

Abstract
Bari elements are members of the Tc1-mariner superfamily of DNA transposons, originally discovered in Drosophila melanogaster, and subsequently identified in silico in 11 sequenced Drosophila genomes and as experimentally isolated in four non-sequenced Drosophila species. Bari-like elements have been also studied for their mobility both in vivo and in vitro. We analyzed 23 Drosophila genomes and carried out a detailed characterization of the Bari elements identified, including those from the heterochromatic Bari1 cluster in D. melanogaster. We have annotated 401 copies of Bari elements classified either as putatively autonomous or inactive according to the structure of the terminal sequences and the presence of a complete transposase-coding region. Analyses of the integration sites revealed that Bari transposase prefers AT-rich sequences in which the TA target is cleaved and duplicated. Furthermore evaluation of transposon’s co-occurrence near the integration sites of Bari elements showed a non-random distribution of other transposable elements. We also unveil the existence of a putatively autonomous Bari1 variant characterized by two identical long Terminal Inverted Repeats, in D. rhopaloa. In addition, we detected MITEs related to Bari transposons in 9 species. Phylogenetic analyses based on transposase gene and the terminal sequences confirmed that Bari-like elements are distributed into three subfamilies. A few inconsistencies in Bari phylogenetic tree with respect to the Drosophila species tree could be explained by the occurrence of horizontal transfer events as also suggested by the results of dS analyses. This study further clarifies the Bari transposon’s evolutionary dynamics and increases our understanding on the Tc1-mariner elements’ biology.
Introduction
Since their identification eukaryotic Transposable Elements (TEs) developed into an important field of genetic and genomics investigation [1]. Nevertheless, recent advances in sequencing technologies offer a unique and somewhat unappreciated opportunity to increase our understanding of several aspects of the TEs biology, e.g. structure, evolution and regulation (see [2] for a review). Besides their detrimental role as an endogenous source of mutations, TEs transposition and accumulation serve as an evolutionary substrate for genes and genomes evolution [3]. Indeed, inactive TEs play a significant role in macroevolution, contributing in chromosomal rearrangements [4] or being recruited to evolve novel functions [5]. In addition, defective elements and ancient relics of autonomous copies are quite informative to trace the evolution of single TEs families.
The Tc1-mariner constitutes one out of 17 super-families of the class II transposons [6]. Tc1-mariner elements are widespread among all life kingdoms and their diffusion is mainly due to their simple transposition mechanism, and their proposed ability for cross-species diffusion by horizontal transfer mechanisms [7, 8]. The Bari family of transposons belongs to the Tc1-mariner superfamily and Bari-like elements have been identified in several species of the Drosophila genus. Based on their structural and evolutionary features Bari-like elements fall into three distinct subfamilies, Bari1, Bari2 and Bari3 after the founder elements discovered in D. melanogaster, D. erecta and D. mojavensis respectively. The Bari1 and Bari3 subfamilies contain autonomous elements able to perform transposition due to the presence of Terminal Inverted Repeats (TIRs) sequences surrounding a central sequence encoding a functional transposase. On the other hand, Bari2-type elements are non-autonomous due to the accumulation of inactivating mutations. Besides the functional criterion, Bari elements can be classified standing to the structural differences of the terminal sequences. Bari1-type elements harbor short TIRs, usually 28 nucleotides long, called Short Inverted Repeats (SIR) while Bari2 and Bari3 possess Long terminal Inverted Repeats (LIR), roughly 250 nucleotides long. Interestingly, non-autonomous Bari1 elements possessing LIRs have been described in D. ananassae, a species of the melanogaster group, suggesting that the ancestor of the Bari1 subfamily had LIRs that were lost during Bari1 evolution [9]. Regardless their length, both SIR- and LIR- containing elements share three highly similar 18 nucleotides long domains, called Direct Repeats (DRs) [9] [10]. DRs are found within the 250 terminal nucleotides at both ends and are responsible for the transposon-transposase interaction, a crucial step in the transposition event [11, 12]. Such interaction has been previously demonstrated for Bari1 [13] and Bari3 [14].
An interesting genomic feature of the Bari1 subfamily is the presence of a heterochromatic array in the D. melanogaster species. It was known from previous studies that at least two distinct clusters exist in the reference genome of D. melanogaster. The first cluster maps to the h39 region [15], a cytological band adjacent to the centromere of the second chromosome of D. melanogaster, the same cytological band where the Responder satellite maps [16]. It contains several tens of Bari1 copies and is interrupted by a MAX [17] insertion. The second cluster contains few copies and had uncertain heterochromatic map location. Heterochromatic Bari1-Bari1 junctions (Right TIR-Left TIR) are characterized by the deletion of the first two nucleotides (CA) in the left terminus of each element. The terminal copies of both clusters have been also previously characterized [18] thus helping in the reconstruction of their origin [18, 19].
Genome-scale comparison studies are an important tool for both understanding the forces that shaped modern forms of transposable elements, and highlight non-mendelian modes of transposons’ transmission [20]. Early investigations on Bari-like transposable elements at the genomic level [9, 21] were essentially performed in 12 Drosophila sequenced genomes available at that time [22, 23]. The availability of 11 additional Drosophila genome draft sequences [24–27] together with the availability of new assembly releases [28] and data from re-sequencing projects [29] prompted us to investigate the genomic distribution of the Bari transposon family in a wider pool of studied and unexplored genomes.
In this study, we performed an extensive annotation of the Bari-like elements in 23 Drosophila genomes analyzed, and uncovered additional structural variability as compared to previous analyses. In addition, we disclosed the presence of MITE-like forms of the Bari transposons in 7 Drosophila species including, interestingly, species apparently devoid of full-length Bari elements. Analyses of the integration site of Bari elements revealed a preference for AT-rich sequences in which the TA target is duplicated upon integration. Furthermore, annotation of unrelated TEs insertions in the proximity of Bari elements revealed significant co-occurrence of other Tc1-mariner elements while class I TEs avoid these regions. Finally, we propose that incongruences revealed by our phylogenetic analyses could be explained by horizontal transfer events. Taken together our results significantly increase our understanding of the evolution of Bari elements.
Materials and Methods
Bari transposon search strategy and sequence analyses
Searches for Bari homologous elements were carried out in Drosophila species listed in S1 Table. A BLAST strategy was applied to identify of Bari-like elements at the NCBI WGS (Whole Genome Shotgun) database (http://www.ncbi.nlm.nih.gov/genbank/wgs/) or at the FlyBase database [30]. Query sequences for tBLASTn searches were either the Bari1 or Bari3 transposases (GenBank CAA47913 and conceptual translation of GenBank accession CH933806 position 6274049–6275068 respectively). Queries for BLASTn searches were performed using either the whole DNA sequence or the 250 terminal nucleotides containing the three DR sequences of Bari1 or Bari3. All BLAST analyses were performed using the default parameters. Subject sequences from tBLASTn searches with E < 10−120 and similarity greater than 75% over the whole transposase length (339 amino acids), were further analyzed. The threshold E-value was set to higher values in BLASTn searches aimed at MITEs identification (E >10−20).
Two criteria were used to identify full-length Bari-like elements; 1) the detection of a high-scoring subject sequence by tBLASTn search, using either the Bari1 or the Bari3 transposase protein as query sequence; 2) the presence of homologous DRs in the terminal sequences, surrounding the coding region of the elements. Terminal inverted repeats and homologous DRs in the transposon’s termini were identified by a combined analysis using the Dot Plot matrix analyses implemented in the DNA Strider package software [31], the Einverted software (http://embossbioinformaticsnl/cgi-bin/emboss/einverted) and by multiple sequence alignment of terminal ends of Bari-like elements as previously described [9].
Elements’ names were assigned according to the binary nomenclature used in Repbase [32, 33], consisting of the subfamily identifier (either Bari1, Bari2 or Bari3) followed by the species identifier (i.e. the first letter of the genus and three letters of the species name to avoid ambiguity for some species). Similarly, MITE’s elements names look like Bari_Dxyz_MITE-# (where “xyz” is a three-letters species identifier) followed by a number (#) to distinguish different MITEs subfamilies, where necessary.
Once a novel full-length element was identified, it was used to identify other full-length copies as well as truncated elements, by BLASTn analyses against species-specific WGS database. Each copy was then carefully annotated (S2 Table). Additional BLASTn searches were performed using as query the mel-ER (consensus), the sim-ER and the sec-ER elements (here named Bari2_Dmel, Bari2_Dsim and Bari2_Dsec in the text) described in [9], three Bari2-type elements in the genomes of D. melanogaster, D. simulans, and D. sechellia respectively. Redundancy can be excluded for all elements except for split elements, i.e. sequences overlapping either the beginning or the end of a genomic scaffold in which they have been identified. These sequences have been annotated as “partial” elements in S2 Table and were counted as elements. No further analysis was performed using partial elements. Redundant elements were removed on the basis of the flanking sequences comparison. Global alignments were performed using LALIGN (http://wwwchembnetorg/software/LALIGN_formhtml). The Open Reading Frames (ORFs) were inferred using ORF Finder (http://wwwncbinlmnihgov/gorf/gorfhtml) or using a custom script (available upon request) written in Biopython (version 1.63) [34]. The similarity with previously reported sequences was established using CENSOR [35] at the RepBase database [36].
dS analysis was performed using the Nei and Gojobori method [37] implemented in MEGA5 [38]. Fisher’s exact test (one-tailed) was conducted using 2 x 2 tables to verify if transposon dS values were statistically lower than those presented by the host genes.
The number of base substitutions per site between sequence pairs was calculated using MEGA 5.0 [38]. Analyses were performed using the Kimura 2-parameter model. Rate variation among sites was modeled with a gamma distribution (shape parameter = 1). All ambiguous positions were removed for each sequence pair. The whole transposon sequence alignment was used to perform Plotcon analyses (http://emboss.bioinformatics.nl/cgi-bin/emboss/help/plotcon) by moving a window of 100 nucleotides along the aligned sequences. Sequences marked as “partial sequence” in S2 Table were excluded from p-distance and Plotcon analyses. Data analysis was carried out with the R System (R version 3.1.0) (https://www.r-project.org). The non-parametric Kruskal-Wallis test and Tukey test were used to assess significance for the observed differences, considered significant at p values<0.05. Hartigan dip test was carried out to assess unimodality/multimodality of the observed similarity values distributions [39].
Insertion sites analyses
WebLogo analyses [40] were performed in those species where it was possible to retrieve at least four flanking sequences of equally oriented Bari elements. A larger scale insertion site analysis was also performed. Regions of 2500 bp flanking downstream and upstream the insertion sites of Bari elements, when available, were analyzed with RepeatMasker-open-4.0.5 (http://www.repeatmasker.org) using the RepBase20.02 dataset. The same analysis was performed, as control, on 1000, non-overlapping, random sequences, 2,5 kb in length, irrespective of their gene content selected from species containing at least four Bari insertions, taking care to avoid regions with internal sequencing gaps. The distance of each masked transposon from the sequence origin (fixed at the first base flanking Bari elements or to the first nucleotide of the randomly-selected scaffold) was calculated and used to create a positions dataset. The detected distances were then classified in 5 distance ranges from the sequence origin, considering a 500 bp cumulative increment (0–500; 0–1000; 0–1500; 0–2000 and 0–2500). Poisson distribution has been applied to the data, given that the repeat frequency in 1000 random regions was considered as the expected value, and the frequency in the Bari flanking regions as the observed value to test. Sequence accessions and coordinates of the control dataset are reported in S1 File. Relative probability was calculated to identify a significant association between Bari and the selected repeat families.
Multiple alignments and phylogenetic analysis
Multiple sequence alignments were performed using ClustalW2 [41] with default parameters. Nucleotide sequences relative either to the Coding Sequence (CDS) or to the terminal sequences were used in the analyses of Bari elements. The Bari1_Dsim and Bari1_Dsec elements were not included in the multiple alignment of terminal sequences and transposase sequences because they are identical in sequence to the Bari1_Dmel element. The D. yakuba element was excluded from the alignment of TIR sequences because it has been re-classified as MITE, and thus included in the MITE elements’ analysis. The BioEdit software was used for alignment editing and visualization [42]. Alignment slices were obtained using the Alignment Slicer tool (http://www.hiv.lanl.gov/content/sequence/Slice_Align/).
Species phylogeny was based on the alignment of nine concatenated orthologous CDSs encoding subunits of the V-ATPase complex, for which orthologous genes could be identified in the 23 species analyzed. FlyBase annotation symbols, GenBank accession of mRNA and length of D. melanogaster CDSs used in this analysis are the following: CG3762 NM_143747 (1845 bp), CG2934 NM_130724 (1054 bp), CG7071 NM_142770 (909 bp), CG34131 NM_001043279 (483 bp), CG12403 NM_001273497 (1845 bp), CG1088 NM_169073(681 bp), CG7007 NM_143753 (639 bp), CG3161 NM_057453 (480 bp), CG6213 NM_058089 (354 bp). The jModelTest program v 2.1.7 [43] [44] [45] was used to select the simplest evolutionary model that fitted adequately the sequence data. A Bayesian Markov chain Monte Carlo (MCMC) method implemented in BEAST package 1.8.0 [46, 47] was used for Bayesian analysis. The MCMC chains were run for at least 50 million generations and, sampled every 5.000 steps. Convergence was assessed on the basis of the effective sampling size after a10% burn-in. Phylogenetic trees were visualized and edited using FigTree 1.4.2 software (http://treebioedacuk/software/figtree/). Paris [48] and S [49] elements were used as outgroups.
Results
Bari elements identification and their structural diversity in 23 sequenced genomes
We applied a BLAST-based search to identify insertions of Bari-like elements in 23 Drosophila genomes (S1 Table). The evolutionary time of the species analyzed in this work spans roughly 40 million year [50]. We used the full-length Bari1 and Bari3 transposase as query in tBLASTn analyses, and the whole transposon DNA sequences in BLASTn analyses against the genome shotgun database or reference genomes. Using this approach, we identified 401 Bari-like elements comprising full-length copies, truncated elements and MITEs. The actual number of Bari insertions annotated in the species analyzed could be slightly inflated because we have also annotated partial sequences (53 out of 249 sequences in S2 Table), i.e. split sequences overlapping either the beginning or the end of a genomic scaffold in which they have been identified, for which we cannot exclude redundancy. Redundancy can be instead excluded in the remaining sequences for which we compared the flanking sequences to establish uniqueness in the dataset. Inflation is particularly relevant in D. pseudoobscura (22/33 total insertions) and in D. miranda (10/16 total insertions). However, inflation could be compensated by the possible underestimation of the Bari copy number due to the draft status of the genome assembly in some species. New Bari-related sequences were annotated in 11 species (namely D. biarmipes, D. bipectinata, D. rhopaloa, D. takahashii, D. kikkawai, D. eugracilis, D. ficusphila, D elegans, D. suzukii, D. miranda, and D. albomicans). For clarity purposes, data concerning full-length and truncated elements are presented separately from data concerning MITEs. The features of representative elements (i.e. the top scoring and the most complete subjects after BLAST search) are summarized in Table 1, while information concerning all the annotated insertions can be found in S2 and S3 Tables.
Table 1. Bari elements features in the analyzed genomes.
| Drosophila species | Length(bp) | TIR structure | Representative sequence (GenBank accession) | Copies detected | 3DRs -containing sequence (bp) | Left vs Right terminal sequences similarity (%) | TNP (aa) | % sim vs Ba1TNP (%) | %sim vs Ba3TNP (%) | Sub-family | Element’s name |
|---|---|---|---|---|---|---|---|---|---|---|---|
| melanogaster | 1726 | SIR-3DR | X67681 | 69 | 254 | 85.2 | 339 | 100 | 79,7 | Bari1 | Bari1_Dmel |
| melanogaster | 1730 | LIR-3DR | reconstructed | 10 | 255 | 92.2 | 334* | 74 | 76 | Bari2 | Bari2_Dmel |
| melanogaster | 116 | SIR-1DR | AE013599.5 | 4 | NA | NA | NA | NA | NA | MITE | Bari_Dmel_MITE1 |
| melanogaster | 692 | SIR-1DR | JSAE01000428.1 | 1 | NA | NA | NA | NA | NA | MITE | Bari_Dmel_MITE2 |
| simulans | 1726 | SIR-3DR | NC_011089.1 | 8 | 254 | 85,2 | 339 | 99 | 79 | Bari1 | Bari1_Dsim |
| simulans | 1222 | ND | NT_167066.1 | 1 | ND | ND | 310* | 61 | 58 | Bari2 | Bari2_Dsim |
| simulans | 77 | SIR-1DR | AASU01036800.1 | 1 | NA | NA | NA | NA | NA | MITE | Bari_Dsim_MITE |
| sechellia | 1727 | SIR-3DR | CH481259 | 7 | 254 | 85,2 | 339* | 97 | 77 | Bari1 | Bari1_Dsec |
| sechellia | 1265 | ND | NW_001999696.1 | 1 | ND | ND | 329* | 56 | 55 | Bari2 | Bari2_Dsec |
| sechellia | 77 | SIR-1DR | AAKO01017469.1 | 24 | NA | NA | NA | NA | NA | MITE | Bari_Dsec_MITE1@ |
| sechellia | 77 | SIR-1DR | AAKO01010824.1 | 25 | NA | NA | NA | NA | NA | MITE | Bari_Dsec_MITE2@ |
| yakuba | 467 | LIR-3DR | AAEU02002077.1 | 1 | NA | NA | NA | NA | NA | MITE | Bari_Dyak_MITE |
| erecta | 1639 | LIR-3DR | Y13853 | 16 | 254 | 93,8 | 325* | 84 | 85 | Bari2 | Bari2_Dere |
| ficusphila | 687 | SIR-1DR | AFFG02006632.1 | 36 | NA | NA | NA | NA | NA | MITE | Bari_Dyak_MITE |
| eugracilis | 1020 | ND-3DR | AFPQ02005153 | 1 | 230 | NA | NA | NA | NA | Bari1 | Bari1_Deug |
| biarmipes | 1728 | LIR-3DR | AFFD02006349 | 9 | 262 | 93,9 | 335* | 88 | 77 | Bari1 | Bari1_Dbia |
| suzukii# | 1689 | LIR-3DR | reconstructed | 4 | 255 | 92,2 | 333* | 82 | 62 | Bari1 | Bari1_Dsuz |
| suzukii | 398 | SIR-1DR | AWUT01015981.1 | 11 | NA | NA | NA | NA | NA | MITE | Bari_Dsuz_MITE |
| takahashii§ | 1825 | LIR-3DR | AFFI02005672 | 2 | 254 | 100 | 373* | 74 | 58 | Bari1 | Bari1_Dtak |
| takahashii | 78 | SIR-1DR | AFFI02008544 | 8 | NA | NA | NA | NA | NA | MITE | Bari_Dtak_MITE |
| elegans | 78 | SIR-1DR | AFFF02008066.1 | 4 | NA | NA | NA | NA | NA | MITE | Bari_Dele_MITE |
| rhopaloa | 1726 | LIR-3DR | AFPP02028090 | 7 | 255 | 100 | 339 | 84 | 65 | Bari1 | Bari1_Drho |
| rhopaloa | 78 | SIR-1DR | AFPP02033070.1 | 37 | NA | NA | NA | NA | NA | MITE | Bari_Drho_MITE |
| kikkawai | 1720 | SIR-3DR | AFFH02005270 | 6 | 247 | 59,5 | 339* | 78 | 62 | Bari1 | Bari1_Dkik |
| ananassae | 1727 | LIR-3DR | AAPP01019820 | 16 | 255 | 93,7 | 337* | 89 | 77 | Bari1 | Bari1_Dana |
| bipectinata# | 1729 | LIR-3DR | Reconstructed | 2 | 255 | 97,6 | 339* | 84 | 65 | Bari1 | Bari1_Dbip |
| pseudoobscura | 1706 | LIR-3DR | AADE01010081 | 33 | 243 | 78,5 | 339 | 81 | 85 | Bari3 | Bari3_Dpse |
| persimilis | 1704 | LIR-3DR | AAIZ01001634 | 18 | 242 | 80,2 | 339 | 81 | 85 | Bari3 | Bari3_Dper |
| miranda | 1704 | LIR-3DR | AJMI02000744 | 15 | 243 | 64,7 | 339 | 67 | 73 | Bari3 | Bari3_Dmir |
| willistoni | 1725 | LIR-3DR | AAQB01008459 | 6 | 254 | 95,7 | 339 | 79 | 78 | Bari3 | Bari3_Dwil |
| mojavensis | 1717 | LIR-3DR | AAPU01011127 | 15 | 256 | 99,6 | 339 | 80 | 100 | Bari3 | Bari3_Dmoj |
| virilis | 1645 | LIR-3DR | AANI01016007 | 3 | 255 | 56,5 | 322* | 78,2 | 78,2 | Bari2 | Bari2_Dvir |
| albomicans£ | 109 | ND-ND | ACVV01103928 | 1 | NA$ | NA | ND | NA | NA | Bari2 | Bari2_Dalb |
| grimshawi | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | NA |
GenBank accessions are provided (column 4) for sequences representing the Bari elements identified in a given species. Left vs Right terminal sequences similarity (column 7) refers to the DRs-containing sequences (roughly 250 bp).
Symbols legend
*: inferred from a reconstructed transposase gene
§: a MITE nested insertion was removed
#: element reconstructed from two different truncated elements
£: sequence identified by BLASTn analysis
$: sequence containing two DRs
@: Described by Dias and Carareto [21].
ND (NA): none detected (not applicable).
A snapshot of the distribution of Bari subfamilies and the presence of potentially active elements in the species analyzed is reported in Fig 1. Details relative to individual insertions identified in this study are reported in S2 and S3 Tables. The presence of Bari-like elements in every genome analyzed, with the exception of D. grimshawi, confirms previous evidence on the widespread diffusion of this transposon family in the Drosophila genus. As can be observed in Fig 1A, distinct Bari subfamilies have colonized specific genomes, with little subfamilies overlaps, as can be observed in the species of the melanogaster complex (D. melanogaster, D. simulans, D. sechellia) for the Bari1 and Bari2 subfamilies.
Fig 1. Overview of the distribution and structure of Bari elements in the species analyzed.

(A). Bayesian phylogenetic tree (GTR+G+I model) of the species analyzed with a schematic structure of the Bari elements identified in each species (not in scale). All nodes are highly supported (posterior probability >0,9). Colored triangles represent subfamily-specific TIRs (see graphical boxed legend). DRs are depicted as black arrowheads. In species containing only truncated elements, elements are depicted using broken boxes. Species harboring only inactive elements due to mutations in CDS are marked with “***”. (B). Histograms recapitulating number and type of Bari elements in each analyzed species (see also S2 and S3 Tables).
Drosophila melanogaster contains the highest number of Bari-like element copies (84 elements annotated) mainly clustered in a specific heterochromatic region, (best described in the next paragraph), followed by D. pseudoobscura (33 insertions detected), while D. albomicans (1 element) virtually lacks Bari elements and D. grimshawii is devoid of Bari elements. A preliminary analysis of all the elements identified suggests that Bari-like elements have variable length ranging from 17 bp (S2 Table, D. melanogaster sheet, element #25) to 4353 bp (S2 Table, D. ananassae sheet, element #12), due to the existence of canonical copies, truncated copies (i.e. transposon chunks and elements overlapping the extremities of the sequence contigs) or other mobile elements nested within Bari elements. The median length of all annotated Bari elements is 696 bp (IQR = 1635 bp; max. length = 4353; min. length = 17). In more details, the median length of full-length and truncated elements (S2 Table) is 1704 bp (IQR = 593; max. length = 4353; min. length = 17), while the median length of MITEs (S3 Table) is 82 bp (IQR = 394; max. length = 939; min. length = 69). Considering only those elements containing two terminal sequences (each one containing three DRs) framing an intervening sequence, irrespective of its coding potential (134 elements) Bari-like elements look more homogeneous (median length = 1723 bp; IQR =; min. length = 746 bp in D. pseudoobscura; max length = 4353 bp in D. ananassae). The TA dinucleotide TSD, a feature of the Tc1-mariner superfamily, was identified in 60 out of these 134 sequences. Twenty-four copies of Bari, distributed in seven species (D. melanogaster, D. simulans, D. rhopaloa, D. persimilis, D. pseudoobscura, D. miranda and D. mojavensis) can be considered as autonomous elements due to the presence of two terminal sequences (either with LIR or SIR structure) bracketing a CDS encoding homologous Bari transposase.
The presence of three conserved DR sequences within the 250 terminal nucleotides bracketing the transposase gene, typical of the Bari family [9], suggest that all the new elements identified are bona fide Bari-like elements (Fig 2). We performed a comparative analysis to estimate the sequence variability of Bari elements and the deterioration profile both at the inter-species and intra-species level. We calculated the pairwise distance (p-distance) relative to the left and right terminal sequences, comprising the DRs, and to the transposase coding sequence. At the inter-species level, no significant differences can be observed among the CDS and the terminal sequences in the three Bari subfamilies (Fig 3A and S4 Table), suggesting the uniform distribution of point mutations in the three regions analyzed. However, some differences were observed by analyzing the same elements at the intra-species level, where the p-distance analysis was also coupled with the determination of the degradation level of the Bari elements in each species. As an example, three representative results obtained are shown in Fig 3 (see S1 Fig for complete results). In two species (D. willistoni and D. simulans, Fig 3A) the p-distances at the terminal sequences are significantly different if compared to the central region (S4 Table), suggesting an increased mutational load at the terminal sequence level. In four species one of the TIRs has a significantly higher distance if compared to the CDS (D. ananassae D. erecta, D. pseudoobscura, D. miranda, Fig 3B). In the remaining three species (D. melanogaster, D. persimilis, D. mojavensis, Fig 3C) no significant difference was observed, suggesting uniform mutation pattern. The respective degradation profiles show that truncated elements are generated in each of the analyzed species by deletion and/or unrelated TEs nested insertions, occurring without any obvious positional preference pattern.
Fig 2. The terminal sequences of Bari elements.

(A) Schematic representation recapitulating the structure of Bari elements. Black arrowheads represent DRs within either the LIRs (green dashed triangles) or SIRs (red dashed triangles) framing a central transposase gene (TNP). See the main text for detailed description of elements belonging to single subfamilies. (B) Alignment slices relative to the conserved blocks of 18 nucleotides (DRs) in the left (Lo, Lm, Li) and right (Ro, Rm, Ri) termini of Bari-like transposons. Note that Bari1_Dmel, Bari2_Dere and Bari3_Dmoj correspond to the previously described Bari1, Bari2 and Bari3 respectively. This analysis was performed using the representative sequences shown in Table 1. Lo, Left outer; Lm, Left middle; Li, Left inner; Ro, Right outer; Rm, Right middle; Ri, Right inner.
Fig 3. Number of base substitutions per site (p-distance) and graphic representation of the deterioration profiles for Bari elements.
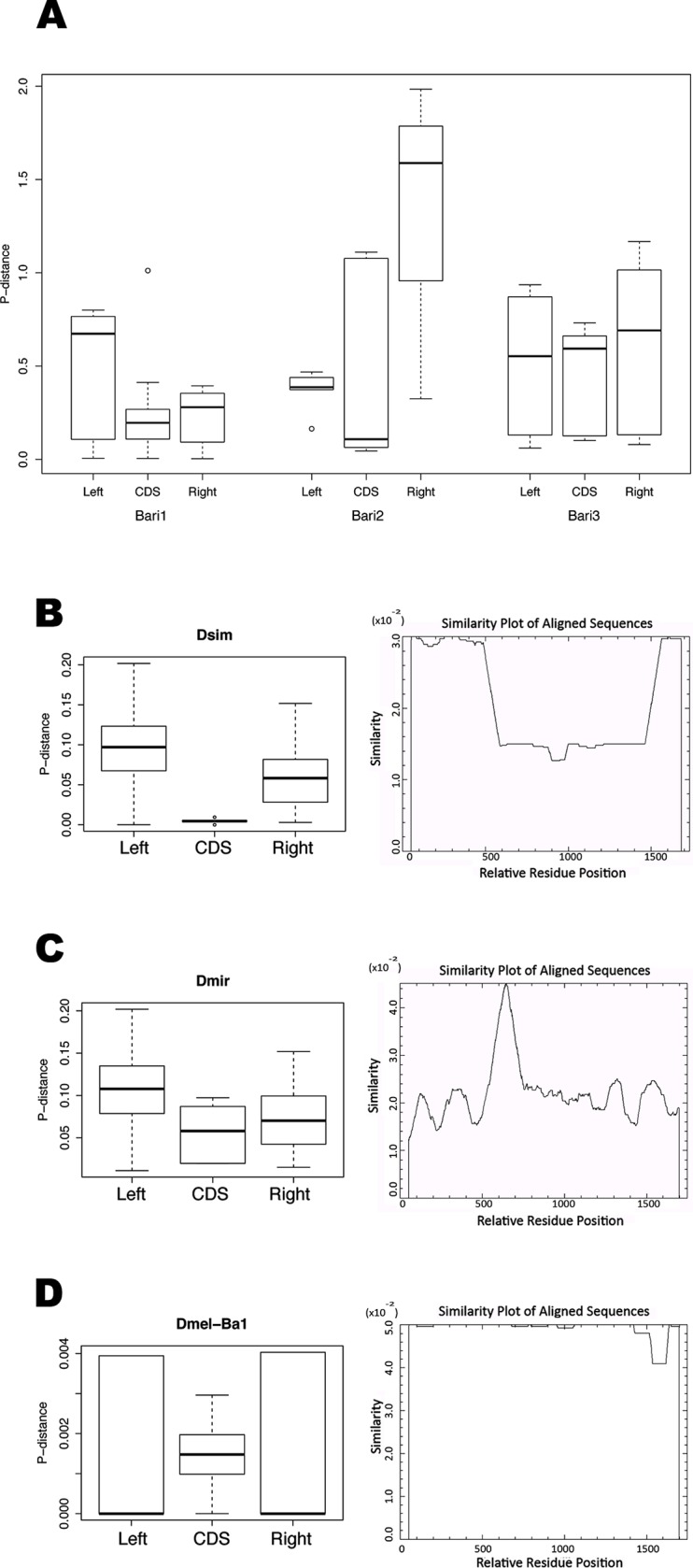
Bottom and top of the boxes represent the first and third quartiles, the line inside each box represent the median value; whiskers mark the data within the 1.5 IQR range. Outliers are depicted as dots outside the whiskers. (A) Inter-species analysis of the three Bari subfamilies. Boxplots report the p-distance relative to the 5’ (Left) and 3’ (Right) sequences containing the DRs (roughly 250 nucleotides analyzed) and to the region homologous to the transposase gene (CDS). Panels B, C, D report examples of intra-species analyses. (B) An example of Bari elements showing a significant divergence of both TIRs if compared to the CDS. (C) An example of Bari elements showing a significant divergence of a single TIR if compared to the CDS. (D) An example of Bari elements not showing a significant TIRs divergence compared to the CDS. The number of sequences used to perform single analyses is reported in S1 Fig. Statistical significance for each analysis is shown in S4 Table.
A description of Bari elements in those genomes containing few copies, and thus not analyzed as described above, is given below.
The genome of D. takahashii contains two inactive elements due to insertions and deletions. The Bari1_Dtak element was reconstructed removing an unreported MITE insertion (not shown). Bari1_Dtak includes a 912 bp-long ORF encoding a protein that shares 81% and 72% similarity with Bari1 and Bari3 transposase respectively framed by two LIRs containing 3 DRs.
The Bari1_D.rho element coding sequence encodes a protein 84% similar to the Bari1 transposase, framed by LIR-type terminal sequences. Bari1-like elements with a similar structure have been identified in the genomes of D. bipectinata, D. biarmipes, D. suzukii, D. takahashii and previously in D. ananassae [9] but all them are inactive, while the Bari1_D.rho can be considered as a putatively active element (although the presence of the very last two nucleotides in the right TIR has not been confirmed, see also S2 Table).
The D. miranda Bari element encodes a Bari3-like transposase (73% similarity, see Table 1) and shows divergent terminal sequences (71.8% similarity, Table 1). A single putatively autonomous element can be found in the current genome assembly of D. miranda along with several defective elements carrying internal deletions (see S2 Table).
In D. albomicans a 109 bp long fragment can be only identified on the basis of its nucleotide similarity (75%) with the Bari2_Dere element (Bari2). This sequence, arbitrarily assumed to be a left terminus, contains the left middle (Lm) and left inner (Li) DRs, that can be easily recognized with multiple alignment analysis (Fig 2) and allow its classification in the Bari family, and specifically its assignment to the Bari2 subfamily (see last paragraph of the Results section).
The putative ancestral sequence of Bari elements of D. suzukii and D. bipectinata element were reconstructed from truncated elements (Bari1_Dsuz element reconstructed from AWUT01004624 and AWUT01009036; Bari1_Dbip element reconstructed from contigs AFFE02004473 and AFFE02005005. See S2 Table for sequence coordinates). Both the Bari1_Dbip and the Bari1_Dsuz elements are members of the Bari1 subfamily and possess LIR-type terminal sequences.
In D. eugracilis a low-scoring sequence contained a truncated copy of a Bari element with single terminal sequences with three DRs (Fig 2 and S2 Table).
While in D. grimshawi we were not able to detected elements related to the Bari family, in D. ficusphila and D. elegans we have identified Bari-related MITEs, described in a specific paragraph. In D. grimshawi the absence of Bari-like elements is possibly due to the actual absence of the transposon or early draft status of the genome sequencing.
New insights into the Bari1 heterochromatic cluster in D. melanogaster
We identified 61 independent heterochromatic Bari1 copies in heterochromatic tandem repeat configuration. The contig JSAE01000772 contains the LTR-retrotransposon MAX embedded in 20 Bari1 copies repeated in tandem. This observation matches with previous findings concerning the discontinuity in the Bari1 repeat [17]. Two additional contigs identified in this study (ac. nos. JSAE01000400 and JSAE01000412) contain respectively 18 and 17 Bari1 copies in tandem repeat configuration. These contigs map to the borders of the Bari1 cluster and contain terminal copies matching also identified in previous studies [18]. We can orient of these contigs with respect to the centromere of the second chromosome considering that: 1) the Responder repeats, representing the major satellite DNA in this heterochromatic region [16] are abundant in the JSAE01000412 contig (388 copies); 2) Responder maps proximally to the centromere with respect to the Bari repeat as demonstrated by previous studies [15, 19]. It can be concluded that contigs JSAE01000400 and JSAE01000412 map, respectively, distally and proximally to the centromere of the second chromosome.
Scaffold JSAE01000412 contains two adjacent truncated Bari1 copies (position 169962–173070, corresponding to elements #31 and #32, in S2 Table). We hypothesized that either inter-monomer recombination or unequal crossing over events involving adjacent or non-adjacent copies of the cluster and occurring at short homologous sequences, as shown in S2 Fig (panel A). Comparison of these two adjacent elements to two canonical adjacent Bari1 heterochromatic copies (i.e. containing two full-length Bari1 elements carrying the deletion of the fist two nucleotides) revealed a 340 bp deletion flanked by short stretches of identical sequences that could mediate recombination leading to the observed deletion (S2 Fig, panel B).
The last Bari1 copy of the cluster toward the centromere of chromosome 2 represents an additional defective Bari1 element. This truncated copy retains only 16 terminal nucleotides of the right TIR, previously described in [18]. A possible origin of the terminal copies of the h39 cluster, linked to an aberrant activity of the transposase has been previously proposed [19].
The last worth-noting scaffold (JSAE01000184) contains six Bari1 copies arranged in a tandem repeat configuration, embedded in more than 200 kb of repetitive DNA. RepeatMasker analyses revealed the presence of transposable elements belonging to the R1 and the R2 class, typically inserted into rDNA sequences as well as few unmasked sequences related to rDNA genes (at least 4 copies). As previously suggested, this short Bari1 cluster could either map in a distinct h39 sub-region or could be located on the X chromosome, in the region of the nucleolar organizer region. Based on the recent findings that no rDNA repeat nor type 1 rDNA insertions exist outside the nucleolar organizer regions D. melanogaster [51], it could be concluded that the small Bari1 cluster may be located in the heterochromatin of the X or Y chromosomes, where the nucleolar organizers map.
MITE elements related to Bari transposons
The tBLASTn strategy described above fails in detecting Bari copies carrying extensive deletions of the transposase gene, as can be observed in MITEs. Dias and Carareto recently described two MITE elements related to Bari1 (msechBari1 and msechBari2, hereafter Bari_Dsec_MITE1 and Bari_Dsec_MITE2) present in multiple copies in the genome of D. sechellia [21], therefore, the existence of Bari-related MITEs in other species cannot be excluded. To test this possibility we performed a BLASTn-based search of Bari-like sequences in the species in which Bari-like elements were not found on the first attempt (namely D. ficusphila, D. elegans and D. grimshawi). The genomes of D. ficusphila and D. elegans harbor 36 and 4 Bari-related MITEs respectively. We, therefore, searched for Bari-related MITEs in all the available sequenced Drosophila genomes. We identified and annotated 152 copies of Bari-related MITEs in nine genomes, including D. sechellia (Table 1 and S3 Table). Names were assigned according to the rule given in the Materials and Methods section. In the genomes of D. simulans, we identified a Bari-related MITE identical in sequence to Bari_Dsec_MITE1 of D. sechellia [21]. However, while Bari_Dsim_MITE is present as single copy element in the genome of D. simulans, more than twenty Bari_Dsec_MITE1 copies are found in D. sechellia ([21] and this study, Table 1). In D. melanogaster, we identified two Bari-related MITE families based on their length. The Bari_Dmel_MITE1 family is an 116 bp long element with nearly identical, 46 bp long, terminal inverted repeats. The Bari_Dmel_MITE2 family contains longer elements (692 bp) and has 50 bp long terminal inverted repeats. D. suzukii harbors a single family of Bari-related MITEs, nearly 500 bp long, sharing high sequence similarity if compared to each other. A retrospective analysis of the single copy element previously identified in D. yakuba [9] led us to conclude that it can be classified as a Bari-related MITE. A peculiar feature of this MITE consists in the presence of two DR sequences in the left terminus while all the other Bari-related MITEs display a single DR in each terminus.
An overall comparison of the terminal sequences of the Bari-related MITEs with the terminal sequences of Bari1_Dmel, Bari2_Dere and Bari3_Dmoj, representative of the Bari1, Bari2 and Bari3 subfamilies respectively, is shown in Fig 4A. All the terminal sequences of Bari-related MITEs are very similar to each other and share similarity with the corresponding full-length elements. Bari-MITE sequences contain an intervening sequence between the two terminal that can be either short or long but with poor sequence similarity if compared to reference elements (S3 Fig), suggesting that complex mechanisms might cause their origin from master elements (Fig 4B). Although the sequence similarity at the terminal sequences level suggests that these MITEs belong to the Bari family, no obvious ancestor can be inferred using this approach. MITEs with identical sequence have been identified in different species (i.e. Bari_Dsec_MITE1 and Bari_Dsim_MITE) while in other cases MITEs from different species have a very similar sequence (Bari_Drho_MITE and Bari_Dtak_MITE share 75 out of 78 nucleotides).
Fig 4. The Bari-derived MITEs.

(A) Multiple alignment slices relative to the left and right terminal sequences of the Bari-related MITEs compared to the canonical elements Bari1, Bari2 and Bari3. Lo and Ro are Left outer and Right outer Direct Repeats respectively. (B) Possible origin of Bari related MITEs through internal deletion of a functional element with breakpoints between the Lo-Lm and Rm-Ro DRs (see arrows). (C) Pairwise nucleotide diversity distribution in Bari-related MITE elements. Species displaying a unimodal distribution of the sequence similarity (Drho, Dfic and Dsec) and species displaying bi- or multi-modal distribution (Dsuz and Dtak) of the sequence similarity are shown. Hartigan dip test for unimodality / multimodality: Drho p = 0,598; Dfic, p = 0,2135; Dsec, p = 0,4119; Dsuz, p = 0,01805; Dtak, p = 0, 01374. H0 is unimodality.
To investigate the mechanisms of MITEs expansion, we postulated that the analyzed MITEs derived from the same ancestor and performed pairwise nucleotide diversities analysis of these elements (Fig 4C). Rogers and Harpending [52] reported that episodes of population growth leave a characteristic signature in the distribution of nucleotide differences between pairs of individuals, and this concept was used to describe the mode of amplification of MITEs in Oryza sativa [53] [54]. We adapted this type of analysis to the Bari-derived MITEs and statistically evaluated whether the wave-like form of the histograms could fit unimodal distribution. Our data do not allow rejection of unimodality in three species (D. sechellia, D. ficusphila, D. rhopaloa; Hartigan dip test for unimodality: Drho p = 0,598; Dfic, p = 0,2135; Dsec, p = 0,4119) suggesting single burst of transposition in these genomes. In the genomes of D. suzukii and D. takahashii, a multimodal distribution of the pairwise differences (Hartigan dip test for unimodality: Dsuz, p = 0,01805; Dtak, p = 0, 01374) suggests that multiple rounds of MITEs transposition occurred.
Bari-like elements and their target site selection preferences
To gain insight into the choice of integration target sites for Bari-like elements, we performed flanking sequences analyses. It is well established that Tc1-mariner elements integrate themselves into the TA target, which is duplicated upon element insertion [10]. Multiple alignment of 15 bp long sequences encompassing the Bari elements insertion sites we identified, showed that this is also true for Bari elements, as also highlighted by the WebLogo analyses (Fig 5A).
Fig 5. Target site preferences of Bari elements.

(A) WebLogos showing the preferred target sequence and the target site duplicated upon integration (TSD) of Bari elements. Results obtained from single species are shown in S4 Fig. The number of flanking sequences analyzed (n) is given. (B) Correlation between Bari elements and members of different transposon super-families. Y-axis indicates the number of genomes in which a given transposon family is significantly found associated (positive values) or is significantly under-represented (negative values) at a given distance (X axis) from the Bari elements. The position of the Bari element is conventionally fixed at the origin of X-axis. Results obtained from single genome analyses are shown in S5 Fig.
Furthermore, the Bari transposase targets AT-rich sequences irrespective of the Bari subfamily and of the host species, as can be observed in S4 Fig.
We further investigated the target site preferences of Bari-like elements on a larger sequence scale by analyzing the sequence context in which Bari transposons insert themselves.
The presence of transposable elements belonging to three LTR-retrotransposons superfamilies (Copia, Gypsy and PAO), three non-LTR retrotransposons superfamilies (LOA, I and Jockey), the Tc1-mariner superfamily and to the Helitron superfamily, was annotated within a 2,5 kb long sequence flanking upstream and/or downstream the Bari elements in each species analyzed. To assess whether the observed distribution of transposable elements within the neighborhoods of Bari, significantly differ from their genomic distribution in the respective genomes, we collected 1000, non-overlapping, random sequences, of the same length (2,5 kb), irrespective of their gene content (S1 File), and recorded the occurrence of tested TEs. This analysis was performed in genomes containing a minimum of 4 Bari insertions (an arbitrarily chosen threshold), resulting in 15 investigated genomes (Fig 5B and S5 Fig). In these genomes, the Bari transposon insertions occur in genomic loci depleted in Copia, LOA and I elements and enriched in Tc1-mariner elements. In 5 genomes we also found a significant tight association (within 500 bp from the Bari elements) with elements of the Jockey and PAO families; however in 7 genomes, taking into account the same sequence range, Bari elements insertions occur in genomic regions significantly depleted in Jockey and PAO elements. Gypsy-like retrotransposons do not display significant enrichment or depletion within the 2,5 kb range considered. Helitron-like elements also significantly co-occur within 2,5 kb from Bari elements.
Evolutionary analyses of Bari transposons
We multi-aligned the transposase-coding sequences from 16 representative Bari copies and built a phylogenetic tree using Bayesian inference methods. The phylogenetic tree created recovered three clades with significant statistical support (Fig 6A, left tree) and corresponding to the previously described Bari1, Bari2 and Bari3 clades [9]. This analysis placed the newly identified Bari elements of D. suzukii, D. takahashii, D. rhopaloa, D. bipectinata, D. biarmipes, D. kikkawai and of D. eugracilis in the Bari1 clade while the element of D. miranda cluster together with other Bari3 elements.
Fig 6. Evolution of the Bari elements.

(A) Bayesian phylogenetic trees of Bari-like elements. The tree on the left was built using the transposase genes alignment and the GTR+G model while the tree on the right was built using the left terminal sequences containing the DRs of elements and the HKY+G model. Asterisks next to the branches denote posterior probability values greater than 0,9. The color code legend used to indicate the Bari three subfamilies is shown between the two trees. B) Dot plot matrix (window = 13; stringency = 11) showing the similarity of two Bari elements in D. biarmipes and D. bipectinata. The percent identity between the two sequences is also shown. C) Comparison of dS values of the Bari transposase-coding region with the dS values of two nuclear genes in D. biarmipes and D. bipectinata. (***; p<0,005).
We also performed phylogenetic analyses based on the terminal sequences of Bari-like elements. Roughly 250 nucleotides containing the three DRs from the left terminal sequence of Bari-like elements were analyzed in a Bayesian tree (Fig 6A, right tree). The phylogenetic tree obtained displays a topology similar to that obtained for the transposase gene-based tree, with three distinct and well-supported clades consisting of the three Bari subfamilies.
A careful inspection of the transposase-based phylogenetic tree revealed an inconsistency in a well-supported Bari1 sub-clade if compared to the phylogeny of species (Fig 1A). The Bari1_Dbia and Bari1_Dbip elements are indeed very close to each other in the transposase tree, while their respective host species, D. biarmipes and D. bipectinata, are more distantly related, being members of the suzukii and ananassae subgroups respectively (Fig 1A). These elements share high sequence similarity (Fig 6B) and are part of a highly supported clade in the tree. In order to discern if the incongruences observed in the phylogeny of Bari-like elements and host species were due to vertical transmission or horizontal transfer, the divergence at synonymous sites (dS), taken as a measure of neutral evolution in the absence of a strong codon usage bias, was compared between TEs and Adh or Gapdh1 genes of the two species. Fisher exact test-based comparisons were performed to verify whether the dS of the Bari-like elements was significantly lower than the dS of nuclear genes [55]. The dS of Bari elements (0,0603) was significantly different (p<0,005) if compared to dS of Adh (0,3281) or Gapdh1 (0,4006) genes in these species (Fig 6C), supporting the hypothesis of horizontal transfer of Bari-like elements between these species.
Discussion
This work aimed to identify Bari transposon insertions in the sequenced Drosophila genomes, a step forward in the annotation of the complete set of TEs insertions in these genomes. We have annotated 401 copies (S2 and S3 Tables) of Bari-like transposon in the genomes of 22 Drosophila species out of 23 available sequenced genomes. Although this data may seem as an overestimation of the actual number of Bari elements in the analyzed species, we expect that elements being missed due to the early draft status of some genome assembly (especially in the heterochromatin) would somewhat compensate any number discrepancies. Importantly, Southern blot experiments testing the genomic distribution of Bari elements in some of the genomes analyzed in this work [56, 57] are in substantial agreement with our sequence analyses, suggesting that Bari is present in low copy number in these genomes and that D. melanogaster is the only species (among those studied) containing heterochromatic clusters.
This work provided a number of novel insights into the biology of the Bari transposon’s family:
high sequence variability of the terminal sequences coupled with high conservation of the DRs and wide diffusion in the Drosophila species’ genomes.
MITE-like forms of this transposon, arising occasionally during the species evolution.
strong target site preference for AT-rich DNA regions, which is also the case for other Tc1-mariner elements, that are instead avoided by class I transposable elements.
occasional horizontal transfer events involving Bari-like elements.
Bari structural diversity
Transposons structural diversity is an obvious evolutionary phenomenon if large families or super-families of mobile elements are taken into account [58]. However, the emergence of structural variants can also be detected in small transposons’ families, such as the Bari family.
The dynamic processes leading to TEs replication, amplification, degradation and elimination from a given genome are difficult to be elucidated. However the graphic representation of the deterioration pattern of TEs proposed by Fernandez Medina et al., [58] can be used to shed light into this task. Our results (Fig 3) suggest that Bari elements have undergone deterioration in different ways depending on the host genome. Point mutations and indels probably weighted differently in producing non-functional Bari copies during evolution, because mutations in the terminal sequences are more tolerated if compared to the coding sequence. Despite some degree of diversification observed at the terminal sequence level (Fig 3 and S1 Fig), a strong conservation of the DR sequences in all elements’ subfamilies from all species, including those containing only MITEs (Fig 2A and Fig 4A) suggests that these sequences may have acquired a potential role during the evolution. The observed differences in the p-values of the terminal sequences can be ascribed either to the transposition activity of Bari elements or to the presence of functional elements in the terminal sequences. For example D. mojavensis contains 8 potentially active and 7 inactive Bari3-like elements and a median p-distance value of 0 for both terminal sequences, while D. miranda contains 1 potentially active and 4 inactive Bari3 elements and a median p-distance values of 0,171 and 0,06 for left and right terminal sequences respectively. Similarly D. melanogaster contains 4 potentially active and 1 inactive Bari1-type elements with a median p-distance value of 0 for both terminal sequences while D. simulans contains two potentially active elements with a median p-distance value of 0,01 for both terminal sequences. In conclusion, it can be speculated that species displaying lower p-distance values host active Bari elements, which produce identical copies upon transposition thus lowering the p-distance. By contrast, species with higher p-distance values contain many dead or almost dead elements, which rapidly accumulate more mutations and become divergent in sequence. However, we have evidence of Bari transposition only in D. melanogaster [13] and in D. mojavensis [14], needing additional studies in order to establish if Bari is active in non-model species. The presence of functional elements could also explain the differences observed in p-distance values between the terminal sequences if compared to the CDS. While both the left and right termini contain DRs able to bind the transposase, the left terminus might carry the promoter or part of it, and the right terminus might contain important signals for the termination of transcription. Interestingly in the context of the terminal sequences the DRs appear strongly conserved in sequence (Fig 2) even in species lacking active Bari elements. Based on this it could be speculated that DRs have acquired a new function (e.g. production of siRNA, binding of different protein partner etc) in the genomes where Bari elements have been completely inactivated.
There is evidence from previous studies [9] suggesting a well-defined structural vs phylogenetic relationship among Bari-like elements. While elements of the inactive Bari2 subfamily contain LIR-type terminal sequences, elements of the Bari1 and Bari3 subfamilies usually harbor SIR and LIR respectively. The non-autonomous Bari1 elements identified in D. ananassae represent an exception as they harbor LIR [9]. In this paper, we show the existence in D. rhopaloa of potentially active Bari1 elements containing identical LIR, which is a previously unreported feature in the Bari1 subfamily that increases the diversity among Bari elements. In addition we identified Bari1-type elements with LIRs in D. bipectinata, D. biarmipes, D. suzukii and D. takahashii, but in these species all the Bari elements are inactive. To date, the Bari1_Drho represents the only potentially active Bari-1 type element with LIRs. It was previously suggested that the identical LIRs might represent the first stage of the evolution of the terminal repeats of Bari-like elements [9]. The LIR structure may subsequently evolve into SIR structures as a consequence of the intrinsic instability associated with the long terminal repeats structure [9]. This hypothesis was best fitting with the Bari3 subfamily, in which the Bari3_Dmoj represents a “young” Bari element having perfect LIR and the Bari3_Dper and Bari3_Dpse elements, representing older elements that are going to lose the similarity between their TIRs. The same hypothesis can be now formulated for the Bari1 subfamily, where SIR- and LIR-containing elements were previously identified. The two TIRs of the Bari_Dana element indeed share 94% similarity and the two TIRs of the Bari1_Drho element reported here are identical to each other. In this view the Bari1_Drho element represents a young Bari1-type element, still awaiting the divergence of its terminal sequences. This conclusion raises further questions concerning the mechanisms that generate Bari1- and Bari3-type elements with perfectly matching terminal sequences, which are still to be identified.
The powerful sequencing technologies developed in the last years facilitate the molecular determination of repeat-rich genomic such as the h39 heterochromatic region of D. melanogaster which hosts roughly 80 clustered copies of Bari1 and the Responder satellite. Our detailed analysis allowed orienting the Bari1 cluster with respect to the centromere of the second chromosome, thanks to the presence of the Responder repeats in one of the sequences flanking the Bari cluster.
It was previously observed that the Rsp satellite displayed an extreme quantitative and structural variability while Bari1 cluster showed remarkable homogeneity [15]. The occurrence of recombination events between clustered copies of Bari1, as inferred from our data, suggests that this transposon cluster could be subjected to expansions or contractions as observed for other complex DNA repeats over evolutionary time. Our sequence analyses of heterochromatic copies of Bari1 in D. melanogaster support the hypothesis that an additional Bari1 cluster might exist outside the h39 region of the mitotic chromosomes, specifically on the X or Y chromosome, since it is associated with DNA sequences specific of the Nucleolar Organizer Region (rDNA and R1 element insertions [51]).
The presence of such kind of clusters in a single Drosophila species (i.e. D. melanogaster) is peculiar and it could be speculated that the reiterated formation of clusters, might depend on species-specific host factors contributing to an error-prone activity of the transposase. In addition resolving the structure of heterochromatic genomic blocks, rich in transposable elements would help understanding important regulatory loci, as reported for the flamenco locus in D. melanogaster [59].
MITEs related to Bari elements
Miniature Inverted-repeat Transposable Elements (MITEs) are non-autonomous, short repeats that mobilize within the host genome even without the potential to encode the key proteins responsible for their mobilization (i.e. the transposase). MITEs are generally smaller than 600 bp with few exceptions, have conserved TIRs, a target site preference, do not display coding potential, are AT-rich and amplified within the host genome [60]. In general, they are supposed to originate by deletions internal to autonomous elements, which leave untouched the TIRs and, sometimes, just portions of the transposase. This origin supports the hypothesis of their mobilization in trans by a transposase encoded by a full-length element [61]. In addition to the previously described Bari-related MITEs in the genome of D. sechellia [21], we identified this element type in eight additional Drosophila species. These novel Bari-related MITE sequences match the definition of MITEs; however some of them (Bari_Dmel_MITE2, Bari_Dsim_MITE, Bari_Dyak_MITE) lack genomic amplification while others show only modest amplification (Bari_Dmel_MITE1, Bari_Dtak_MITE, Bari_Dele_MITE). By contrast Bari_Dsec_MITE1, Bari_Dsec_MITE2 [21], Bari_Dfic_MITE and Bari_Drho_MITE are quite abundant in the respective genomes. It is possible that MITEs in D. melanogaster, D. simulans and D. takahashi could be in a very initial stage of their amplification. Alternatively, they might represent the product of abortive amplification.
Wallau et al., [62] recently described 27 independent sub-lineages of mariner-derived MITEs in 20 Drosophila species, which have internal sequences and TIRs similar to the sequences of the full-length copies and a typical size of 900–1000 bp with few exceptions. Bari-related MITEs are shorter than the mariner-related and possess internal regions not easily comparable with autonomous elements, reflecting a more complex rearrangement differing from abortive gap repair [63]. Furthermore, Bari-derived MITEs can be short (less than 120 bp) or long (greater than 120 bp and less than 700 bp) in sequence, and the intervening sequence between the TIRs, where present, is apparently unrelated to Bari elements (S3 Fig). Contrarily to the mariner MITEs [62], Bari-related MITEs apparently originated by deletions in the same point, with the possible exception of the Bari_Dyak_MITE. It can be hypothesized that Bari-related MITEs originated through internal deletion of a master element, with the deletion breakpoints occurring between the Lo-Lm and between Rm-Ro direct repeats (Fig 4B), which has been possibly followed either by direct junction of the broken extremities or by the addition of unrelated sequences (Fig 4B). Finally, as possible explanations for the presence of MITEs in those species lacking full-length elements (namely D. yakuba, D. ficusphila and D. elegans), it is plausible that these MITEs derived from the ancestral transposition of full-length elements, followed by generation of MITEs and elimination of the original founders. Thus, the single elements identified are just the relic of this elimination. Expansion of MITEs has probably occurred one or multiple times within the host genomes as suggested by sequence similarity distribution (Fig 4C). Notably, the D. sechellia, D. suzukii D. ficusphila and D. takahashii genomes lack autonomous Bari elements, so it would be of particular interest to know if transposition events involving MITEs occurred after the elimination of autonomous elements, resulting in transposition mediated by unrelated transposases.
Bari target site preferences and the physical relationships with other mobile elements
It is widely accepted that mobile elements do not integrate themselves randomly [64]. Primary and secondary DNA structure, as well as chromatin status, could affect the target site selection. Our results point out a strong preference of Bari-like elements for AT-rich sequences. Similar results were obtained for other Tc1-like transposons, like SB which has a preference for a palindromic AT-repeat (ATATATAT) in which the central AT is the cleaved and duplicated target [65].
In addition to the above-described analysis we have performed a larger-scale analysis aiming to the identification of TEs that preferentially insert, or preferentially avoid, the DNA neighborhood of Bari elements. We found that loci in which Bari transposons are inserted are also populated by other Tc1-mariner elements (p<0,05). The significant association between Bari elements and other transposons of the same class lead us to hypothesize a possible correlation between the DNA (or chromatin) structure and the insertion preferences of transposons belonging to the same superfamily. On the other hand, the members of the copia, Jockey and I families are significantly under-represented in the range of 2,5 kb around Bari elements in the analyzed species. Helitron elements also display a preference for sites near Bari insertions, while gypsy–like elements do not show significant preference/avoidance respect to Bari elements insertion sites suggesting that gypsy-like elements have a random genomic distribution. Besides its importance in identifying the structural organization of the single loci in which Bari elements lie, this kind of analyses would hopefully help understanding how a DNA domain is built during evolution. This is especially interesting for heterochromatic DNA blocks, which are largely composed of transposons’ arrays and can develop to master loci producing small regulatory RNAs [66, 67].
Bari phylogeny inconsistencies and possible horizontal transfer events
The evolution of Bari elements has been extensively studied in previous works [9, 19, 57]. The non-uniform distribution of the three subfamilies across the species of the Drosophila and Sophophora subgenera was observed by Moschetti et al., [9] and explained with a distinct evolutionary history in different genomes. The overlap observed here, between the Bari1-Bari2 subfamilies in some species (D. melanogaster, D. simulans, D. sechellia), might be explained hypothesizing two independent waves of genomic invasion: the first, most ancient, by Bari2 elements which probably occurred in the ancestor of the melanogaster subgroup (comprising the melanogaster, simulans, sechellia and erecta species analyzed in this work), the second, more recent, by Bari1 which have invaded the ancestor of the melanogaster complex (including the melanogaster simulans and sechellia species). Assuming only vertical transmission, Bari2 elements have been inactivated while Bari1 elements are still active in D. melanogaster and, potentially, in D. simulans. After these invasions, Bari2 elements were probably inactivated resulting in the absence of active copies in these genomes. It would be interesting to know if Bari2 elements are also present in non-sequenced genomes of the Drosophila subgenus, or if Bari2 subfamily is restricted to species of the Sophophora subgenus.
The emergence of transposable elements in a genome can occur in three ways: de novo emergence, horizontal transfer and introgression [68]. Patchy TEs distribution, incongruent TE vs host species phylogeny and the presence of highly similar sequences in distantly related species [69] are used as proofs in support of TEs horizontal transfer. Several horizontal transfer events involving Tc1-mariner elements have been described so far [70]. In a recent paper, Dupeyron and collaborators described horizontal transfer events between terrestrial isopod crustaceans and hexapods involving Tc1-mariner elements [7]. Horizontal transfer events were also suggested for Bari-like elements between the sibling species D. melanogaster and D. simulans as reported by former studies [71] [69]. Here we present evidence of horizontal transfer events involving Bari-like elements between D. bipectinata and D. biarmipes. Although these two species diverged at least 27 million years ago [25], Bari elements within the respective genomes are nearly identical. Analysis of synonymous substitutions differences between transposases and host genes suggest that horizontal transfer occurred, which explain the incongruences observed in the phylogenetic trees and the high similarity between the Bari elements in these species (Fig 6A and 6B). The geographic distribution of the Drosophila species involved in transposon horizontal transfer events supports this possibility, as these two species coexist in the Indian subcontinent. Very recently Wallau et al., reported the same horizontal transfer event described here by applying a sophisticated and statistically supported method, called VHICA, [72] which used 50 orthologous, vertically transmitted genes, as a reference set to infer horizontal transposon transfer of Bari between D. biarmipes and D. bipectinata. Other horizontal transposon transfer events involving different species have been also detected using this method [72] suggesting that complex evolutionary mechanisms have originated the actual distribution of Bari elements, complicating the inference of their evolutionary history.
Conclusions
Our annotation and analyses of 401 insertions unveiled sequence and structure variability of Bari-like elements in the sequenced genomes of Drosophila species. Besides the dynamic structure of the TIRs and the presence of active and inactive elements in the three Bari subfamilies, we detected the presence of MITEs derived from Bari elements in 9 Drosophila species, suggesting that the generation of such inactive form can be considered as a common event in this family. The analysis of genomic sites targeted by Bari transposition showed that the same sites are also preferred or avoided by other mobile elements, and this may be important to understand how transposons model genomic domains. Finally our phylogenetic analysis showed that three subfamilies (Bari1, Bari2 and Bari3) can be recognized both in TIR- and transposase-based phylogenetic trees and that a previously unreported horizontal transfer event has probably occurred between D. biarmipes and D. bipectinata.
Supporting Information
(TIF)
A. Possible molecular mechanism generating the observed adjacent heterochromatic copies carrying deletions of terminal sequences. (B) Global alignment (Needleman-Wunsch) of two adjacent canonical Bari1 heterochromatic copies and the detected copies carrying a deletion (elements #31 and 31, S2 Table). The sequences of the two adjacent monomers are shown in red and blue color fonts. Deletion breakpoints, corresponding also to homologous sequences involved in inter-monomer recombination events, are highlighted in yellow.
(PDF)
(PDF)
Fifty bp upstream or downstream the Bari elements in 8 Drosophila species were analyzed. The number of sequences analyzed is reported (n).
(TIF)
The red-boxed area in each plot indicates area of significant (p<0,05) enrichment (positive values) or depletion (negative values) of the analyzed transposable elements’ super-families in the proximity of Bari elements in a sequence range of 2,5 kb. X axes report p value. Y axes report the distance from origin in bp.
(PDF)
(XLSX)
(PDF)
(XLSX)
(XLSX)
(PDF)
Acknowledgments
We gratefully acknowledge Dr. Kostantinos Lefkimmiatis for critical discussion and language editing of the manuscript.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
The authors received no specific funding for this work.
References
- 1.Mc Clintock B. The origin and behavior of mutable loci in maize. Proc Natl Acad Sci U S A. 1950;36(6):344–55. Epub 1950/06/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mardis ER. Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif). 2013;6:287–303. Epub 2013/04/09. 10.1146/annurev-anchem-062012-092628 . [DOI] [PubMed] [Google Scholar]
- 3.Pardue ML. Transposable elements: friends, foes, or merely fellow travelers? Trends Genet. 2000;16(4):155–6. Epub 2000/06/10. . [DOI] [PubMed] [Google Scholar]
- 4.Lonnig WE, Saedler H. Chromosome rearrangements and transposable elements. Annu Rev Genet. 2002;36:389–410. Epub 2002/11/14. 10.1146/annurev.genet.36.040202.092802 040202.092802 [pii]. . [DOI] [PubMed] [Google Scholar]
- 5.Sinzelle L, Izsvak Z, Ivics Z. Molecular domestication of transposable elements: from detrimental parasites to useful host genes. Cell Mol Life Sci. 2009;66(6):1073–93. Epub 2009/01/10. 10.1007/s00018-009-8376-3 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuan YW, Wessler SR. The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proc Natl Acad Sci U S A. 2011;108(19):7884–9. Epub 2011/04/27. 10.1073/pnas.1104208108 1104208108 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dupeyron M, Leclercq S, Cerveau N, Bouchon D, Gilbert C. Horizontal transfer of transposons between and within crustaceans and insects. Mob DNA. 2014;5(1):4 10.1186/1759-8753-5-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oliveira SG, Bao W, Martins C, Jurka J. Horizontal transfers of Mariner transposons between mammals and insects. Mob DNA. 2012;3(1):14 10.1186/1759-8753-3-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moschetti R, Chlamydas S, Marsano RM, Caizzi R. Conserved motifs and dynamic aspects of the terminal inverted repeat organization within Bari-like transposons. Mol Genet Genomics. 2008;279(5):451–61. Epub 2008/02/06. 10.1007/s00438-008-0324-7 . [DOI] [PubMed] [Google Scholar]
- 10.Plasterk RH, Izsvák Z, Ivics Z. Resident aliens: the Tc1/mariner superfamily of transposable elements. Trends Genet. 1999;15(8):326–32. . [DOI] [PubMed] [Google Scholar]
- 11.Vos JC, De Baere I, Plasterk RH. Transposase is the only nematode protein required for in vitro transposition of Tc1. Genes Dev. 1996;10(6):755–61. . [DOI] [PubMed] [Google Scholar]
- 12.Lampe DJ, Churchill ME, Robertson HM. A purified mariner transposase is sufficient to mediate transposition in vitro. Embo J. 1996;15(19):5470–9. [PMC free article] [PubMed] [Google Scholar]
- 13.Palazzo A, Marconi S, Specchia V, Bozzetti MP, Ivics Z, Caizzi R, et al. Functional characterization of the Bari1 transposition system. PLoS One. 2013;8(11):e79385 Epub 2013/11/19. 10.1371/journal.pone.0079385 PONE-D-13-21853 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palazzo A, Moschetti R, Caizzi R, Marsano RM. The Drosophila mojavensis Bari3 transposon: distribution and functional characterization. Mob DNA. 2014;5:21 Epub 2014/08/06. 10.1186/1759-8753-5-21 1759-8753-5-21 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caizzi R, Caggese C, Pimpinelli S. Bari-1, a new transposon-like family in Drosophila melanogaster with a unique heterochromatic organization. Genetics. 1993;133(2):335–45. Epub 1993/02/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu CI, Lyttle TW, Wu ML, Lin GF. Association between a satellite DNA sequence and the Responder of Segregation Distorter in D. melanogaster. Cell. 1988;54(2):179–89. Epub 1988/07/15. doi: 0092-8674(88)90550-8 [pii]. . [DOI] [PubMed] [Google Scholar]
- 17.Marsano RM, Marconi S, Moschetti R, Barsanti P, Caggese C, Caizzi R. MAX, a novel retrotransposon of the BEL-Pao family, is nested within the Bari1 cluster at the heterochromatic h39 region of chromosome 2 in Drosophila melanogaster. Mol Genet Genomics. 2004;270(6):477–84. Epub 2003/11/25. 10.1007/s00438-003-0947-7 . [DOI] [PubMed] [Google Scholar]
- 18.Marsano RM, Moschetti R, Barsanti P, Caggese C, Caizzi R. A survey of the DNA sequences surrounding the Bari1 repeats in the pericentromeric h39 region of Drosophila melanogaster. Gene. 2003;307:167–74. Epub 2003/04/23. . [DOI] [PubMed] [Google Scholar]
- 19.Marsano RM, Milano R, Minervini C, Moschetti R, Caggese C, Barsanti P, et al. Organization and possible origin of the Bari-1 cluster in the heterochromatic h39 region of Drosophila melanogaster. Genetica. 2003;117(2–3):281–9. Epub 2003/05/02. . [DOI] [PubMed] [Google Scholar]
- 20.Wallau GL, Ortiz MF, Loreto EL. Horizontal transposon transfer in eukarya: detection, bias, and perspectives. Genome Biol Evol. 2012;4(8):689–99. Epub 2012/07/17. 10.1093/gbe/evs055 evs055 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dias ES, Carareto CM. msechBari, a new MITE-like element in Drosophila sechellia related to the Bari transposon. Genet Res (Camb). 2011;93(6):381–5. Epub 2011/12/23. 10.1017/S0016672311000371 S0016672311000371 [pii]. . [DOI] [PubMed] [Google Scholar]
- 22.Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287(5461):2185–95. Epub 2000/03/25. doi: 8392 [pii]. . [DOI] [PubMed] [Google Scholar]
- 23.Richards S, Liu Y, Bettencourt BR, Hradecky P, Letovsky S, Nielsen R, et al. Comparative genome sequencing of Drosophila pseudoobscura: chromosomal, gene, and cis-element evolution. Genome Res. 2005;15(1):1–18. Epub 2005/01/06. doi: 15/1/1 [pii] 10.1101/gr.3059305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen ZX, Sturgill D, Qu J, Jiang H, Park S, Boley N, et al. Comparative validation of the D. melanogaster modENCODE transcriptome annotation. Genome Res. 2014;24(7):1209–23. 10.1101/gr.159384.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ometto L, Cestaro A, Ramasamy S, Grassi A, Revadi S, Siozios S, et al. Linking genomics and ecology to investigate the complex evolution of an invasive Drosophila pest. Genome Biol Evol. 2013;5(4):745–57. 10.1093/gbe/evt034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou Q, Bachtrog D. Sex-specific adaptation drives early sex chromosome evolution in Drosophila. Science. 2012;337(6092):341–5. 10.1126/science.1225385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou Q, Zhu HM, Huang QF, Zhao L, Zhang GJ, Roy SW, et al. Deciphering neo-sex and B chromosome evolution by the draft genome of Drosophila albomicans. BMC Genomics. 2012;13:109 10.1186/1471-2164-13-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoskins RA, Carlson JW, Wan KH, Park S, Mendez I, Galle SE, et al. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res. 2015. 10.1101/gr.185579.114 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiu JC, Jiang X, Zhao L, Hamm CA, Cridland JM, Saelao P, et al. Genome of Drosophila suzukii, the spotted wing drosophila. G3 (Bethesda). 2013;3(12):2257–71. 10.1534/g3.113.008185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.St Pierre SE, Ponting L, Stefancsik R, McQuilton P. FlyBase 102—advanced approaches to interrogating FlyBase. Nucleic Acids Res. 2014;42(Database issue):D780–8. Epub 2013/11/16. 10.1093/nar/gkt1092 gkt1092 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marck C. 'DNA Strider': a 'C' program for the fast analysis of DNA and protein sequences on the Apple Macintosh family of computers. Nucleic Acids Res. 1988;16(5):1829–36. Epub 1988/03/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110(1–4):462–7. Epub 2005/08/12. 10.1159/000084979 . [DOI] [PubMed] [Google Scholar]
- 33.Kapitonov VV, Jurka J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat Rev Genet. 2008;9(5):411–2; author reply 4. Epub 2008/04/19. 10.1038/nrg2165-c1 . [DOI] [PubMed] [Google Scholar]
- 34.Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25(11):1422–3. 10.1093/bioinformatics/btp163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics. 2006;7:474 Epub 2006/10/27. 10.1186/1471-2105-7-474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jurka J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 2000;16(9):418–20. Epub 2000/09/06. . [DOI] [PubMed] [Google Scholar]
- 37.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3(5):418–26. Epub 1986/09/01. . [DOI] [PubMed] [Google Scholar]
- 38.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. 2011. Epub 2011/05/07. 10.1093/molbev/msr121 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartigan JA, Hartigan PM. The Dip Test of Unimodality. Ann Stat. 1985;13(1):70–84. 10.1214/Aos/1176346577 . [DOI] [Google Scholar]
- 40.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–90. Epub 2004/06/03. 10.1101/gr.849004 14/6/1188 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–8. Epub 2007/09/12. doi: btm404 [pii] 10.1093/bioinformatics/btm404 . [DOI] [PubMed] [Google Scholar]
- 42.Hall TA. BioEdit: A user-friendly biological sequence alignment, editor and analysis program for Windows 95/98 NT. Nucl Acids Symp Ser 1999;41:95–8. [Google Scholar]
- 43.Posada D, Buckley TR. Model selection and model averaging in phylogenetics: advantages of akaike information criterion and bayesian approaches over likelihood ratio tests. Syst Biol. 2004;53(5):793–808. Epub 2004/11/17. doi: REHE4656RGAP72AK [pii] 10.1080/10635150490522304 . [DOI] [PubMed] [Google Scholar]
- 44.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772 Epub 2012/08/01. 10.1038/nmeth.2109 nmeth.2109 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704. Epub 2003/10/08. doi: 54QHX07WB5K5XCX4 [pii]. . [DOI] [PubMed] [Google Scholar]
- 46.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. Epub 2007/11/13. doi: 1471-2148-7-214 [pii] 10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 2005;22(5):1185–92. 10.1093/molbev/msi103 . [DOI] [PubMed] [Google Scholar]
- 48.Wallau GL, Kaminski VL, Loreto EL. The role of vertical and horizontal transfer in the evolution of Paris-like elements in drosophilid species. Genetica. 2011;139(11–12):1487–97. Epub 2012/04/25. 10.1007/s10709-012-9648-7 . [DOI] [PubMed] [Google Scholar]
- 49.Merriman PJ, Grimes CD, Ambroziak J, Hackett DA, Skinner P, Simmons MJ. S elements: a family of Tc1-like transposons in the genome of Drosophila melanogaster. Genetics. 1995;141(4):1425–38. Epub 1995/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Russo CA, Takezaki N, Nei M. Molecular phylogeny and divergence times of drosophilid species. Mol Biol Evol. 1995;12(3):391–404. Epub 1995/05/01. . [DOI] [PubMed] [Google Scholar]
- 51.He B, Caudy A, Parsons L, Rosebrock A, Pane A, Raj S, et al. Mapping the pericentric heterochromatin by comparative genomic hybridization analysis and chromosome deletions in Drosophila melanogaster. Genome Res. 2012;22(12):2507–19. Epub 2012/06/30. 10.1101/gr.137406.112 gr.137406.112 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rogers AR, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol. 1992;9(3):552–69. Epub 1992/05/01. . [DOI] [PubMed] [Google Scholar]
- 53.Lu C, Chen J, Zhang Y, Hu Q, Su W, Kuang H. Miniature inverted-repeat transposable elements (MITEs) have been accumulated through amplification bursts and play important roles in gene expression and species diversity in Oryza sativa. Mol Biol Evol. 2012;29(3):1005–17. Epub 2011/11/19. 10.1093/molbev/msr282 msr282 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen J, Lu C, Zhang Y, Kuang H. Miniature inverted-repeat transposable elements (MITEs) in rice were originated and amplified predominantly after the divergence of Oryza and Brachypodium and contributed considerable diversity to the species. Mob Genet Elements. 2012;2(3):127–32. Epub 2012/10/13. 10.4161/mge.20773 2012MGE0015R1 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ludwig A, Valente VL, Loreto EL. Multiple invasions of Errantivirus in the genus Drosophila. Insect Mol Biol. 2008;17(2):113–24. Epub 2008/03/21. 10.1111/j.1365-2583.2007.00787.x . [DOI] [PubMed] [Google Scholar]
- 56.Caggese C, Pimpinelli S, Barsanti P, Caizzi R. The distribution of the transposable element Bari-1 in the Drosophila melanogaster and Drosophila simulans genomes. Genetica. 1995;96(3):269–83. Epub 1995/01/01. . [DOI] [PubMed] [Google Scholar]
- 57.Moschetti R, Caggese C, Barsanti P, Caizzi R. Intra- and interspecies variation among Bari-1 elements of the melanogaster species group. Genetics. 1998;150(1):239–50. Epub 1998/09/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fernandez-Medina R, Ribeiro JM, Carareto CM, Velasque L, Struchiner C. Losing identity: structural diversity of transposable elements belonging to different classes in the genome of Anopheles gambiae. BMC Genomics. 2012;13(1):272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zanni V, Eymery A, Coiffet M, Zytnicki M, Luyten I, Quesneville H, et al. Distribution, evolution, and diversity of retrotransposons at the flamenco locus reflect the regulatory properties of piRNA clusters. Proc Natl Acad Sci U S A. 2013;110(49):19842–7. Epub 2013/11/20. 10.1073/pnas.1313677110 1313677110 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feschotte C, Jiang N, Wessler SR. Plant transposable elements: where genetics meets genomics. Nat Rev Genet. 2002;3(5):329–41. Epub 2002/05/04. 10.1038/nrg793 . [DOI] [PubMed] [Google Scholar]
- 61.Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–68. 10.1146/annurev.genet.40.110405.090448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wallau GL, Capy P, Loreto E, Hua-Van A. Genomic landscape and evolutionary dynamics of mariner transposable elements within the Drosophila genus. BMC Genomics. 2014;15:727 Epub 2014/08/29. 10.1186/1471-2164-15-727 1471-2164-15-727 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rubin E, Levy AA. Abortive gap repair: underlying mechanism for Ds element formation. Mol Cell Biol. 1997;17(11):6294–302. Epub 1997/10/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Craig NL. Target site selection in transposition. Annu Rev Biochem. 1997;66:437–74. Epub 1997/01/01. 10.1146/annurev.biochem.66.1.437 . [DOI] [PubMed] [Google Scholar]
- 65.Vigdal TJ, Kaufman CD, Izsvak Z, Voytas DF, Ivics Z. Common physical properties of DNA affecting target site selection of sleeping beauty and other Tc1/mariner transposable elements. J Mol Biol. 2002;323(3):441–52. Epub 2002/10/17. doi: S0022283602009919 [pii]. . [DOI] [PubMed] [Google Scholar]
- 66.Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, et al. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007;128(6):1089–103. Epub 2007/03/10. 10.1016/j.cell.2007.01.043 . [DOI] [PubMed] [Google Scholar]
- 67.Goriaux C, Theron E, Brasset E, Vaury C. History of the discovery of a master locus producing piRNAs: the flamenco/COM locus in Drosophila melanogaster. Front Genet. 2014;5:257 Epub 2014/08/20. 10.3389/fgene.2014.00257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Capy P. Dynamics and evolution of transposable elements New York; London: Springer; 1998. [Google Scholar]
- 69.Loreto EL, Carareto CM, Capy P. Revisiting horizontal transfer of transposable elements in Drosophila. Heredity. 2008;100(6):545–54. Epub 2008/04/24. 10.1038/sj.hdy.6801094 . [DOI] [PubMed] [Google Scholar]
- 70.Robertson HM, Soto-Adames FN, Walden KKO, Avanchi RMP, Lampe DJ. The mariner transposons of animals: horizontally jumping genes. 1998. p. 268–84.
- 71.Dias ES, Carareto CM. Ancestral polymorphism and recent invasion of transposable elements in Drosophila species. BMC Evol Biol. 2012;12:119 Epub 2012/07/25. 10.1186/1471-2148-12-119 1471-2148-12-119 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wallau GL, Capy P, Loreto E, Le Rouzic A, Hua-Van A. VHICA, a New Method to Discriminate between Vertical and Horizontal Transposon Transfer: Application to the Mariner Family within Drosophila. Mol Biol Evol. 2016;33(4):1094–109. Epub 2015/12/20. 10.1093/molbev/msv341 msv341 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
A. Possible molecular mechanism generating the observed adjacent heterochromatic copies carrying deletions of terminal sequences. (B) Global alignment (Needleman-Wunsch) of two adjacent canonical Bari1 heterochromatic copies and the detected copies carrying a deletion (elements #31 and 31, S2 Table). The sequences of the two adjacent monomers are shown in red and blue color fonts. Deletion breakpoints, corresponding also to homologous sequences involved in inter-monomer recombination events, are highlighted in yellow.
(PDF)
(PDF)
Fifty bp upstream or downstream the Bari elements in 8 Drosophila species were analyzed. The number of sequences analyzed is reported (n).
(TIF)
The red-boxed area in each plot indicates area of significant (p<0,05) enrichment (positive values) or depletion (negative values) of the analyzed transposable elements’ super-families in the proximity of Bari elements in a sequence range of 2,5 kb. X axes report p value. Y axes report the distance from origin in bp.
(PDF)
(XLSX)
(PDF)
(XLSX)
(XLSX)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.