

Abstract
RNA has become an increasingly important target for therapeutic interventions and for chemical probes that dissect and manipulate its cellular function. Emerging targets include human RNAs that have been shown to directly cause cancer, metabolic disorders, and genetic disease. In this review, we describe various routes to obtain bioactive compounds that target RNA, with a particular emphasis on the development of small molecules. We use these cases to describe approaches that are being developed for target validation, which include target-directed cleavage, classic pull-down experiments, and covalent cross-linking. Thus, tools are available to design small molecules to target RNA and to identify the cellular RNAs that are their targets.
Introduction
Nucleic acids have critical functions in cellular biology: They form genetic material and the machinery that converts genetic material into protein. RNA is a particularly attractive target as it folds into complex three-dimensional structures that result in its diverse cellular functions (1, 2). Further, RNA is dysregulated or mutated in disease (3, 4). Taken together, RNA is an important target for lead therapeutics and chemical probes of function. There are two main classes of RNA-targeting modalities: oligonucleotides (5) and small molecules (6–8).
Historically, RNAs have been most commonly targeted with oligonucleotides, due to their ease of design using Watson–Crick base-pairing rules (Figure 1). Oligonucleotides are high–molecular weight compounds that are not inherently cell permeable. However, various modifications have been developed that allow this class of compound to traverse cell membranes and disease-affected tissues. For example, gapmer oligonucleotides—oligonucleotides that recruit ribonuclease H (RNase H) to cleave the RNA strand in an RNA–DNA hybrid—have proven efficacious in cellular and animal models (9). Alternatively, modified oligonucleotides that do not induce RNase H cleavage can modulate RNA function by sterically blocking translation, inhibiting the formation of toxic RNA-protein interactions, or covering up disease-associated cryptic splice sites (Figure 2) (10).
Figure 1.
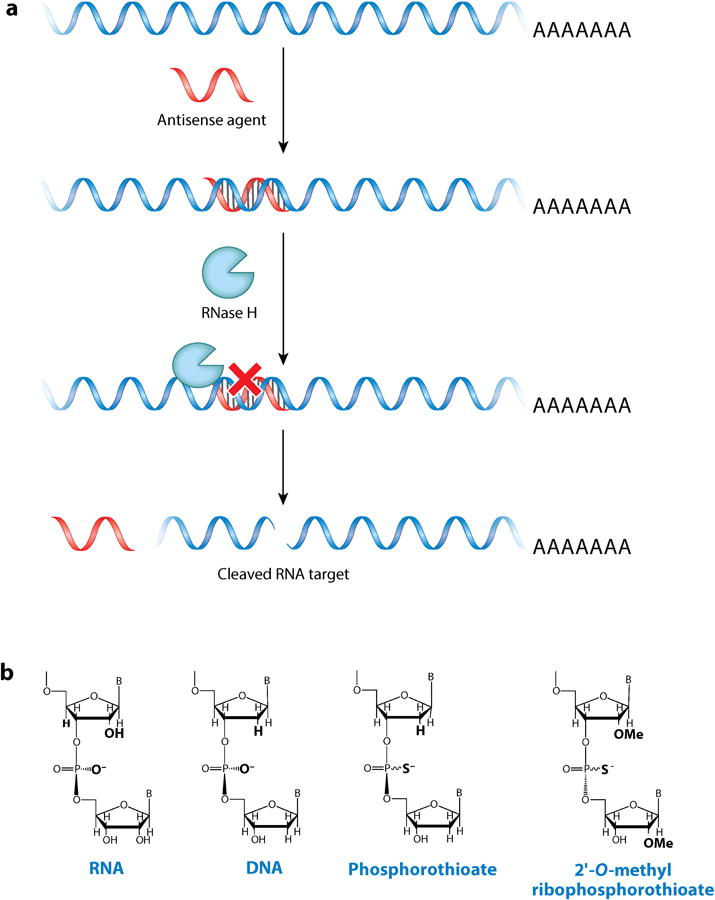
Antisense oligonucleotides and their general mode of action in cells. (a) An antisense oligonucleotide recruits ribonuclease H (RNase H), which cleaves the RNA strand and decreases RNA abundance. (b) RNA sugar and backbone modifications have been used to enhance the effect of oligonucleotides in cells. Note that phosphorothioate backbones are chiral.
Figure 2.
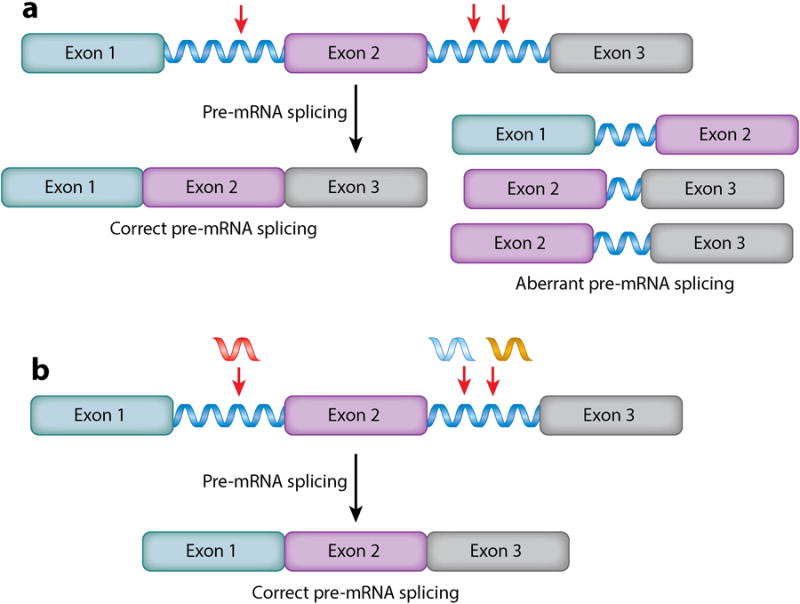
Modulation of RNA function by oligonucleotides that do not recruit ribonuclease H (RNase H). Such oligonucleotides can be potently bioactive, affecting precursor messenger RNA (pre-mRNA) splicing outcomes, for example. 2′-O-methyl phosphorothioates do not induce RNase H–dependent cleavage of their RNA targets; however, they can be used to target single nucleotide polymorphisms (SNPs) that cause pre-mRNA splicing defects. (a) Normal pre-mRNA splicing of β-globin pre-mRNA and aberrant pre-mRNA splicing when SNPs are present. Red arrows indicate positions of SNPs that activate cryptic splice sites. (b) Oligonucleotides that bind SNPs cover up cryptic splice sites and direct splicing patterns back to wild type.
More recently, various groups have shown that RNA is druggable with small molecules despite the inherent difficulty (Figure 3) (6–8). Although most of the collective work has been focused on targeting the bacterial ribosome (11–13), there is emerging activity targeting human RNAs (6). For example, small molecules have been designed to target RNA repeats involved in human orphan genetic diseases (Figure 4) (14–20) and microRNA (miRNA) precursors involved in many diseases, including cancer (21–23).
Figure 3.
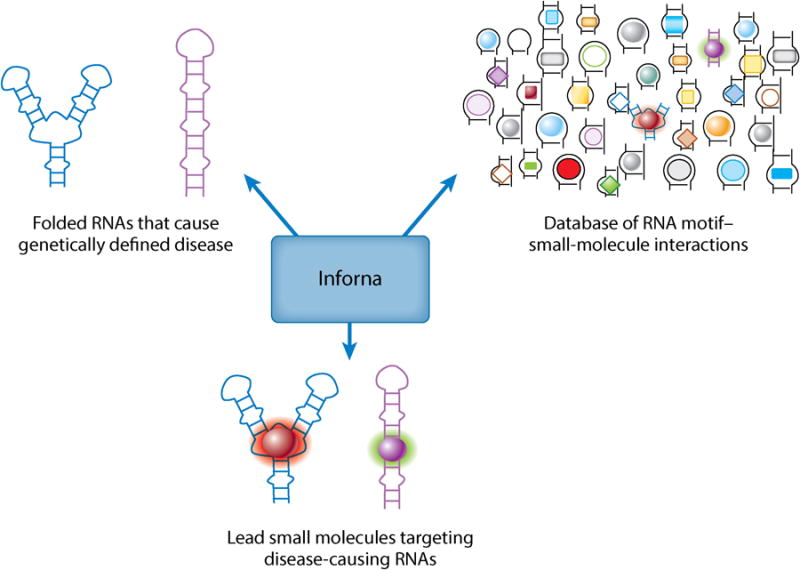
Inforna facilitates the design of small molecules to target RNA. This approach uses RNA secondary structure to inform rational design of small molecules. Lead small molecules are generated by computationally mining the motifs in an RNA target and comparing them to an annotated database of RNA motif–small-molecule interactions. These leads serve as starting points for chemical probe and lead therapeutic design.
Figure 4.
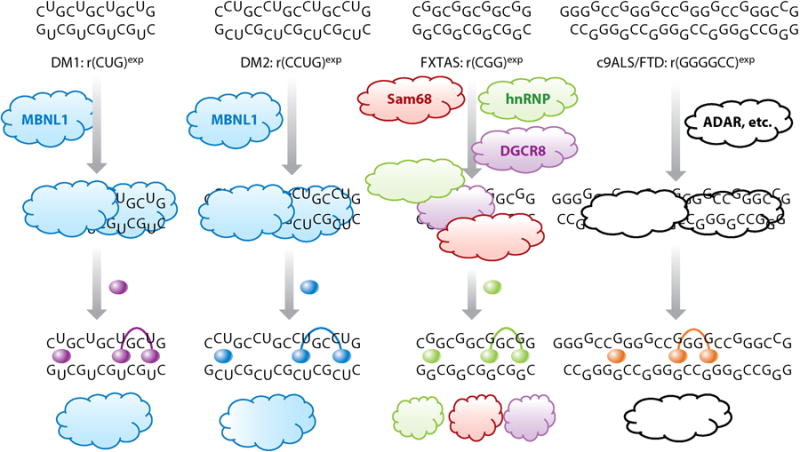
RNA repeat expansions cause microsatellite disorders. The secondary structures of these RNAs are typically extended hairpins that bind to and sequester proteins involved in RNA biogenesis (clouds). Small-molecule leads (spheres) that target motifs within these expanded RNA structures can be used to design monomeric or multivalent compounds that displace or inhibit protein binding. Release of sequestered proteins improves disease-associated defects, including alternative precursor messenger RNA splicing defects, formation of inclusions, production of toxic repeat-associated non-ATG (RAN) proteins, and RNA-mediated DNA silencing. Abbreviations: ADAR, adenosine deaminase acting on RNA; c9ALS/FTD, C9ORF72-related amyotrophic lateral sclerosis and frontal temporal dementia; DGCR8, DiGeorge syndrome chromosomal region 8; hnRNP, heterogeneous nuclear ribonucleoprotein; MBNL1, muscleblind-like 1 protein; Sam68, Src-associated substrate in mitosis of 68 kDa.
In this review, we highlight oligonucleotide-based modalities, emerging approaches to design and study low–molecular weight small molecules that target RNA, and the development of strategies to validate the targets and pathways that these RNA-targeting compounds modulate. Strategies to decipher the effects of these compounds in cells include functional assays, deep sequencing (RNA-seq) and quantitative real-time polymerase chain reaction (qRT–PCR) (24); noncovalent target pull-down assays (18); and chemical cross-linking (19, 25). Each case demonstrates that RNA is druggable via rational and predictable methods and that small molecules can bind to the desired RNA targets in cellulo (22).
Targeting Rna with Oligonucleotides
Development of Oligonucleotides and Important Considerations
Antisense oligonucleotides were first developed by Zamecnik's (26, 27) group to target Rous sarcoma virus. They used complementary DNAs to bind to the virus and affect the production of its RNA. Subsequent studies showed that DNA oligonucleotides form DNA–RNA hybrids in cells, which recruit RNase H and result in cleavage of the RNA strand (Figure 1) (28). Since then, there has been intensive effort to expand oligonucleotide-based targeting agents. For example, phosphorothioate-containing oligonucleotides (Figure 1), in which the nonbridging oxygen atom in a phosphodiester backbone is replaced with a sulfur atom, were developed to improve stability against endogenous cellular nucleases while maintaining RNase H–dependent activity (29). Other modified oligonucleotides were developed with site-specific modifications to both the backbone and the sugar, which dramatically improved the potential of these compounds as preclinical molecules to treat human disease (30–32).
Deep investigations into sites that can be targeted with antisense oligonucleotides revealed, not surprisingly, that unstructured regions are more accessible to oligonucleotide binding than structured ones (33). Because antisense oligonucleotides must form a duplex upon target recognition, self-structure in the target RNA can impede binding. In contrast, small molecules generally target structured regions of RNA (vide infra); thus these two modalities could be synergistic.
Specific Applications of Oligonucleotides: Target Modulation and Identification
In one of many initial examples of the potential of oligonucleotide-based therapeutics, phosphorothioate oligonucleotides were developed to target human C-raf kinase as anticancer compounds (34). The oligonucleotides were active in vivo and efficacy was correlated with ability to hybridize with their target messenger RNA (mRNA). Target engagement and validation were determined by using Northern blot analysis to study the oligonucleotide's effect on the target RNA's abundance; nanomolar concentrations were sufficient to see near-complete or partial knockdown of the target mRNA. The effect on tumor growth in animal models correlated well with in cellulo knockdown data. One advantage of antisense oligonucleotides is that target validation is relatively straightforward as the extent of target engagement is correlated with target abundance.
In addition to targeting mRNAs, oligonucleotides have also been employed to target RNAs with single nucleotide polymorphisms (SNPs) that lead to disease. Various diseases are caused by SNPs that activate cryptic splice sites and dysregulate alternative precursor mRNA (pre-mRNA) splicing (Figure 2). Kole's (10, 35, 36) group targeted cryptic splice sites caused by SNPs in the β-globin pre-mRNA (A110G in intron 1; U705G and C654U in intron 2) that cause β-thalassemia (Figure 2). By using 2′-O-methyl ribooligonucleotides to target these sites (Figure 1), pre-mRNA splicing was directed toward normal intron–exon junctions. Although this first study demonstrated the utility of this approach in vitro, numerous follow-up studies have shown that the approach also works in vivo. Interestingly, a green fluorescent protein (GFP) reporter construct has been developed, which generates GFP when pre-mRNA splicing defects are corrected (37). This reporter has been used to study cellular activity and tissue permeability of oligonucleotides.
Another class of RNA targets to which oligonucleotides have been applied is RNA repeating transcripts (Figure 4) (38–40). Expanded RNA repeats are associated with various diseases including myotonic dystrophy types 1 and 2 (DM1 and DM2, respectively) (41, 42); C9ORF72-related amyotrophic lateral sclerosis and frontal temporal dementia (c9ALS/FTD) (43, 44); and Huntington's disease (HD) (45). To date, a DM1-targeting oligonucleotide (46) is showing efficacy in clinical trials, which has broad implications for other microsatellite disorders (38–40).
DM1 is caused by an expanded r(CUG) repeat [r(CUG)exp] present in the 3′ untranslated region (UTR) of the dystrophia myotonica protein kinase (DMPK) mRNA (Figure 4). When the repeat is of sufficient (pathogenic) length, it binds to and sequesters proteins such as muscleblind-like 1 protein (MBNL1), a pre-mRNA splicing regulator. Thus, alternative pre-mRNA splicing is dysregulated in DM1. Initial studies using oligonucleotides showed that they act as steric blockers of protein binding. A 2′-O-methyl phosphorothioate (47) and a morpholino oligonucleotide (48) improved DM1-associated splicing defects in patient-derived cells and a mouse model. The latter oligonucleotide backbone is attractive because morpholinos exhibit little toxicity and have uncharged backbones. In both of these cases, target engagement was inferred by knockdown of the target RNA. Although the exact mechanism of this decrease is unclear, it is likely not mediated by RNase H, as morpholino backbones are known not to trigger RNase H cleavage. A gapmer oligonucleotide, consisting of DNA residues (which support RNase H activity), flanked by 2′-O-methoxyethyl residues on both the 5′ and 3′ ends (which confer nuclease stability), was used to target the DMPK mRNA coding sequence. The oligonucleotide, which is currently in clinical trials, strongly knocked down DMPK mRNA abundance and, remarkably, improved pre-mRNA splicing defects one year after treatment was discontinued (46).
Oligonucleotides have also been used to decrease expression of mutant huntingtin protein (HTT), which contributes to HD (49). The mutant huntingtin protein contains polyglutamine, which is encoded by an expanded r(CAG) repeat. Peptide nucleic acids, complementary to both a sequence 5′ to the repeat and the r(CAG) repeats themselves, suppress expression of the mutant, but not wild-type, huntingtin protein at certain concentrations and do not affect mutant HTT mRNA abundance (50). These studies are a boon for the development of compounds that selectively inhibit the translation of toxic proteins found in many human diseases.
Antisense oligonucleotides have also been developed to destroy the r(G4C2) RNA repeat expansion [r(G4C2)exp] that causes c9ALS/FTD (19, 43, 44, 51, 52). These oligonucleotides bind a nonrepeating region of C9ORF72 mRNA (53, 54). Similar to the RNAs that cause DM1 and HD, r(G4C2)exp causes toxicity in two ways: by sequestering nuclear proteins and producing toxic proteins generated by repeat-associated non-ATG (RAN) translation (52, 55–57). (In RAN translation, the repeats serve as internal ribosome entry sites and are translated without a start codon.) In various cellular and animal systems, it has been shown that antisense oligonucleotides knock down expression of C9ORF72 mRNA, inhibit production of toxic RAN proteins, and have no inherent toxicity.
Development of Strategies to Design Small Molecules to Target Structured RNAs
In the previous section, methods for targeting RNA with oligonucleotides were described. One of the major advantages of this approach is that oligonucleotide design follows simple Watson–Crick base-pairing rules. The rules for targeting RNA with small molecules are much more complex and are only now beginning to emerge, as RNA has traditionally been recalcitrant to small-molecule intervention (Figure 3). Challenges include (a) nonspecific interactions of cationic drugs due to RNA's negatively charged backbone; (b) perceived structural redundancy in cellular RNAs due to the fact that RNA has only four building blocks, which could limit selectivity; (c) structural dynamics of RNA, which can hinder the use of computational methods, such as docking, to define or refine RNA binders; and (d) limited suitability of small-molecule screening collections for RNA binding, as most were developed for protein targets. Screening hit rates for nucleic acids are often much lower than hit rates for proteins.
One major advantage of targeting RNA, however, is that RNA folds into diverse structures (Figures 3 and 4). For example, structured RNAs are composed of base pairs and noncanonically paired regions such as hairpins, internal loops, and bulges. These latter regions can be unique to a single RNA, or a small subset, whereas base-paired regions are generally common to all RNAs. Furthermore, there are various methods available to gain insight into the secondary structural fold of a given RNA (58–63).
We recently reported a lead identification strategy named Inforna (21, 22) that compares secondary structural elements in a target RNA to known RNA motif–small-molecule interactions that are highly selective (Figure 3). These interactions were identified using a library-versus-library strategy in which a library of small molecules (chemical microarray) is screened for binding to a library of RNA motifs such as hairpins, internal loops, and bulges (64, 65). Inforna has informed the design of lead small molecules that target RNAs responsible for microsatellite disorders and miRNAs associated with cancer (Figures 4 and 5).
Figure 5.
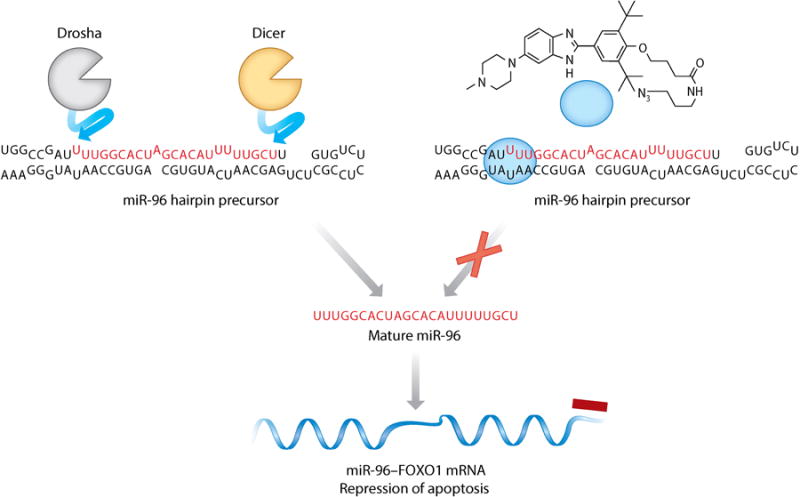
Inforna can be used to design small molecules that target microRNA (miRNA) precursors. A target agnostic approach was used in which the results of two-dimensional combinatorial screening determine the optimal target. This approach has identified multiple bioactive partners, including a small-molecule benzimidazole (blue sphere) that binds to the Drosha nuclease processing site in the microRNA-96 (miR-96) hairpin precursor. The small molecule selectively modulates the activity of this RNA, increases production of forkhead box protein O1 (FOXO1), and triggers apoptosis.
Other methods have also been used to design small molecules to target RNA, including structure-aided drug design, which has been particularly useful given the explosion of ribosome structures that were refined in the early 2000s (11–13, 66, 67). These structures identified specific contact sites and orientations of antibacterials with their RNA target, facilitating the development of improved compounds (68). Structure-aided design has also proven useful for the design of small molecules that mimic the binding of proteins to RNA. For example, various peptidomimetics have been designed to target the HIV trans-activation response element (HIV TAR), providing compounds that are bioactive in cell culture (69–74). Another important approach is docking of small molecules into RNA dynamic ensembles generated using a combination of NMR spectroscopy and molecular dynamics simulations (75, 76). Such studies identified various small molecules that target HIV TAR.
Selective Targeting of RNA Repeating Transcripts
In general, RNA repeating transcripts fold into a hairpin structure that contains a repeating motif in the stem (Figure 4). We have therefore utilized Inforna to identify small-molecule modules that target these repeating motifs. We have also implemented a multivalent approach to custom assemble RNA-binding modules so that they mimic the periodicity of the targetable motifs in the transcript (77). If RNAs cause disease by sequestering protein, as in the case of DM1, then the small molecule could displace RNA-bound protein in cellulo or in vivo and improve cellular defects caused by sequestration. Indeed, monomeric and multivalent compounds have been developed against the RNAs that cause DM1, DM2, c9ALS/FTD, and fragile X-associated tremor ataxia syndrome (FXTAS) (18, 78, 79). In general, bioactive compounds improve disease-associated defects in cellulo in the low- to mid-micromolar range (18, 78, 79).
In Cellulo Synthesis of Inhibitors of Repeating Transcripts
One potential liability of multivalent compounds, despite their improved affinities, selectivities, and potencies, is their high molecular weights, which could decrease cellular and tissue permeability (Figure 6). Thus, we sought to develop novel strategies to allow custom synthesis of multivalent compounds in cellulo to exploit the favorable properties of both monomeric (permeability) and multivalent ligands (potency, affinity, selectivity). Repeating transcripts are ideal targets for this approach as they are modular, like the compounds they template.
Figure 6.
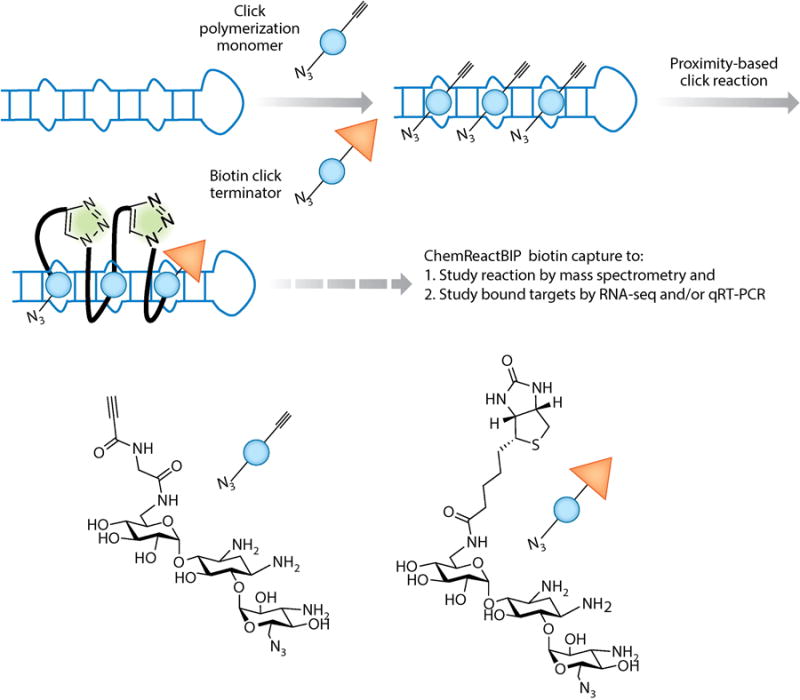
Using a disease-affected cell to synthesize its own drug at the required site of action. This approach is enabled by using click chemistry in which azide and alkyne modules are added to a compound (blue spheres) at positions that are brought into close proximity upon binding to a cellular target, thereby affording a multivalent compound. Here, we illustrate the approach using r(CCUG)exp, the causative agent of myotonic dystrophy type 2 (DM2). ChemReactBIP uses a biotin terminator to identify the cellular catalyst(s) of the click polymerization reaction by qRT-PCR and the extent of polymerization by mass spectrometry. The biotin terminator terminates the click reaction and allows pull down of the bound catalyst with streptavidin beads. Abbreviations: ChemReactBIP, chemical reactivity and binding isolated by pull down; qRT-PCR, quantitative real-time polymerase chain reaction.
Click chemistry enabled our approach; azide and alkyne modules were site-selectively added to small molecules that bind RNA repeats (80–83). In particular, we modeled binding of a bioactive dimeric compound to the r(CCUG) repeats that cause DM2 to inform positioning of azide and alkyne functional groups (78). The two groups are unreactive unless brought into close proximity (by binding to adjacent sites on the RNA) or exposed to a metal catalyst. Indeed, the toxic DM2 RNA is the catalyst for chemical transformation in cellulo, and the bifunctional small molecule is a nanomolar inhibitor of r(CCUG)exp dysfunction (84). In cellulo synthesis of multivalent compounds, and catalytic activity of r(CCUG)exp, was confirmed using chemical reactivity and binding isolated by pull down (ChemReactBIP; described below in the section Methods for Target Validation of Small Molecules) (84). This click chemistry approach is likely broadly applicable to the development of precise medicines to target repeating RNAs (Figure 6). The use of low–molecular weight building blocks that assemble in cellulo may be particularly important for this class of RNA as microsatellite diseases are associated with brain dysfunction.
Targeting MicroRNAs Involved in Cancer
Inforna has also been used to agnostically identify disease-associated miRNA precursor hairpins that can be targeted with small molecules (Figure 5). In this iteration of the rational design approach, the sequences of all human miRNA hairpin precursors were folded computationally by free energy minimization to obtain their secondary structures (Figure 5). The RNA secondary structures for the Dicer and Drosha cleavage sites were then compared to our database of RNA motif–small-molecule interactions to identify lead compounds that might inhibit miRNA processing (22).
This analysis yielded over 20 lead interactions of which >40% were shown to inhibit production of the mature miRNA in cell lines. Of these interactions, the binding of a benzimidazole was shown to avidly and selectively inhibit the biogenesis of microRNA-96 (miR-96) hairpin precursor in cells (22). Importantly, the compound boosted production of a downstream transcription factor, forkhead box protein O1 (FOXO1), triggering apoptosis in breast cancer cells.
The targets of this benzimidazole compound were validated using various molecular biology tools. For example, qRT-PCR was used to determine target selectivity by measuring the effect of the compound on the abundance of other mature miRNAs. The selectivity observed for this cell-permeable small molecule rivaled that observed for oligonucleotides. Furthermore, a short interfering (si)RNA functional assay was used to decipher how selectively the compound induced apoptosis via the FOXO1–miR-96 pathway. Ablation of FOXO1 was expected to render cells insensitive to the apoptotic effects of the compound, and indeed this was the case, illustrating that the compound is a selective modulator of apoptosis (76).
Other compounds, namely aminoglycosides or derivatives thereof, have been identified that target miRNAs and affect biogenesis. These compounds were typically identified by screening libraries of compounds for inhibition of miRNA cleavage (23). However, a miR-10b modulator was identified by using the bottom-up approach enabled by Inforna (21).
Methods for Target Validation of Small Molecules
One of the challenges associated with developing small molecules to target RNA is the lack of in cellulo target validation methods. Target engagement for antisense oligonucleotides can be inferred by changes in RNA abundance, but such effects may not be intrinsically available for small molecules. A variety of methods have been used to identify whether small molecules bind to RNAs in vitro and the corresponding binding sites. A classic example of this is the work of the Noller group (85, 86) to identify sites within an RNA where small molecules protect against modification by Gilbert-Maxam-type sequencing reagents such as dimethyl sulfate, kethoxal, and 1-cyclohexyl-(2-morpholinoethyl)carbodiimide metho-p-toluene sulfonate. The positions of modification and protection by small molecules were determined by using reverse transcriptase and primer extension, with chemical modification inhibiting extension. These studies identified the binding sites of aminoglycosides within the bacterial ribosome (85, 86). Additionally, the protection of RNA from nuclease cleavage can be used to determine binding sites (22, 62). Lastly, in-line probing can be used to identify binding sites and alterations in RNA structure upon binding of ligands by using the general instability of RNA phosphodiester bonds; ligand binding alters the rates of cleavage in and around the binding site (87). Below, we describe emerging methods to measure target engagement of small molecules in cellulo.
Pull Down of RNA Targets
One of the first methods to identify the cellular targets of small molecules involved a pull-down experiment, analogous to immunoprecipitation, which identifies the cellular protein targets of ligands (88). This approach has been used by the Wong group (89) to gain insight into cellular RNAs that are bound by aminoglycosides, thereby representing potential off-targets. Binding was determined by applying the RNAs pulled down by aminoglycosides to DNA microarrays (89). In an analogous approach, we appended biotin to a bioactive compound and created an affinity matrix using streptavidin beads (Figure 7) (18). Total RNA was then incubated with the beads, and RNAs that bound the small molecule were identified by qRT-PCR. Indeed, this approach was used to confirm that small molecules can be designed to bind a desired target (18, 79). In one study, we showed that a small molecule designed to bind the r(CGG) repeat that causes FXTAS pulls down more of the desired target than an oligonucleotide complementary to the repeat (18).
Figure 7.
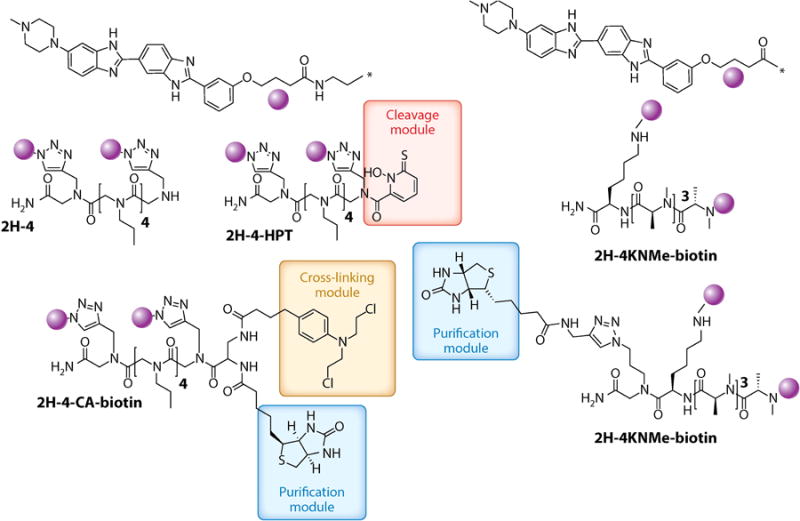
Structures of multivalent compounds that have been designed to target the expanded r(CUG) repeat RNA [r(CUG)exp] that causes myotonic dystrophy type 1 (purple spheres). The RNA-binding modules were identified by querying a database of RNA motif–small-molecule interactions to identify r(CUG)exp-binding modules. These modules were then systematically assembled onto various polyvalent scaffolds, such as peptoids (2H-4) or N-methyl peptides (2H-4KNMe), which allow precise control of RNA-binding module spacing and valency. Because of their modular nature, these compounds can be easily functionalized for target identification, for example, biotin, cross-linking agents [chlorambucil (CA)], and hydroxyl radical–producing moieties that cleave RNA such as N-hydroxylthiopyridine (HPT).
Chemical Cross-Linking and Isolation by Pull Down to Identify Targets in Cellulo
Although the above pull-down studies are informative, the small molecule pulls down targets outside of cells (Figures 7 and 8), which can cause biases in target recognition. Thus, we developed an in cellulo pull-down approach, chemical cross-linking and isolation by pull down (Chem-CLIP; Figure 8) (25). In Chem-CLIP, compounds are appended with a nucleic acid–reactive module such as chlorambucil (CA) and a purification tag (biotin). When these compounds are applied to cells, they react (cross-link) with their cellular targets, effectively biotinylating them (Figure 7). The cells are then lysed and the lysate is incubated with streptavidin beads. The captured targets can be analyzed by qRT-PCR (25) or RNA-seq. To control for nonselective cross-linking of the reactive module, a method called Competitive Chem-CLIP (C-Chem-CLIP) (19) was developed in which the reactive compound and its unreactive parent are co-administered (Figure 8). Bona fide sites for the noncovalent compound are thus depleted in the pulled-down fraction. Chem-CLIP and C-Chem-CLIP approaches have been applied to various RNA-targeting small-molecule campaigns. These include profiling new chemical entities that bind r(CUG)exp and validating that designer small molecules indeed bind to r(G4C2)exp in c9ALS/FTD. Interestingly, the reactive compound that forms a cross-link with r(CUG)exp is greater than 1,000-fold more potent than the parent compound from which it was derived (25). If reactivity could be controlled, then covalent modifiers could have utility in greatly enhancing the potency of designer compounds.
Figure 8.
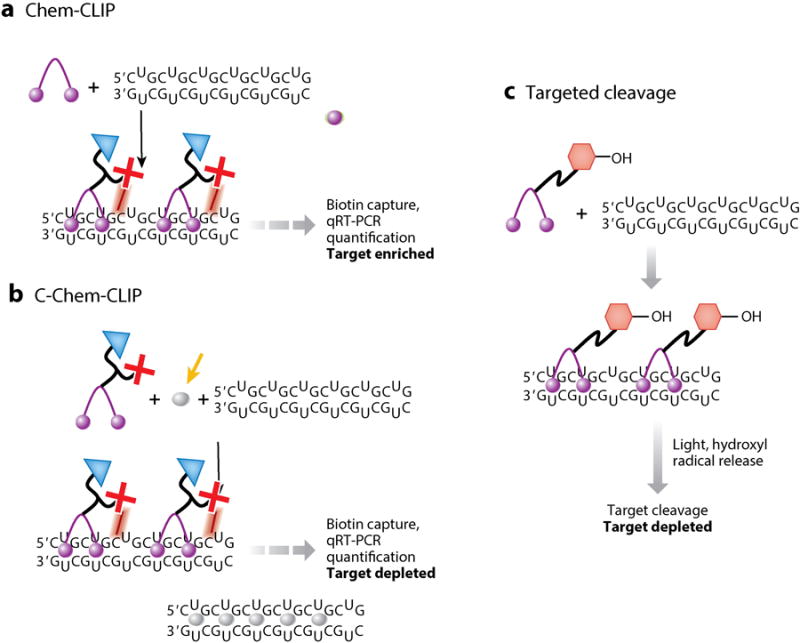
Schemes of chemical approaches used for target validation in cellulo. (a) In Chem-CLIP, a small molecule (purple sphere) is appended with a nucleic acid–reactive moiety, which cross-links to targets that it binds in cellulo, and biotin for facile isolation of small-molecule–RNA adducts. (b) In C-Chem-CLIP, a Chem-CLIP experiment is completed in the presence of an unreactive small molecule (gray sphere), which competes with the covalent probe for RNA binding, to infer the cellular targets of the small molecule in question. (c) In direct cleavage of RNA targets, small molecules are appended with modules that allow the targeted destruction of RNA in cellulo to validate a target. One example module is HPT (hexagons), which generates hydroxyl radicals that cleave RNA targets to which the small molecules bind. Abbreviations: Chem-CLIP, chemical cross-linking and isolation by pull down; C-Chem-CLIP, competitive chemical cross-linking and isolation by pull down; HPT, hydroxylthiopyridine; qRT-PCR, quantitative real-time polymerase chain reaction.
Covalent approaches have also been used to define the cellular targets of small molecules that are used clinically or to study specificity (90–95). For example, a variety of methods have been used to detect the RNAs that react with cisplatin. These important studies have shown that the ribosome is a target of these tried-and-true anticancer compounds (94).
Targeted Cleavage as a Means to Validate and Modulate Cellular Targets
In addition to Chem-CLIP, we developed another approach to validate targets of small molecules, which involves cleavage of RNAs that are bound by small molecules in cellular systems (Figures 7 and 8). Thus, target validation can be inferred by a reduction in the amount of a given RNA, akin to the effects measured for antisense agents. In this approach, we appended a modularly assembled compound (2H-4) that targets r(CUG)exp to N-hydroxylthiopyridine, which generates hydroxyl radicals that cleave RNA upon irradiation (Figure 7) (24). Indeed, the designed compound induced targeted cleavage of r(CUG)exp in cellulo and improved DM1-associated pre-mRNA splicing defects. Although this approach requires light for cleavage, the platform is useful to identify the cellular targets of a compound and could be further developed so that light is not required.
Chemical Reactivity and Binding Isolated by Pull Down
Above we described in cellulo synthesis of inhibitors of the RNA that causes DM2, r(CCUG)exp, using click chemistry (84). This strategy was developed into a target validation approach named chemical reactivity and binding isolated by pull down (ChemReactBIP; Figure 6). ChemReactBIP uses a derivative of the clickable compounds in which one of the reactive groups is switched to a biotin purification tag. Cells are co-treated with a click polymerization monomer (with both an azide and alkyne group) and a click terminator (in which biotin replaces the alkyne). This allows the reaction products and their cellular targets to be isolated from cells using streptavidin beads. Indeed, when this approach was applied to r(CCUG)exp, reaction of the clickable compounds occurred only in cells that expressed r(CCUG)exp and the products pulled down the desired RNA. Thus, these studies show that disease-affected cells can synthesize their own inhibitor and suggest that r(CCUG)exp RNA is the catalyst for the chemical transformation. This approach could be developed for other RNA targets, but will likely require optimization.
Conclusions & Outlook
There has been a rapid expansion in our understanding of the roles that RNA plays in both healthy and disease-affected cells. Thus, there is increased interest in the development of precise medicines that target RNA and affect cellular and in vivo function. Two methods were presented in this review: the use of oligonucleotides and emerging approaches that use small molecules. Indeed, some RNAs are likely more amendable to oligonucleotide-based targeting modalities (for example, largely unstructured RNAs), whereas others will be more amenable to small-molecule targeting (for example, highly structured RNAs). For small molecules, it is also important to develop approaches for target validation. Various emerging strategies have been presented, including pull down, Chem-CLIP, and ChemReactBIP. It is likely that these approaches will expand in the short and intermediate terms as advances are made in understanding how small molecules target RNA.
Acknowledgments
The work presented herein was funded by the National Institutes of Health (R01-GM097455) and the Muscular Dystrophy Association (254929) to M.D.D.
Footnotes
Disclosure Statement: The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Contributor Information
Jessica L. Childs-Disney, Email: jdisney@scripps.edu.
Matthew D. Disney, Email: disney@scripps.edu.
References
- 1.Batey RT, Rambo RP, Doudna JA. Tertiary motifs in RNA structure and folding. Angew Chem Int Ed Engl. 1999;38:2326–43. doi: 10.1002/(sici)1521-3773(19990816)38:16<2326::aid-anie2326>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 2.Doudna JA. Structural genomics of RNA. Nat Struct Biol. 2000;7(Suppl):954–56. doi: 10.1038/80729. [DOI] [PubMed] [Google Scholar]
- 3.Sicot G, Gomes-Pereira M. RNA toxicity in human disease and animal models: from the uncovering of a new mechanism to the development of promising therapies. Biochim Biophys Acta. 2013;1832:1390–409. doi: 10.1016/j.bbadis.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Visone R, Croce CM. MiRNAs and cancer. Am J Pathol. 2009;174:1131–38. doi: 10.2353/ajpath.2009.080794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swayze EE, Bhat B. The medicinal chemistry of oligonucleotides. In: Crooke ST, editor. Antisense Drug Technology: Principles, Strategies, and Applications. Boca Raton, FL: CRC; 2008. pp. 143–82. [Google Scholar]
- 6.Guan L, Disney MD. Recent advances in developing small molecules targeting RNA. ACS Chem Biol. 2012;7:73–86. doi: 10.1021/cb200447r. [DOI] [PubMed] [Google Scholar]
- 7.Gallego J, Varani G. Targeting RNA with small-molecule drugs: therapeutic promise and chemical challenges. Acc Chem Res. 2001;34:836–43. doi: 10.1021/ar000118k. [DOI] [PubMed] [Google Scholar]
- 8.Thomas JR, Hergenrother PJ. Targeting RNA with small molecules. Chem Rev. 2008;108:1171–224. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 9.Nesterova M, Cho-Chung YS. Killing the messenger: antisense DNA and siRNA. Curr Drug Targets. 2004;5:683–89. doi: 10.2174/1389450043345137. [DOI] [PubMed] [Google Scholar]
- 10.Sierakowska H, Agrawal S, Kole R. Antisense oligonucleotides as modulators of pre-mRNA splicing. Methods Mol Biol. 2000;133:223–33. doi: 10.1385/1-59259-215-5:223. Medline. [DOI] [PubMed] [Google Scholar]
- 11.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–48. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 12.Poehlsgaard J, Douthwaite S. The bacterial ribosome as a target for antibiotics. Nat Rev Microbiol. 2005;3:870–81. doi: 10.1038/nrmicro1265. [DOI] [PubMed] [Google Scholar]
- 13.Yonath A, Bashan A. Ribosomal crystallography: Initiation, peptide bond formation, and amino acid polymerization are hampered by antibiotics. Annu Rev Microbiol. 2004;58:233–51. doi: 10.1146/annurev.micro.58.030603.123822. Abstract. [DOI] [PubMed] [Google Scholar]
- 14.Gareiss PC, Sobczak K, McNaughton BR, Palde PB, Thornton CA, Miller BL. Dynamic combinatorial selection of molecules capable of inhibiting the (CUG) repeat RNA-MBNL1 interaction in vitro: discovery of lead compounds targeting myotonic dystrophy (DM1) J Am Chem Soc. 2008;130:16254–61. doi: 10.1021/ja804398y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jahromi AH, Nguyen L, Fu Y, Miller KA, Baranger AM, Zimmerman SC. A novel CUGexp·MBNL1 inhibitor with therapeutic potential for myotonic dystrophy type 1. ACS Chem Biol. 2013;8:1037–43. doi: 10.1021/cb400046u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Childs-Disney JL, Hoskins J, Rzuczek SG, Thornton CA, Disney MD. Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. ACS Chem Biol. 2012;7:856–62. doi: 10.1021/cb200408a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar A, Parkesh R, Sznajder LJ, Childs-Disney JL, Sobczak K, Disney MD. Chemical correction of pre-mRNA splicing defects associated with sequestration of muscleblind-like 1 protein by expanded r(CAG)-containing transcripts. ACS Chem Biol. 2012;7:496–505. doi: 10.1021/cb200413a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tran T, Childs-Disney JL, Liu B, Guan L, Rzuczek S, Disney MD. Targeting the r(CGG) repeats that cause FXTAS with modularly assembled small molecules and oligonucleotides. ACS Chem Biol. 2014;9:904–12. doi: 10.1021/cb400875u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron. 2014;83:1043–50. doi: 10.1016/j.neuron.2014.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pushechnikov A, Lee MM, Childs-Disney JL, Sobczak K, French JM, et al. Rational design of ligands targeting triplet repeating transcripts that cause RNA dominant disease: application to myotonic muscular dystrophy type 1 and spinocerebellar ataxia type 3. J Am Chem Soc. 2009;131:9767–79. doi: 10.1021/ja9020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Velagapudi SP, Disney MD. Two-dimensional combinatorial screening enables the bottom-up design of a microRNA-10b inhibitor. Chem Commun. 2014;50:3027–29. doi: 10.1039/c3cc00173c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Velagapudi SP, Gallo SM, Disney MD. Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat Chem Biol. 2014;10:291–97. doi: 10.1038/nchembio.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bose D, Jayaraj G, Suryawanshi H, Agarwala P, Pore SK, et al. The tuberculosis drug streptomycin as a potential cancer therapeutic: inhibition of miR-21 function by directly targeting its precursor. Angew Chem Int Ed Engl. 2012;51:1019–23. doi: 10.1002/anie.201106455. [DOI] [PubMed] [Google Scholar]
- 24.Guan L, Disney MD. Small molecule-mediated cleavage of RNA in living cells. Angew Chem Int Ed Engl. 2013;52:1462–65. doi: 10.1002/anie.201206888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guan L, Disney MD. Covalent small molecule-RNA complex formation enables cellular profiling of small molecule-RNA interactions. Angew Chem Int Ed Engl. 2013;52:10010–13. doi: 10.1002/anie.201301639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stephenson ML, Zamecnik PC. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. PNAS. 1978;75:285–88. doi: 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zamecnik PC, Stephenson ML. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. PNAS. 1978;75:280–84. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller W, Crouch R. Degradation of DNA RNA hybrids by ribonuclease H and DNA polymerases of cellular and viral origin. PNAS. 1972;69:3360–64. doi: 10.1073/pnas.69.11.3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agrawal S. Importance of nucleotide sequence and chemical modifications of antisense oligo-nucleotides. Biochim Biophys Acta. 1999;1489:53–68. doi: 10.1016/s0167-4781(99)00141-4. [DOI] [PubMed] [Google Scholar]
- 30.Arechavala-Gomeza V, Khoo B, Aartsma-Rus A. Splicing modulation therapy in the treatment of genetic diseases. Appl Clin Genet. 2014;7:245–52. doi: 10.2147/TACG.S71506. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farooqi AA, Rehman ZU, Muntane J. Antisense therapeutics in oncology: current status. Onco Targets Ther. 2014;7:2035–42. doi: 10.2147/OTT.S49652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lorenzer C, Dirin M, Winkler AM, Baumann V, Winkler J. Going beyond the liver: progress and challenges of targeted delivery of siRNA therapeutics. J Control Release. 2015;203:1–15. doi: 10.1016/j.jconrel.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 33.Lima WF, Monia BP, Ecker DJ, Freier SM. Implication of RNA structure on antisense oligo-nucleotide hybridization kinetics. Biochemistry. 1992;31:12055–61. doi: 10.1021/bi00163a013. [DOI] [PubMed] [Google Scholar]
- 34.Monia BP, Sasmor H, Johnston JF, Freier SM, Lesnik EA, et al. Sequence-specific antitumor activity of a phosphorothioate oligodeoxyribonucleotide targeted to human C-raf kinase supports an antisense mechanism of action in vivo. PNAS. 1996;93:15481–84. doi: 10.1073/pnas.93.26.15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lacerra G, Sierakowska H, Carestia C, Fucharoen S, Summerton J, et al. Restoration of hemoglobin A synthesis in erythroid cells from peripheral blood of thalassemic patients. PNAS. 2000;97:9591–96. doi: 10.1073/pnas.97.17.9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suwanmanee T, Sierakowska H, Fucharoen S, Kole R. Repair of a splicing defect in erythroid cells from patients with β-thalassemia/HbE disorder. Mol Ther. 2002;6:718–26. doi: 10.1006/mthe.2002.0805. [DOI] [PubMed] [Google Scholar]
- 37.Sazani P, Gemignani F, Kang SH, Maier MA, Manoharan M, et al. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nat Biotechnol. 2002;20:1228–33. doi: 10.1038/nbt759. [DOI] [PubMed] [Google Scholar]
- 38.La Spada AR, Paulson HL, Fischbeck KH. Trinucleotide repeat expansion in neurological disease. Ann Neurol. 1994;36:814–22. doi: 10.1002/ana.410360604. [DOI] [PubMed] [Google Scholar]
- 39.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. Abstract. [DOI] [PubMed] [Google Scholar]
- 40.Ranum LP, Cooper TA. RNA-mediated neuromuscular disorders. Annu Rev Neurosci. 2006;29:259–77. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- 41.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–67. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 42.Mankodi A, Logigian E, Callahan L, McClain C, White R, et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–73. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 43.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–56. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–68. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gusella JF, Wexler NS, Conneally PM, Naylor SL, Anderson MA, et al. A polymorphic DNA marker genetically linked to Huntington's disease. Nature. 1983;306:234–38. doi: 10.1038/306234a0. [DOI] [PubMed] [Google Scholar]
- 46.Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, et al. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature. 2012;488:111–15. doi: 10.1038/nature11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mulders SA, van den Broek WJ, Wheeler TM, Croes HJ, van Kuik-Romeijn P, et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. PNAS. 2009;106:13915–20. doi: 10.1073/pnas.0905780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, et al. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science. 2009;325:336–39. doi: 10.1126/science.1173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu J, Matsui M, Gagnon KT, Schwartz JC, Gabillet S, et al. Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat Biotechnol. 2009;27:478–84. doi: 10.1038/nbt.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gagnon KT, Watts JK, Pendergraff HM, Montaillier C, Thai D, et al. Antisense and antigene inhibition of gene expression by cell-permeable oligonucleotide-oligospermine conjugates. J Am Chem Soc. 2011;133:8404–7. doi: 10.1021/ja200312y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013;126:829–44. doi: 10.1007/s00401-013-1192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–46. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–28. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. PNAS. 2013;110:E4530–39. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126:385–99. doi: 10.1007/s00401-013-1149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014;128:485–503. doi: 10.1007/s00401-014-1329-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mori K, Weng SM, Arzberger T, May S, Rentzsch K, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–38. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 58.Turner DH, Sugimoto N, Freier SM. RNA structure prediction. Annu Rev Biophys Biophys Chem. 1988;17:167–92. doi: 10.1146/annurev.bb.17.060188.001123. Abstract. [DOI] [PubMed] [Google Scholar]
- 59.Deigan KE, Li TW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA structure determination. PNAS. 2009;106:97–102. doi: 10.1073/pnas.0806929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. PNAS. 2004;101:7287–92. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Woese CR, Magrum LJ, Gupta R, Siegel RB, Stahl DA, et al. Secondary structure model for bacterial 16S ribosomal RNA: phylogenetic, enzymatic and chemical evidence. Nucleic Acids Res. 1980;8:2275–93. doi: 10.1093/nar/8.10.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gutell RR, Weiser B, Woese CR, Noller HF. Comparative anatomy of 16-S-like ribosomal RNA. Prog Nucleic Acid Res Mol Biol. 1985;32:155–216. doi: 10.1016/s0079-6603(08)60348-7. [DOI] [PubMed] [Google Scholar]
- 64.Childs-Disney JL, Wu M, Pushechnikov A, Aminova O, Disney MD. A small molecule microarray platform to select RNA internal loop-ligand interactions. ACS Chem Biol. 2007;2:745–54. doi: 10.1021/cb700174r. [DOI] [PubMed] [Google Scholar]
- 65.Disney MD, Labuda LP, Paul DJ, Poplawski SG, Pushechnikov A, et al. Two-dimensional combinatorial screening identifies specific aminoglycoside-RNA internal loop partners. J Am Chem Soc. 2008;130:11185–94. doi: 10.1021/ja803234t. [DOI] [PubMed] [Google Scholar]
- 66.Auerbach T, Mermershtain I, Davidovich C, Bashan A, Belousoff M, et al. The structure of ribosome-lankacidin complex reveals ribosomal sites for synergistic antibiotics. PNAS. 2010;107:1983–88. doi: 10.1073/pnas.0914100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Borovinskaya MA, Pai RD, Zhang W, Schuwirth BS, Holton JM, et al. Structural basis for aminoglycoside inhibition of bacterial ribosome recycling. Nat Struct Mol Biol. 2007;14:727–32. doi: 10.1038/nsmb1271. [DOI] [PubMed] [Google Scholar]
- 68.Ippolito JA, Kanyo ZF, Wang D, Franceschi FJ, Moore PB, et al. Crystal structure of the oxazolidinone antibiotic linezolid bound to the 50S ribosomal subunit. J Med Chem. 2008;51:3353–56. doi: 10.1021/jm800379d. [DOI] [PubMed] [Google Scholar]
- 69.Moehle K, Athanassiou Z, Patora K, Davidson A, Varani G, Robinson JA. Design of β-hairpin peptidomimetics that inhibit binding of α-helical HIV-1 Rev protein to the Rev response element RNA. Angew Chem Int Ed Engl. 2007;46:9101–4. doi: 10.1002/anie.200702801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Athanassiou Z, Patora K, Dias RL, Moehle K, Robinson JA, Varani G. Structure-guided peptidomimetic design leads to nanomolar β-hairpin inhibitors of the Tat-TAR interaction of bovine immunodeficiency virus. Biochemistry. 2007;46:741–51. doi: 10.1021/bi0619371. [DOI] [PubMed] [Google Scholar]
- 71.Leeper TC, Athanassiou Z, Dias RL, Robinson JA, Varani G. TAR RNA recognition by a cyclic peptidomimetic of Tat protein. Biochemistry. 2005;44:12362–72. doi: 10.1021/bi0510532. [DOI] [PubMed] [Google Scholar]
- 72.Davidson A, Patora-Komisarska K, Robinson JA, Varani G. Essential structural requirements for specific recognition of HIV TAR RNA by peptide mimetics of Tat protein. Nucleic Acids Res. 2011;39:248–56. doi: 10.1093/nar/gkq713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lalonde MS, Lobritz MA, Ratcliff A, Chamanian M, Athanassiou Z, et al. Inhibition of both HIV-1 reverse transcription and gene expression by a cyclic peptide that binds the Tat-transactivating response element (TAR) RNA. PLOS Pathog. 2011;7:e1002038. doi: 10.1371/journal.ppat.1002038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davidson A, Leeper TC, Athanassiou Z, Patora-Komisarska K, Karn J, et al. Simultaneous recognition of HIV-1 TAR RNA bulge and loop sequences by cyclic peptide mimics of Tat protein. PNAS. 2009;106:11931–36. doi: 10.1073/pnas.0900629106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stelzer AC, Kratz JD, Zhang Q, Al-Hashimi HM. RNA dynamics by design: biasing ensembles towards the ligand-bound state. Angew Chem Int Ed Engl. 2010;49:5731–33. doi: 10.1002/anie.201000814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stelzer AC, Frank AT, Kratz JD, Swanson MD, Gonzalez-Hernandez MJ, et al. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat Chem Biol. 2011;7:553–59. doi: 10.1038/nchembio.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee MM, Pushechnikov A, Disney MD. Rational and modular design of potent ligands targeting the RNA that causes myotonic dystrophy 2. ACS Chem Biol. 2009;4:345–55. doi: 10.1021/cb900025w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Childs-Disney JL, Yildirim I, Park H, Lohman JR, Guan L, et al. Structure of the myotonic dystrophy type 2 RNA and designed small molecules that reduce toxicity. ACS Chem Biol. 2014;9:538–50. doi: 10.1021/cb4007387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rzuczek SG, Gao Y, Tang ZZ, Thornton CA, Kodadek T, Disney MD. Features of modularly assembled compounds that impart bioactivity against an RNA target. ACS Chem Biol. 2013;8:2312–21. doi: 10.1021/cb400265y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Agnew HD, Rohde RD, Millward SW, Nag A, Yeo WS, et al. Iterative in situ click chemistry creates antibody-like protein-capture agents. Angew Chem Int Ed Engl. 2009;48:4944–48. doi: 10.1002/anie.200900488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed Engl. 2001;40:2004–21. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 82.Lewis WG, Green LG, Grynszpan F, Radic Z, Carlier PR, et al. Click chemistry in situ: acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array of building blocks. Angew Chem Int Ed Engl. 2002;41:1053–57. doi: 10.1002/1521-3773(20020315)41:6<1053::aid-anie1053>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 83.Millward SW, Henning RK, Kwong GA, Pitram S, Agnew HD, et al. Iterative in situ click chemistry assembles a branched capture agent and allosteric inhibitor for Akt1. J Am Chem Soc. 2011;133:18280–88. doi: 10.1021/ja2064389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rzuczek SG, Park H, Disney MD. A toxic RNA catalyzes the in cellulo synthesis of its own inhibitor. Angew Chem Int Ed Engl. 2014;53:10956–59. doi: 10.1002/anie.201406465. [DOI] [PubMed] [Google Scholar]
- 85.Stern S, Moazed D, Noller HF. Structural analysis of RNA using chemical and enzymatic probing monitored by primer extension. Methods Enzymol. 1988;164:481–89. doi: 10.1016/s0076-6879(88)64064-x. [DOI] [PubMed] [Google Scholar]
- 86.Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature. 1987;327:389–94. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- 87.Regulski EE, Breaker RR. In-line probing analysis of riboswitches. Methods Mol Biol. 2008;419:53–67. doi: 10.1007/978-1-59745-033-1_4. [DOI] [PubMed] [Google Scholar]
- 88.Lomenick B, Olsen RW, Huang J. Identification of direct protein targets of small molecules. ACS Chem Biol. 2011;6:34–46. doi: 10.1021/cb100294v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liang FS, Greenberg WA, Hammond JA, Hoffmann J, Head SR, Wong CH. Evaluation of RNA-binding specificity of aminoglycosides with DNA microarrays. PNAS. 2006;103:12311–16. doi: 10.1073/pnas.0605264103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rijal K, Bao X, Chow CS. Amino acid-linked platinum(II) analogues have altered specificity for RNA compared to cisplatin. Chem Commun. 2014;50:3918–20. doi: 10.1039/c3cc49035a. [DOI] [PubMed] [Google Scholar]
- 91.Rijal K, Chow CS. A new role for cisplatin: probing ribosomal RNA structure. Chem Commun. 2008;1:107–9. doi: 10.1039/b816633a. [DOI] [PubMed] [Google Scholar]
- 92.Hostetter AA, Osborn MF, DeRose VJ. RNA-Pt adducts following cisplatin treatment of Saccharomyces cerevisiae. ACS Chem Biol. 2012;7:218–25. doi: 10.1021/cb200279p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Moghaddam AD, White JD, Cunningham RM, Loes AN, Haley MM, DeRose VJ. Convenient detection of metal-DNA, metal-RNA, and metal-protein adducts with a click-modified Pt(II) complex. Dalton Trans. 2015;44:3536–39. doi: 10.1039/c4dt02649g. [DOI] [PubMed] [Google Scholar]
- 94.Osborn MF, White JD, Haley MM, DeRose VJ. Platinum-RNA modifications following drug treatment in S. cerevisiae identified by click chemistry and enzymatic mapping. ACS Chem Biol. 2014;9:2404–11. doi: 10.1021/cb500395z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.White JD, Osborn MF, Moghaddam AD, Guzman LE, Haley MM, DeRose VJ. Picazoplatin, an azide-containing platinum(II) derivative for target analysis by click chemistry. J Am Chem Soc. 2013;135:11680–83. doi: 10.1021/ja402453k. [DOI] [PMC free article] [PubMed] [Google Scholar]