly residues in the MS1 template. To choose residues for the mutation sites, we used the large amount of mutational data from SPOT arrays and side-chain scanning of BH3 peptides.7,8,32 We also analyzed structures of Mcl-1 bound to different BH3 variants using Bioluminate (version 1.9, Schrödinger, LLC, New York, NY, 2015) to select non-natural side chains for the library. We incorporated non-natural functionalities that we predicted could make contacts with Mcl-1 that cannot be satisfied by natural side chains. LB was sorted for binding to Mcl-1 in two rounds of competition screening in which beads were selected that could bind to Mcl-1 in the presence of 12.5- or 50-fold higher concentration of Bcl-xL.
LB was initially screened under conditions optimized for library LA: 0.2 μM BioMcl-1 in presence of 2.5 μM myc-tagged Bcl-xL (mycBcl-xL). However, for LB, we used a large-particle flow cytometer that permits fluorescence-based sorting of up to 300 beads/s. The 5% of beads with the highest fluorescence intensities were sorted into 96-well plates (Figure S3). Beads were washed and then re-screened using more stringent conditions: 0.05 μM BioMcl-1 plus 2.5 μM mycBcl-xL. After incubation with streptavidin coated quantum dots, beads were washed and visualized using a fluorescence microscope. The 170 brightest beads from a pool of ~1,000 were isolated manually. MALDI mass spectrometry confirmed that the selected beads included 6 different sequences out of the possible 108 stapled BH3 peptides (Table 1). Sequences M1d (32 beads), M2d (48 beads), and M3d (38 beads) were chosen for further analysis. Interestingly, M2d and M3d incorporate previously untested unnatural side chains at positions 2e and 3a (Table 1). These positions are highly conserved in known BH3 motifs as alanine/glycine and leucine, respectively. M2d and M3d were tested in solution in competition with a fluorescently labeled Bim BH3 peptide for binding to five human Bcl-2 paralogs (Figure 1b,c and Figure S4). The competition experiments indicated that M2d and M3d are both highly selective for Mcl-1 over 4 other anti-apoptotic family members and are both tighter binders of Mcl-1 than is M1d (Figure 1b,c). These peptides competed with fluoresceinated Bim 21mer for Mcl-1 binding with IC50 values of 106 ± 12 nM and 72 ± 11 nM, for M2d and M3d respectively (compare with 350 nM for M1d and 811 nM for MS1).
Position 2e is usually conserved as small (alanine, glycine, serine) in natural BH3 sequences. However, previous studies have shown that Mcl-1 can bind BH3 peptides with bulkier threonine or leucine residues at this site.8,33 Our results show that, likewise, introducing the larger, branched 2-aminoisobutyric acid (Aib) at the 2e position in M2d and M3d is not only well tolerated but increases binding affinity for Mcl-1. A model of M3d-peptide bound to Mcl-1, built using Bioluminate and based on PDB structure 3MK8,33 shows how two methyl groups can form contacts with Mcl-1 (Figure S5). Peptide M3d also includes Cha at 3a. The selection of Cha at 3a was not easily anticipated because leucine is universally conserved at this site in native BH3 domains and substitution with any residue other than Ile is highly destabilizing according to SPOT array experiments.7 Comparing our molecular model of M3d to the binding of a 21-mer BH3 peptide derived from Mcl-1 (PDB ID: 3MK8) demonstrates that the Cha side chain can fill more of the p2 binding pocket of Mcl-1 than can the conserved Leu, which occupies only the upper part of this pocket (Figure 1e). This pocket in Mcl-1 has been observed to accommodate small molecule ligands,45–47 however, these molecules do not make the additional contacts that can be achieved by a BH3 peptide, including interactions made by hydrophobic side chains at 3d and 4a. The improved contacts of M3d result in increased potency, providing a 5-fold decrease in IC50 for Mcl-1 binding compared to M1d, and an ~11-fold decrease compared to the original MS1 peptide (Figure 1b).45
We used biolayer interferometry to study the binding of M2d and M3d to Mcl-1 (Figure 1f, S6 and Table S2). We generated biotinylated stapled peptides and attached these to a streptavidin-modified probe surface. Quantification of binding and dissociation kinetics established that enhanced binding of M2d and M3d to Mcl-1 arises from slower off-rates compared to M1d (6- and 16-fold, respectively). Interestingly, the observed trend for binding affinities was paralleled by the measured helicity (Figure 1a). M2d and M3d are more helical than M1d when unbound, with mean residue ellipticities (MREs) at 222 nm of 15,450 and 21,000 deg cm2 dmol−1, respectively, compared to 11,590 for M1d. Therefore, increases in helicity can be attributed to both non-natural amino-acid substitutions: Aib at 2e and Cha at 3a.48,49
All of the stapled MS1 variants that we tested were more protease resistant than un-metathesized M1d, which is a key pharmacologic advantage of the stapling approach. Unstapled M1d has a half-life 2–6-fold shorter than the corresponding stapled peptides (Figure 1d). Notably, the protease resistance analysis revealed longer half-lives for both M2d and M3d compared to M1d, indicating the utility of introducing helix-promoting and/or bulky non-natural amino acids into a stapled peptide for maximizing protease resistance.
Finally, we performed BH3 profiling to test the function of stapled variants of MS1 in cells. A whole-cell BH3 profiling experiment quantifies the dependence of cancer cell mitochondrial integrity on specific anti-apoptotic proteins and can be predictive of cellular responses to chemotherapy.50,51 In this assay, permeabilized cells are stained with dye JC1 to monitor mitochondrial membrane integrity in response to increasing doses of BH3 peptides. We used this assay to test the specificity of our Mcl-1-binding stapled peptides in cell lines with different established dependencies on Bcl-xL and Mcl-1. Mcl-1/Myc 2640 is an engineered murine leukemia cell line overexpressing murine Mcl-1 and Myc, and MDA-MB 231 is a human breast cancer cell line with a primed Bcl-xL-dependent profile.50,52 By BH3 profiling using native BH3 peptides from Bad and NoxaA, we confirmed cell line dependencies on these anti-apoptotic proteins (Figure S7). Both M1d and M2d showed Bax/Bak independent activity in Bax/Bak deficient cells, indicating non-specific toxicity (Figure S8). However, peptide M3d showed no activity in Bax/Bak negative cells, consistent with an on-target mechanism,[17] and was remarkably potent when tested on Mcl-1/Myc 2640, with an EC50 value of 27 nM (Figure 3). Thus, the Mcl-1-dependent cell line was much more sensitive to M3d than to MS1 and the natural BH3 NoxaA (Figures 3). The specificity of M3d was confirmed by the much higher EC50 observed for the Bcl-xL dependent cell line MDA-MB 231 (no detectable deplorization up to 400 nM, Figure S8a).
Figure 3.

Depolarization of the mitochondrial membrane of Mcl-1 2640 cells in response to treatment with different BH3 peptides.
In conclusion, we demonstrated new library synthesis chemistry that can be used to make diverse stapled helical peptides that are suitable for on-bead testing. By applying this powerful method, we tailored highly stable, potent and selective Mcl-1 inhibitors. Using competition screening of stapled peptides, we identified novel molecules with low nanomolar binding affinity for Mcl-1 and greater than 1000-fold selectivity over other Bcl-2 paralogs. Our tightest-binding peptide, M3d, including hydrophobic and helix-inducing unnatural moieties, showed potent activity only in an Mcl-1 dependent cell line. We demonstrated the feasibility of combining peptide post-modification with one-bead-one-compound synthesis, which accelerates the design and optimization of novel chemically modified peptidic inhibitors. We anticipate that this approach will find utility in other stapled peptide discovery projects that target important biomedical proteins. For example, exciting progress identifying stabilized peptides that bind to G-protein-coupled receptors53, apoptosis regulators MDM2/MDMX54, and epidermal growth factor receptor tyrosine kinases55 provide examples of cases where our method can be used to drive further improvements in binding.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences under award number R01GM110048 (peptide library work), and by the Koch Institute/MIT - Dana-Farber/Harvard Cancer Center Bridge Project (cell studies). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. A German research foundation fellowship to R.R.A. supported early development of the library methodology. We acknowledge the Biophysical Instrumentation Facility for use of the Octet BioLayer Interferometry System (NIH S10 OD016326). The authors thank G. Bird and L. Walensky for helpful discussions and technical advice, V. Potapov and V. Xue for calculating the possible non-identical masses for the synthetic libraries, R. Cook for use of the MALDI mass spectrometer, A. Jasanoff for access to chemistry facilities and fluorescence microscopy, P. Rogers for arranging access to COPAS bead sorting, and members of the Keating lab for many helpful comments.
Footnotes
ASSOCIATED CONTENT
This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Stockwin LH, Holmes S. Antibodies as therapeutic agents: vive la renaissance! Expert Opin Biol Ther. 2003;3:1133–1152. doi: 10.1517/14712598.3.7.1133. [DOI] [PubMed] [Google Scholar]
- 2.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 3.Verdine GL, Walensky LD. The Challenge of Drugging Undruggable Targets in Cancer: Lessons Learned from Targeting BCL-2 Family Members. Clin Cancer Res. 2007;13:7264–7270. doi: 10.1158/1078-0432.CCR-07-2184. [DOI] [PubMed] [Google Scholar]
- 4.Foight GW, Keating AE. Comparison of the peptide binding preferences of three closely related TRAF paralogs: TRAF2, TRAF3, and TRAF5. Protein Sci. 2016 doi: 10.1002/pro.2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tonikian R, Zhang Y, Sazinsky SL, Currell B, Yeh JH, Reva B, Held HA, Appleton BA, Evangelista M, Wu Y, Xin X, Chan AC, Seshagiri S, Lasky LA, Sander C, Boone C, Bader GD, Sidhu SS. A Specificity Map for the PDZ Domain Family. PLoS Biol. 2008;6:e239. doi: 10.1371/journal.pbio.0060239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reich L “Luther”, Dutta S, Keating AE. SORTCERY—A High–Throughput Method to Affinity Rank Peptide Ligands. J Mol Biol. 2015;427:2135–2150. doi: 10.1016/j.jmb.2014.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dutta S, Gullá S, Chen TS, Fire E, Grant RA, Keating AE. Determinants of BH3 Binding Specificity for Mcl-1 versus Bcl-xL. J Mol Biol. 2010;398:747–762. doi: 10.1016/j.jmb.2010.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foight GW, Ryan JA, Gullá SV, Letai A, Keating AE. Designed BH3 Peptides with High Affinity and Specificity for Targeting Mcl-1 in Cells. ACS Chem Biol. 2014;9:1962–1968. doi: 10.1021/cb500340w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lau YH, de Andrade P, Wu Y, Spring DR. Peptide stapling techniques based on different macrocyclisation chemistries. Chem Soc Rev. 2015;44:91–102. doi: 10.1039/c4cs00246f. [DOI] [PubMed] [Google Scholar]
- 10.Heinis C, Rutherford T, Freund S, Winter G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat Chem Biol. 2009;5:502–507. doi: 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- 11.Tjhung KF, Kitov PI, Ng S, Kitova EN, Deng L, Klassen JS, Derda R. Silent Encoding of Chemical Post-Translational Modifications in Phage-Displayed Libraries. J Am Chem Soc. 2016;138:32–35. doi: 10.1021/jacs.5b10390. [DOI] [PubMed] [Google Scholar]
- 12.Morimoto J, Kodadek T. Synthesis of a large library of macrocyclic peptides containing multiple and diverse N-alkylated residues. Mol Biosyst. 2015;11:2770–2779. doi: 10.1039/c5mb00308c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dewan V, Liu T, Chen KM, Qian Z, Xiao Y, Kleiman L, Mahasenan KV, Li C, Matsuo H, Pei D, Musier-Forsyth K. Cyclic Peptide Inhibitors of HIV-1 Capsid-Human Lysyl-tRNA Synthetase Interaction. ACS Chem Biol. 2012;7:761–769. doi: 10.1021/cb200450w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu T, Liu Y, Kao HY, Pei D. Membrane Permeable Cyclic Peptidyl Inhibitors against Human Peptidylprolyl Isomerase Pin1. J Med Chem. 2010;53:2494–2501. doi: 10.1021/jm901778v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joo SH, Xiao Q, Ling Y, Gopishetty B, Pei D. High-Throughput Sequence Determination of Cyclic Peptide Library Members by Partial Edman Degradation/Mass Spectrometry. J Am Chem Soc. 2006;128:13000–13009. doi: 10.1021/ja063722k. [DOI] [PubMed] [Google Scholar]
- 16.Giudicessi SL, Gurevich-Messina JM, Martínez-Ceron MC, Erra-Balsells R, Albericio F, Cascone O, Camperi Sa. Friendly strategy to prepare encoded one bead-one compound cyclic peptide library. ACS Comb Sci. 2013;15:525–529. doi: 10.1021/co400039a. [DOI] [PubMed] [Google Scholar]
- 17.Cromm PM, Spiegel J, Grossmann TN. Hydrocarbon Stapled Peptides as Modulators of Biological Function. ACS Chem Biol. 2015;10:1362–1375. doi: 10.1021/cb501020r. [DOI] [PubMed] [Google Scholar]
- 18.Henchey LK, Jochim AL, Arora PS. Contemporary Strategies for the Stabilization of Peptides in the α-Helical Conformation. Curr Opin Chem Biol. 2008;12:692–697. doi: 10.1016/j.cbpa.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pelay-Gimeno M, Glas A, Koch O, Grossmann TN. Structure-Based Design of Inhibitors of Protein–Protein Interactions: Mimicking Peptide Binding Epitopes. Angew Chemie Int Ed. 2015;54:8896–8927. doi: 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang DCS, Strasser A. BH3-Only Proteins—Essential Initiators of Apoptotic Cell Death. Cell. 2000;103:839–842. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- 21.Adams JM, Cory S. The Bcl-2 Protein Family: Arbiters of Cell Survival. Sci. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 22.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 23.Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW. BH3-only proteins: Orchestrators of apoptosis. Biochim Biophys Acta - Mol Cell Res. 2011;1813:508–520. doi: 10.1016/j.bbamcr.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 24.Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DCS, Xiong H, Cui Y, Busman TA, McKeegan EM, Krivoshik AP, Enschede SH, Humerickhouse R. Substantial Susceptibility of Chronic Lymphocytic Leukemia to BCL2 Inhibition: Results of a Phase I Study of Navitoclax in Patients With Relapsed or Refractory Disease. J Clin Oncol. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, Chu Q, Giaccone G, Khaira D, Ramalingam SS, Ranson MR, Dive C, McKeegan EM, Chyla BJ, Dowell BL, Chakravartty A, Nolan CE, Rudersdorf N, Busman TA, Mabry MH, Krivoshik AP, Humerickhouse RA, Shapiro GI, Gandhi L. Phase II Study of Single-Agent Navitoclax (ABT-263) and Biomarker Correlates in Patients with Relapsed Small Cell Lung Cancer. Clin Cancer Res. 2012;18:3163–3169. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, Yang X, Zhang H, Fesik S, Rosenberg SH, Elmore SW. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 27.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 28.Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL, Bataille R, Amiot M. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-xL is an essential survival protein of human myeloma cells. Blood. 2002;100:194–199. doi: 10.1182/blood.v100.1.194. [DOI] [PubMed] [Google Scholar]
- 29.Ding Q, He X, Xia W, Hsu JM, Chen CT, Li LY, Lee DF, Yang JY, Xie X, Liu JC, Hung MC. Myeloid Cell Leukemia-1 Inversely Correlates with Glycogen Synthase Kinase-3β Activity and Associates with Poor Prognosis in Human Breast Cancer. Cancer Res. 2007;67:4564–4571. doi: 10.1158/0008-5472.CAN-06-1788. [DOI] [PubMed] [Google Scholar]
- 30.Zhou T, Li G, Cao B, Liu L, Cheng Q, Kong H, Shan C, Huang X, Chen J, Gao N. Downregulation of Mcl-1 through inhibition of translation contributes to benzyl isothiocyanate-induced cell cycle arrest and apoptosis in human leukemia cells. Cell Death Dis. 2013;4:e515. doi: 10.1038/cddis.2013.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Certo M, Moore VDG, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 32.Boersma MD, Sadowsky JD, Tomita YA, Gellman SH. Hydrophile scanning as a complement to alanine scanning for exploring and manipulating protein–protein recognition: Application to the Bim BH3 domain. Protein Sci. 2008;17:1232–1240. doi: 10.1110/ps.032896.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stewart ML, Fire E, Keating AE, Walensky LD. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat Chem Biol. 2010;6:595–601. doi: 10.1038/nchembio.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grossmann TN, Yeh JTH, Bowman BR, Chu Q, Moellering RE, Verdine GL. Inhibition of oncogenic Wnt signaling through direct targeting of β-catenin. Proc Natl Acad Sci. 2012;109:17942–17947. doi: 10.1073/pnas.1208396109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verdine GL, Hilinski GJ. Stapled Peptides for Intracellular Drug Targets. In: Enzymology KDW, GLVBT-M, editors. Protein Engineering for Therapeutics, Part B. Academic Press; 2012. pp. 3–33. [DOI] [PubMed] [Google Scholar]
- 36.Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. Reactivation of the p53 Tumor Suppressor Pathway by a Stapled p53 Peptide. J Am Chem Soc. 2007;129:2456–2457. doi: 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edwards AL, Gavathiotis E, LaBelle JL, Braun CR, Opoku-Nsiah KA, Bird GH, Walensky LD. Multimodal Interaction with BCL-2 Family Proteins Underlies the Pro-Apoptotic Activity of PUMA BH3. Chem Biol. 2013;20:888–902. doi: 10.1016/j.chembiol.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Activation of Apoptosis in Vivo by a Hydrocarbon-Stapled BH3 Helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bird GH, Gavathiotis E, LaBelle JL, Katz SG, Walensky LD. Distinct BimBH3 (BimSAHB) Stapled Peptides for Structural and Cellular Studies. ACS Chem Biol. 2014;9:831–837. doi: 10.1021/cb4003305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao W, Bononi FC, Townsend J, Li Y, Liu R, Lam KS. Immobilized OBOC combinatorial bead array to facilitate multiplicative screening. Comb Chem High Throughput Screen. 2013;16:441–448. doi: 10.2174/1386207311316060004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim YW, Grossmann TN, Verdine GL. Synthesis of all-hydrocarbon stapled [alpha]-helical peptides by ring-closing olefin metathesis. Nat Protoc. 2011;6:761–771. doi: 10.1038/nprot.2011.324. [DOI] [PubMed] [Google Scholar]
- 42.Schafmeister CE, Po J, Verdine GL. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J Am Chem Soc. 2000;122:5891–5892. [Google Scholar]
- 43.DeBartolo J, Taipale M, Keating AE. Genome-Wide Prediction and Validation of Peptides That Bind Human Prosurvival Bcl-2 Proteins. PLoS Comput Biol. 2014;10:e1003693. doi: 10.1371/journal.pcbi.1003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kodadek T, Bachhawat-Sikder K. Optimized protocols for the isolation of specific protein-binding peptides or peptoids from combinatorial libraries displayed on beads. Mol Biosyst. 2006;2:25–35. doi: 10.1039/b514349g. [DOI] [PubMed] [Google Scholar]
- 45.Burke JP, Bian Z, Shaw S, Zhao B, Goodwin CM, Belmar J, Browning CF, Vigil D, Friberg A, Camper DV, Rossanese OW, Lee T, Olejniczak ET, Fesik SW. Discovery of Tricyclic Indoles That Potently Inhibit Mcl-1 Using Fragment-Based Methods and Structure-Based Design. J Med Chem. 2015;58:3794–3805. doi: 10.1021/jm501984f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Friberg A, Vigil D, Zhao B, Daniels RN, Burke JP, Garcia-Barrantes PM, Camper D, Chauder BA, Lee T, Olejniczak ET, Fesik SW. Discovery of Potent Myeloid Cell Leukemia 1 (Mcl-1) Inhibitors Using Fragment-Based Methods and Structure-Based Design. J Med Chem. 2013;56:15–30. doi: 10.1021/jm301448p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen M, Cencic R, Ertel F, Bernier C, Pelletier J, Roulston A, Silvius J, Shore G. Obatoclax is a direct and potent antagonist of membrane-restricted Mcl-1 and is synthetic lethal with treatment that induces Bim. BMC Cancer. 2015;15:568. doi: 10.1186/s12885-015-1582-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Armstrong KM, Fairman R, Baldwin RL. The (i,i+4) Phe-His Interaction Studied in an Alanine-based α-Helix. J Mol Biol. 1993;230:284–291. doi: 10.1006/jmbi.1993.1142. [DOI] [PubMed] [Google Scholar]
- 49.Venkatraman J, Shankaramma SC, Balaram P. Design of Folded Peptides. Chem Rev. 2001;101:3131–3152. doi: 10.1021/cr000053z. [DOI] [PubMed] [Google Scholar]
- 50.Ryan JA, Brunelle JK, Letai A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc Natl Acad Sci. 2010;107:12895–12900. doi: 10.1073/pnas.0914878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ryan J, Letai A. BH3 profiling in whole cells by fluorimeter or FACS. Methods. 2013;61:156–164. doi: 10.1016/j.ymeth.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brunelle JK, Ryan J, Yecies D, Opferman JT, Letai A. MCL-1–dependent leukemia cells are more sensitive to chemotherapy than BCL-2–dependent counterparts. J Cell Biol. 2009;187:429–442. doi: 10.1083/jcb.200904049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shemesh R, Toporik A, Levine Z, Hecht I, Rotman G, Wool A, Dahary D, Gofer E, Kliger Y, Soffer MA, Rosenberg A, Eshel D, Cohen Y. Discovery and Validation of Novel Peptide Agonists for G-protein-coupled Receptors. J Biol Chem. 2008;283:34643–34649. doi: 10.1074/jbc.M805181200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biderman L, Manley JL, Prives C. Mdm2 and MdmX as Regulators of Gene Expression. Genes Cancer. 2012;3:264–273. doi: 10.1177/1947601912455331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roengvoraphoj M, Tsongalis GJ, Dragnev KH, Rigas JR. Epidermal growth factor receptor tyrosine kinase inhibitors as initial therapy for non-small cell lung cancer: Focus on epidermal growth factor receptor mutation testing and mutation-positive patients. Cancer Treat Rev. 2013;39:839–850. doi: 10.1016/j.ctrv.2013.05.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.