

Abstract
AIM: To investigate the effect of Tenascin C (TNC) on the expression of pro-inflammatory cytokines and matrix metalloproteinases in human cardiac myofibroblasts (CMF).
METHODS: CMF were isolated and cultured from patients undergoing coronary artery bypass grafting. Cultured cells were treated with either TNC (0.1 μmol/L, 24 h) or a recombinant protein corresponding to different domains of the TNC protein; fibrinogen-like globe (FBG) and fibronectin type III-like repeats (TNIII 5-7) (both 1 μmol/L, 24 h). The expression of the pro-inflammatory cytokines; interleukin (IL)-6, IL-1β, TNFα and the matrix metalloproteinases; MMPs (MMP1, 2, 3, 9, 10, MT1-MMP) was assessed using real time RT-PCR and western blot analysis.
RESULTS: TNC increased both IL-6 and MMP3 (P < 0.01) mRNA levels in cultured human CMF but had no significant effect on the other markers studied. The increase in IL-6 mRNA expression was mirrored by an increase in protein secretion as assessed by enzyme-linked immunosorbant assay (P < 0.01). Treating CMF with the recombinant protein FBG increased IL-6 mRNA and protein (P < 0.01) whereas the recombinant protein TNIII 5-7 had no effect. Neither FBG nor TNIII 5-7 had any significant effect on MMP3 expression. The expression of toll-like receptor 4 (TLR4) in human CMF was confirmed by real time RT-PCR, western blot and immunohistochemistry. Pre-incubation of cells with TLR4 neutralising antisera attenuated the effect of both TNC and FBG on IL-6 mRNA and protein expression.
CONCLUSION: TNC up-regulates IL-6 expression in human CMF, an effect mediated through the FBG domain of TNC and via the TLR4 receptor.
Keywords: Tenascin C, Matrix metalloproteinase, Toll-like receptor, Interleukin-6, Cardiac fibroblasts
Core tip: Tenascin C (TNC) is transiently expressed in cardiac tissue following acute myocardial infarction (MI) and MI patients with higher serum TNC levels have worse long term prognosis. This suggests that TNC is important in ventricular remodelling, although a functional role in this process is unclear. We report that TNC stimulates interleukin-6 synthesis in cardiac myofibroblasts, an effect mediated by toll-like receptor 4. As a growing body of evidence suggests that prolongation of the post-infarction inflammatory response results in worse remodelling and dysfunction this important observation may in part explain the mechanism by which TNC induces maladaptive ventricular remodelling following MI.
INTRODUCTION
A number of cardiac pathologies including acute myocardial infarction (MI), ischaemia/reperfusion (I/R) injury, hypertensive heart disease and myocarditis are associated with activation of pro-inflammatory mediators in the heart. Sustained expression of pro-inflammatory cytokines such as tumor necrosis factor-α (TNFα), interleukin (IL)-1 and IL-6 by infiltrating/resident inflammatory cells and cardiac fibroblasts is associated with increased matrix metalloproteinase (MMP) production, adverse cardiac remodelling leading to fibrosis, left ventricular (LV) dysfunction and heart failure[1]. The mechanisms that drive the inflammatory response in the heart are not fully understood.
The matricellular protein Tenascin C (TNC) is an extracellular glycoprotein that is highly expressed during embryonic development but is absent from healthy adult tissue. It is re-expressed in adult tissue during wound healing, inflammation and cancer invasion. In the adult myocardium TNC is up-regulated in pathological conditions that are closely associated with inflammation and extensive tissue remodelling such as MI[2], myocarditis[3] and dilated cardiomyopathy[4]. TNC is synthesised by interstitial fibroblasts and is up-regulated by various pro-inflammatory cytokines (e.g., IL-1, IL-4, IL-13) and growth factors, as well as by oxidative and mechanical stress[5-11]. Evidence suggests that TNC may promote wound healing by recruiting cardiac myofibroblasts (CMF) during tissue repair[12]. However, it may also contribute to adverse ventricular remodelling as it can upregulate MMP production leading to excessive extracellular matrix (ECM) degradation. This in turn could weaken the adhesion of cardiomyocytes to the ECM, leading to cardiomyocyte slippage, LV dilation and reduction in contractile function[13]. Recent studies using synovial fibroblasts demonstrated that TNC was an endogenous activator of the toll-like receptor 4 (TLR4) pathway in the arthritic joint[14].
TLRs play a key role in driving inflammation and ECM turnover. They promote innate and adaptive immune responses including induction of pro-inflammatory cytokines and MMPs[15-18]. TLR4 has been identified on cardiac myocytes and TLR4 signalling is involved in the expression of cytokines in the myocardium[19-21]. Moreover, the TLR4 signalling pathway has been implicated in maladaptive ventricular remodelling[1] and in cardiac dysfunction after global I/R[22].
In the present study we investigated the effect of TNC on IL-6, IL-1β and MMP expression in a key cell type involved in the myocardial remodelling process, namely the human CMF[23,24]. In particular we investigated the interplay between TNC, TLR4 and the pro-inflammatory cytokine IL-6.
MATERIALS AND METHODS
Reagents
All cell culture reagents were purchased from Invitrogen (Paisley, Scotland, United Kingdom), except foetal calf serum (FCS) which was from Biosera (Ringmer, East Sussex, United Kingdom). Native human TNC was obtained from AbD Serotec (#8640-0502, Oxford, United Kingdom) and recombinant IL-1α from Life Technologies (Paisley, Scotland, United Kingdom). Lipopolysaccharide (LPS) was obtained from Sigma (Poole, United Kingdom).
Purification of human TNC
Purified human TNC protein (CC065, Millipore) from the human glioma cell line U251 was used in the in vitro experiments. Endotoxin levels were measured using the ToxinSensor Chromogenic LAL Endotoxin Assay Kit (Genscript). The TNC protein was taken through an endotoxin removal process using Detoxi-Gel Endotoxin Removal Columns (Thermoscientific) following the manufacturer’s instructions. Commercial TNC, which was initially found to have an LPS concentration of approximately 15.8 pg/μg protein by LAL test, was column purified and found thereafter to be almost devoid of contamination (i.e., < 0.1 pg/μg protein, which equates to < 3 pg/mL per reaction). The levels of LPS contamination of the TNC recombinant proteins [fibrinogen-like globe (FBG) and TNIII 5-7] was less than 10 pg/mL as described earlier[14]. These levels of contamination were more than 300-fold lower than that required (i.e., 1 ng/mL) to stimulate IL-6 mRNA expression in human CMF. Nevertheless, polymyxin B was added in our experiments to block the biological effects of LPS. There was no evidence that polymyxin B alone could trigger IL-6 mRNA expression.
Recombinant TNC fragments
Recombinant TNC fragments corresponding to the FBG and fibronectin type III-like repeats (TNIII 5-7) regions of the TNC protein were synthesised and purified as described previously[14].
Cell culture
Right atrial appendage biopsies from patients undergoing elective coronary artery bypass surgery at the Leeds General Infirmary were obtained following local ethical committee approval (reference number: 01/040) and informed patient consent. All investigations conformed to the principles outlined in the Declaration of Helsinki, 1997. Human cardiac fibroblasts were harvested, characterised and cultured as we have previously described[25,26]. Cells exhibit a myofibroblast phenotype as determined by positive staining for both α-smooth muscle actin and vimentin at passage 1 through to at least passage 5[26]. Experiments were performed on cells from passage 3-5 obtained from multiple donors. Cells were serum starved for 24 h before performing experiments in basal medium (DMEM) supplemented with 0.4% FCS and polymyxin B (50 μg/mL), an LPS neutralising agent, to ensure that any residual LPS in the TNC could not elicit a signal. Cells were treated with either IL-1α (10 ng/mL, 24 h), TNC (0.1 μmol/L, 24 h) or TNC recombinant fragments (FBG, TNIII 5-7, 1 μmol/L, 24 h). Concentrations and time point were based on preliminary dose response and time course experiments (data not shown). In TLR4 neutralising experiments, cells were pre-treated with TLR4 neutralising antibody (25 μg/mL, #AF1478, R and D Systems, Minneapolis, United States) for 1 h prior to TNC addition.
Quantitative RT-PCR
Cellular RNA was extracted from cells at the end of the incubation period and cDNA was prepared as described previously[27]. Real time RT-PCR was performed in duplicate using the Applied Biosystems 7500 Real-Time PCR System. Intron spanning primers and Taqman probes for human IL-1β (Hs00174097_m1), IL-6 (Hs00174131_m1), TNFα (Hs00174128_m1), MMP-1 (Hs00233958_m1), MMP-2 (Hs00234422_m1), MMP-3 (Hs00233962_m1), MMP-9 (Hs00234579_m1), MMP-10 (Hs00233987_m1), MT1-MMP (Hs00237119_m1), TLR2 (Hs01872448_s1) and TLR4 (Hs00152939_m1) were from Applied Biosystems (www.appliedbiosystems.com). Data are presented as a relative percentage of expression of the endogenous control GAPDH (Hs99999905_m1 primers) using the formula 2-∆CT × 100 in which CT is the cycle threshold number.
Real time RT-PCR array for TLR expression
RNA was extracted from human CMF from 3 different donors using the Aurum Total RNA kit (BioRad). Equivalent RNA samples from each of the 3 donors were pooled before preparing cDNA and measuring expression levels of TLRs as part of a SYBR Green-based real-time PCR array (RT2 Profiler Human Innate and Adaptive Immune Response Array, SABiosciences, Qiagen). ∆CT values for the target genes were calculated by subtracting the mean CT value (threshold cycle number) of the 5 housekeeping (HK) genes on the array (β2-microglobulin, hypoxanthine phosphoribosyltransferase 1, ribosomal protein L13A, β-actin and GAPDH) from the CT value of the target genes. Data are expressed relative to the mean of HK genes using the formula 2-∆CT.
Western blot analysis for TLR4
Whole cell homogenates were prepared from human CMF and cultured human saphenous vein smooth muscle cells, as described previously[28]. Proteins (10 μg) were resolved by SDS-PAGE and immunoblotting performed as described previously[28] with TLR4-specific primary antibody raised in rabbit (#sc10741, Santa Cruz Biotechnology, CA, United States) and horseradish peroxidase-conjugated donkey anti-rabbit secondary antibody (#NA934V, GE Health Care, United Kingdom). Immunolabelled bands were visualised on X-ray film by SuperSignal West Pico chemiluminescence kit (Perbio, Cramlington, United Kingdom).
Enzyme-linked immunosorbant assay
Cells cultured in 48-well plates were serum-starved for 48 h before exposure to appropriate stimuli for 24 h in a volume of 0.25 mL. Conditioned media were collected and centrifuged to remove cellular debris, and samples were stored at -40 °C for subsequent analysis. Enzyme-linked immunosorbant assay for IL-6 was performed according to the manufacturer’s instructions (R and D Systems, Abingdon, United Kingdom) using samples diluted 1:100.
Immunocytochemistry for TLR4
Human CMF were fixed in paraformaldehyde (40 g/L) before permeabilising with Triton X-100 (1 mL/L) in PBS. Cells were blocked with bovine serum albumin and incubated with goat primary antibody to human TLR4 (sc-8694, Santa Cruz Biotechnology, CA, United States) followed by Cy3-conjugated donkey anti-goat secondary antibody (1:1000, Stratech Scientific, Soham, Cambridgeshire, United Kingdom). Labelled cells were visualised using a Zeiss Imager. Z1 Apotome microscope and Axiovision image analysis software (Zeiss, Hamburg, Germany). Cells were mounted in Vectashield with DAPI (H-1200, Vector Laboratories, CA, United States). Antibody specificity was confirmed by pre-absorption of primary antibody with TLR4 protein (8 μg/mL for 24 h, sc-8694P, Santa Cruz Biotechnology, CA, United States).
MI model
All procedures involving animals were carried out in accordance with the Home Office Animals (Scientific Procedures) Act 1986 and the University of Leeds Animal Welfare and Ethical Committee. Experiments were performed on C57BL/6 mice (25-30 g, University of Leeds). Animals were maintained at 22 °C on a 12 h light and dark cycle with ad libitum access to food and water. The animal protocol was designed to minimise pain or discomfort to the animal. Mice were anaesthetised with isofluorane. Under a dissecting microscope, a left thoracotomy was performed at the level of the 4th intercostal space to allow the left anterior descending coronary artery to be visualised. This was ligated at the edge of the left atrium with 8-0 prolene suture. Occlusion was confirmed by observation of pallor of the anterior wall of the LV.
TNC, TRL4 and α-smooth muscle actin immunohistochemistry
Three days following MI, animals were perfused with formaldehyde, hearts were removed and wax-embedded. Tissue sections (3 μm) were cut and stained for both TNC (10337, Immuno-Biological Laboratories Co. Ltd, Japan) and TLR4 (sc-10741, Santa Cruz Biotechnology, CA, United States) using the MenaPath X-Cell Plus Multiplex Double Stain Detection Kit 2 (A.MENARINI Diagnostics, Berkshire, United Kingdom) and a Mouse on Mouse Polymer IHC Kit (Abcam, Cambridge, United Kingdom). Sections were also stained for α smooth muscle actin (clone 1A4, code no. M851; Dakopatts, Glostrup, Denmark) and TLR4 in similar fashion. Tissue sections were counterstained with haematoxylin and imaged using an Axioplan Zeiss microscope and AxioVision 4.8 software (Carl Zeiss Inc.).
Statistical analysis
Results are mean ± SEM with n representing the number of experiments on cells from different patients. Data were analysed using the student’s t-test or repeated measures one-way ANOVA and Bonferroni multiple comparison post hoc test (GraphPad Prism software, www.graphpad.com). P < 0.05 was considered statistically significant. The statistical methods of this study were reviewed by Dr. Emma Spary from the University of Leeds.
RESULTS
Increased expression of TNC and TLR4 in the infarcted ventricle
Three days following LAD ligation, in an experimental mouse model of MI, positive interstitial TNC immunoreactivity (brown) was observed in the infarcted side of the left ventricle but not in the remote non-infarcted regions (Figure 1A and D). Light diffuse TLR4 staining (pink) was noted throughout the myocardium on myocytes and interstitial cells (Figure 1D). On the infarcted side the TLR4 staining was more evident with scattered foci of intense staining visible on myocytes, a feature consistent with previous reports[11] as well as on CMF (Figure 1B and C).
Figure 1.
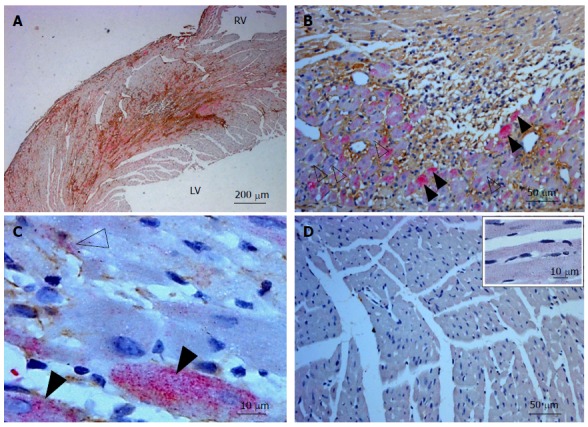
Tenascin C and toll-like receptor 4 staining in the murine left ventricle following infarction. A: Low power view of the infarcted mouse LV showing the proximity of TNC (brown) and TLR4 (pink) 3 d following the occlusion of the left anterior coronary artery; B: Diffuse TLR4 staining (open arrows) in myocytes and interstitial cells and intense TLR4 staining (solid arrows) in some myocytes. Interstitial TNC staining (brown) is evident around these cells; C: High powered view of TLR4 (pink) and alpha smooth muscle actin (brown) staining of cells in the infarcted LV. Intense TLR4 staining can be seen in some myocytes (solid arrow) with more diffuse staining seen in some cardiac myofibroblasts (labelled both pink and brown, open arrow); D: Low power view of the non-infarcted side of the mouse myocardium stained for TNC (brown) and TLR4 (pink). An absence of TNC staining and light diffuse TLR4 staining of the cells is observed. Inset image: a high powered view of cells in this area. In each image cell nuclei were stained with Mayer’s Haematoxylin. RV: Right ventricle. LV: Left ventricle; TNC: Tenascin C; TLR4: Toll-like receptor 4.
TNC up-regulates IL-6 mRNA and protein expression in human CMF
To determine whether TNC could stimulate MMP or pro-inflammatory cytokine synthesis in vitro, human CMF were incubated with TNC (0.1 μmol/L TNC or vehicle for 24 h) and mRNA levels were assessed using RT-PCR. A significant increase (14 fold, P < 0.01) in IL-6 mRNA expression from basal levels was observed in response to TNC (Figure 2). TNC also induced an approximately 25-fold increase in MMP3 levels (P < 0.01), whereas neither IL-1β nor any of the other MMPs analysed by real-time RT-PCR showed significant changes in mRNA expression in response to TNC (Figure 2). Basal TNFα mRNA expression in human CMF was minimal (0.002% GAPDH) and TNC had no effect on its expression (data not shown).
Figure 2.
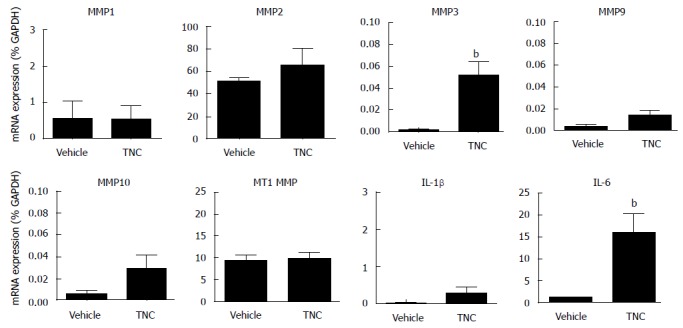
Changes in cytokine and matrix metalloproteinase mRNA expression in human cardiac myofibroblasts following incubation with Tenascin C. Effect of Tenascin C (TNC) (0.1 μmol/L, 24 h) on MMP1, MMP2, MMP3, MMP9, MMP10, MT1-MMP, IL-1β and IL-6 mRNA expression was assessed in human cardiac myofibroblasts (n = 4-6 donors). A significant increase in the expression of MMP3 and IL-6 was observed. Data are expressed as mean ± SEM. bP < 0.01 vs vehicle (student’s t-test). MMP: Matrix metalloproteinase; IL: Interleukin.
TLR4 is expressed in human CMF
We examined the mRNA expression of TLR4 in human CMF by RT-PCR and TLR4 protein by both western blot analysis and immunocytochemistry. Real-time RT-PCR array analysis of TLRs 1-9 revealed that TLR4 was by far the most highly expressed TLR in human CMF at the mRNA level (Figure 3A). Lower levels of TLR6 > TLR3 > TLR7 > TLR1 were also observed, but TLR2 was not detected. Follow-up studies with Taqman RT-PCR (different primers) on cells from a further 4 donors confirmed the TLR4 and TLR2 data (Figure 3B). Western blot analysis revealed a single 95 kDa band corresponding to TLR4 in protein lysates isolated from human CMF from 4 different donors (Figure 3C). Immunocytochemistry with primary antibodies directed against human TLR4 revealed a pattern of punctate staining in the cytoplasm and nucleus of human CMF (Figure 3D1). Staining was not evident if the primary antibody was pre-incubated with TLR4 protein (Figure 3D2). To assess the effect of inflammatory cytokines on TLR4 mRNA expression, human CMF were incubated with IL-1α. Real-time RT-PCR analysis of TLR4 revealed a 2.5 fold increase of expression after 6 h of treatment, falling back towards basal levels after 24 h (Figure 3E).
Figure 3.
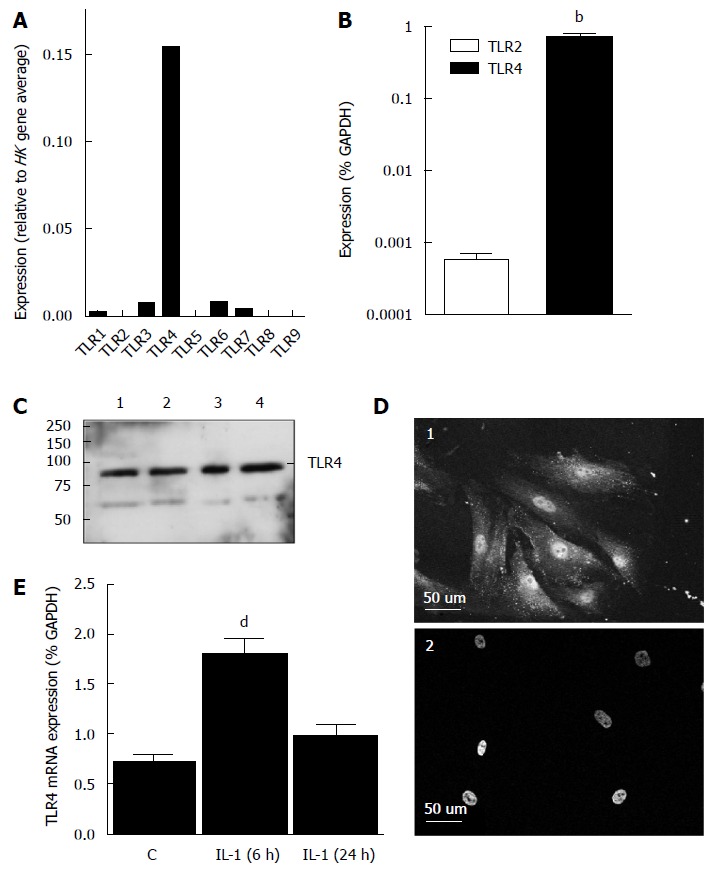
Toll-like receptor 4 expression in human cardiac myofibroblasts. A: Data from RT-PCR array showing abundance of TLR mRNA in a pooled sample of human CMF (n = 3 donors). Data expressed relative to 5 housekeeping genes; B: Taqman RT-PCR analysis of TLR2 and TLR4 mRNA levels in human CMF (n = 4 donors). Note log scale. bP < 0.001 (paired t-test); C: Western blot of CMF homogenates from 4 donors probed with anti-TLR4 antibody. Fifteen micrograms protein per lane. Molecular size (kDa) on left. TLR4 = 95 kDa; D1: Immunocytochemical localisation of TLR4 in human CMF using a primary antibody to human TLR4 and a Cy3-conjugated secondary antibody; D2: Effect of pre-absorption of primary antibody with TLR4 peptide (8 μg/mL) prior to immunostaining. Nuclei are labelled with DAPI. Loss of immunostaining confirms specificity of the antibody. Scale bar 50 μm; E: Effect of IL-1α (10 ng/mL, 6 and 24 h) on TLR4 mRNA expression in human CMF (n = 4 donors). Data are expressed as mean ± SEM. dP < 0.01 vs vehicle (ANOVA post-hoc). TLR: Toll-like receptor.
TLR4 mediates TNC up-regulation of IL-6 in human CMF
To determine whether the effects of TNC on IL-6 mRNA expression in human CMF were mediated by TLR4, cells from 5 donors were pre-incubated with TLR4 neutralising antibodies prior to TNC treatment (100 nmol/L, 24 h). TLR4 neutralisation had no significant effect on basal IL-6 mRNA expression but did prevent TNC-stimulated expression of IL-6 (Figure 4A). These changes in IL-6 mRNA expression were mirrored by changes in IL-6 protein secretion (Figure 4B). The specificity of the TLR4 antisera for the TLR4 receptor in our in vitro studies was confirmed by demonstrating that pre-treating cells with TLR4 neutralising antibodies had no effect on the IL-1α induced IL-6 mRNA expression (Figure 4C).
Figure 4.
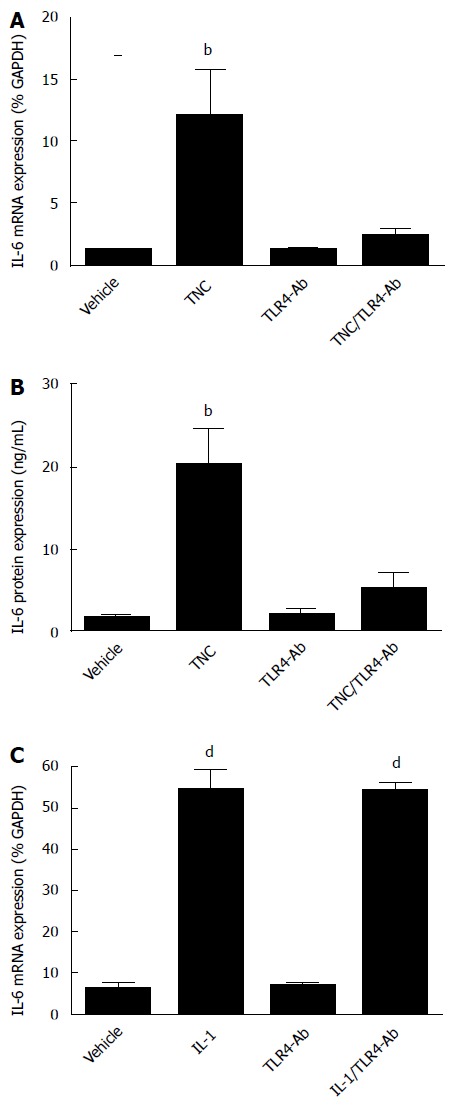
Tenascin C upregulates interleukin-6 expression in human cardiac myofibroblasts via toll-like receptor 4. A and B: Effect of TNC (0.1 μmol/L, 24 h) with and without 1 h pre-incubation in TLR4 antisera (25 μg/mL) on IL-6 mRNA (A) and IL-6 protein expression (B) in CMF (4-5 donors). Data expressed as mean ± SEM. bP < 0.01 vs vehicle (ANOVA post-hoc). TNC: Stimulation with TNC alone; TLR4-Ab: Pre-incubation with TLR4 neutralising antisera; TNC/TLR4-Ab: Stimulation with TNC following TLR4 pre-incubation; C: Effect of 1 h pre-incubation in TLR4 antisera (25 μg/mL) on IL-1α (10 ng/mL, 24 h)-stimulated IL-6 mRNA expression in CMF (n = 3 donors). The TLR4 neutralising antisera had no effect on IL-1α induced IL-6mRNA expression. Data expressed as mean ± SEM. dP < 0.0001 vs vehicle (ANOVA post-hoc). IL-1: Stimulation with IL-1α alone; TLR4-Ab: Pre-incubation with TLR4 neutralising antisera; IL-1/TLR4-Ab: Stimulation with IL-1α following TLR4 pre-incubation; TNC: Tenascin C; CMF: Cardiac myofibroblasts; IL: Interleukin; TLR: Toll-like receptor.
FBG domain upregulates IL-6 mRNA expression in human CMF
Previous studies have implicated the FBG domain of TNC in the promotion of cytokine production in synovial fibroblasts[14]. To determine whether a similar effect was evident in human CMF, cells were incubated with recombinant FBG protein (1 μmol/L, 24 h) and IL-6 mRNA expression assessed. FBG induced a 6-fold increase in IL-6 mRNA expression (P < 0.01), whereas incubation with the TNIII 5-7 recombinant TNC fragment had no effect, endorsing the suggestion that the FBG domain was crucial for promoting IL-6 production (Figure 5A). Neither FBG nor TNIII 5-7 recombinant proteins significantly increased MMP-3 expression in human CMF (Figure 5B).
Figure 5.
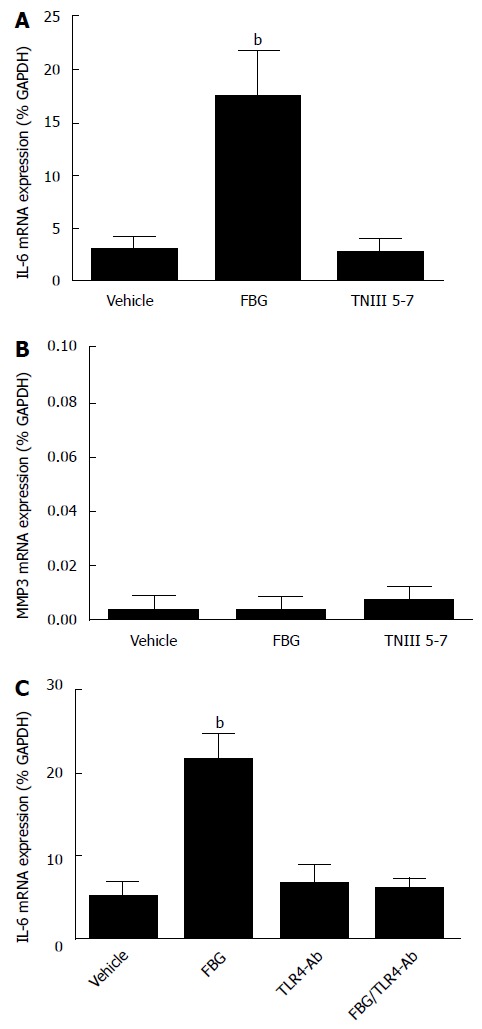
Fibrinogen-like globe domain of Tenascin C up-regulates interleukin-6 mRNA expression in human cardiac myofibroblasts via toll-like receptor 4. A and B: Effect of 1 μmol/L (24 h) recombinant protein, corresponding to either the FBG domain or the fibronectin type III domain (TNIII 5-7) of TNC, on IL-6 mRNA (A) and MMP3 mRNA (B) in human CMF (n = 3-5 donors). Data expressed as mean ± SEM. bP < 0.01 vs vehicle (ANOVA post-hoc); C: Effect of 1 h pre-incubation in TLR4 neutralising antisera (25 μg/mL) alone on IL-6 mRNA expression and on FBG (1 μmol/L, 24 h) stimulated IL-6 mRNA expression in CMF (n = 3 donors). Data expressed as mean ± SEM. bP < 0.01 vs vehicle (ANOVA post-hoc). FBG: Stimulation with FBG recombinant alone; TLR4-Ab: Pre-incubation with TLR4 neutralising antisera; FBG/TLR4-Ab: Stimulation with FBG following pre-incubation with TLR4 neutralising antisera. FBG: Fibrinogen-like globe; TNC: Tenascin C; IL: Interleukin; CMF: Cardiac myofibroblasts.
TLR4 mediates FBG up-regulation of IL-6 in human CMF
To determine whether the effects of FBG on IL-6 mRNA expression in human CMF were mediated by TLR4 pathways, cells from 3 donors were pre-incubated with TLR4 neutralising antibodies prior to FBG treatment (1 μmol/L, 24 h). TLR4 antibodies had no significant effect on basal IL-6 mRNA expression but did prevent FBG -stimulated expression of IL-6 (Figure 5C).
DISCUSSION
The main finding of the present study is that TNC up-regulates IL-6 expression in human CMF and that this effect is mediated through its FBG domain and the TLR4 receptor.
Inflammation and matrix turnover are important features in cardiac remodelling post infarction. If unchecked, these can lead to chronic maladaptive LV remodelling, characterised by progressive ventricular dilatation, myocardial hypertrophy, fibrosis, cardiac dysfunction and failure. TNC is an ECM glycoprotein that is re-expressed in the heart under pathological conditions such as MI[29,30], myocarditis[3,31,32] and dilated cardiomyopathy[4,33] and is closely associated with tissue injury and inflammation[3,31,34]. Following MI, TNC has been reported to appear during the acute stage at the interface between the infarcted and intact myocardium where tissue remodelling is at its most active[2]. There is evidence that TNC is a sensitive marker for active inflammation in acute myocarditis[3]. Moreover, serum levels of TNC correlate with the severity of heart failure, LV dysfunction and remodelling in patients with dilated cardiomyopathy[35]. These observations suggest that TNC expression is maintained in cardiac pathologies in which there is ongoing inflammation and remodelling.
We previously reported that IL-1α up-regulates TNC expression in human CMF[11]. IL-1 has been identified as one of the initial stimuli that drive the acute inflammatory response following MI[36-39]. Increased levels of IL-1 have also been implicated in the inflammatory response and adverse LV remodelling seen in heart failure[40]. The observation that TNC up-regulates IL-6 expression in our present study supports the previous notion that after its initial induction TNC could drive the inflammatory response further[14].
IL-6 has previously been shown to have important roles in inflammation and remodelling in the heart following injury by promoting leukocyte infiltration and activation, and by modulating fibroblast function[23,41-43]. In addition to its pro-inflammatory and fibrogenic properties, there is also evidence that IL-6 regulates ECM remodelling by enhancing cardiac fibroblast MMP expression and modulating tissue inhibitor of MMPs (TIMP) expression levels[23,42,44]. Our observations are consistent with a previous report in synovial fibroblasts, where an up-regulation of IL-6 expression following TNC stimulation was purported to play a role in the chronic inflammatory response seen in the arthritic joint[14]. In that study, the TLR4 pathway was identified as a mediator of this effect with the TLR4 receptor being activated by the FBG domain of TNC. Importantly, our study suggests that this notable mechanism may also play a role in the inflammatory response in the heart. As TLR4 signalling has long been implicated in a range of cardiac pathologies, the confirmation of such a mechanism in cardiac cells contributes to our understanding of the process of inflammation that occurs in the heart.
A regulatory role for TLR4 signalling in inflammation and ECM turnover has been established and its involvement in post-infarct maladaptive remodelling in the heart has been reported[1]. Moreover, TLR4 signalling has been implicated in myocarditis[45] and in the myocardial inflammatory response following regional or global ischemia/reperfusion[20,22,46,47]. Although TLR4 signalling contributes to cardiac dysfunction after MI in part through its influence on myocardial production of cytokines, the mechanism by which TLR4 is activated in these circumstances remains to be defined[22]. Evidence that TNC is up-regulated following oxidative stress suggests it may be a candidate ligand responsible for TLR4 signalling following ischemic injury[48]. Furthermore, our observations that the inflammatory cytokine, IL-1α, upregulates both TNC[11] and TLR4 expression (Figure 3E) in CMF and that both TNC and TLR4 are upregulated in the infarcted ventricle (Figure 1) lends support to their involvement in the cardiac inflammatory response following MI[2,14,49].
With the exception of MMP3, neither IL-1β nor any of the MMPs analysed by real-time RT-PCR showed significant changes in mRNA expression in response to TNC. In the case of MMP3, although a significant increase from basal expression was indeed observed with TNC, the mean induced MMP3 levels were only 0.05% that of the HK gene GAPDH. Nevertheless, this is equivalent to the mRNA level observed in response to a low concentration of IL-1 (0.1 ng/mL) in these cells which resulted in detectable MMP3 protein secretion[50]. We found no evidence, however, that FBG was responsible for the increase in MMP3 observed. The possibility that a different domain of this molecule, other than FBG or TNIII5-7, plays a role in MMP3 induction remains to be tested. Moreover, the recent demonstration that some of the effects of TNC are mediated via αVβ3 integrin, suggests that the induction of MMP3 by TNC may occur via a different receptor subtype[51].
The up-regulation of MMP3 by TNC would be consistent with TNC’s role in the degradation of ECM following MI and the promotion of a de-adhesive state that facilitates migration of fibroblasts and other granulation tissue cells to the site of injury[52]. TNC has previously been reported to induce cell-specific increases in MMP levels including cultured human smooth muscle cells, murine mammary cancer cells, macrophages and synovial fibroblasts[53-56].
Finally, the lack of induction of IL-1β and TNFα by TNC is consistent with previous studies which have reported that stimulation of synovial fibroblasts with TNC resulted in augmented gene expression of some pro-inflammatory cytokines (e.g., IL-1α and IL-6), but not others (e.g., interferon-γ, TNF-α and IL-1β)[14,56].
In conclusion, we have demonstrated that TNC up-regulates MMP3 and the pro-inflammatory cytokine IL-6 in human CMF. The latter effect of TNC is mediated via the TLR4 receptor and the FBG domain of the TNC. Targeting the FBG domain of TNC may provide a novel therapeutic option to counteract aberrant inflammation and maladaptive cardiac remodelling.
ACKNOWLEDGMENTS
We are grateful to Mr David J O’Regan for providing right atrial appendage biopsy tissue for this study and to Phil Warburton for primary cell culture.
COMMENTS
Background
Adverse ventricular remodelling following cardiac injury is a key determinant of heart failure. Despite major advances in the field, the number of heart failure deaths is increasing steadily. A better understanding of the processes involved in the remodelling process would enable the development of novel therapeutics.
Research frontiers
The importance of inflammation in cardiac remodelling that occurs after myocardial injury is unequivocal. The inflammatory process can cause myocardial damage, while inflammatory agents contribute to the worsening and progression of heart failure. A detailed understanding of the underlying inflammatory processes involved in cardiac remodelling, with the aim to develop better therapeutics, remains an important area in heart failure research.
Innovation and breakthroughs
Tenascin C (TNC) is highly expressed during embryogenesis but is virtually absent in the adult heart. It is re-expressed in the heart following injury where its expression correlates with the extent of inflammation and myocardial remodelling. The continued expression of TNC in pathologies associated with cardiac inflammation (e.g., myocarditis, heart failure) had been recognised yet its functional significance remained elusive. This study is the first to demonstrate that TNC is able to stimulate the expression of the inflammatory cytokine interleukin (IL)-6 in human cardiac myofibroblasts (CMF) in a toll-like receptor 4-dependent fashion. This action is similar to that reported in the arthritic joint where TNC contributes to the persistent inflammation observed. As IL-6 plays an important role in myocardial inflammation and fibrosis, identification of this notable mechanism in CMF will further our understanding of the inflammatory processes occurring after myocardial stress or injury.
Application
The results provide further insight into the underlying mechanism(s) involved in cardiac inflammation and may help identify novel therapeutic targets to attenuate this in disease states.
Peer-review
This manuscript reports the effects of TNC on the expression of the pro-inflammatory cytokines in human CMF. The findings are of interest and suitable for the publication by the journal.
Footnotes
Supported by The British Heart Foundation, No. CH/92005.
Institutional review board statement: All work on human tissue was carried out with local ethical committee approval (reference number: 01/040 - documents attached) and informed patient consent. All investigations conformed to the principles outlined in the Declaration of Helsinki). All procedures involving animals were carried out in accordance with the Home Office Animals (Scientific Procedures) Act 1986 and the University of Leeds Animal Welfare and Ethical Committee.
Institutional animal care and use committee statement: All procedures involving animals were carried out in accordance with the Home Office Animals (Scientific Procedures) Act 1986 and the University of Leeds Animal Welfare and Ethical Committee.
Conflict-of-interest statement: None.
Data sharing statement: Complete dataset and statistical analyses available from the corresponding author at cvsam@leeds.ac.uk.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: March 4, 2016
First decision: March 22, 2016
Article in press: April 18, 2016
P- Reviewer: Cheng TH, Guo ZS, Sipos F S- Editor: Ji FF L- Editor: A E- Editor: Jiao XK
References
- 1.Timmers L, Sluijter JP, van Keulen JK, Hoefer IE, Nederhoff MG, Goumans MJ, Doevendans PA, van Echteld CJ, Joles JA, Quax PH, et al. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res. 2008;102:257–264. doi: 10.1161/CIRCRESAHA.107.158220. [DOI] [PubMed] [Google Scholar]
- 2.Imanaka-Yoshida K, Hiroe M, Nishikawa T, Ishiyama S, Shimojo T, Ohta Y, Sakakura T, Yoshida T. Tenascin-C modulates adhesion of cardiomyocytes to extracellular matrix during tissue remodeling after myocardial infarction. Lab Invest. 2001;81:1015–1024. doi: 10.1038/labinvest.3780313. [DOI] [PubMed] [Google Scholar]
- 3.Morimoto S, Imanaka-Yoshida K, Hiramitsu S, Kato S, Ohtsuki M, Uemura A, Kato Y, Nishikawa T, Toyozaki T, Hishida H, et al. Diagnostic utility of tenascin-C for evaluation of the activity of human acute myocarditis. J Pathol. 2005;205:460–467. doi: 10.1002/path.1730. [DOI] [PubMed] [Google Scholar]
- 4.Tamura A, Kusachi S, Nogami K, Yamanishi A, Kajikawa Y, Hirohata S, Tsuji T. Tenascin expression in endomyocardial biopsy specimens in patients with dilated cardiomyopathy: distribution along margin of fibrotic lesions. Heart. 1996;75:291–294. doi: 10.1136/hrt.75.3.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rettig WJ, Erickson HP, Albino AP, Garin-Chesa P. Induction of human tenascin (neuronectin) by growth factors and cytokines: cell type-specific signals and signalling pathways. J Cell Sci. 1994;107(Pt 2):487–497. [PubMed] [Google Scholar]
- 6.Chiquet M. Regulation of extracellular matrix gene expression by mechanical stress. Matrix Biol. 1999;18:417–426. doi: 10.1016/s0945-053x(99)00039-6. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto K, Dang QN, Kennedy SP, Osathanondh R, Kelly RA, Lee RT. Induction of tenascin-C in cardiac myocytes by mechanical deformation. Role of reactive oxygen species. J Biol Chem. 1999;274:21840–21846. doi: 10.1074/jbc.274.31.21840. [DOI] [PubMed] [Google Scholar]
- 8.Järvinen TA, Józsa L, Kannus P, Järvinen TL, Hurme T, Kvist M, Pelto-Huikko M, Kalimo H, Järvinen M. Mechanical loading regulates the expression of tenascin-C in the myotendinous junction and tendon but does not induce de novo synthesis in the skeletal muscle. J Cell Sci. 2003;116:857–866. doi: 10.1242/jcs.00303. [DOI] [PubMed] [Google Scholar]
- 9.Chiquet M, Sarasa-Renedo A, Tunç-Civelek V. Induction of tenascin-C by cyclic tensile strain versus growth factors: distinct contributions by Rho/ROCK and MAPK signaling pathways. Biochim Biophys Acta. 2004;1693:193–204. doi: 10.1016/j.bbamcr.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 10.Ogawa K, Ito M, Takeuchi K, Nakada A, Heishi M, Suto H, Mitsuishi K, Sugita Y, Ogawa H, Ra C. Tenascin-C is upregulated in the skin lesions of patients with atopic dermatitis. J Dermatol Sci. 2005;40:35–41. doi: 10.1016/j.jdermsci.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 11.Maqbool A, Hemmings KE, O’Regan DJ, Ball SG, Porter KE, Turner NA. Interleukin-1 has opposing effects on connective tissue growth factor and tenascin-C expression in human cardiac fibroblasts. Matrix Biol. 2013;32:208–214. doi: 10.1016/j.matbio.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Tamaoki M, Imanaka-Yoshida K, Yokoyama K, Nishioka T, Inada H, Hiroe M, Sakakura T, Yoshida T. Tenascin-C regulates recruitment of myofibroblasts during tissue repair after myocardial injury. Am J Pathol. 2005;167:71–80. doi: 10.1016/S0002-9440(10)62954-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imanaka-Yoshida K, Hiroe M, Yoshida T. Interaction between cell and extracellular matrix in heart disease: multiple roles of tenascin-C in tissue remodeling. Histol Histopathol. 2004;19:517–525. doi: 10.14670/HH-19.517. [DOI] [PubMed] [Google Scholar]
- 14.Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, Drexler S, Sofat N, Kashiwagi M, Orend G, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15:774–780. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- 15.Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 16.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 17.Baumgarten G, Knuefermann P, Nozaki N, Sivasubramanian N, Mann DL, Vallejo JG. In vivo expression of proinflammatory mediators in the adult heart after endotoxin administration: the role of toll-like receptor-4. J Infect Dis. 2001;183:1617–1624. doi: 10.1086/320712. [DOI] [PubMed] [Google Scholar]
- 18.Monaco C, Gregan SM, Navin TJ, Foxwell BM, Davies AH, Feldmann M. Toll-like receptor-2 mediates inflammation and matrix degradation in human atherosclerosis. Circulation. 2009;120:2462–2469. doi: 10.1161/CIRCULATIONAHA.109.851881. [DOI] [PubMed] [Google Scholar]
- 19.Frantz S, Kobzik L, Kim YD, Fukazawa R, Medzhitov R, Lee RT, Kelly RA. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J Clin Invest. 1999;104:271–280. doi: 10.1172/JCI6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chong AJ, Shimamoto A, Hampton CR, Takayama H, Spring DJ, Rothnie CL, Yada M, Pohlman TH, Verrier ED. Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J Thorac Cardiovasc Surg. 2004;128:170–179. doi: 10.1016/j.jtcvs.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 21.Shimamoto A, Chong AJ, Yada M, Shomura S, Takayama H, Fleisig AJ, Agnew ML, Hampton CR, Rothnie CL, Spring DJ, et al. Inhibition of Toll-like receptor 4 with eritoran attenuates myocardial ischemia-reperfusion injury. Circulation. 2006;114:I270–I274. doi: 10.1161/CIRCULATIONAHA.105.000901. [DOI] [PubMed] [Google Scholar]
- 22.Cha J, Wang Z, Ao L, Zou N, Dinarello CA, Banerjee A, Fullerton DA, Meng X. Cytokines link Toll-like receptor 4 signaling to cardiac dysfunction after global myocardial ischemia. Ann Thorac Surg. 2008;85:1678–1685. doi: 10.1016/j.athoracsur.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 24.Turner NA, Porter KE. Function and fate of myofibroblasts after myocardial infarction. Fibrogenesis Tissue Repair. 2013;6:5. doi: 10.1186/1755-1536-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porter KE, Turner NA, O’Regan DJ, Ball SG. Tumor necrosis factor alpha induces human atrial myofibroblast proliferation, invasion and MMP-9 secretion: inhibition by simvastatin. Cardiovasc Res. 2004;64:507–515. doi: 10.1016/j.cardiores.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 26.Mughal RS, Warburton P, O’Regan DJ, Ball SG, Turner NA, Porter KE. Peroxisome proliferator-activated receptor gamma-independent effects of thiazolidinediones on human cardiac myofibroblast function. Clin Exp Pharmacol Physiol. 2009;36:478–486. doi: 10.1111/j.1440-1681.2008.05088.x. [DOI] [PubMed] [Google Scholar]
- 27.Turner NA, Mughal RS, Warburton P, O’Regan DJ, Ball SG, Porter KE. Mechanism of TNFalpha-induced IL-1alpha, IL-1beta and IL-6 expression in human cardiac fibroblasts: effects of statins and thiazolidinediones. Cardiovasc Res. 2007;76:81–90. doi: 10.1016/j.cardiores.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 28.Turner NA, Ball SG, Balmforth AJ. The mechanism of angiotensin II-induced extracellular signal-regulated kinase-1/2 activation is independent of angiotensin AT(1A) receptor internalisation. Cell Signal. 2001;13:269–277. doi: 10.1016/s0898-6568(01)00135-8. [DOI] [PubMed] [Google Scholar]
- 29.Smoak KA, Aloor JJ, Madenspacher J, Merrick BA, Collins JB, Zhu X, Cavigiolio G, Oda MN, Parks JS, Fessler MB. Myeloid differentiation primary response protein 88 couples reverse cholesterol transport to inflammation. Cell Metab. 2010;11:493–502. doi: 10.1016/j.cmet.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willems IE, Arends JW, Daemen MJ. Tenascin and fibronectin expression in healing human myocardial scars. J Pathol. 1996;179:321–325. doi: 10.1002/(SICI)1096-9896(199607)179:3<321::AID-PATH555>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 31.Imanaka-Yoshida K, Hiroe M, Yasutomi Y, Toyozaki T, Tsuchiya T, Noda N, Maki T, Nishikawa T, Sakakura T, Yoshida T. Tenascin-C is a useful marker for disease activity in myocarditis. J Pathol. 2002;197:388–394. doi: 10.1002/path.1131. [DOI] [PubMed] [Google Scholar]
- 32.Sato M, Toyozaki T, Odaka K, Uehara T, Arano Y, Hasegawa H, Yoshida K, Imanaka-Yoshida K, Yoshida T, Hiroe M, et al. Detection of experimental autoimmune myocarditis in rats by 111In monoclonal antibody specific for tenascin-C. Circulation. 2002;106:1397–1402. doi: 10.1161/01.cir.0000027823.07104.86. [DOI] [PubMed] [Google Scholar]
- 33.Tsukada B, Terasaki F, Shimomura H, Otsuka K, Otsuka K, Katashima T, Fujita S, Imanaka-Yoshida K, Yoshida T, Hiroe M, et al. High prevalence of chronic myocarditis in dilated cardiomyopathy referred for left ventriculoplasty: expression of tenascin C as a possible marker for inflammation. Hum Pathol. 2009;40:1015–1022. doi: 10.1016/j.humpath.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 34.Nishioka T, Suzuki M, Onishi K, Takakura N, Inada H, Yoshida T, Hiroe M, Imanaka-Yoshida K. Eplerenone attenuates myocardial fibrosis in the angiotensin II-induced hypertensive mouse: involvement of tenascin-C induced by aldosterone-mediated inflammation. J Cardiovasc Pharmacol. 2007;49:261–268. doi: 10.1097/FJC.0b013e318033dfd4. [DOI] [PubMed] [Google Scholar]
- 35.Terasaki F, Okamoto H, Onishi K, Sato A, Shimomura H, Tsukada B, Imanaka-Yoshida K, Hiroe M, Yoshida T, Kitaura Y, et al. Higher serum tenascin-C levels reflect the severity of heart failure, left ventricular dysfunction and remodeling in patients with dilated cardiomyopathy. Circ J. 2007;71:327–330. doi: 10.1253/circj.71.327. [DOI] [PubMed] [Google Scholar]
- 36.Dinarello CA, Pomerantz BJ. Proinflammatory cytokines in heart disease. Blood Purif. 2001;19:314–321. doi: 10.1159/000046960. [DOI] [PubMed] [Google Scholar]
- 37.Deten A, Volz HC, Briest W, Zimmer HG. Cardiac cytokine expression is upregulated in the acute phase after myocardial infarction. Experimental studies in rats. Cardiovasc Res. 2002;55:329–340. doi: 10.1016/s0008-6363(02)00413-3. [DOI] [PubMed] [Google Scholar]
- 38.Turner NA, Das A, Warburton P, O’Regan DJ, Ball SG, Porter KE. Interleukin-1alpha stimulates proinflammatory cytokine expression in human cardiac myofibroblasts. Am J Physiol Heart Circ Physiol. 2009;297:H1117–H1127. doi: 10.1152/ajpheart.00372.2009. [DOI] [PubMed] [Google Scholar]
- 39.Turner NA. Effects of interleukin-1 on cardiac fibroblast function: relevance to post-myocardial infarction remodelling. Vascul Pharmacol. 2014;60:1–7. doi: 10.1016/j.vph.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 40.Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res. 2002;91:988–998. doi: 10.1161/01.res.0000043825.01705.1b. [DOI] [PubMed] [Google Scholar]
- 41.Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol. 2005;175:3463–3468. doi: 10.4049/jimmunol.175.6.3463. [DOI] [PubMed] [Google Scholar]
- 42.Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, Frangogiannis NG. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008;173:57–67. doi: 10.2353/ajpath.2008.070974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nowell MA, Williams AS, Carty SA, Scheller J, Hayes AJ, Jones GW, Richards PJ, Slinn S, Ernst M, Jenkins BJ, et al. Therapeutic targeting of IL-6 trans signaling counteracts STAT3 control of experimental inflammatory arthritis. J Immunol. 2009;182:613–622. doi: 10.4049/jimmunol.182.1.613. [DOI] [PubMed] [Google Scholar]
- 44.Kossakowska AE, Edwards DR, Prusinkiewicz C, Zhang MC, Guo D, Urbanski SJ, Grogan T, Marquez LA, Janowska-Wieczorek A. Interleukin-6 regulation of matrix metalloproteinase (MMP-2 and MMP-9) and tissue inhibitor of metalloproteinase (TIMP-1) expression in malignant non-Hodgkin’s lymphomas. Blood. 1999;94:2080–2089. [PubMed] [Google Scholar]
- 45.Fairweather D, Yusung S, Frisancho S, Barrett M, Gatewood S, Steele R, Rose NR. IL-12 receptor beta 1 and Toll-like receptor 4 increase IL-1 beta- and IL-18-associated myocarditis and coxsackievirus replication. J Immunol. 2003;170:4731–4737. doi: 10.4049/jimmunol.170.9.4731. [DOI] [PubMed] [Google Scholar]
- 46.Oyama J, Blais C, Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–789. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 47.Stapel H, Kim SC, Osterkamp S, Knuefermann P, Hoeft A, Meyer R, Grohé C, Baumgarten G. Toll-like receptor 4 modulates myocardial ischaemia-reperfusion injury: Role of matrix metalloproteinases. Eur J Heart Fail. 2006;8:665–672. doi: 10.1016/j.ejheart.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 48.Taki J, Inaki A, Wakabayashi H, Imanaka-Yoshida K, Ogawa K, Hiroe M, Shiba K, Yoshida T, Kinuya S. Dynamic expression of tenascin-C after myocardial ischemia and reperfusion: assessment by 125I-anti-tenascin-C antibody imaging. J Nucl Med. 2010;51:1116–1122. doi: 10.2967/jnumed.109.071340. [DOI] [PubMed] [Google Scholar]
- 49.Turner NA. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs) J Mol Cell Cardiol. 2016;94:189–200. doi: 10.1016/j.yjmcc.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 50.van Nieuwenhoven FA, Hemmings KE, Porter KE, Turner NA. Combined effects of interleukin-1α and transforming growth factor-β1 on modulation of human cardiac fibroblast function. Matrix Biol. 2013;32:399–406. doi: 10.1016/j.matbio.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 51.Shimojo N, Hashizume R, Kanayama K, Hara M, Suzuki Y, Nishioka T, Hiroe M, Yoshida T, Imanaka-Yoshida K. Tenascin-C may accelerate cardiac fibrosis by activating macrophages via the integrin αVβ3/nuclear factor-κB/interleukin-6 axis. Hypertension. 2015;66:757–766. doi: 10.1161/HYPERTENSIONAHA.115.06004. [DOI] [PubMed] [Google Scholar]
- 52.Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol. 2010;48:504–511. doi: 10.1016/j.yjmcc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wallner K, Li C, Shah PK, Wu KJ, Schwartz SM, Sharifi BG. EGF-Like domain of tenascin-C is proapoptotic for cultured smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:1416–1421. doi: 10.1161/01.ATV.0000134299.89599.53. [DOI] [PubMed] [Google Scholar]
- 54.Kalembeyi I, Inada H, Nishiura R, Imanaka-Yoshida K, Sakakura T, Yoshida T. Tenascin-C upregulates matrix metalloproteinase-9 in breast cancer cells: direct and synergistic effects with transforming growth factor beta1. Int J Cancer. 2003;105:53–60. doi: 10.1002/ijc.11037. [DOI] [PubMed] [Google Scholar]
- 55.Khan KM, Falcone DJ. Role of laminin in matrix induction of macrophage urokinase-type plasminogen activator and 92-kDa metalloproteinase expression. J Biol Chem. 1997;272:8270–8275. doi: 10.1074/jbc.272.13.8270. [DOI] [PubMed] [Google Scholar]
- 56.Kanayama M, Kurotaki D, Morimoto J, Asano T, Matsui Y, Nakayama Y, Saito Y, Ito K, Kimura C, Iwasaki N, et al. Alpha9 integrin and its ligands constitute critical joint microenvironments for development of autoimmune arthritis. J Immunol. 2009;182:8015–8025. doi: 10.4049/jimmunol.0900725. [DOI] [PubMed] [Google Scholar]