

Abstract
CuA is a binuclear electron transfer (ET) center found in cytochrome c oxidases (CcOs), nitrous oxide reductases (N2ORs), and nitric oxide reductase (NOR). In these proteins, the CuA centers facilitate efficient ET (kET > 104 s−1) under low thermodynamic driving forces (10–90 mV). While the structure and functional properties of CuA are well understood, a detailed mechanism of copper incorporation into the protein and the identity of the intermediates formed during the CuA maturation process are still lacking. Previous studies of the CuA assembly mechanism in vitro using a biosynthetic model CuA center in azurin (CuAAz) identified a novel intermediate X (Ix) during reconstitution of the binuclear site. However, due to the instability of Ix and the coexistence of other Cu centers, such as CuA’ and type 1 copper centers, the identity of this intermediate could not be established. Here, we report the mechanism of CuA assembly using variants of Glu114XCuAAz (X=Gly, Ala, Leu, Gln), the backbone carbonyl of which acts as a ligand to the CuA site, with a major focus on characterization of the novel intermediate Ix. We show that CuA assembly in these variants proceeds through several types of Cu centers, such as mononuclear red type 2 Cu, the novel intermediate Ix, and blue type 1 Cu. Our results show that the backbone flexibility of the Glu114 residue is an important factor in determining the rates of T2Cu→Ix formation, suggesting that the CuA formation is facilitated by swinging of the ligand loop, which internalizes the T2Cu capture complex to the protein interior. The kinetic data further suggests that the nature of the Glu114 side chain influences the timescales on which these intermediates are formed, the wavelengths of the absorption peaks, and how cleanly one intermediate is converted to another. Through careful understanding of these mechanism and optimization of the conditions, we have obtained Ix in ~80–85% population in these variants, which allowed us to employ UV-Vis, EPR, and EXAFS spectroscopic techniques to identify the Ix as a mononuclear Cu(Cys)2(His) complex. Since some of the intermediates have been proposed to be involved in the assembly of native CuA, these results shed light on the structural features of the important intermediates and mechanism of CuA formation.
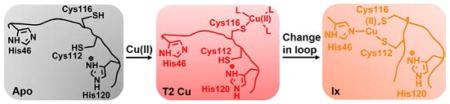
Graphical abstract
Introduction
Electron transfer (ET) is essential for many biological processes, such as photosynthesis, respiration, and dinitrogen fixation.1 To facilitate these processes, copper proteins involved in redox and ET process, called cupredoxins, are often utilized, and they share a common Greek-key-β-barrel fold, where the majority of copper-coordinating ligands occur in a loop.2–9 A Cys thiolate is a ligand common in cupredoxins, which makes these proteins intense in color, due to SCys→Cu charge transfer (CT) transitions. Three types of copper sites are typically found in cupredoxins: the blue mononuclear type 1 copper (T1Cu) with distorted trigonal geometry, the red mononuclear type 2 copper (T2Cu) with square pyramidal geometry, and the purple binuclear CuA with a diamond core geometry.3, 10–12 While the T1Cu and CuA sites facilitate ET, the T2Cu sites are generally involved in catalysis.10
One important class of these copper sites is the binuclear purple CuA, which is found in enzymes involved in aerobic and anaerobic respiration, such as cytochrome c oxidases (CcOs),13–15 quinol oxidase (SoxH),16, 17 nitrous oxide reductases (N2ORs),18, 19 and nitric oxide reductase (qCuANOR).20 In these proteins, the CuA site acts as a terminal ET center by accepting electrons from donors, such as cytochromes c, and transferring them to redox partners with high efficiency (e.g. ET rate, kET > 104 s−1), despite low thermodynamic driving forces (10–90 mV) for such ET. The CuA centers are characterized by a mixed valence [Cu(+1.5)-Cu(+1.5)] site, containing a weak Cu-Cu bond, in which one electron is delocalized between the two copper ions.21–23 The two copper ions are coordinated by two Cys residues to form a rigid “diamond” core, and each metal ion is also coordinated by a His residue (Fig. 1A). These structural features of the CuA site also bestow them with unique spectroscopic signatures. The UV-vis of CuA sites have two SCys→Cu(II) CT at ~480 and 530 nm (ε ~3000–4000 M−1cm−1), and a broad peak due to a Cu-based (ψ→ψ*) transition centered at ~800 nm (ε ~2000 M−1cm−1), giving the CuA a strong purple color.16, 21, 24–27 The electron paramagnetic resonance (EPR) spectrum consists of a seven line hyperfine splitting pattern due to valence delocalization of the unpaired d electron between the two Cu nuclei, each having a nuclear spin I = 3/2.22 On the other hand, the T1Cu center also displays intense blue color, due to strong absorbance at ~600 nm (ε ~2000–6000 M−1cm−1) that arises from SCys(π)→Cu(II) CT transition. It also differs from the typical T2 Cu center by a small 4-line EPR hyperfine splitting pattern, which is due to valence delocalization of the unpaired d electron onto the mononuclear Cu. In general, the CuA center displays even smaller hyperfine splitting (Az ~30–40 x 10−4 cm−1) than T1Cu site (Az ~30–90 x 10−4 cm−1) as the hyperfine splitting is divided into two copper atoms.10 The red T2Cu center, in contrast, exhibits an intense absorption ~360–400 nm, due to a SCys(σ)→Cu(II) CT transition. It also displays 4-line EPR hyperfine splitting pattern with a much larger hyperfine splitting (Az ~130–180 x 10−4 cm−1) than those in CuA or T1Cu centers, reflecting a lesser extent of covalency.28, 29 From phylogenetic and sequence analyses, it has been suggested that the blue T1Cu, red T2Cu, and purple CuA sites have evolved from a common ancestor.30 All three types of Cu centers were observed in a study involving in vitro reconstitution of the CuA site of N2OR from Paracoccus denitrificans.31
Figure 1.

A. Orientation of the Glu114 side chain in CuAAz in between the two hydrophobic amino acids L86 and Y72, shown as sticks. CuA ligands are shown as lines, Cu atoms as purple spheres, water molecules that form hydrogen bonding interactions with the Glu114 side chain are shown as red spheres. Protein backbone is shown as cartoon. B. Cartoon representation of CuAAz (purple), and blue Cu Az (blue) showing how the placement of Glu114 side chain in CuAAz sterically pushes the two helices that house L86, and Y72, compared to the blue Cu Az. Figures were generated in PyMol.
Due to the presence of multiple cofactors in the proteins containing CuA centers (hemes in CcOs and qCuANOR, tetranuclear CuZ in N2ORs), studying the CuA sites in these proteins has been hindered by intense and overlapping spectral features.16, 17, 19, 20 One strategy that has been employed to study CuA sites involves expressing the soluble CuA domain from CcO and reconstituting the CuA site in vitro.16, 25–27, 32–36 In a second strategy the CuA ligands have been engineered in another cupredoxin protein followed by in vitro reconstitution of the binuclear site.37–40 By replacing the Cu binding loop from T1Cu proteins with the CuA loop from CcO, the CuA site was successfully engineered into CyoA domain of a copper-less quinol oxidase (called CuA-CyoA) and blue T1Cu proteins amicyanin (called CuAAmi) and azurin (Az, called CuAAz).38,37, 40–44
While the CuA center has been well studied, one of the remaining challenges in understanding this class of copper center is the mechanism of its formation, particularly how the mixed valence species [Cu(+1.5)-Cu(+1.5)] is formed from either Cu(I) or Cu(II) ions. The red T2Cu Sco1 protein from the inner mitochondrial membrane, which exhibits Cu(Cys)2His ligation, has been implicated to be the chaperone responsible for Cu delivery to the CuA site.45–50 Quite interestingly, binding of both Cu(II) and Cu(I) to Sco1 has been proposed to be important for normal functioning of this protein.51–53 Alternatively, it has been proposed that Sco1 acts as disulfide reductase to keep the active site Cys of CuA in the reduced state or to maintain a proper redox buffering of metal ions. The latter inference stems from the fact that Sco1 possesses the thioredoxin fold.50, 54, 55 In Bacillus subtilis, another protein, YpmQ, which is the yeast homologue of Sco1, and has a Cu(Cys)2His ligation, has also been proposed to be required for CuA assembly. Furthermore, the periplasmic protein PCuAC, found in several prokaryotes has been shown to deliver Cu(I) to apo CuA, forming the binuclear Cu(I)-CuA site.54
In order to gain insight into CuA assembly, studies have been undertaken using both soluble domain of Thermus thermophilus (Tt) CuA56, 57 and biosynthetic models of CuA in azurin (called CuAAz).58, 59 A single intermediate was observed as a rapidly-formed green Cu center in the soluble domain of Tt CuA and this intermediate was identified as a mononuclear Cu(II)(Cys)2(His) complex, based on UV-vis, EPR and X-ray absorption spectroscopy (XAS) studies.56 On the other hand, addition of 10-fold excess CuSO4 to the apo CuAAz, a red T2Cu intermediate was formed within 10 ms, followed by formation of the purple CuA site at longer times.58 Recently, it has been shown that under sub-equivalent Cu(II) addition, the formation of binuclear CuA center is an intricate process, such that in addition to the formation of the red T2Cu intermediate, a novel green copper intermediate, Ix, and a blue T1Cu intermediate was observed prior to CuA formation.59 The proposed mechanism invoked the formation of an initial red T2Cu involving Cys116 that is close to the protein exterior. The next step of the kinetic process is then branched into a) the formation of binuclear CuA’, a CuA lacking His120 ligation, and b) a conformational change of the metal binding loop, leading to reorientation of the capture complex toward the protein interior and accompanying ligation by Cys112 and His46, forming Ix. Through comparison to the spectral properties of Cu(II)-substituted horse liver alcohol dehydrogenase (HLADH),60–62 a Cu(II)-dithiolate complex was proposed as the identity of Ix. However, the exact ligand environment of Ix could not be established, as it was a short-lived intermediate present in a mixture with other species. The next step of the mechanism proposed the formation of a T1Cu complex from oxidation of the reduced Cu(I) product of Ix. The T1Cu complex was finally converted to the valence-delocalized CuA site via incorporation of in situ reduced Cu(I).
While the above results are valuable, they do not fully clarify the mechanism of the CuA formation. A critical missing piece of this mechanistic puzzle is the identity of the Ix intermediate. To identify this Ix, we examine the X-ray structure of CuAAz, which reveals that the side chain of Glu114, whose backbone carbonyl is very close to one of the Cu ions (2.2 Å), is positioned in between two hydrophobic amino acids, Leu86 and Tyr72 (Fig. 1A).63 We thus hypothesize that the Glu114 side chain is likely more solvent exposed in the apo form due to the negative charge on the Glu carboxylate at neutral pH. In the holo form, the coordination of the backbone carbonyl group of Glu114 to one of the copper ions likely requires a specific orientation of the Glu114 side chain in between the two hydrophobic amino acids. As a result of this steric effect, the helices housing these two amino acids are pushed away from each other (Fig. 1B, purple). Interestingly, no such steric repulsion of the helices in blue T1Cu Az is observed (Fig. 1B, blue). In contrast, the corresponding Glu side chain, found in the majority (80%) of native CcOs is oriented towards the membrane interior, where it is coordinated to a Mg(II) ion.14
Herein, we investigate the effect of the Glu114 side chain on the sterics of the Cu binding loop and on the CuA assembly, by systematic substitution of this side chain with other side chains such as Gly (lacking a side chain), Ala (having a smaller, more hydrophobic methyl group), Leu (same chain length but more hydrophobic), and Gln (same chain length, but with no charge). Thus, the Gly, Ala, and Leu mutations would allow us to test the effect of increasing side chain bulk, while the Leu and Gln mutations should address the role of side chain polarity on the mechanism of the CuA formation. The specific issues we try to address in this report are: a) whether these mutations have any effect on the overall mechanism of CuA formation, in particular the rate of the T2Cu→Ix step of the mechanism, as this step was proposed to involve a conformational change in the metal binding loop, and b) whether these mutations affect the formation and decay kinetics of these intermediates. Of particular interest was to obtain the Ix in higher purity compared to the original CuAAz and determine its identity.
Materials and Methods
Protein Expression and Purification
Plasmids of the CuAAz variants Glu114Gly/Ala/Leu/Gln containing a periplasmic leader sequence from Pseudomonas aeruginosa were cloned into pET-9a vector and expressed in E.coli BL21*(DE3) (Invitrogen) competent cells. Expression and purification was done as described previously,38, 44, 58, 59 with some modifications. Bacterial cultures were grown in 2XYT media at 25°C until an OD of ~0.6–1.0 was reached. Protein expression was induced with Isopropyl β-D-1-thiogalactopyranoside (IPTG) (~200mg/2L) and allowed to grow for 4 hours. Cells were harvested and re-suspended in 20% sucrose, 1 mM Ethylenediaminetetraacetic acid (EDTA), 30 mM 2-Amino-2-hydroxymethyl-propane-1,3-diol (Tris).HCl solution. After centrifugation the periplasmic membranes were lysed by osmotic shock solution containing 4 mM NaCl and 1 mM Dithiothreitol (DTT). Following centrifugation, the supernatant was treated with 500 mM NaOAc pH 4.1 to precipitate the impurities. The solution was centrifuged one final time and the supernatant was applied to SP Sepharose cation exchange column (GE Healthcare) pre-equilibrated with 50 mM NH4OAc pH 4.1, and the protein was eluted using a shallow gradient of 50 mM NH4OAc pH 6.35. The fractions containing the apo protein were pooled, pH adjusted to 6.3 and further purified by HiTrap Q-Sepharose anion exchange column (GE Healthcare). The colorless apo protein was then concentrated to ~10 mL and further subjected to a size exclusion column (Sephacryl S100, GE Healthcare) to separate misfolded protein. 50 mM NH4OAc pH 6.35 was used as the elution buffer. The purity and the identity of the properly folded apo protein were verified by electrospray ionization mass spectrometry. Aliquots of apo proteins were flash frozen and stored at −80°C until further use. Whenever required, the apo protein was thawed, exchanged into Universal Buffer (UB) containing 40 mM 2-(N-morpholino)ethanesulfonic acid (MES), 3-(N-morpholino)propanesulfonic acid (MOPS), Tris, and, N-cyclohexyl-2-hydroxyl-3-aminopropanesulfonic acid (CAPS), 50 mM NaOAc, and 100 mM NaNO3 at the appropriate pH using Sephadex G-25 desalting columns and concentrated to the required concentration using Amicon Ultra filtration membranes (Millipore). Protein concentrations were determined using ε280 = 8440 M−1cm−1.
Stopped flow UV-vis Absorption Spectroscopy
Stopped flow UV-Vis absorbance data were collected on an Applied Photophysics Ltd. (Leatherhead, U.K.) SX18.MV stopped flow spectrophotometer equipped with a 256 element photodiode array detector. Equal volumes of 0.5 mM apo protein in UB pH 7 (UB 7) and 0.2 mM CuSO4 solutions (1.0 : 0.4 eq. protein : CuSO4 ratio) were mixed by two syringes and spectra were collected over 1000 s on logarithmic scale containing 200 data points with 1 ms sampling time. The final concentrations of protein and CuSO4 were 0.25 mM and 0.1 mM, respectively. A water bath connected to the instrument was used to maintain the temperature at 15°C. For anaerobic stopped-flow experiments the protein and the buffer were degassed on a Schlenk line by three freeze-pump-thaw cycles. Prior to mixing, the stopped-flow instrument was washed with degassed buffer. Similarly, for O2-rich experiments, the buffer was made O2-saturated by purging with O2 for ~15 minutes prior to the experiments. Raw data were subjected to global analysis using SpecFit/32 (Spectrum Software Associates, Inc.).
Global Fit Analysis of the Stopped flow Data
The raw stopped-flow data were processed using SpecFit/32 in order to deconvolute the complex spectral features and multiple kinetic processes. The data analysis is based on Single Value Decomposition (SVD) and non-linear least square fits of the experimental data in an iterative process. During the fitting process a set of kinetic models that seem most consistent with the experimental data were input into the software and fit globally. Using the information from the initial fitting process, the kinetic models were modified accordingly until satisfactory fit results were obtained based on statistics and kinetic parameters.
Kinetic UV-vis Absorption Spectroscopy
Kinetic UV-vis absorption data of samples containing 0.25 mM apo protein in UB 7 and 0.1 mM CuSO4 were collected with constant stirring at 10°C on an Agilent 8453 photodiode array spectrophotometer connected with a water bath cooling system. A lower temperature (10°C) was chosen in the kinetic UV-vis absorption experiment than that (15°C) in the stopped-flow UV-Vis absorption experiment, because of concerns of potential stability issue of the proteins at much longer time scale in the kinetic UV-vis absorption experiment than that in the stopped-flow experiment. Because of this temperature difference, the kinetic rate constants (see Table 1) were obtained only from the stopped-flow data. For these experiments, 1000 °L of 0.5 mM apo protein in UB 7 was taken on a 1cm x 1cm cuvette with stirring and the instrument was set to collect data for 3600 s with scan time of 0.5 s incremented by 5% after 30 s to reduce the number of data points. After collecting a few spectra, 1000 μL of 0.2 mM CuSO4 was added to the apo protein and the cap of the cuvette was sealed with parafilm. The final concentrations of protein and CuSO4 were 0.25 mM and 0.1 mM. After 3600 s, another experiment was started to continue for additional 10–24 hours using a longer scan time of 600 s as the later kinetic processes were slow. In other instances, similar experiments were performed with solutions containing equal volume of 3 mM apo protein in UB 7 and 1.2 mM CuSO4, amounting to a final concentration of 1.5 mM apo protein and 0.6 mM CuSO4. These experiments were performed at the concentrations similar to those of target EPR experiments, so as to determine the time points at which different intermediates formed with maximum population in order to characterize them by EPR. Due to fast formation and decay of the red T2 Cu intermediate (~100 ms), we focused on characterizing the later intermediates, in particular Ix, as this intermediate was not thoroughly characterized previously. From inspection of the UV-vis absorbance data from these experiments, Ix formed with maximum population at ~50–60 s.
Table 1.
Kinetic models used to fit the stopped-flow data shown in Fig. 2. Rate constants obtained from global analysis using Specfit are shown.
| CuA Variant | Kinetic Model | Rate Constants |
|---|---|---|
| Glu114GlyCuAAz | k1 = 11.28 ± 0.35 s−1 | |
| k2 = 1.95 ± 0.03 s−1 | ||
| k3 = (0.78 ± 0.04) × 10−3 s−1 | ||
| Glu114AlaCuAAz | CuSO4 → T2 Cu T2 Cu → Ix Ix → T1 Cu |
k1 = 13.52 ± 0.52 s−1 |
| k2 = 0.46 ± 0.01 s−1 | ||
| k3 = (1.75 ± 0.05) × 10−3 s−1 | ||
| Glu114LeuCuAAz | k1 = 13.12 ± 0.98 s−1 | |
| k2 = 0.34 ± 0.01 s−1 | ||
| k3 = (1.31 ± 0.05) × 10−3 s−1 | ||
| Glu114GlnCuAAz | k1 = 9.36 ± 0.66 s−1 | |
| k2 = 0.31 ± 0.01 s−1 | ||
| k3 = (2.37 ± 0.06) × 10−3 s−1 |
Electron Paramagnetic Resonance Spectroscopy
EPR data were collected using an X-band Varian E-122 spectrometer at the Illinois EPR Research Center (IERC) at 30 K using liquid He and an Air Products Helitran cryostat. Magnetic fields were calibrated with a Varian NMR Gaussmeter while the microwave frequencies were measured with an EIP frequency counter. Based on the results of the kinetic UV-vis experiments, EPR samples were flash frozen in liquid N2 with 22% glycerol as glassing agent at a broad range of time points after CuSO4 addition. The time points were chosen so as to obtain as much information as possible about the discrete kinetic processes and the species present therein at that time point. The earliest time points were determined to capture the Ix in each mutant. The earliest time point (50s-60s) samples were prepared by quick mixing of equal volumes of 3 mM apo protein in UB 7 and 1.2 mM CuSO4 premixed with glycerol to give a final concentration of 1.5 mM protein, 0.6 mM CuSO4. For longer time point samples (5 min – 24 hours), a pool of apo protein and CuSO4 was mixed together, stirred at 10°C while aliquots from the mixture were frozen at desired time points to monitor either the decay of Ix, or formation of T1Cu intermediate or the formation of the final CuA form, after adding glycerol to a final concentration of 22%. All samples for longer time points had the same starting concentrations of apo protein and CuSO4 as the samples frozen at the earliest time points, to be consistent. Pure holo CuA forms of each variant were prepared by adding a solution of premixed 0.8 eq. Cu(I) and 0.8 eq. CuSO4 to the apo protein in UB 7. Sub-stoichiometric Cu was added to ensure no excess Cu is present in solution. Cu(I) was added in the form of tetrakis(acetonitrile)copper(I)hexafluorophosphate. Experimental EPR data were simulated using SIMPOW6.64 All the spectra were fit simultaneously to obtain relative populations and EPR parameters of various species so as to minimize the total rms difference between experimental and simulated spectra. For spin quantification, CuSO4 standards were prepared at concentrations ranging from 2 – 0.0625 mM. EPR data for the standards and the protein samples were collected at a constant temperature and power of 30K, and 0.2 mW, respectively. Instrument gain was normalized across all samples. Standard curve was obtained by plotting the double integration area as a function of CuSO4 concentration. Cu(II) concentration in protein samples were calculated from the standard curve.
X-ray Absorption Spectroscopy
The Extended X-ray Absorption Fine Structure (EXAFS) spectra were collected at beam line X3B, National Synchrotron Light Source with a sagittally focused Si (111) crystal monochromator. Samples were cooled to 10K before scanning and kept cold during data collection. Kα fluorescence was collected by a Canberra 31-element Ge detector. A Cu foil was used as reference to calibrate energy. A 6μm Ni filter was placed before detector to reduce elastic scattering. Six scans of each sample were collected. These samples were prepared using similar procedure as described in the EPR section. Samples contained 5.2 – 5.8 mM apo protein in UB 7, 2.08–2.32 mM CuSO4 [1:0.4 ratio of apo protein to Cu(II)] to a final concentration and flash frozen in liquid N2 at various time points (50 s – 10 hours) after adding 20% ethylene glycol as the glassing agent. For earliest time points (50s – 60s) ethylene glycol and CuSO4 were premixed prior to adding to the apo protein. Data reduction and background subtraction were performed with EXAFSPAK65 and data was fitted with EXCURVE 9.2.66
Results and Discussions
Kinetics of CuA Reconstitution in Glu114Gly/Ala/Leu/GlnCuAAz
The kinetic processes of CuA reconstitution in Glu114Gly/Ala/Leu/GlnCuAAz were monitored by stopped flow and standard UV-vis absorption spectroscopy in the presence of sub-equivalent amounts of CuSO4. Addition of 0.4 eq. CuSO4 to 0.25 mM Glu114GlyCuAAz led to the formation, within 100 ms, of a species displaying an intense peak at 390 nm and a weak absorption peak at ~625 nm (Fig. 2A). The intense absorption centered at 390 nm is characteristic of the SCys(σ)→Cu(II) charge transfer (CT) transition of a red T2Cu species.10, 59 Over the next 50s, an intermediate species formed exhibiting two intense SCys→Cu CT transitions at 413 nm (sharp) and 616 nm (broad), plus a broad peak at 762 nm. These features are similar to the previously reported intermediate X (Ix).59 The 413 nm peak formed faster than the 616 nm peak over the next 14 minutes, and also decayed faster. At the end of 14 minutes, the 413 nm peak red shifted to 420 nm, and the 616 nm peak blue shifted to ~610 nm, indicating the formation of blue T1Cu species. Over the next 5 hours, as monitored using separate kinetic UV-vis experiments (see materials and methods for details) the intense absorption bands at 420 and 610 nm attributable to T1Cu decayed, and the peaks for CuA at 478 nm, 534 nm, and 795 nm (Fig. 2A, inset) fully developed. Unlike other variants (vide infra), the kinetic processes of Glu114GlyCuAAz did not display any isosbestic point at any time point. Analysis of the stopped flow data (1000 s) was performed using a kinetic model involving the formation of the early red T2Cu intermediate followed by the formation of Ix and the T1Cu (Fig. S1, Table 1).
Figure 2.

Stopped-flow spectra obtained after mixing 0.5 mM apo Glu114GlyCuAAz (A), Glu114AlaCuAAz (B), Glu114LeuCuAAz (C), and Glu114GlnCuAAz (D) in UB 7 buffer, with 0.2 mM CuSO4. Insets show separate kinetic UV-vis experiments under the same condition, monitored for longer time. Isosbestic points are shown as asterisks (*). Unique UV-vis signatures for individual Ix (Ix1, and Ix2) as seen by EPR (vide infra) cannot be deconvoluted due to spectral overlap and are labeled as Ix. Up and down arrows indicate the formation and decay of various species.
Next we investigated the kinetics of Cu incorporation into the Glu114AlaCuAAz variant. Similar to Glu114GlyCuAAz, an early red T2Cu intermediate formed within 134 ms with an absorption band occurring at ~362 nm (Fig. 2B). A new intermediate species, corresponding to Ix, formed within the next 50 s, with intense peaks at 412 nm and 624 nm, and a broad feature at ~774 nm. This process was accompanied by an isosbestic point occurring at 390 nm. The intensity of the 412 nm peak was higher than that of the 624 nm peak. A small amount of CuA was also present. In the next ~900 s, the 412 nm peak decreased in intensity and red shifted to 426 nm, while the 624 nm peak blue shifted to 616 nm, with a concomitant increase in intensity. The new features are attributed to a blue T1Cu species that developed with isosbestic conversion from Ix. The 426 and 616 nm peaks of the T1Cu intermediate decayed over the next 10 hours, while absorptions in the regions typical of CuA increased, with isosbestic points occurring at 460 nm, and 556 nm (Fig. 2B, inset). The kinetic processes within the first 1000 s were modeled (Fig. S1, Table 1) similar to that of Glu114GlyCuAAz.
For Glu114LeuCuAAz, a red T2Cu intermediate with an intense absorption peak appearing at ~360 nm was also formed within 100 ms of mixing, (Fig. 2C), which decayed to Ix within 60 s with isosbestic point at 387 nm. The Ix displayed two intense absorption peaks at 413 nm, 470 nm, and a broad feature at ~793 nm. The intensity of the 413 nm peak was higher than that of the 470 nm peak. The Ix started to decay after 85 s, and was converted to a blue T1Cu intermediate with peaks occurring at 467 nm, 614 nm and a weak band at 793 nm and isosbestic points at 560 nm, and 680 nm. The T1Cu was fully formed within 40 min and started to decay to CuA with an isosbestic point at 560 nm over 10 hours (Fig. 2C, inset). The stopped flow data were modeled in the same way as other variants (Fig. S1, Table 1).
Finally, in Glu114GlnCuAAz, a red T2Cu intermediate displaying an intense peak at ~385 nm was observed within 100 ms of mixing (Fig. 2D). Within 50 s the Ix formed displaying two intense absorption bands at 409 nm, 470 nm, and a broad peak at ~790 nm with an isosbestic point at 390 nm. The 409 nm peak is slightly higher in intensity than the 470 nm peak. The Ix decayed to a blue T1Cu intermediate with isosbestic points at 572 nm and 653 nm. The T1Cu intermediate having peaks at 467 nm, 601 nm, and ~790 nm fully developed within 20 min. After 30 min from mixing, CuA started to form having a weak peak at ~351 nm, two intense peaks at 479 nm, 541 nm, and a broad feature at ~760 nm, formed with isosbestic points at 434 nm, and 531 nm. The full CuA formation was observed within 10 hours (Fig. 2D, inset). The kinetic processes in the first 1000 s were modeled as others (Fig. S1, Table 1).
Since no external reductant was added and the formation of the mixed valence CuA site requires Cu(I), the reducing equivalents must be supplied by the active site cysteines. To investigate the effect of oxygen on the overall kinetic processes, we performed stopped-flow experiments under O2-free and O2-rich conditions. The results showed that, in the absence of O2, no T1Cu intermediate was formed in any of the mutants, whereas under O2-rich condition, the rate of T1Cu formation from Ix was faster compared to normal atmospheric O2 levels (Fig. S2, Table S1). These observations suggest a role of O2 in the formation of the T1Cu species. The absorption at ~625 nm for Ix of Glu114Gly/Ala/CuAAz bears a strong resemblance to the T1Cu species that forms at latter time points. However, this peak is also present under anaerobic conditions, where the T1Cu species does not form, suggesting that this peak does indeed belong to the Ix of the G and A variants.
Effect of Glu114 Substitutions on the Rates of Formation and the Characteristics of the Intermediates
It has been previously proposed that the formation of the initial capture complex for Cu ion involves Cys116 as the Cu ligand close to the exterior of the protein.59 Our results indicate that the initial rates of formation of the red T2Cu capture complex are quite similar in all the four variants (Table 1, Fig. 3A), suggesting that substitution of the Glu114 side chain has minimal effect on the formation of the capture complex. However, the rate of the formation of Ix from the red T2Cu complex is fastest in Glu114GlyCuAAz (Fig. 3B, Table 1) among the four variants. In the previous study,59 the formation of Ix from the T2Cu capture complex was proposed to involve a conformational change of the CuA loop, where the Cu-bound Cys116 reoriented from the protein exterior to the interior. Our results herein support this hypothesis, as the conformational change of the CuA loop required for the formation of Ix would be most favorable in Glu114GlyCuAAz, because Gly has the least steric side chain and most flexible backbone conformation in the Ramachandran plot.67 The presence of longer side chain groups in Ala, Leu, Gln or Glu would accordingly disfavor such a conformational change, owing to steric encumbrance from these side chains and less backbone flexibility. Taken together, these results suggest that the backbone flexibility is an important factor in determining the rates of T2Cu→Ix formation. In a related study involving Tt CuA the presence of a hydrophobic patch involving Leu155 in the metal binding loop was shown to be important in facilitating ET between cytochrome c552 and the Tt CuA.68
Figure 3.

A) Rates of CuSO4→T2Cu, and B) T2Cu→Ix conversion of CuA variants obtained from the Stopped-flow data. Rates in A) are similar in all variants. In B) highest rate is observed for Glu114GlyCuAAz which supports the hypothesis that the T2Cu→Ix conversion involves a conformation change of the CuA ligand loop as Gly has the most flexible backbone conformation, and thus favors such conformation change compared to the other amino acid side chains.
The kinetic data further suggest that the nature of the Glu114 side chain influences the timescales on which these intermediates are formed, the wavelengths of the absorption peaks, and how cleanly one intermediate is converted to another. For example, in Glu114GlyCuAAz, no isosbestic point was observed during any of the kinetic processes, whereas, in the other three variants, well-defined isosbestic points were observed for conversions between different species. The cleanest conversion from one intermediate to another was observed in Glu114LeuCuAAz, and Glu114GlnCuAAz, where minimal spectral overlap among different species was present. In contrast, a strong overlap between the Ix, and T1Cu intermediates was observed in the other two variants. Interestingly, the split peaks for the Ix at 410, and 470 nm were observed only in Glu114LeuCuAAz, and Glu114GlnCuAAz. Additionally, the relative intensities of these two peaks are reversed in Glu114LeuCuAAz, and Glu114GlnCuAAz, where the 410 nm peak is more intense in the former, whereas the 470 nm peak has higher intensity in the latter variant. These observations, therefore, indicate that Glu114 side chain plays a role in dictating the characteristics of the intermediates.
EPR Studies of the Ix species
To further understand the nature of the intermediates, in particular, Ix, we collected X-band EPR data of two representative variants, the Glu114GlyCuAAz that did not display kinetic isosbestic point and Glu114GlnCuAAz that did. The EPR spectrum of Glu114GlyCuAAz collected at 50s (the time point at which the Ix formed with maximum population) (Fig. 4a) was simulated as two mononuclear Cu(II) species in almost equal population (38% and 46%, respectively). From the simulations, the gz and Az values for the two Ix were extracted to be 2.152, 101×10−4 cm−1, for Ix1, and 2.185, 90×10−4 cm−1 (Table 2) for Ix2, respectively. While the Z-components of hyperfine couplings (Az) are similar in both Ix1 and Ix2 that fall in between those of T1Cu and T2Cu complexes, their gz values differ significantly from each other.10, 69 A smaller gz value in Ix1 suggests that the strength of the thiolate-Cu interaction is slightly stronger in Ix1 in comparison to Ix2. Both the gz and Az for Ix1 is similar to the Sco protein (gz=2.150).45 In addition, the Az value is similar to the intermediate observed in Tt CuA (Az = 109 × 10−4 cm−1).56 In both of these systems, copper is present as a bis-thiolate Cu(Cys)2His complex. By analogy, we assign the Ix1 having a similar ligand environment to these two Cu(II) centers. The Ix2 with a gz of 2.185 is lower than most T1Cu sites (gz ~2.190–2.300).10 The Ix2 can be best assigned as having a slightly different ligand coordination/geometry around the metal site relative to that of Ix1.
Figure 4.

X-band EPR spectra of samples containing 1.5 mM Glu114GlyCuAAz (a), and Glu114GlnCuAAz (b), and 0.6 Mm CuSO4, prepared at 50 s post-mixing. Experimental parameters: H = 9.053 GHz, T=30 K, Modulation = 4G, Microwave power = 0.2–0.5 mW. Simulated spectra are shown in black.
Table 2.
EPR parameters extracted from simulation of the early time points EPR spectra shown in Fig. 4.
| CuA Variant | Parameters | Populationsa | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Ix1 | Ix2 | Time | Ix1 | Ix2 | ||
| Glu114GlyCuAAz | gx | 2.034 | 2.027 | 50 s | 38% | 46% |
| gy | 2.044 | 2.020 | ||||
| gz | 2.152 | 2.185 | ||||
| |Ax|×10−4 cm−1 | 10 | 23 | ||||
| |Ay|×10−4 cm−1 | 6 | 22 | ||||
| |Az|×10−4 cm−1 | 101 | 90 | ||||
| Glu114GlnCuAAz | gx | 2.011 | 1.991 | 50 s | 68% | 18% |
| gy | 2.035 | 2.092 | ||||
| gz | 2.151 | 2.249 | ||||
| |Ax|×10−4 cm−1 | 20 | 8 | ||||
| |Ay|×10−4 cm−1 | 14 | 41 | ||||
| |Az|×10−4 cm−1 | 82 | 133 | ||||
| CuAAz59 | gx | 2.007 | ||||
| gy | 2.056 | |||||
| gz | 2.234 | |||||
| |Ax|×10−4 cm−1 | 9 | |||||
| |Ay|×10−4 cm−1 | 0.3 | |||||
| |Az|×10−4 cm−1 | 115 | |||||
| Tt CuA56 | gx | 2.013 | ||||
| gy | 2.052 | |||||
| gz | 2.134 | |||||
| |Ax|×10−4 cm−1 | 16 | |||||
| |Ay|×10−4 cm−1 | 16 | |||||
| |Az|×10−4 cm−1 | 109 | |||||
Only Ix present in the earliest time points are shown. See Table S2 for time-dependent populations of various species and their EPR parameters. Based on higher gz of CuAAz, the Ix can be categorized as Ix2s in the variants.
For Glu114GlnCuAAz, two mononuclear Cu(II) species were also observed at 50 s (time point at which the Ix formed with maximum population), but with unequal populations of 68% and 18%, respectively (Fig. 4b). The first species has a gz of 2.151 and Az of 82×110−4 cm−1 (Table 2), which is indicative of a species with a strong thiolate-Cu interaction and can be assigned as Ix1 with Cu(Cys)2His ligation, using a similar analogy to the Ix1 of Glu114GlyCuAAz. The second species, Ix2, present in 18% population, has a gz of 2.249 and Az of 133×10−4 cm−1. Although both the gz and Az values are higher than the Ix2 of Glu114GlyCuAAz, these parameters are similar to the Cu(Cys)2(His)L (L=pyrazole, NADH) complex of HLADH (gz=2.200, Az=115 × 10−4 cm−1).60–62 Based on this similarity, we assign the Ix2 of Glu114GlnCuAAz as having a similar Cu(Cys)2(His)L (L=weak axial ligand) ligation/geometry.
These EPR data also suggest that a small amount of CuA is formed along with the Ix (Table S2). Subsequently, the Ix decayed and more T1Cu and CuA formed (see the SI). The pure CuA forms of each variant show the expected well-resolved seven-line hyperfine pattern, as well as the characteristic EPR parameters (Fig. S3 Table S3).
X-ray Absorption Spectroscopy
To probe the ligand and coordination environment of the Ix, Cu k edge X-ray absorption spectroscopy was used. The EXAFS data for the Ix of Glu114GlyCuAAz was best simulated as 2 Cu-SCys ligands at 2.17 Å, and 1 Cu-NHis ligand at 1.97 Å (Fig. 5A, Table 3). For Glu114GlnCuAAz, the EXAFS data (Fig. 5B) for the Ix was best simulated as 1 Cu-NHis at 1.93 Å, and 2 Cu-SCys at 2.20 Å (Table 3). According to EPR simulation, two Ix species are present at this time point with slightly different ligand geometry (vide supra). However, as EXAFS measures average Cu-ligand distances, the individual Cu(II) species cannot be deconvoluted from the EXAFS data.
Fig. 5.

Fourier transform and EXAFS (inset) data of 50s samples of Glu114GlyCuAAz (A), and Glu114GlnCuAAz (B). Experimental data are shown as solid black lines and simulation are shown as dashed red lines. Parameters used to fit data are shown in Table 3.
Table 3.
Parameters extracted from fitting of the EXAFS data of Ix.
| Sample/Fit | F | Cu-S (Cys) | Cu-N (His) | Cu-Cua | E0 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| N | R(Å) | DW(Å2) | N | R(Å2) | DW(Å) | N | R(Å) | DW(Å2) | |||
| Glu114GlyCuAAz | 0.53 | 2 | 2.17 | 0.016 | 1 | 1.97 | 0.02 | 0.15 | 2.412 | 0.0035 | −0.76 |
| Glu114GlnCuAAz | 0.60 | 2 | 2.20 | 0.012 | 1 | 1.93 | 0.005 | 0.1 | 2.340 | 0.005 | 1.52 |
Small amounts of the binuclear CuA forms present at the earliest time point samples.
In the Ix of these variants the Cu(Cys)2His ligand coordination is similar to what was observed for the green intermediate in Tt CuA as well as for the Sco protein.45, 56, 59 The EXAFS spectra and data analysis of the holo CuA samples of the Glu114XCuAAz variants are consistent with other reported CuA systems (Fig. S4, Table S4).56
Proposed Mechanism of CuA Formation
The combined UV-vis, EPR, and EXAFS data presented in this work has allowed us to propose a mechanism of CuA formation in these CuAAz variants (Scheme 1). Upon addition of sub-equivalent Cu(II) to the apo proteins, an initial red T2Cu capture complex is formed within ~100 ms. Previous results from CuAAz established that the T2Cu capture complex is formed close to the exterior of the protein with Cys116 as the only protein-derived ligand.44, 59 The next step of the mechanism is bifurcated (Scheme 1): conversion of T2Cu to Ix, and formation of a small population of CuA in a direct pathway from the T2Cu complex. Previously, it was hypothesized that T2Cu to Ix formation involved a conformation change of the CuA loop by which the capture complex is internalized from the protein exterior. Our results in this work show that T2Cu to Ix formation is fastest in Glu114GlyCuAAz compared to the other mutants. As Gly has the most flexible backbone conformation, this observation, therefore, supports the hypothesis that indeed T2Cu to Ix formation is facilitated by swinging of the CuA loop, which brings the Cu to the protein interior.
Scheme 1.

Proposed mechanism of CuA formation in Glu114XCuAAz variants. Identity of the residues in the intermediates, and the protonation state of His120 have not been experimentally verified for the variants, and are proposed from what is known about CuAAz.
In the next step the Ix decay and T1Cu complexes form within ~15 minutes. The formation of T1Cu complex is dependent on the presence of O2, as the T1Cu species does not form in the absence of O2 (Fig. S2). Since, in all cases, the decay of Ix is accompanied by the formation of the T1Cu species in the presence of O2, it can be inferred that the T1Cu complex is a result of the oxidation of the decay product of Ix. EPR spin quantification shows that the Ix of Glu114GlyCuAAz decay with time (Fig. S5), indicating spin loss due to the formation of the EPR silent Cu(I) species. Therefore, the T1 Cu is a product of the oxidation of Cu(I) formed during the decay of Ix. At even longer time scale, the T1Cu complex decays and CuA start to form (Fig. 2, insets). Formation of the binuclear CuA from T1Cu complexes must be achieved by reaction of the T1Cu species with either free Cu(I) generated in situ or with Cu(I)-CuAAz.
The formation of a small population of CuA from T2Cu as observed here must be achieved by reaction of free Cu(I) with the Cu(II)-loaded protein. As no external reductant was added, the Cu(I) must be generated in situ by the reduction of Cu(II) to Cu(I) by the active site cysteines of an apo CuA site. As the oxidation of cysteines to cystine is a 2e− process, more than one Cu(II)-bound proteins must be interacting with each other to receive the reducing equivalents from the cysteine thiols. Alternatively, Cu(I) may also be generated by the reaction of free Cu(II) with the Ix, as suggested in the case of Tt CuA system.56 It is highly unlikely that nature only uses Cu(II) to metallate the CuA sites as it would be a significant waste of apo proteins to supply the reducing equivalents. Using both Cu(II) and Cu(I) would make the metallation process much more efficient as also shown in Tt CuA.56 The yield of holo CuA in the Glu114XCuAAz variants significantly increased while using a mixture of Cu(II) and Cu(I) (Table S3).
Identity of Intermediate X (Ix)
A major focus of this study is to determine the ligand and coordination environment of the Ix using the Glu114XCuAAz variants. In the previous kinetic study of CuAAz, at the time when the Ix was formed with maximum population (~55% at ~30s), significant populations of other types of Cu species (CuA’, T1Cu) were present.59 As a result, the identity of this species could not be experimentally determined. In contrast, the Ix in these CuA variants presented here can be obtained with a maximum of ~80–85% combined population, with only a minor percentage of CuA coexisting with the Ix. The electronic absorption data of the Glu114XCuAAz variants indicate two sharp peaks in the 410–600 nm region and a broad feature at ~700–800 nm, similar to the pyrazole-substituted HLADH, which displays two peaks at ~400–500 nm, and a broad feature ~600–800 nm.60–62 The EPR results indicate the presence of mononuclear T2Cu species with thiolate ligands. In all the variants, the EPR data suggest two mononuclear Cu complexes with minor differences in ligand coordination/geometry. Comparing the EPR parameters of the Ix1 to the green intermediate in Tt CuA and Cu(II)-Sco proteins (Table 2), the Ix1 intermediate observed in the Glu114GlyCuAAz, and Glu114GlnCuAAz variants can be best described as Cu(Cys)2(His) complexes in trigonal geometry, while the Ix2 can be best described as Cu(Cys)2(His)L (L = weak axial ligand) complex in distorted tetrahedral geometry. The proposed ligands and geometries are consistent with EXAFS data, which can be modeled as 3-coordinate mononuclear Cu(Cys)2His complex. Therefore, a combined UV-Vis, EPR, and EXAFS data indicate the presence of Cu-bisthiolato complexes with slightly different ligation/geometry as the identity of these intermediates (Fig. 6).
Fig. 6.

Schematic representation of the proposed identity of different Ix in the CuA variants. Two mononuclear Ix with 3-coordinate Cu(Cys)2(His) ligation for Ix1s(A), and as Cu(Cys)2(His)L (L= weak axial ligand) complex in distorted tetrahedral geometry for Ix2s (B) are proposed. Absolute residue numbering has not been experimentally assigned.
Conclusions
We have investigated the influence of a key CuA ligand (Glu114) in CuA assembly as well as probed the nature of intermediates formed during in vitro reconstitution of the CuA center from Cu(II) ion and apo-forms of biosynthetic models in azurin (CuAAz). Our results show that the mechanism of CuA formation in variants of Glu114XCuAAz (X=Gly, Ala, Leu, Gln) proceeds via an initial formation of a red T2Cu center followed by another mononuclear intermediate X (Ix), then a T1Cu intermediate and finally the binuclear CuA center. The results showed that the Glu114 backbone flexibility is an important factor in determining the rates of T2Cu→Ix formation, and suggest that CuA formation is facilitated by swinging of the ligand loop, which brings the external T2Cu complex to the protein interior. The kinetic data further suggest that the nature of the Glu114 side chain influences the timescales on which these intermediates are formed, the wavelengths of the absorption peaks, and how cleanly one intermediate is converted to another. This mechanistic investigation allowed us to find optimal conditions to obtain a high population of the Ix intermediate (80–85%), facilitating the use of UV-vis, EPR and EXAFS to elucidate the structure of Ix, which is best described as Cu(Cys)2(His) complex in trigonal geometry. While similar intermediates and mechanisms have been identified in native CuA, the exact structural features and pathways for the CuA formation are still not well understood. Our findings here of the mechanistic steps and the identification of key mononuclear intermediates during the assembly of the biosynthetic models of CuA in azurin and its variants have shed light into the mechanism of CuA formation.
Supplementary Material
Acknowledgments
Funding
This work was supported by the U.S. National Science Foundation (CHE-1413328 to Y.L. and NSF Graduate Research Fellowship DGE-0925180 to K.N.C.) and U. S. National Institutes of Health (GM054803 to N.J.B. and 5T32-GM070421 to J.R.), and the Howard Hughes Medical Institute Exceptional Research Opportunities Program (to M.J.P.). The EXAFS data were collected at beamline X3B of the National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory. Beamline X3B at NSLS is operated by the Case Center for Synchrotron Biosciences, supported by NIH NIBIB grant P30-EB-009998. NSLS is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH
We wish to thank Dr. Arnab Mukherjee for helpful discussions.
Footnotes
The authors declare no competing financial interest.
Stopped flow data of Glu114XCuAAz variants under O2-rich and anaerobic conditions, full EPR parameters of all intermediates, Specfit derived spectra of intermediates, EPR and EXAFS data of holo Glu114XCuAAz variants, and spin quantification of Glu114GlyCuAAz. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Liu J, Chakraborty S, Hosseinzadeh P, Yu Y, Tian S, Petrik I, Bhagi A, Lu Y. Metalloproteins Containing Cytochrome, Iron–Sulfur, or Copper Redox Centers. Chem Rev. 2014;114:4366–4469. doi: 10.1021/cr400479b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gray HB, Malmström BG, Williams RJP. Copper coordination in blue proteins. J Biol Inorg Chem. 2000;5:551. doi: 10.1007/s007750000146. [DOI] [PubMed] [Google Scholar]
- 3.Vila AJ, Fernandez CO. Copper in Electron Transfer Proteins. In: Bertini I, Sigel A, Sigel H, editors. Handbook on Metalloproteins. Marcel Dekker; New York, NY: 2001. p. 813. [Google Scholar]
- 4.Wilson TD, Yu Y, Lu Y. Understanding Copper-thiolate Containing Electron Transfer Centers by Incorporation of Unnatural Amino Acids and the CuA Center into the Type 1 Copper Protein Azurin. Coord Chem Rev. 2012;257:260–276. [Google Scholar]
- 5.Lu Y, Chakraborty S, Miner KD, Wilson TD, Mukherjee A, Yu Y, Liu J, Marshall NM. Metalloprotein Design. In: Reedijk J, Poeppelmeier K, editors. Comprehensive Inorganic Chemistry II. Elsevier; Amsterdam: 2013. pp. 565–593. [Google Scholar]
- 6.Chakraborty S, Hosseinzadeh P, Lu Y. Metalloprotein Design and Engineering. In: Scott RA, editor. Encyclopedia of Inorganic and Bioinorganic Chemistry. John Wiley and Sons, Ltd; Chichester: 2014. pp. 1–51. [Google Scholar]
- 7.Farver O, Pecht I. Prog Inorg Chem. John Wiley & Sons, Inc; 2008. Elucidation of Electron- Transfer Pathways in Copper and Iron Proteins by Pulse Radiolysis Experiments; pp. 1–78. [Google Scholar]
- 8.Savelieff MG, Lu Y. Cu(A) centers and their biosynthetic models in azurin. J Biol Inorg Chem. 2010;15:461–483. doi: 10.1007/s00775-010-0625-2. [DOI] [PubMed] [Google Scholar]
- 9.Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Copper active sites in biology. Chem Rev. 2014;114:3659–3853. doi: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Y. Electron transfer: Cupredoxins. In: Que JL, Tolman WB, editors. Biocoordination Chemistry. Elsevier; Oxford, UK: 2004. pp. 91–122. [Google Scholar]
- 11.Solomon EI. Spectroscopic Methods in Bioinorganic Chemistry: Blue to Green to Red Copper Sites. Inorg Chem. 2006;45:8012–8025. doi: 10.1021/ic060450d. [DOI] [PubMed] [Google Scholar]
- 12.Solomon EI, Xie X, Dey A. Mixed valent sites in biological electron transfer. Chem Soc Rev. 2008;37:623–638. doi: 10.1039/b714577m. [DOI] [PubMed] [Google Scholar]
- 13.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Structures of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8 A. Science (Washington, DC, United States) 1995;269:1069–1074. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 14.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 15.Wikstrom M. Cytochrome c oxidase: 25 years of the elusive proton pump. Biochim Biophys Acta. 2004;1655:241–247. doi: 10.1016/j.bbabio.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 16.Komorowski L, Anemuller S, Schafer G. First expression and characterization of a recombinant CuA-containing subunit II from an archaeal terminal oxidase complex. J Bioenerg Biomembr. 2001;33:27–34. doi: 10.1023/a:1005668522801. [DOI] [PubMed] [Google Scholar]
- 17.Komorowski L, Schafer G. Sulfocyanin and subunit II, two copper proteins with novel features, provide new insight into the archaeal SoxM oxidase supercomplex. FEBS Lett. 2001;487:351–355. doi: 10.1016/s0014-5793(00)02343-7. [DOI] [PubMed] [Google Scholar]
- 18.Brown K, Tegoni M, Prudencio M, Pereira AS, Besson S, Moura JJ, Moura I, Cambillau C. A novel type of catalytic copper cluster in nitrous oxide reductase. Nat Struct Biol. 2000;7:191–195. doi: 10.1038/73288. [DOI] [PubMed] [Google Scholar]
- 19.Zumft WG, Kroneck PMH. Respiratory transformation of nitrous oxide (N2O) to dinitrogen by Bacteria and Archaea. Adv Microb Physiol. 2007;52:107–227. doi: 10.1016/S0065-2911(06)52003-X. [DOI] [PubMed] [Google Scholar]
- 20.Suharti Strampraad MJ, Schröder I, de Vries S. A Novel Copper A Containing Menaquinol NO Reductase from Bacillus azotoforman s. Biochemistry. 2001;40:2632–2639. doi: 10.1021/bi0020067. [DOI] [PubMed] [Google Scholar]
- 21.Gamelin DR, Randall DW, Hay MT, Houser RP, Mulder TC, Canters GW, de VS, Tolman WB, Lu Y, Solomon EI. Spectroscopy of Mixed-Valence CuA-Type Centers: Ligand-Field Control of Ground-State Properties Related to Electron Transfer. J Am Chem Soc. 1998;120:5246–5263. [Google Scholar]
- 22.Neese F, Zumft WG, Antholine WE, Kroneck PMH. The Purple Mixed-Valence CuA Center in Nitrous-oxide Reductase: EPR of the Copper-63-, Copper-65-, and Both Copper-65- and [15N]Histidine-Enriched Enzyme and a Molecular Orbital Interpretation. J Am Chem Soc. 1996;118:8692–8699. [Google Scholar]
- 23.Olsson MH, Ryde U. Geometry, reduction potential, and reorganization energy of the binuclear Cu(A) site, studied by density functional theory. J Am Chem Soc. 2001;123:7866–7876. doi: 10.1021/ja010315u. [DOI] [PubMed] [Google Scholar]
- 24.Hulse CL, Averill BA. Isolation of a high specific activity pink, monomeric nitrous oxide reductase from Achromobacter cycloclastes. Biochem Biophys Res Commun. 1990;166:729–735. doi: 10.1016/0006-291x(90)90870-s. [DOI] [PubMed] [Google Scholar]
- 25.Lappalainen P, Aasa R, Malmstrom BG, Saraste M. Soluble CuA-binding domain from the Paracoccus cytochrome c oxidase. J Biol Chem. 1993;268:26416–26421. [PubMed] [Google Scholar]
- 26.Slutter CE, Sanders D, Wittung P, Malmstrom BG, Aasa R, Richards JH, Gray HB, Fee JA. Water-soluble, recombinant CuA-domain of the cytochrome ba3 subunit II from Thermus thermophilus. Biochemistry. 1996;35:3387–3395. doi: 10.1021/bi9525839. [DOI] [PubMed] [Google Scholar]
- 27.von Wachenfeldt C, de Vries S, van der Oost J. The CuA site of the caa3-type oxidase of Bacillus subtilis is a mixed-valence binuclear copper center. FEBS Lett. 1994;340:109–113. doi: 10.1016/0014-5793(94)80182-7. [DOI] [PubMed] [Google Scholar]
- 28.Arciero DM, Hooper AB, Rosenzweig AC. Crystal Structure of a Novel Red Copper Protein from Nitrosomonas europaea. Biochemistry. 2001;40:5674–5681. doi: 10.1021/bi0102611. [DOI] [PubMed] [Google Scholar]
- 29.Arciero DM, Pierce BS, Hendrich MP, Hooper AB. Nitrosocyanin, a red cupredoxin-like protein from Nitrosomonas europaea. Biochemistry. 2002;41:1703–1709. doi: 10.1021/bi015908w. [DOI] [PubMed] [Google Scholar]
- 30.Ryden LG, Hunt LT. Evolution of protein complexity: the blue copper-containing oxidases and related proteins. J Mol Evol. 1993;36:41–66. doi: 10.1007/BF02407305. [DOI] [PubMed] [Google Scholar]
- 31.Savelieff MG, Wilson TD, Elias Y, Nilges MJ, Garner DK, Lu Y. Experimental evidence for a link among cupredoxins: red, blue, and purple copper transformations in nitrous oxide reductase. Proc Natl Acad Sci. 2008;105:7919–7924. doi: 10.1073/pnas.0711316105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farrar JA, Lappalainen P, Zumft WG, Saraste M, Thomson AJ. Spectroscopic and mutagenesis studies on the CuA centre from the cytochrome-c oxidase complex of Paracoccus denitrificans. Eur J Biochem. 1995;232:294–303. doi: 10.1111/j.1432-1033.1995.tb20811.x. [DOI] [PubMed] [Google Scholar]
- 33.Maneg O, Ludwig B, Malatesta F. Different Interaction Modes of Two Cytochrome-c Oxidase Soluble CuA Fragments with Their Substrates. J Biol Chem. 2003;278:46734–46740. doi: 10.1074/jbc.M307594200. [DOI] [PubMed] [Google Scholar]
- 34.Paumann M, Lubura B, Regelsberger G, Feichtinger M, Koellensberger G, Jakopitsch C, Furtmueller PG, Peschek GA, Obinger C. Soluble CuA Domain of Cyanobacterial Cytochrome c Oxidase. J Biol Chem. 2004;279:10293–10303. doi: 10.1074/jbc.M308903200. [DOI] [PubMed] [Google Scholar]
- 35.Slutter CE, Langen R, Sanders D, Lawrence SM, Wittung P, Di Bilio AJ, Hill MG, Fee JA, Richards JH, et al. Electron-transfer studies with the CuA domain of Thermus thermophilus cytochrome ba3. Inorg Chim Acta. 1996;243:141–145. [Google Scholar]
- 36.Williams PA, Blackburn NJ, Sanders D, Bellamy H, Stura EA, Fee JA, McRee DE. The CuA domain of Thermus thermophilus ba3-type cytochrome c oxidase at 1.6 Å resolution. Nat Struct Biol. 1999;6:509–516. doi: 10.1038/9274. [DOI] [PubMed] [Google Scholar]
- 37.Dennison C, Vijgenboom E, de Vries S, van der Oost J, Canters GW. Introduction of a CuA site into the blue copper protein amicyanin from Thiobacillus versutus. FEBS Lett. 1995;365:92–94. doi: 10.1016/0014-5793(95)00429-d. [DOI] [PubMed] [Google Scholar]
- 38.Hay M, Richards JH, Lu Y. Construction and characterization of an azurin analog for the purple copper site in cytochrome c oxidase. Proc Natl Acad Sci. 1996;93:461. doi: 10.1073/pnas.93.1.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones LH, Liu A, Davidson VL. An Engineered CuA Amicyanin Capable of Intermolecular Electron Transfer Reactions. J Biol Chem. 2003;278:47269–47274. doi: 10.1074/jbc.M308863200. [DOI] [PubMed] [Google Scholar]
- 40.van der Oost J, Lappalainen P, Musacchio A, Warne A, Lemieux L, Rumbley J, Gennis RB, Aasa R, Pascher T, Malmstrom BG. Restoration of a lost metal-binding site: construction of two different copper sites into a subunit of the E. coli cytochrome o quinol oxidase complex. EMBO J. 1992;11:3209–3217. doi: 10.1002/j.1460-2075.1992.tb05398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Farver O, Lu Y, Ang MC, Pecht I. Enhanced rate of intramolecular electron transfer in an engineered purple CuA azurin. Proc Natl Acad Sci. 1999;96:899–902. doi: 10.1073/pnas.96.3.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hay MT, Ang MC, Gamelin DR, Solomon EI, Antholine WE, Ralle M, Blackburn NJ, Massey PD, Wang X, Kwon AH, Lu Y. Spectroscopic Characterization of an Engineered Purple CuA Center in Azurin. Inorg Chem. 1998;37:191–198. [Google Scholar]
- 43.Hwang HJ, Berry SM, Nilges MJ, Lu Y. Axial Methionine Has Much Less Influence on Reduction Potentials in a CuA Center than in a Blue Copper Center. J Am Chem Soc. 2005;127:7274–7275. doi: 10.1021/ja0501114. [DOI] [PubMed] [Google Scholar]
- 44.Hwang HJ, Nagraj N, Lu Y. Spectroscopic Characterizations of Bridging Cysteine Ligand Variants of an Engineered Cu2(SCys)2 CuA Azurin. Inorg Chem. 2006;45:102–107. doi: 10.1021/ic051375u. [DOI] [PubMed] [Google Scholar]
- 45.Andruzzi L, Nakano M, Nilges MJ, Blackburn NJ. Spectroscopic studies of metal binding and metal selectivity in Bacillus subtilis BSco, a homologue of the yeast mitochondrial protein Sco1p. J Am Chem Soc. 2005;127:16548–16558. doi: 10.1021/ja0529539. [DOI] [PubMed] [Google Scholar]
- 46.Banci L, Bertini I, Cavallaro G, Ciofi-Baffoni S. Seeking the determinants of the elusive functions of Sco proteins. FEBS J. 2011;278:2244–2262. doi: 10.1111/j.1742-4658.2011.08141.x. [DOI] [PubMed] [Google Scholar]
- 47.Horng YC, Cobine PA, Maxfield AB, Carr HS, Winge DR. Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome C oxidase. J Biol Chem. 2004;279:35334–35340. doi: 10.1074/jbc.M404747200. [DOI] [PubMed] [Google Scholar]
- 48.Nittis T, George GN, Winge DR. Yeast Sco1, a Protein Essential for Cytochrome cOxidase Function Is a Cu (I)-binding Protein. J Biol Chem. 2001;276:42520–42526. doi: 10.1074/jbc.M107077200. [DOI] [PubMed] [Google Scholar]
- 49.Balatri E, Banci L, Bertini I, Cantini F, Ciofi-Baffoni S. Solution structure of Sco1: a thioredoxin-like protein involved in cytochrome c oxidase assembly. Structure. 2003;11:1431–1443. doi: 10.1016/j.str.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 50.Banci L, Bertini I, Calderone V, Ciofi-Baffoni S, Mangani S, Martinelli M, Palumaa P, Wang S. A hint for the function of human Sco1 from different structures. Proc Natl Acad Sci. 2006;103:8595–8600. doi: 10.1073/pnas.0601375103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Horng YC, Leary SC, Cobine PA, Young FB, George GN, Shoubridge EA, Winge DR. Human Sco1 and Sco2 function as copper-binding proteins. J Biol Chem. 2005;280:34113–34122. doi: 10.1074/jbc.M506801200. [DOI] [PubMed] [Google Scholar]
- 52.Siluvai GS, Nakano M, Mayfield M, Blackburn NJ. The essential role of the Cu (II) state of Sco in the maturation of the CuA center of cytochrome oxidase: evidence from H135Met and H135SeM variants of the Bacillus subtilis Sco. J Biol Inorg Chem. 2011;16:285–297. doi: 10.1007/s00775-010-0725-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siluvai GS, Nakano MM, Mayfield M, Nilges MJ, Blackburn NJ. H135A controls the redox activity of the Sco copper center. Kinetic and spectroscopic studies of the His135Ala variant of Bacillus subtilis Sco. Biochemistry. 2009;48:12133–12144. doi: 10.1021/bi901480g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abriata LA, Banci L, Bertini I, Ciofi-Baffoni S, Gkazonis P, Spyroulias GA, Vila AJ, Wang S. Mechanism of CuA assembly. Nat Chem Biol. 2008;4:599–601. doi: 10.1038/nchembio.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siluvai GS, Mayfield M, Nilges MJ, DeBeer George S, Blackburn NJ. Anatomy of a red copper center: spectroscopic identification and reactivity of the copper centers of Bacillus subtilis Sco and its Cys-to-Ala variants. J Am Chem Soc. 2010;132:5215–5226. doi: 10.1021/ja910759v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chacón KN, Blackburn NJ. Stable Cu (II) and Cu (I) mononuclear intermediates in the assembly of the CuA center of Thermus thermophilus cytochrome oxidase. J Am Chem Soc. 2012;134:16401–16412. doi: 10.1021/ja307276z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ghosh MK, Basak P, Mazumdar S. Mechanism of Copper Incorporation in Subunit II of Cytochrome c Oxidase from Thermus thermophilus: Identification of Intermediate Species. Biochemistry. 2013;52:4620–4635. doi: 10.1021/bi400101g. [DOI] [PubMed] [Google Scholar]
- 58.Wang X, Ang MC, Lu Y. Kinetics of Copper Incorporation into an Engineered Purple Azurin. J Am Chem Soc. 1999;121:2947–2948. [Google Scholar]
- 59.Wilson TD, Savelieff MG, Nilges MJ, Marshall NM, Lu Y. Kinetics of Copper Incorporation into a Biosynthetic Purple CuA Azurin: Characterization of Red, Blue, and a New Intermediate Species. J Am Chem Soc. 2011;133:20778–20792. doi: 10.1021/ja205281t. [DOI] [PubMed] [Google Scholar]
- 60.Farrar JA, Formicka G, Zeppezauer M, Thomson AJ. Magnetic and optical properties of copper-substituted alcohol dehydrogenase: a bisthiolate copper(II) complex. Biochem J. 1996;317:447–456. doi: 10.1042/bj3170447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maret W, Dietrich H, Ruf HH, Zeppezauer M. Active Site-Specifc Reconstituted Copper (II) Horse Liver Alcohol Dehydrogenase: A Biological Model for Type 1 Cu2+ and its Changes Upon Ligand Binding and Confnrmational Transitions. J Inorg Biochem. 1980;12:241–252. doi: 10.1016/s0162-0134(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 62.Maret W, Kozlowski H. Biochim Biophys Acta. 1987;912:329. doi: 10.1016/0167-4838(87)90036-7. [DOI] [PubMed] [Google Scholar]
- 63.Robinson H, Ang MC, Gao Y-G, Hay MT, Lu Y, Wang AHJ. Structural Basis of Electron Transfer Modulation in the Purple CuA Center. Biochemistry. 1999;38:5677–5683. doi: 10.1021/bi9901634. [DOI] [PubMed] [Google Scholar]
- 64.Nilges MJ, Matteson K, Belford RL. SIMPOW6: A software package for the simulation of ESR powder-type spectra. In: Hemminga MA, Berliner L, editors. ESR Spectroscopy in Membrane Biophysics. Vol. 27. Springer; New York: 2006. [Google Scholar]
- 65.George GN. EXAFSPAK. Stanford Synchrotron Radiation Laboratory; Menlo Park, CA: 1995. [Google Scholar]
- 66.Gurman S, Binsted N, Ross I. A rapid, exact, curved-wave theory for EXAFS calculations. II The multiple-scattering contributions. J Phys C: Solid State Phys. 1986;19:1845. [Google Scholar]
- 67.Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol. 1963;7:95–99. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- 68.Ghosh MK, Rajbongshi J, Basumatary D, Mazumdar S. Role of the Surface-Exposed Leucine 155 in the Metal Ion Binding Loop of the CuA Domain of Cytochrome c Oxidase from Thermus thermophilus on the Function and Stability of the Protein. Biochemistry. 2012;51:2443–2452. doi: 10.1021/bi2017574. [DOI] [PubMed] [Google Scholar]
- 69.Solomon EI, Szilagyi RK, DeBeer George S, Basumallick L. Electronic Structures of Metal Sites in Proteins and Models: Contributions to Function in Blue Copper Proteins. Chem Rev. 2004;104:419–458. doi: 10.1021/cr0206317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.