

Abstract
The serial founder model of modern human origins predicts that the phylogeny of ancestries exhibits bifurcating, tree-like behavior. Here, we tested this prediction using three methods designed to investigate gene flow in autosome-wide genotype data from 3,528 unrelated individuals from 163 global samples. Specifically, we investigated whether Cushitic ancestry has an East African or Middle Eastern origin. We found evidence for non-tree-like behavior in the form of four migration events. First, we found that Cushitic ancestry is a mixture of ancestries closely related to Arabian ancestry and Nilo-Saharan or Omotic ancestry. We found evidence for additional migration events in the histories of: 1) Indian and Arabian ancestries, 2) Kalash ancestry, and 3) Native American and Northern European ancestries. These findings, based on analysis of ancestry of present-day humans, reveal migration in the distant past and provide new insights into human history.
Genetic data provide insight into the migratory history and geographic structuring of modern human populations. The recent origin of modern humans reflects migration from sub-Saharan Africa, with the oldest divergence event at approximately 140,000 years ago evident from analysis of Y DNA haplogroups1,2 and autosomal markers3. Subsequently, at least 19 ancestries arose as humans migrated across the continents3,4. These ancestries reflect shared history at a level higher than populations, tribes, or ethnic groups. The divergence of ancestries is mainly due to random genetic drift subsequent to a barrier to random mating, with two common barriers being geographic and linguistic, following serial founder effects as modern humans peopled the world5.
Although most ancestries showed topological consistency in phylogenetic relationships assuming a strictly bifurcating or tree-like process, Cushitic ancestry showed unstable placement between East African and Middle Eastern ancestries3,4,6 and Melanesian ancestry showed an unexpectedly long branch3. These findings suggested the presence of non-tree-like behavior, i.e., some ancestries may have formed due to a mixture process rather than a splitting process. Here, we investigate this possibility using three different approaches: analysis of the distance matrix using split decomposition analysis7, analysis of ancestry-specific allele frequencies using f3 and f4 statistics8, and analysis of ancestry-specific allele frequencies using a graph-based model of gene flow9. We find evidence for non-tree-like behavior in the form of four migration events.
Results
We previously analyzed autosomal genotype from 3,528 individuals (Supplementary Table S1) and identified 19 ancestries3. Phylogenetic trees based on these ancestries assume a strictly bifurcating process. To investigate this assumption, we first employed split decomposition analysis of the distance matrix based on pairwise FST estimates between ancestries. This analysis revealed network-like behavior, with Cushitic ancestry appearing the most non-tree-like (Fig. 1). As measured by the Q-residual score, Cushitic ancestry indeed contributed the most to network-like behavior (Table 1).
Figure 1. Split decomposition network of ancestries.
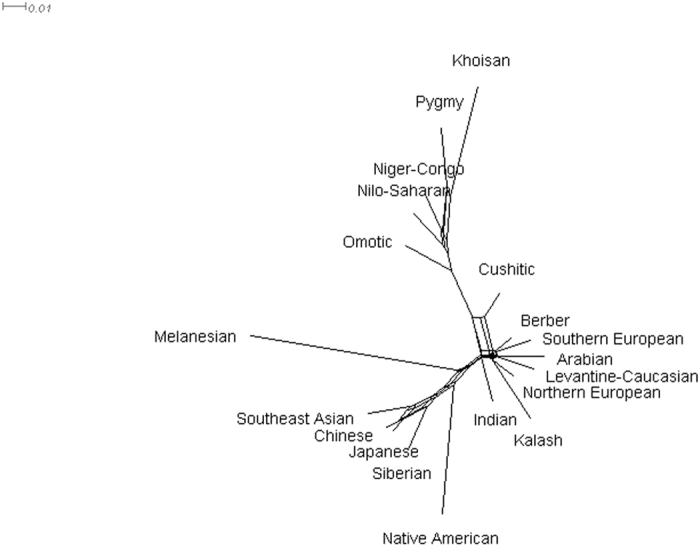
The underlying distance matrix is based on pairwise FST.
Table 1. Non-tree-like behavior among ancestries.
| Excluded Ancestry | Q-residual score |
|---|---|
| Cushitic | 0.0073 |
| Berber | 0.0084 |
| Northern European | 0.0095 |
| Arabian | 0.0096 |
| Southern European | 0.0098 |
| Levantine-Caucasian | 0.0100 |
| Indian | 0.0103 |
| Kalash | 0.0103 |
| Southeast Asian | 0.0104 |
| Niger-Congo | 0.0105 |
| Nilo-Saharan | 0.0106 |
| Japanese | 0.0106 |
| Chinese | 0.0106 |
| Siberian | 0.0107 |
| Native American | 0.0108 |
| Omotic | 0.0108 |
| Melanesian | 0.0110 |
| Pygmy | 0.0110 |
| Khoisan | 0.0115 |
The Q-residual score measures deviation from tree-like behavior. The graph including all 19 ancestries had a Q-residual score of 0.0102. We excluded each ancestry one at a time and recalculated the Q-residual score. A lower score indicates that the excluded ancestry contributed more to non-tree-like behavior, as the remaining ancestries are more tree-like. Thus, Cushitic ancestry contributed the most to non-tree-like behavior.
Networks can represent splits that are incompatible due to several factors, including misspecification in the evolutionary model (e.g., substitution probabilities) and misspecification of the topology (e.g., due to gene flow). To further investigate network-like behavior in our data set, we used two approaches based on ancestry-specific allele frequencies. First, we estimated all 2,907 possible combinations of the f3 statistic8. A negative f3 statistic is consistent with admixture, as well as isolation by distance. Of the 2,907 combinations, 273 were significantly negative after Bonferroni correction (Supplementary Table S2). These results further support the existence of network-like behavior. Of the 273 significant f3 statistics, 41 were consistent with an admixed origin of Cushitic ancestry, with one parent being one of five sub-Saharan ancestries and the other parent being one of the other 13 ancestries (Table 2).
Table 2. f3 statistics testing for admixture in Cushitic ancestry.
| Parent Ancestry A | Parent Ancestry B | Z-score | P-value |
|---|---|---|---|
| Niger-Congo | Southern European | −124.39 | 0 |
| Khoisan | Southern European | −102.48 | 0 |
| Niger-Congo | Arabian | −97.76 | 0 |
| Nilo-Saharan | Southern European | −97.58 | 0 |
| Niger-Congo | Levantine-Caucasian | −95.69 | 0 |
| Khoisan | Levantine-Caucasian | −83.61 | 0 |
| Pygmy | Southern European | −80.98 | 0 |
| Khoisan | Arabian | −77.93 | 0 |
| Niger-Congo | Northern European | −77.75 | 0 |
| Nilo-Saharan | Arabian | −74.33 | 0 |
| Khoisan | Northern European | −71.98 | 0 |
| Nilo-Saharan | Levantine-Caucasian | −69.53 | 0 |
| Pygmy | Arabian | −61.39 | 0 |
| Pygmy | Levantine-Caucasian | −60.49 | 0 |
| Nilo-Saharan | Northern European | −57.05 | 0 |
| Pygmy | Northern European | −49.18 | 0 |
| Niger-Congo | Native American | −33.52 | 1.04 × 10−246 |
| Niger-Congo | Kalash | −33.11 | 1.23 × 10−240 |
| Khoisan | Native American | −28.86 | 1.89 × 10−183 |
| Khoisan | Chinese | −28.02 | 5.18 × 10−173 |
| Khoisan | Berber | −25.09 | 3.45 × 10−139 |
| Niger-Congo | Melanesian | −24.60 | 6.96 × 10−134 |
| Khoisan | Kalash | −22.81 | 1.80 × 10−115 |
| Niger-Congo | Chinese | −21.20 | 4.72 × 10−100 |
| Omotic | Southern European | −19.20 | 1.89 × 10−82 |
| Nilo-Saharan | Kalash | −17.24 | 6.38 × 10−67 |
| Nilo-Saharan | Native American | −15.08 | 1.11 × 10−51 |
| Niger-Congo | Siberian | −13.19 | 4.86 × 10−40 |
| Pygmy | Kalash | −9.72 | 1.27 × 10−22 |
| Khoisan | Japanese | −9.67 | 2.02 × 10−22 |
| Pygmy | Berber | −9.46 | 1.53 × 10−21 |
| Niger-Congo | Berber | −8.91 | 2.57 × 10−19 |
| Khoisan | Southeast Asian | −8.63 | 3.05 × 10−18 |
| Niger-Congo | Japanese | −8.28 | 5.92 × 10−17 |
| Omotic | Arabian | −8.17 | 1.52 × 10−16 |
| Khoisan | Indian | −7.85 | 2.08 × 10−15 |
| Khoisan | Siberian | −7.72 | 5.97 × 10−15 |
| Nilo-Saharan | Berber | −6.65 | 1.47 × 10−11 |
| Niger-Congo | Southeast Asian | −6.62 | 1.82 × 10−11 |
| Pygmy | Native American | −6.21 | 2.73 × 10−10 |
| Nilo-Saharan | Melanesian | −4.44 | 4.46 × 10−6 |
Second, we used the graph-based model implemented in TreeMix9 to estimate migration events from the ancestry-specific allele frequencies. The likelihood function in TreeMix is a composite likelihood, not a maximum likelihood, and cannot be used for formal significance testing. In particular, as we increased the number of migration events fit by the model, log-likelihoods became positive, raising a concern of model misspecification or over-fitting. Also, the directions of migration events are not identifiable by the model; directions are assigned by assuming gene flow from the edge with the largest weight, with weights related to mixture proportions. Conditional on no migration events, two trees accounted for 98% of the search space. The tree with the best log-likelihood was found 75% of the time and contained the subtree (Arabian, (Levantine-Caucasian, (Northern European, Southern European))) (Fig. 2). The tree with the second best log-likelihood was found 23% of the time and contained the subtree (Northern European, (Levantine-Caucasian, (Southern European, Arabian))) (Fig. 3).
Figure 2. TreeMix analysis with no migration events.
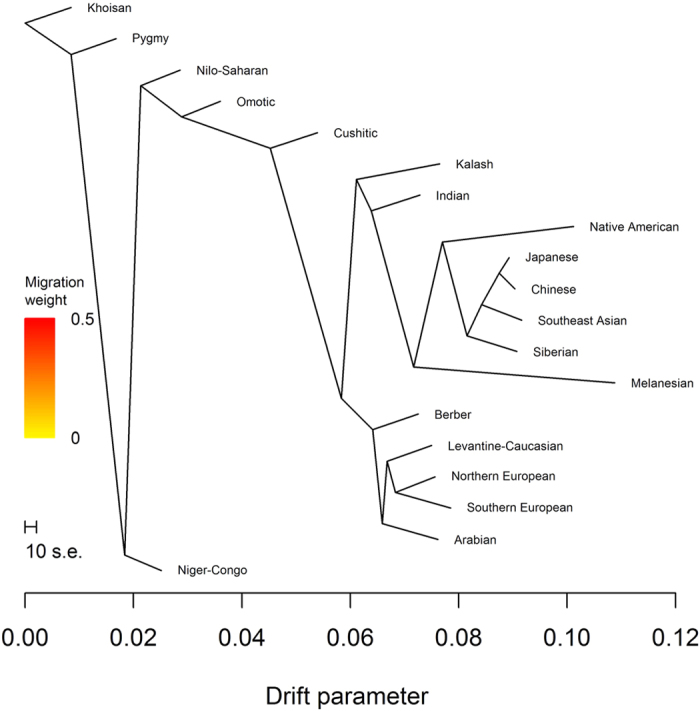
The most frequently found tree with no migration events.
Figure 3. TreeMix analysis with no migration events.
The second most frequently found tree with no migration events.
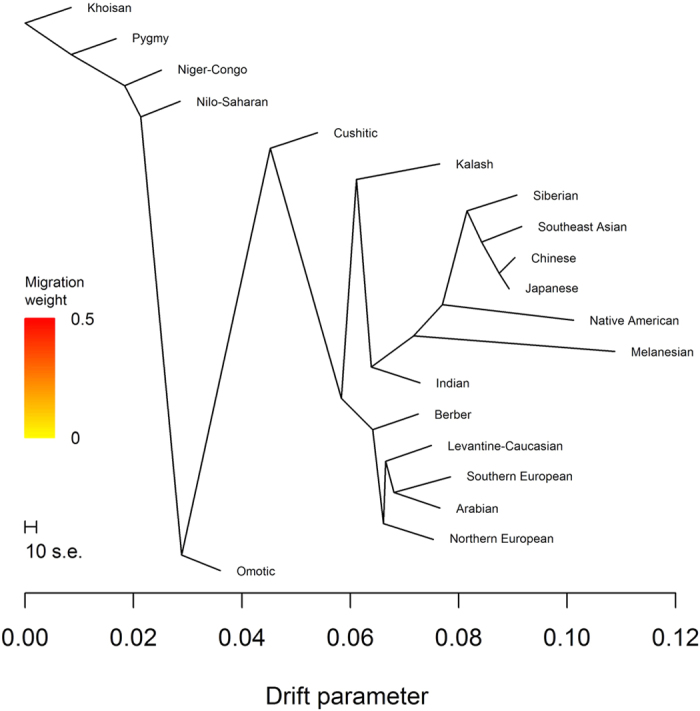
Conditional on one migration event, we observed the tree containing the (Northern European, Southern European) subtree 71% of the time (Fig. 4) and the tree containing the (Southern European, Arabian) subtree 27% of the time (Fig. 5). The same migration event was inferred for both trees: gene flow between Kalash ancestry, after it diverged from Indian ancestry, and the internal node representing the common ancestor of Arabian, Levantine-Caucasian, Northern European, and Southern European ancestries. Four f3 statistics supported Kalash ancestry as significantly admixed (Supplementary Table S2). Using the f4 statistics (Southern European, Khoisan; Kalash, Indian) and (Southern European, Khoisan; Northern European, Indian), we estimated a median mixture proportion of 0.490 (IQR 1.604), indicating that Kalash ancestry is a mixture of 49.0% Northern European and 51.0% Indian ancestries. Similarly, we estimated mixtures of 37.1% Arabian and 62.9% Indian ancestries or 49.4% Levantine-Caucasian and 50.6% Indian ancestries. It is possible that Arabian, Levantine-Caucasian, Northern European, and Southern European ancestries are proxies for one ancestry. However, the distribution of Y DNA haplogroups in the Kalash people consists of 20.5% H and 25.0% L, common in India and South Asia, respectively, mixed with 18.2% G, 9.1% J, and 18.2% R1a, common in the Levant and the Caucasus, the Middle East, and Northern Europe, respectively10. Thus, our results based on autosomal data are consistent with Y DNA haplogroup data indicating multi-way admixture in the history of Kalash ancestry.
Figure 4. TreeMix analysis with one migration event.
The most frequently found tree with one migration event.
Figure 5. TreeMix analysis with one migration event.
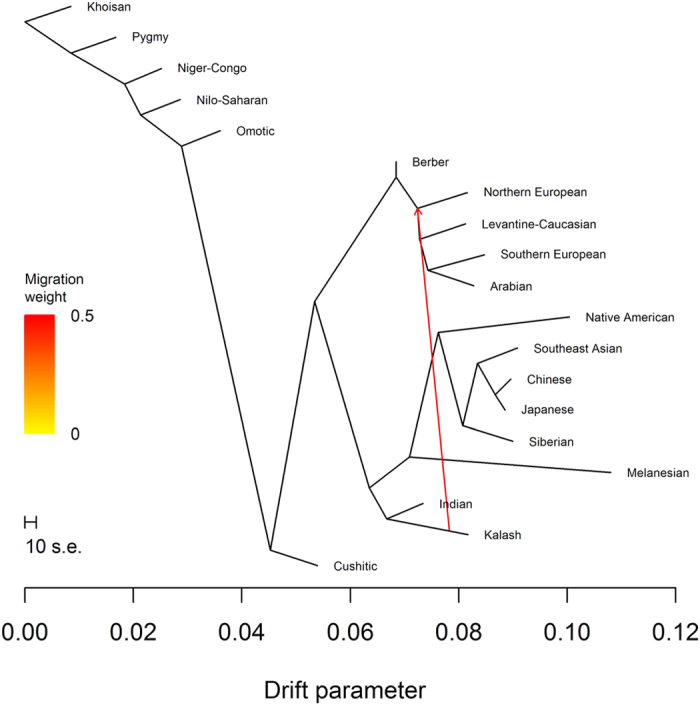
The second most frequently found tree with one migration event.
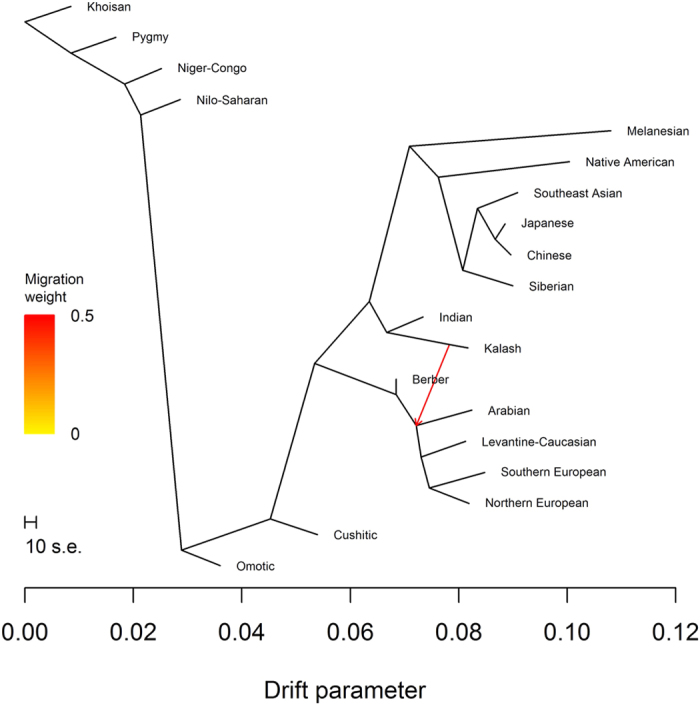
Conditional on two migration events, we observed the tree containing the (Northern European, Southern European) subtree 69% of the time (Fig. 6). The second migration event indicated gene flow between the common ancestor of Arabian, Levantine-Caucasian, Northern European, and Southern European ancestries and Cushitic ancestry, which grouped with Nilo-Saharan and Omotic ancestries. The weights suggested 39.9% ancestry closely related to Arabian ancestry and 60.1% ancestry closely related to Nilo-Saharan and/or Omotic ancestry. We observed the tree containing the (Southern European, Arabian) subtree 30% of the time (Fig. 7). For this tree, Cushitic ancestry no longer grouped with the other sub-Saharan African ancestries but was most closely related to Arabian ancestry, with gene flow from ancestry closely related to Nilo-Saharan and/or Omotic ancestry. The weights suggested 46.0% ancestry closely related to Nilo-Saharan and/or Omotic ancestry and 54.0% ancestry closely related to Arabian ancestry. Weighting these weights by the frequencies of the trees yielded estimates of 55.8% ancestry closely related to Nilo-Saharan and/or Omotic ancestry and 44.2% ancestry closely related to Arabian ancestry. Using the f4 statistics (Niger-Congo, Khoisan; Cushitic, Arabian) and (Niger-Congo, Khoisan; Nilo-Saharan, Arabian), we estimated a median mixture proportion of 0.412 (IQR 1.024), indicating 41.2% Nilo-Saharan and 58.8% Arabian ancestry. The f4 statistics (Niger-Congo, Khoisan; Cushitic, Arabian) and (Niger-Congo, Khoisan; Omotic, Arabian) yielded 41.7% Omotic ancestry and 58.3% Arabian ancestry, suggesting that Nilo-Saharan and Omotic ancestries are nearly equally good proxies for one parent of Cushitic ancestry.
Figure 6. TreeMix analysis with two migration events.
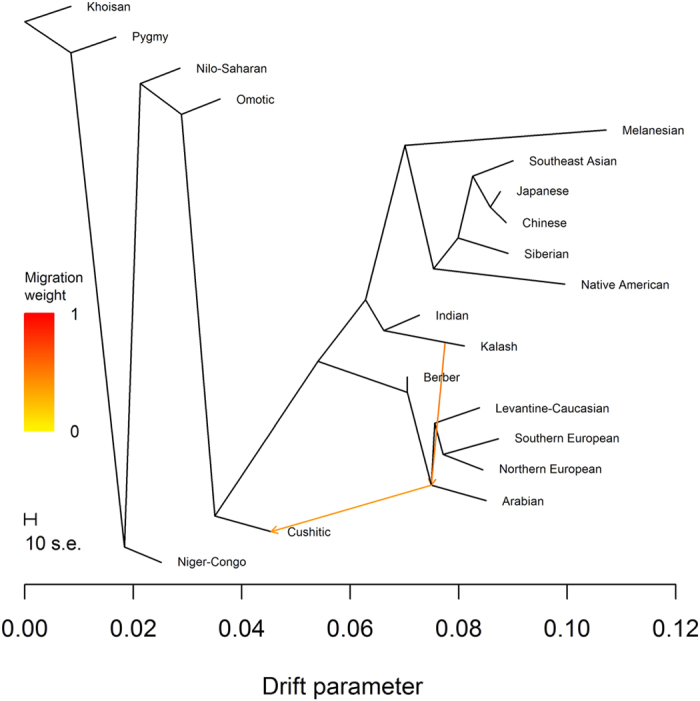
The most frequently found tree with two migration events.
Figure 7. TreeMix analysis with two migration events.
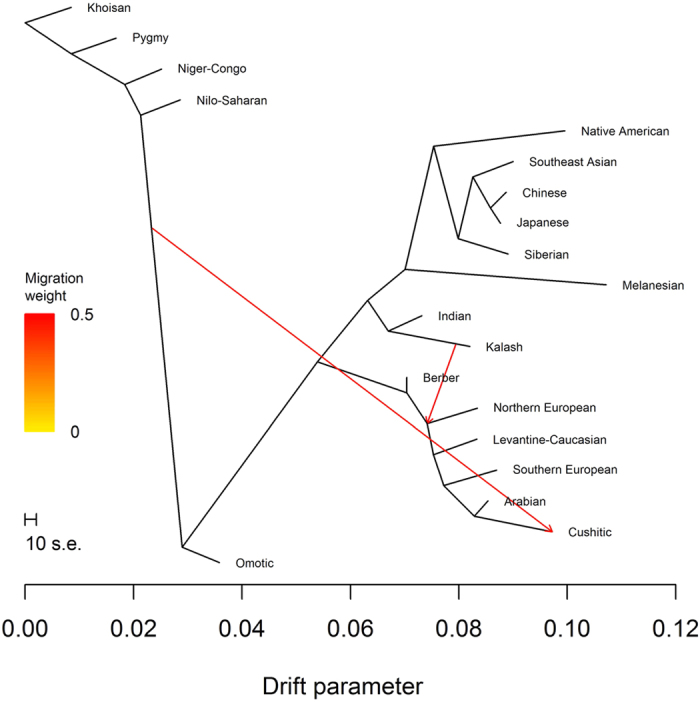
The second most frequently found tree with two migration events.
Conditional on three migration events, we observed the tree containing the (Northern European, Southern European) subtree 71% of the time (Fig. 8). The third migration event indicated gene flow involving 15.5% Native American ancestry and 84.5% Northern European ancestry. The migration edge connected the tips of the Native American and Northern European branches, implying an event more recent than the divergence of Y DNA haplogroups Q and R. Using (Levantine-Caucasian, Khoisan; Northern European, Native American) and (Levantine-Caucasian, Khoisan; Southern European, Native American), we estimated a median mixture proportion of 0.776 (IQR 0.916), indicating 22.4% Native American ancestry. Four f3 statistics supported Northern European ancestry as significantly admixed (Supplementary Table S2). The remaining trees showed no well-supported topologies and were inconsistent with the first two migration events so we do not discuss them further.
Figure 8. TreeMix analysis with three migration events.
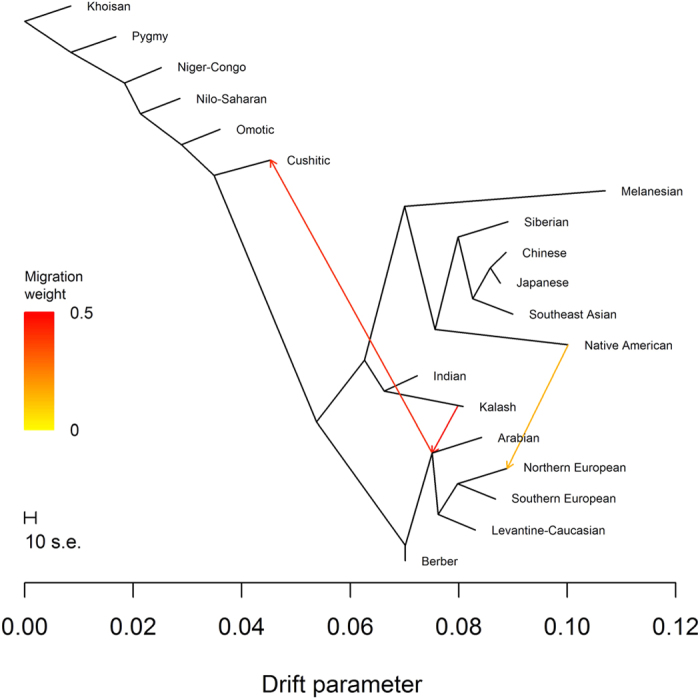
The most frequently found tree with three migration events.
Conditional on four migration events, we observed the tree containing the (Northern European, Southern European) subtree 79% of the time (Fig. 9). The fourth migration event indicated a mixture involving 42.1% Indian ancestry and 57.9% of the common ancestor of Arabian, Levantine-Caucasian, Northern European, and Southern European ancestries. Using (Southern European, Khoisan; Arabian, Indian) and (Southern European, Khoisan; Levantine-Caucasian, Indian), we estimated a median mixture proportion of 0.728 (IQR 1.582), indicating 27.2% Indian ancestry. It is possible that the second and fourth migration events are part of the same global-scale event, connecting India and East Africa. The migration event involving Kalash ancestry was also redefined to indicate specifically a mixture involving Northern European ancestry.
Figure 9. TreeMix analysis with four migration events.
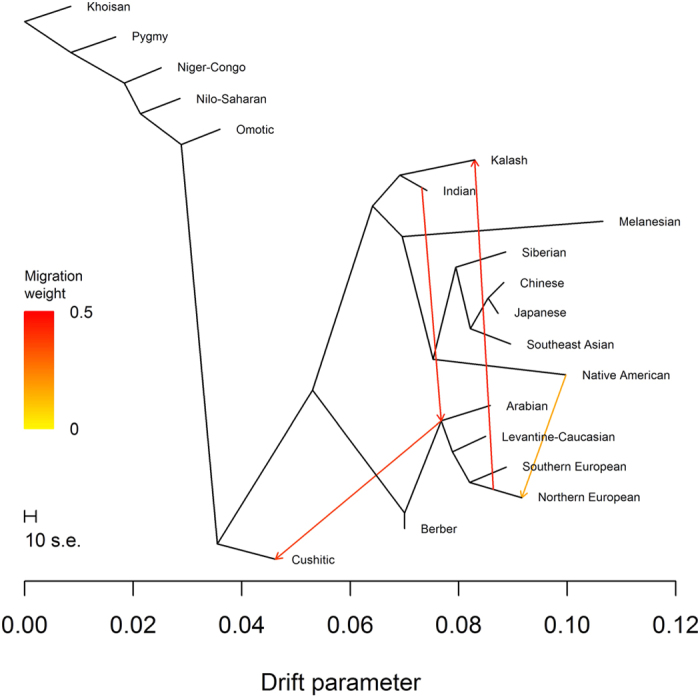
The most frequently found tree with four migration events.
Starting with five migration events, we observed positive log-likelihoods and the range of residuals stopped decreasing (Supplementary Figures S1 and S2). Despite low confidence, we report three additional migration events that may be real: the fifth migration event connects sub-Saharan Africa to an internal node ancestral to Chinese, Japanese, Melanesian, Native American, Siberian, and Southeast Asian ancestries; the sixth migration event connects Siberian and Northern European ancestries; and the seventh migration event connects the Kalash and Levantine-Caucasian ancestries (Supplementary Figure S2).
Discussion
Analyses of ancestries of modern humans typically assume a divergence under isolation model. Here, we performed analyses of human ancestry explicitly allowing for gene flow. We found evidence for four migration events, including one that reconciles the origin of Cushitic ancestry that has been the subject of debate in the literature.
Topologically, the distributions of graphs found by TreeMix were not unimodal. In the primary mode, Northern and Southern European ancestries were siblings, consistent with the distribution and genealogical relationship of Y DNA haplogroups R1a and R1b, respectively11. In the secondary mode, Southern European and Arabian ancestries were siblings, consistent with the distribution and genealogical relationship of Y DNA haplogroups I2 and J, respectively11.
The first migration event we detected is consistent with the distribution of Y DNA haplogroups in the Kalash people: H and L, common in India and South Asia, respectively, mixed with G, J, and R1a, common in the Levant and the Caucasus, the Middle East, and Northern Europe, respectively10. The mixture of Indian/South Asian, Levantine-Caucasian, and Northern European ancestries in the Kalash people has also been observed in several neighboring peoples, including the Baloch, Brahui, Burusho, Makrani, Pashtun, and Sindhi peoples in Pakistan3. Ayub et al.12 reported evidence of admixture approximately 100 generations ago between people with ancestry similar to the Chamars, the Kol, or Gujarati and people with ancestry similar to Armenians or Adygei. The presence of five Y DNA haplogroups in the Kalash people indicates at least four mixing events. The fourth migration event also redefined the first event to involve Kalash and Northern European ancestries, whereas the seventh event involved Levantine-Caucasian ancestry, thus accounting for two of the mixing events.
The second migration event indicates that Cushitic ancestry is a mixture of ancestries closely related to Nilo-Saharan/Omotic and Arabian ancestries. This migration event resolves the observed unstable placement between East African and Middle Eastern ancestries3,4,6: Cushitic ancestry has both East African and Middle Eastern origins. The fourth migration event indicated gene flow between Indian ancestry and the common ancestor of Arabian, Levantine-Caucasian, Northern European, and Southern European ancestries. In conjunction with the second migration event, this event connects India to East Africa. Taken together, these two events are consistent with the presence among East Africans of the mitochondrial DNA haplogroup M1, possibly reflecting migration from India to East Africa following the Last Glacial Maximum13,14,15. In the Nilotic Maasai, the presence of 50% Y DNA haplogroup E1b1b and the absence of Y DNA haplogroups J and T16 suggest a depletion of male Arabian ancestry, raising the possibility that Cushitic ancestry formed as a mixture of male-biased Nilo-Saharan/Omotic ancestry and female-biased Arabian ancestry.
To interpret the third migration event, we note that the main Y DNA haplogroups consistent with Native American ancestry (detected as the major ancestry in Karitiana, Surui, Pima, Colombian, Maya, and MXL samples3) and Northern European ancestry (detected as the major ancestry in FIN, Lithuania, Russian, and Belorussia samples3) are Q17 and R1a11, respectively, which are siblings descended from haplogroup P. Given this relationship, the observation that Native American and Northern European ancestries do not group together suggests that more shared autosomal ancestry is female-derived rather than male-derived and that the migration event therefore represents male-biased gene flow. This event is unlikely to reflect migration of Yamnaya steppe herders during the Bronze Age because Yamnaya males are associated predominantly with Y DNA haplogroup R1b and, to a lesser extent, I2a18,19,20. Also, this event does not reflect migration of Siberians into Northeast Europe21, which is captured by the sixth migration event. Siberian ancestry, detected as the major ancestry in a sample of Yakut people, is distinct from both Native American and Northern European ancestries3 and is associated with Y DNA haplogroup N22. In contrast, this event is consistent with a low frequency of Y DNA haplogroup Q observed in Romania and Hungary23 and Bulgaria24. Given that the migration edge connected the external nodes of both ancestries, we hypothesize that the event was relatively recent and therefore involved Central or North Asians rather than Native Americans.
TreeMix potentially inferred migration involving sub-Saharan and Melanesian ancestries as the fifth migration event. One edge of this event was placed between Khoisan/Pygmy ancestries and Niger-Congo/Nilo-Saharan ancestries. Based on distributions of Y DNA haplogroups, this event falls between haplogroups A/B and E25. Y DNA haplogroup C is presently found in populations in Japan, Oceania, Mongolia and Siberia, Australia, and India26. Y DNA haplogroup D is presently found in Japan, Tibet, and the Andaman Islands. Thus, this event might be capturing elements of Y DNA haplogroups C and/or D remaining from the Out of Africa migrations. It is therefore possible that the long branch we previously observed for Melanesian ancestry resulted from gene flow of K haplogroups (and/or its descendants M, O, and S) into a pre-existing population with predominantly haplogroup C27.
In light of these findings, we re-evaluated ancestral divergence times3, excluding Cushitic, Kalash, and Melanesian ancestries. In one subtree, a divergence event occurred approximately 72,800 years ago between the ancestor of Khoisan and Pygmy ancestries and the ancestor of Omotic, Niger-Congo, and Nilo-Saharan ancestries. A second divergence event occurred approximately 71,500 years ago between the ancestor of Native American, Siberian, Southeast Asian, Japanese, and Chinese ancestries and the ancestor of Indian, Kalash, Arabian, Berber, Levantine-Caucasian, Northern European, and Southern European ancestries. These times are consistent with migration out of Africa involving Y DNA haplogroup DE followed by a second migration involving haplogroup CF28. Temporally, both of these events coincide with the beginning of marine isotope stage 4, a 1,000-year stadial or cold glacial period29.
Our current findings provide genetic evidence for four migration events in the distant past without requiring ancient DNA, although the absence of genotype data for ancestries limits our ability to make inferences, particularly about dates. Additionally, comparison of autosomal data with distributions of Y and mitochondrial DNA haplogroups suggests sex-biased gene flow. Taken together, analyses based on ancestries rather than present-day samples provide new insights into the migratory history of modern humans in the distant past.
Methods
We performed collection and quality control of genome-wide genotype data from 3,528 unrelated individuals from 163 global samples (Supplementary Table S1), as previously described3. These data are freely available at http://crggh.nih.gov/resources.cfm under Ancestry. Unsupervised ancestry analysis was performed using ADMIXTURE30, with the optimal value of the number of ancestral components K determined to be 19 by five-fold cross-validation, averaged over three runs with different starting seeds.
We used SplitsTree version 4.13.17 to perform split decomposition analysis. This analysis utilized the distance matrix based on pairwise FST between ancestries as reported by ADMIXTURE. We used the Q-residual score to assess deviation from tree-like behavior31.
We calculated the f3 statistic per marker as described in the Supplementary Text. We also derived the theoretical variance of the f3 statistic (Supplementary Text). Using unbiased means and variances, we calculated marker-specific z-scores, which we then combined using equal weighting into a single z-score (Supplementary Text). We tested all 2,907 possible combinations of the 19 ancestries, using the allele frequencies reported by ADMIXTURE. For each ancestry, the sum of the estimated proportions for each individual is an estimate of the effective sample size; hence, twice this sum is an estimate of the total allele count.
Using f4 statistics8,32, the mixture proportion can be estimated using the ratio 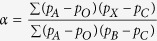 , with allele frequencies pX, pC, pB, pA, and pO in the putatively admixed ancestry X, proxies C and B of the putative parental ancestries, an ancestry A that is a sibling to ancestry B, and outgroup ancestry O, respectively. Both summations are over all markers. The ratio α is the mixture proportion of B and 1 − α is the mixture proportion of C. The f4 statistic follows the normal product distribution. Since the f4 statistic has finite mean and variance, both the numerator and denominator sums are normally distributed, by the Central Limit Theorem. Hence, α is a ratio of dependent normal variables. The distribution of the ratio of dependent normal variables follows a Cauchy-like distribution, which does not have finite moments33. Consequently, the sample estimator described by Reich et al.8 and Patterson et al.32 is inconsistent, as the sample mean is undefined and the sample variance is infinite. Instead, we calculated the ratio per marker and reported sample medians and interquartile ranges across markers. For a Cauchy distribution, the sample median is an estimate of the location parameter and the interquartile range is twice the scale parameter. Note that
, with allele frequencies pX, pC, pB, pA, and pO in the putatively admixed ancestry X, proxies C and B of the putative parental ancestries, an ancestry A that is a sibling to ancestry B, and outgroup ancestry O, respectively. Both summations are over all markers. The ratio α is the mixture proportion of B and 1 − α is the mixture proportion of C. The f4 statistic follows the normal product distribution. Since the f4 statistic has finite mean and variance, both the numerator and denominator sums are normally distributed, by the Central Limit Theorem. Hence, α is a ratio of dependent normal variables. The distribution of the ratio of dependent normal variables follows a Cauchy-like distribution, which does not have finite moments33. Consequently, the sample estimator described by Reich et al.8 and Patterson et al.32 is inconsistent, as the sample mean is undefined and the sample variance is infinite. Instead, we calculated the ratio per marker and reported sample medians and interquartile ranges across markers. For a Cauchy distribution, the sample median is an estimate of the location parameter and the interquartile range is twice the scale parameter. Note that 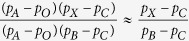 , with equality if pA − pO ≠ 0, in which the right-hand side is the estimator of the mixing proportion assuming αpB + (1 − α)pC = pX (i.e., A and O are unnecessary).
, with equality if pA − pO ≠ 0, in which the right-hand side is the estimator of the mixing proportion assuming αpB + (1 − α)pC = pX (i.e., A and O are unnecessary).
We converted ADMIXTURE output for TreeMix9 by rounding the allele counts to the nearest integer, using the allele frequencies and total allele counts as described above. We assigned Khoisan ancestry as the root. We set the number of migration events from 0 to 7. For each number of migration events, we ran 100 random input orders.
Ethics
This project was determined to be excluded from IRB Review by the National Institutes of Health Office of Human Subjects Research Protections, Protocol #12183.
Additional Information
How to cite this article: Shriner, D. et al. Ancient Human Migration after Out-of-Africa. Sci. Rep. 6, 26565; doi: 10.1038/srep26565 (2016).
Supplementary Material
Acknowledgments
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official view of the National Institutes of Health. This research was supported by the Intramural Research Program of the Center for Research on Genomics and Global Health (CRGGH). The CRGGH is supported by the National Human Genome Research Institute, the National Institute of Diabetes and Digestive and Kidney Diseases, the Center for Information Technology, and the Office of the Director at the National Institutes of Health (Z01HG200362).
Footnotes
Author Contributions D.S. designed the study, performed the analyses and drafted the manuscript. D.S., F.T.-A., A.A. and C.N.R. interpreted the results and edited the manuscript.
References
- Cruciani F. et al. A revised root for the human Y chromosomal phylogenetic tree: the origin of patrilineal diversity in Africa. Am. J. Hum. Genet. 88, 814–818 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poznik G. D. et al. Sequencing Y chromosomes resolves discrepancy in time to common ancestor of males versus females. Science 341, 562–565 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriner D., Tekola-Ayele F., Adeyemo A. & Rotimi C. N. Genome-wide genotype and sequence-based reconstruction of the 140,000 year history of modern human ancestry. Sci. Rep. 4, 6055 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff S. A. et al. The genetic structure and history of Africans and African Americans. Science 324, 1035–1044 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. Z. et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319, 1100–1104 (2008). [DOI] [PubMed] [Google Scholar]
- Hodgson J. A., Mulligan C. J., Al-Meeri A. & Raaum R. L. Early back-to-Africa migration into the Horn of Africa. PLoS Genet. 10, e1004393 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson D. H. & Bryant D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2006). [DOI] [PubMed] [Google Scholar]
- Reich D., Thangaraj K., Patterson N., Price A. L. & Singh L. Reconstructing Indian population history. Nature 461, 489–494 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell J. K. & Pritchard J. K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, e1002967 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firasat S. et al. Y-chromosomal evidence for a limited Greek contribution to the Pathan population of Pakistan. Eur. J. Hum. Genet. 15, 121–126 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikipedia. Y-DNA haplogroups in European populations. (2015) Available at: https://en.wikipedia.org/wiki/Y-DNA_haplogroups_in_European_populations. Accessed 12 August 2015.
- Ayub Q. et al. The Kalash genetic isolate: ancient divergence, drift, and selection. Am. J. Hum. Genet. 96, 775–783 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maca-Meyer N., González A. M., Larruga J. M., Flores C. & Cabrera V. M. Major genomic mitochondrial lineages delineate early human expansions. BMC Genet. 2, 13 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- González A. M. et al. Mitochondrial lineage M1 traces an early human backflow to Africa. BMC Genomics 8, 223 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark P. U. et al. The Last Glacial Maximum. Science 325, 710–714 (2009). [DOI] [PubMed] [Google Scholar]
- Wood E. T. et al. Contrasting patterns of Y chromosome and mtDNA variation in Africa: evidence for sex-biased demographic processes. Eur. J. Hum. Genet. 13, 867–876 (2005). [DOI] [PubMed] [Google Scholar]
- Wikipedia. Y-DNA haplogroups in indigenous peoples of the Americas. (2015) Available at: https://en.wikipedia.org/wiki/Y-DNA_haplogroups_in_indigenous_peoples_of_the_Americas. Accessed 12 August 2015.
- Allentoft M. E. et al. Population genomics of Bronze Age Eurasia. Nature 522, 167–172 (2015). [DOI] [PubMed] [Google Scholar]
- Haak W. et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 522, 207–211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson I. et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 528, 499–503, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der Sarkissian C. et al. Ancient DNA reveals prehistoric gene-flow from Siberia in the complex human population history of North East Europe. PLoS Genet. 9, e1003296 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tambets K. et al. The western and eastern roots of the Saami–the story of genetic “outliers” told by mitochondrial DNA and Y chromosomes. Am. J. Hum. Genet. 74, 661–682 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Cruz B. et al. Y-chromosome analysis in individuals bearing the Basarab name of the first dynasty of Wallachian kings. PLoS ONE 7, e41803 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karachanak S. et al. Y-chromosome diversity in modern Bulgarians: new clues about their ancestry. PLoS ONE 8, e56779 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikipedia. Y-DNA haplogroups by populations of Sub-Saharan Africa. (2015) Available at: https://en.wikipedia.org/wiki/Y-DNA_haplogroups_by_populations_of_Sub-Saharan_Africa. Accessed 12 August 2015.
- Wikipedia. Haplogroup C-M130. (2015) Available at: https://en.wikipedia.org/wiki/Haplogroup_C-M130. Accessed 12 August 2015.
- Wikipedia. Y-DNA haplogroups in Oceanian populations. (2015) Available at: https://en.wikipedia.org/wiki/Y-DNA_haplogroups_in_Oceanian_populations. Accessed 12 August 2015.
- Underhill P. A. & Kivisild T. Use of Y chromosome and mitochondrial DNA population structure in tracing human migrations. Annu. Rev. Genet. 41, 539–564 (2007). [DOI] [PubMed] [Google Scholar]
- Lisiecki L. E. & Raymo M. E. A Pliocene-Pleistocene stack of 57 globally distributed benthic d18O records. Paleoceanography 20, PA1003 (2005). [Google Scholar]
- Alexander D. H., Novembre J. & Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R. D., Bryant D. & Greenhill S. J. On the shape and fabric of human history. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365, 3923–3933 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N. et al. Ancient admixture in human history. Genetics 192, 1065–1093 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedilnik A., Košmelj K. & Blejec A. The Distribution of the Ratio of Jointly Normal Variables. Metodološki zvezki 1, 99–108 (2004). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.