

Abstract
High mobility group protein 1 (HMGB1) is a multifunctional protein that interacts with DNA and chromatin to influence the regulation of transcription, DNA replication and repair and recombination. We show that HMGB1 alters the structure and stability of the canonical nucleosome (N) in a nonenzymatic, adenosine triphosphate-independent manner. As a result, the canonical nucleosome is converted to two stable, physically distinct nucleosome conformers. Although estrogen receptor (ER) does not bind to its consensus estrogen response element within a nucleosome, HMGB1 restructures the nucleosome to facilitate strong ER binding. The isolated HMGB1-restructured nucleosomes (N’ and N’’) remain stable and exhibit a number of characteristics that are distinctly different from the canonical nucleosome. These findings complement previous studies that showed (1) HMGB1 stimulates in vivo transcriptional activation at estrogen response elements and (2) knock down of HMGB1 expression by siRNA precipitously reduced transcriptional activation. The findings indicate that a major facet of the mechanism of HMGB1 action involves a restructuring of aspects of the nucleosome that appear to relax structural constraints within the nucleosome. The findings are extended to reveal the differences between ER and the other steroid hormone receptors. A working proposal outlines mechanisms that highlight the multiple facets that HMGB1 may utilize in restructuring the nucleosome.
Keywords: Nucleosome dynamics, Estrogen receptor, High mobility group protein 1, Conformational dynamics, Energy landscape
Core tip: Response elements or target sites for transcription factors in DNA are often found veiled within a nucleosome in a chromatin milieu and in many cases are not accessible. Although the nucleosome/chromatin network is generally repressive to transcription, there are now a number of enzymatic strategies that have now been recognized that remodel the nucleosome to facilitate transcription factor access. We recently showed that estrogen receptor does not bind to a canonical nucleosome. However, we have discovered that high mobility group protein 1 (HMGB1) restructures the nucleosome in a nonenzymatic manner to facilitate strong estrogen receptor binding. This review will provide background for this work and outline our findings, characterize the HMGB1-restructured nucleosomes and propose a working model to account for the HMGB1 restructuring activity.
INTRODUCTION
Life is dynamic and ever changing by its very nature. We sense immediate changes in our environment and react to the challenges to best deal with them with the resources available to us. And so it is also within the trillions of individual cells that make up our bodies and the communication network that coordinates these efforts. As diverse extracellular conditions and challenges fluctuate endlessly, the cells sense and integrate these signals to react to and regulate the expression of the appropriate genes to address these diverse conditions. Herein lies the heart of cellular phenotypic plasticity[1-5].
Hormones released from an endocrine gland provide an excellent example of molecular signals that can impinge on the dynamic character within the cell and result in extraordinary changes in cellular activities. The lipophilic steroid hormones are a subgroup of the nuclear hormone superfamily that includes estrogens, androgens, progestins, glucocorticoids, and mineralocorticoids[6].
The action by estrogen on its ligand activated receptor, the estrogen receptor (ER), and the role of high mobility group protein 1 (HMGB1) in the subsequent transcriptional activation is the focus of this minireview. ER plays a dominant role in a number of different tissues and in a host of physiological and pathophysiological activities that include sexual development, atherosclerosis, osteoporosis, dementia and cancer, to mention just a few of its major influences. The effects of estrogen can be derived from two avenues of action: (1) non-genomic, associated with signal transduction networks and is independent of transcription; and (2) genomic, in which the action occurs on the genome at the transcriptional level[7-9].
BASIC VIEW OF EUKARYOTIC TRANSCRIPTION
Eukaryotic transcription of protein-coding genes is carried out by RNA polymerase II (RNA pol II) in multiple steps. A sequence-specific activator or transcription factor (TF) binds to its response element (upstream promoter or enhancer elements). Upon TF binding, coregulators are recruited to the site and this complex must then communicate with the pre-initiation complex (PIC) at the proximal promoter. The complex at this site is made up of RNA pol II, the Mediator complex and a collection of general transcription factors that include TFIIA, TFIIB, TFIID [TATA binding protein (TBP) and associated TAFs], TFIIE, TFIIF and TFIIH to initiate transcription[10]. The enormous Mediator complex appears to be a global participant in this communication network, with its most distinguishing characteristic being the central “hub” in regulating most, if not all RNA pol II transcription. Mediator communicates between a spectrum of different sequence-specific transcription factors (bound at upstream promoters or enhancer elements) and the PIC. Thus, it is not surprising that its subunit composition is variable since it must display conformational, structural and functional flexibility, all of which is consistent with the demands that it function for different genes, different physiological conditions and in different cell types[11-13].
However, this process is more complex in the context of the cell since the DNA is complexed within a nucleosome/chromatin network and many of the response elements are inaccessible to TFs as a result.
CHROMATIN BASICS
Chromatin presents a formidable barrier for all manipulations on the genetic material - be it DNA replication, DNA repair, transcription or genetic recombination. The first level of DNA packaging is the nucleosome in which 147 bps of DNA makes approximately 1 ¾ left-handed superhelical turns around the outside surface of an octamer of core histone proteins, which includes two copies each of four highly conserved core histones - H3, H4, H2A and H2B. The histone octamer binds the DNA at 14 superhelical locations, making greater than 120 direct contacts with the histone interactions. These contacts are exclusively in the minor groove with the phosphate groups of the DNA backbone, with few, if any, contacts to the base[14,15]. The DNA structure differs considerably from the classical B form DNA in that it is highly bent, with regions of DNA that are over- or under-wound, which may affect the successful binding of many transcription factors to their cognate sites in nucleosomal DNA[16-20]. Adjacent nucleosome cores are interconnected by an additional length of DNA (about 20-80 bps of linker DNA) that are less associated with the histone octamer, which together make up the complete repeating nucleosome structural unit.
The N-terminal tails of the core histones, which make up about 30% of the proteins, extend out from the core nucleosome and are considered unstructured and mobile since they have not been resolved in the crystal structure of the nucleosome[21,22]. The positively charged residues in the tails have been implicated in interacting with DNA, are subject to post-translational modifications, the status of which can influence the higher-order chromatin structure and clearly participate in regulating gene expression[23]. Of the many post-translational modifications and their role in the epigenetic marking system, acetylation of the tails appears important here since, at least some of its effects, may have parallels and implication on a potential role for HMGB1 in altering histone-DNA interactions[24]. Nucleosomes can then cluster together to form a secondary level of chromatin structure, the 30 nm fiber, that is a tight compaction of nucleosomes and is stabilized by the core histone tails and the linker histone H1, which binds to linker DNA. Studies show that removal of the core histone tails inhibits compaction and opens up chromatin structure, even in the presence of the linker histone H1[25].
The 30 nm fiber can be further condensed into a more highly compacted state and is generally inaccessible to active processes on DNA[26,27].
NUCLEOSOME DYNAMICS AND TRANSCRIPTIONAL ACTIVITY
The nucleosome, even though an enormous structure (250 kDa), is inherently a stable structure due to the constraints from multiple weak histone-DNA interactions. Not withstanding this fundamental stability, the structure of the nucleosome is continuously manipulated to accommodate changes for “local” gene expression and other DNA transactions. For our purposes, because selective gene expression within the cell is both temporal and dynamic, the structure and/or the position of the nucleosome within the chromatin milieu are likewise dynamic.
Key elements in the control of gene expression are regulating the access and binding affinity of transcription factors to their response elements and the successful assembly of multifunctional complexes to link the activation event to the transcriptional machinery at the promoter. Response elements in the genome can be regarded as interacting on the surface of the nucleosome with the histone octamer, in which the sequences are either accessible or inaccessible, or alternatively, they may reside in a nucleosome-free region[16]. Notably, if the sites are inaccessible, eukaryotes have evolved a number of strategies to regulate its dynamic nature in selected genomic stretches to facilitate factors to gain access to them, with the focus here on the action of two major types of complexes and their activities[23,28].
The first class of proteins includes a collection of histone modifying enzymes that post-translationally recognize and reversibly modify specific residues in the histone tails that extend on the outside surface of the nucleosome. These enzymes covalently add or remove small groups (referred to as writing, reading and erasing the message) including acetyl, phosphate and other groups. One subset of such enzymes is the histone acetyltransferase that adds acetyl groups to selected lysines in histones H3 and H4, with such modified nucleosomes being generally correlated with transcriptional activation. On the other hand, the removal of these acetyl groups by histone deacetylases is correlated with transcriptionally inactive regions of chromatin. The distinctive combination of these directed chromatin “marks” leads a key role in defining the transcriptional activity of a gene since these modifications on specific nucleosomes help direct the transcriptional activity or program at selected genes in a “sea” of thousands of other genes within the vast genome. The nature of these progressive histone modifications has been called the “histone code hypothesis”. These “marks” are thought to not only influence the dynamic nature of the nucleosome, but also act as epitopes for the recruitment of additional regulatory proteins[24,29-31]. These inheritable changes in gene function, epigenetic markers, are not associated with alterations in the gene sequence. So, even at this first level of nucleosome structure, there is clearly a heterogeneous family of nucleosome states[32].
The second class of complexes affecting nucleosome dynamics is the ATP-dependent chromatin remodeling complexes (CRCs). They represent multisubunit complexes that contain a core ATPase catalytic subunit that is an energetic engine for altering the nature of the nucleosome. Depending on which of the five families of CRCs, this activity can either alter the position of the nucleosome or lead to nucleosome instability by a number of routes. In some cases, regions may require the collaborative effect of more than one chromatin remodelers to regulate TF accessibility[33-35]. It has become increasingly evident that different remodeling complexes generate different remodeled substrates that depend on the DNA-dependent ATPase present in the complex and perhaps the model system used to study the activities[36-38].
In addition to these enzymatic CRCs, different multisubunit complexes found in human and S. cerevisiae cells, referred to as facilitates chromatin transcription (FACT), interact with nucleosomes, reorganize them in an ATP-independent manner and therefore utilize a nonenzymatic mechanism of action that differs from the ATP-dependent CRCs. The FACT complex contains an HMG box subunit that is essential for activity and both in vitro and in vivo studies reinforce the hypothesis that FACT destabilizes nucleosomes without altering its position by disrupting interactions between DNA and the H2A/H2B dimers. This prevents histone eviction during this process and promotes nucleosome recovery afterwards[39-42].
ESTROGEN RECEPTOR
The ER (classified as NR3A1), like all the nuclear hormone receptors, is a ligand-activated transcription factor[43]. The NHRs have a common modular domain structure that includes the N-terminal transactivation domain, a well-characterized DNA binding domain (DBD), followed immediately by a stretch of amino acids referred to as the C-terminal extension (CTE), which extends into the hinge region, with the ligand binding domain at the C-terminal end, at which the hormone and other agonist and antagonists may bind. The CTE was shown to interact with HMGB1 and is thought to provide additional stability to the ER/consensus estrogen response element (cERE) interaction[44,45] (Figure 1).
Figure 1.
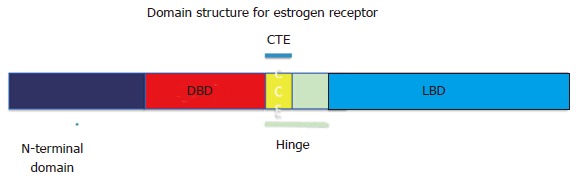
Domain structure for estrogen receptor. The structural and functional domains include the N-terminal domain, DBD, CTE, the flexible hinge region and LBD. DBD: DNA binding domain; CTE: C-terminal extension; LBD: Ligand binding domain.
Upon estrogen binding to ER within the cell, the receptor dimerizes to a functional homodimer and binds to the canonical 15 bp cERE (cERE = AGGTCAnnnTGACCT). Each ER monomer unit uses its DBD to make direct contacts in the major groove to each of the 6 bp consensus ERE half-sites (cHERE) that are separated by 3 unspecified bps. The bound receptor then recruits coactivators that then communicate with the PIC to initiate the subsequent steps in the transcriptional process.
Although the ER dimer does not stably bind to a single cERE half-site, the presence of HMGB1 facilitates strong binding to a half-sites (cHERE), in addition to binding to direct repeats and everted repeats, which have been shown to functionally influence specific estrogen-responsive genes[46-48].
It has been shown that the rotational and translational positioning of a response element in nucleosomal DNA is crucial in determining the level at which the factor gains access to the site[17,49-51]. Within the human mammalian cell line, MCF-7, the chromatin structure of the estrogen-responsive pS2 promoter was mapped to single nucleotide resolution. This included the ERE (in nucleosome E) and the TATA box (nucleosome T), which are each rotationally phased and translationally positioned at the edge of the nucleosome. The rotational phasing showed that the major groove of the ERE was facing outward from the histone octamer and suggested that the ERE was probably accessible to ER binding since there was no nucleosome displacement, even upon maximum transcriptional activation[52].
HMGB1 (ALSO RECOGNIZED AS AMPHOTERIN) AND MELANOMA-ASSOCIATED FACTOR
HMGB1 appears to be multifunctional - a protein “for all seasons”. It is involved in replication, DNA repair, transcription and recombination within the nucleus, while acting outside the cell as a cytokine and proinflammatory mediator[53-62]. As a result, HMGB1 is associated with many aspects of health and disease[63-66].
HMGB1 is a small (about 25 kDa), highly conserved, ubiquitous and abundant nuclear protein that binds in a non-sequence specific manner in the minor groove of DNA. It is considered an “architectural” protein because HMGB1 binding facilitates the assembly of a wide variety of nucleoprotein complexes as noted above[67].
From both a structural and functional perspective, HMGB1 is best described as having three modular domains. It contains tandem HMG boxes, the N-terminal A-box and the central B-box, both having 80-90 residues and are highly basic in nature. These boxes bind to B-form DNA to produce large bends as large as 90 degrees, but bind preferentially to bent, kinked or underwound DNA (Figure 2). The HMGB1 interactions and the bending interactions are dynamic and result in an increase in flexibility or flexure in the DNA that permits DNA to accommodate a broader spectrum of protein interaction in nucleoprotein structures[68,69]. In this regard, HMGB1 has been shown to enhance the binding of a number of basal and regulatory transcription factors to their cognate DNA binding sites, including the human TBP, p53, p73, HoxD9, Oct 1 and 2, nuclear factor kappa beta, sterol regulatory element-binding protein-1, c-Rel proteins, the Epstein Barr viral activator, ZEBRA, the steroid hormone receptors. In many cases, the interaction of HMGB1 with these “HMGB1-sensitive transcription factors” influences the activation of transcription[37,46-48,70-79].
Figure 2.
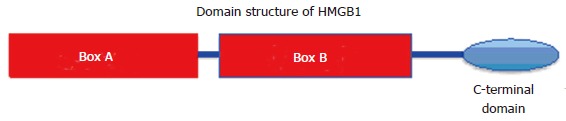
The structural and functional domains of high mobility group protein 1 protein. The tripartite structure includes the A and B boxes which are basic and the acidic C-terminal domain made up entirely of acidic (asp/glu) residues. HMGB1: High mobility group protein 1.
The C-terminal domain of HMGB1 is composed completely of about 30 acidic residues that modulates the interactions with DNA and interacts directly with TBP, the core histones and plays a role in transcriptional activation[67,68,70,80-83]. Both in vitro and in vivo evidence shows that HMGB1 binds to the histone H3. The in vitro study showed that the interaction was between the C-terminal domain of HMGB1 and the H3 tail. It was proposed that this interaction may serve to localize HMGB1 binding near the nucleosome dyad, where the HMGB1 boxes would interact with the DNA to induce further bending and untwisting of the DNA, destabilizing the nucleosome and increasing accessibility[83-85]. In addition, HMGB1 was shown to increase the kinetics of the ATP-dependent CRC, ACF/CHRAC, which facilitates nucleosome sliding. The acidic C-terminal domain of HMGB1 was shown to be required for this in vitro activity[86].
Numerous reports find that the binding of histone H1 and HMGB1 compete in vitro with one another at the entry point of the nucleosome or at linker DNA. Both proteins influence nucleosome dynamics, but in opposite ways. Histone H1 decreases nucleosome accessibility, while HMGB1 decrease compactness and is thought to increase accessibility[70,80,87].
The most detailed model for the steps in estrogen-responsive transcriptional activation that clearly implicates a role for HMGB1 comes from a study that focused on the pS2 promoter in MCF-7 cell. Initial ERE binding by the estrogen-bound ER leads to the recruitment of a TopoIIβ/PARP-1 complex [PARP-1 = poly(adenosine diphosphate-ribose) polymerase-1] to the promoter, which produces a transient, site specific ds DNA cleavage by TopoIIβ. This results in activation of PARP-1 enzymatic activity, which drives the binding of HMGB1/2, with the concomitant release of the previously bound histone H1 and a resulting change in the local chromatin conformation. Importantly, HMGB1 has been identified as an essential component in the transcriptional activation at the pS2 promoter. Further experiments suggested that this mechanism for transcriptional activation may well apply more generally and certainly to other nuclear hormone receptors, including the androgen receptor (AR), retinoic acid receptor (RAR) and thyroid receptor (T3R)[88,89].
Interestingly, for the first time, histone H1 and HMGB1 were found in the novel situation of simultaneously residing on the tumor necrosis factor alpha (TNF-α) promoter to produce endotoxin-mediated silencing of proinflammatory TNF-α activity[90].
Collectively, it is clear that in eukaryotic transcription, HMGB1 interacts with TBP and competes with TFIIA and TFIIB in the PIC at the promoter, enhances regulatory factors binding at their response elements, increases CRC (ACF/CHRAC) activity and stimulates transcriptional activation[36,46-48,67,68,70-83,88-92].
CURRENT FINDINGS: ER BINDING TO RESPONSE ELEMENTS IN DNA
ER binding to the cERE in DNA exhibited a Kd about 10 nmol/L, while the Kd for ER binding in the presence of 400 nmol/L HMGB1 was about 4 nmol/L, increasing ER binding by approximately 2-3-fold. On the other hand, ER does not bind to a single half-site (cHERE) of cERE. However, the presence of HMGB1 facilitates strong ER binding to a single half-site, with ER essentially having the same binding affinity as observed when ER binds to cERE in the absence of HMGB1. In addition, ER binds to a spectrum of noncanonical binding sites including direct repeats, everted repeats and inverted repeats of the cHERE with a variety of spacer DNA between the half-sites. The presence of HMGB1 further enhances ER binding[46,47].
ER BINDING AND TRANSCRIPTIONAL REPORTER ASSAYS FOR ERES WITHIN U2OS CELLS
It was of interest to evaluate the extent to which cERE and these in vitro noncanonical ER binding sites could actually serve as functional EREs and drive transcription within a cell. Using a subset of these binding sites described above - canonical cHERE and cERE, multiple tangent cHEREs and cEREs, direct repeats and everted repeats of the cHERE - a transient transfection assay involving a luciferase reporter assay was used to determine if they could drive transcriptional activity and the effect of exogenous HMGB1 had on the level of expression. The results indicated that ERE half-sites in multiple arrangements do drive luciferase expression and that HMGB1 enhances the level of expression. With the use of siRNA strategies, HMGB1 was shown to play an essential role in enhancing the level of transcription[48].
These results led to the focal question of this minireview. Is the effect of HMGB1 activity associated with restructuring of the nucleosome to facilitate ER/cERE binding[93]?
ER DOES NOT BIND TO THE OPTIMALLY OREINTED CERE IN NUCLEOSOMAL DNA
Figure 3 shows the schematic diagram for the 161 bp DNA used in the binding studies. Although the DNA binding site in a nucleosome could potentially take on many different orientations, the DNA was engineered to define the cERE in a specific rotational and translational position within a nucleosome for optimum binding access. The nucleosome is defined as translationally positioned if the nucleosome occupies a fixed position relative to the underlying DNA sequence. The DNA is rotationally phased as well when an orientation of a DNA sequence in the (DNA) helix is fixed relative to the histone octamer surface. The cERE resides in the center of the DNA and will be positioned at the dyad axis (red in Figure 3) within the nucleosome. With the incorporation of four nucleosome-positioning sequences, two on each side of the cERE (yellow in Figure 3), the cERE is rotationally phased so that the two binding sites, the major grooves in the cERE, for each of the monomers in the ER dimer, are sterically unobstructed and positioned outward from the histone octamer for optimum binding. The same DNA construct, with the replacement of the cERE with a cGRE (consensus glucocorticoid response element) and the designed nucleosome were previously used in analogous studies to investigate glucocorticoid receptors (GR) binding to cGRE. Using the same construct permits a direct comparison of the binding of ER/cERE with GR/cGRE under the same nucleosomal constraints[49].
Figure 3.
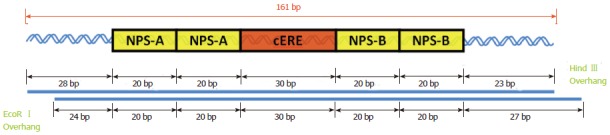
A schematic drawing of the 161 bp DNA containing four nucleosome positioning sequences (yellow boxes), that straddle the 30 bp consensus estrogen response element (red box) at the dyad axis. The 161 bp DNA was excised by EcoRI and HindIII digestion of the pGEM-Q2-2E2 plasmid. cERE: Consensus estrogen response element.
The initial EMSA binding studies showed that, with this most ideal orientation of cERE, ER exhibited no detectable binding to the cERE up to about 80 nmol/L ER, with less than 20% binding up to about 200 nmol/L ER (Kd estimated about 300 nmol/L)[94]. This is in contrast to a Kd about 10 nmol/L for ER binding to DNA[46]. Therefore, although the cERE was rotationally phased for optimum binding at the dyad in the nucleosome, the cERE was inaccessible to ER and it binding affinity in nucleosomal DNA was reduced by about 30-fold from that on DNA[51].
PRELIMINARY FINDINGS: HMGB1 INFLUENCES ER BINDING TO CERE IN NUCLEOSOMAL DNA
ER binding was then tested with the nucleosome incubated at a number of HMGB1 levels (400, 800 and 1600 nmol/L). In each case, the EMSA showed that ER bound, with the EMSA band becoming increasing broad as the levels of HMGB1 was increased. This prompted us to explore the effect of HMGB1 on the character of the nucleosome under more exacting conditions.
CHARACTERIZATION OF THE HMGB1 RESTRUCTURED NUCLEOSOMES (N’ and N’’)
The canonical nucleosome was treated with 1600 nmol/L HMGB1 and then the nucleosomes fractionated on a 5%-20% sucrose gradient. The HMGB1-treated nucleosomes sedimented as a single well-defined band, in the same fraction as previously found for the (untreated) canonical nucleosomes. However, the EMSA of the contents of this band revealed two discrete bands that both had electrophoretic mobilities distinctly different (lower mobility) than that for the canonical nucleosome. The two nucleosome states, referred to as N’ and N’’, which were at comparable levels, were not further separated, so that experiments done on the HMGB1-restructured nucleosomes refer to these species. The EMSA results clearly indicated that HMGB1 treatment changed, or restructured, the nature of the canonical nucleosome to convert it into two stable, physically distinct non-canonical nucleosome conformers or structures.
Although the canonical nucleosomes were treated with 1600 nmol/L HMGB1, the sedimentation would be expected to reduce the level of HMGB1 in the isolated fraction. The determination of the level of HMGB1 in the (N’/N’’) fraction indicated HMGB1 remained with these nucleosomes, but at a greatly reduced level - the level being approximately 2 HMGB1/nucleosome.
The presence of the four core histones in the restructured nucleosomes was verified by a supershift of the nucleosome EMSA band by each of the individual histone antibodies. However, antibodies to HMGB1 did not produce a corresponding supershift, indicating that HMGB1 was not a stable component of N’/N’’. This is consistent with the finding that HMGB1 interactions are transient and HMGB1 functions generally by a “hit-and-run” mechanism on DNA and isolated nucleosomes[46].
An inherent characteristic of canonical nucleosomes is a DNase I cutting pattern, in which the DNase I cuts in the exposed minor groove, having a periodicity about every 10 bp. This periodicity is disrupted for nucleosome remodeling by the ATP-dependent human SWI/SNF complex, presumably as a result of translocation of the DNA and altered rotational phasing, with the resulting pattern tending to look more like that of DNA[17,95]. In contrast, the DNase I cutting pattern for N’/N’’ exhibited the same 10 bp periodicity as observed on the canonical nucleosome, with the additional of a few extra weak bands. This suggested that the general nature of the DNA wrapping around the histone octamer was not changed significantly in the HMGB1-restructured nucleosomes and that the restructuring by HMGB1 was distinctly different than for the hSWI/SNF complex. The Exonuclease III (Exo III) digestion pattern was also essentially the same for N and N’/N’’, indicating that there was no translocation of the DNA as a result of the restructuring. In addition, the invariance of the Exo III pattern indicates that the presence of HMGB1, which has been suggested to bind at the linker DNA region, did not alter the accessibility of the ends of the DNA to this exonuclease.
To determine if the HMGB1 facilitated access to the isolated nucleosomal DNA, ER was reacted with N’/N’’. ER bound strongly, with a Kd about 50 nmol/L and the DNase I footprint verified that ER bound to the cERE in a sequence-specific manner. This Kd value is 6-fold stronger than the estimated Kd value (about 300 nmol/L) for the canonical nucleosome and at about the same value as that for the canonical nucleosomes in the presence of 400 nmol/L HMGB1. In addition, ER binds to N’/N’’ only five times weaker than on naked DNA. HMGB1 has no known enzymatic activity and the nucleosome restructuring activity was unchanged by the presence of ATP. Therefore, the effect of HMGB1 on the nucleosome is nonenzymatic, with the activity being independent of an ATPase activity and so must operate by an different mechanism than CRCs that require ATP hydrolysis to drive nucleosome remodeling. These findings led us to characterize any differences between the restructured nucleosomes (N’, N’’) and the canonical nucleosomes (N) and how these differences might support possible mechanisms by which HMGB restructures the nucleosomes to facilitate ER access to, and binding to cERE.
Previous studies that used atomic force microscopy (AFM) to characterize the mononucleosomes that were remodeled by the SWI/SNF complex showed that the mononucleosomes were converted into altered dimers or a dinucleosome unit[95,96]. To determine if similar dinucleosomes were produced as a result of HMGB1 restructuring on mononucleosomes, analysis of the N’/N’’ fraction by AFM showed only mononucleosomes, with no evidence of dinucleosomes. This further indicates that the HMGB1-restructured nucleosomes are distinctly different than canonical nucleosomes remodeled by hSWI/SNF.
One of the key characteristics of the HMGB1-restructured nucleosomes (N’/N’’) is that they are uniquely stable for extended periods of time at -20 °C - for at least a few months. This is a unique and extremely important property and, as far as I know, is unlike the relative instability (about 30 min or less) for remodeled nucleosomes in which times of stability were reported[97-99].
Nucleosome stability as a function of temperature was examined first. It was shown that the canonical nucleosomes remained stable overnight at 37 °C. Although the N’/N’’ nucleosomes were stable over this same time period at 4 °C, on incubation at 37 °C, the N’’ form was unstable and was converted into a mixture of N’ and N. The N’ form remained stable in an overnight incubation. Therefore the relative thermal stability of the nucleosome states is N about N’ > N’’, with the restructured N’ having a thermal stability comparable to that of the canonical nucleosome.
The influence of electrostatic interactions on stability was tested by challenging the N’’/N’ states with increasing levels of NaCl. At low levels of NaCl, about 0.25 mmol/L, N’’ was unstable, converting into a mixture of N’/N, with increasing NaCl levels leading to the complete conversion of the N’ state into the canonical nucleosome at about 200 mmol/L. These data indicate that there is a differential stability in N’ and N’’, again with N’ being more stable than the N’’ state. This may occur as a result of the restructured nucleosome states having electrostatic interactions that are much more sensitive to NaCl than is the canonical nucleosome and/or that increasing levels of NaCl may weaken, or effectively eliminate HMGB1 interactions with the nucleosomes.
When challenged with low levels of competitor DNA, N‘/N’’ converted to N, with N’’ making the transition at lower levels than did N’. Finally, above about a few hundred nmol/L DNA, only the canonical nucleosome was stable. These findings suggest that the competitor DNA acted as a “HMGB1-sink”, interfering with its interaction with the nucleosomes and leading the conversion to the canonical state. Importantly (see later), all three nucleosome states were found to be stable simultaneously and present in equilibrium from about 25-50 mmol/L NaCl and about 12-50 nmol/L DNA.
It should be noted that in all cases, there was no disruption of the nucleosome as would have been indicated by the presence of a DNA band. We also noted that the supershift experiment indicated that all four different core histones were in the stable complex. The fact that the restructured nucleosomes reverted faithfully to the canonical state also indicates that the stoichiometry of the histones remained the same in all nucleosome states and that the “core” nucleosome forces remained intact and exhibited no significant irreversible disruption.
ER BINDS STRONGLY TO TAILLESS NUCLEOSOMES IN THE ABSENCE OF HMGB1
There were a number of findings that led us to investigate the possible role of the histone tails on ER accessibility to the cERE and how the tails might impact the activity of HMGB1. It was observed that the HMGB1-restructured nucleosomes did not disrupt the DNase I cutting pattern that occurs every 10 bp. Similar to our finding, this same DNase I cutting pattern was also retained on nucleosomes that were made “tailless” by trypsin cleavage of the tails of the core histones. Likewise, the presence of the histone tails plays an important role in their thermal stability[100,101]. The basic residues in the tails are specific targets for post-translational modification, including reversible acetylation and phosphorylation that play key roles in regulatory aspects of transcription. For example, acetylation of the tail domains is important in recruiting coactivators to the nucleosome and influences the accessibility and transcriptional activity of nucleosomes[102-104].
The interaction of the acidic C-terminal domain of HMGB1 has likewise been proposed to interact with the histone tails and therefore possibly provide an additional mechanism to reduce their electrostatic interaction with the DNA[84,85]. As a result, tailless nucleosomes, in the absence of HMGB1, were prepared to determine their influence on ER binding affinity. ER binds strongly to the tailless nucleosomes, with a Kd value of about 45 nmol/L (Table 1). This Kd value is a comparable binding affinity for that reported for both (1) the canonical (tailed) nucleosomes in the presence of 400 nmol/L HMGB1 and with (2) N’/N’’, and in marked contrast to the lack of ER binding to the canonical nucleosomes. This strongly supports the notion that the major effect of the HMGB1 interactions with the nucleosome is to disrupt one or more of the histone tail interactions with the nucleosomal DNA. The Kd value for the tailless nucleosome in 400 nmol/L HMGB1 or the (sucrose gradient) isolated restructured tailless nucleosome was about 30 nmol/L. This suggests that the effect of HMGB1 may extend somewhat beyond disrupting the DNA-tail interactions and is consistent with weakening the DNA-core histone interactions as well. Table 1 summarizes the Kd values for the various nucleosome states.
Table 1.
Estrogen receptor binding affinity to consensus estrogen response element
| Target | Kd (nmol/L) |
| N | Approximately 300 |
| N/400 nmol/L HMGB1 | 52 |
| (N’, N’’)/10 nmol/L HMGB1 | 50 |
| N tailless | 45 |
| N Tailless/400 nmol/L HMGB1 | Approximately 30 |
| Restructured tailless N | Approximately 30 |
| DNA | 10 |
HMGB1: High mobility group protein 1.
DISCUSSION
ER does not bind to a rotationally phased and translationally positioned cERE centered at the dyad axis in the canonical nucleosome. The cERE is rotationally phased such that the major groove of the cERE faces outward from the surface of the histone octamer to facilitate the most optimum orientation for the ER dimer to bind in nucleosomal DNA. A combination of experimental strategies collectively demonstrates that HMGB1 restructures the canonical nucleosome and that ER binds strongly to the HMGB1-restructured nucleosome states. This supports the contention that by HMGB1 changing the forces within the nucleosome, it drives the dynamic change to restructure a novel state of the nucleosome that is linked to the functional significance of ER binding.
SIMILARITIES OF THE CANONICAL AND HMGB1-RESTRUCTURED NUCLEOSOMES
The HMGB1-restructured nucleosomes (N’, N’’), in many ways, are not unlike the canonical nucleosome (N). In all cases, the different nucleosome states exhibit the same hydrodynamic property, have the same stoichiometry of core histones and sediment in the same fraction in a sucrose gradient. This is not unexpected since the core histone tails have no significant effect on the frictional coefficient and the hydrodynamic properties of the core particle[101].
The lack of any significant alteration in the characteristic DNase I 10 bp pattern suggests that no major changes in the DNA interactions with the inner histone octamer occurs and that the wrapping of the DNA around the histone octamer (rotational phasing) experiences little or no change as a result of the presence of HMGB1. The fact that cleaving off the histone tail domains with trypsin also does not affect the DNase I 10 bp pattern in a “tailless” nucleosome and that ER binds strongly to the “tailless” nucleosomes in the absence of HMGB1 supports the view that HMGB1 interactions interfere with the tail-DNA interactions and have a significant contribution to the mechanism by which HMGB1 alters the character of the nucleosome structure and enhances ER binding affinity. The further increase in the ER binding affinity upon addition of HMGB1 reinforces the interpretation that HMGB1 most likely does weaken, to some extent, the DNA-histone interactions within the “core” nucleosome and goes beyond just disrupting the interactions between DNA and the histone tails. The Exo III digestion pattern is likewise not changed in the HMGB1-restructured nucleosomes. This indicates that the HMGB1 interactions do not obstruct Exo III from the DNA ends or impede or enhance Exo III activity. Furthermore, it would indicate that the activity of HMGB1 does not produce any nucleosome translocation. This is not unanticipated since HMGB1 exhibits no ATPase activity, in contrast to the ATP-dependent CRCs, which utilize ATPase activity to power translocation[105].
The supershift data indicate that all the core histones remain in the restructured nucleosomes, while HMGB1 is not a stable component. The latter finding is consistent with previous findings that HMGB1 exhibits a transient “hit-and-run” character (important exceptions discussed later) on DNA and isolated nucleosomes.
DIFFERENCES BETWEEN THE CANONICAL AND HMGB1-RESTRUCTURED NUCLEOSOMES
Although all three states of the nucleosome (N, N’ and N’’) sediment identically in a sucrose gradient, each nucleosome state exhibits a distinctly different electrophoretic mobility in EMSA, with the order of the mobilities being N > N’ > N’’.
However, the most striking effect of the nucleosome restructuring is that ER binds strongly to the (N’, N’’) states. The Kd value for ER binding at the cERE when positioned at the dyad axis is about 50 nmol/L, which is about 5 times weaker that on DNA, yet about 6 times stronger than the corresponding binding on the canonical nucleosome, with an estimated Kd value about 300 nmol/L. The 6-fold increase in the ER binding affinity by HMGB1 on nucleosomal DNA is about three times the effect that HMGB1 has on ER binding to DNA[46].
It should be noted that the Kd value for ER binding at cERE appears to be independent of its location in the nucleosomal DNA. Although unpublished, the Kd values for ER binding at -20 bps and -40 bps from the nucleosome dyad axis was essentially the same as found at the dyad axis, Kd about 50 nmol/L. ER binding to a similar extent at distant and separate locations in the nucleosome raises questions about whether the binding of ER to the HMGB1-restructured nucleosomes are in accord with the “site exposure model”[106-108].
This model postulates that the ends of the DNA are in a rapid dynamic equilibrium between being histone bound (fully wrapped) and unbound (partially unwrapped) and that a binding factor can gain access to and bind stably to its binding site in the unbound state. The DNA is effectively “lifted off” the nucleosome, with the “lifting” of sequences nearer the ends being energetically less costly to dissociate from the histone octamer and therefore have a greater opportunity to bind factors than those sequences near or at the dyad. The experimental basis for the model comes from a kinetic analysis of restriction endonuclease cleavage on DNA that showed that the cleavage sites at the dyad of nucleosomal DNA were protected to a far greater extent than those near the ends[106-108].
If the energetics needed to disrupt histone-DNA interactions to “expose” the cERE at the dyad, which is up to 80 bps from the DNA ends of DNA, can be considered significantly greater than to disrupt decidedly fewer interactions up to a cERE 20 bps or 40 bps from the dyad, different levels of access for the sites could reasonably be expected and therefore different Kd values for ER binding at these distant sites would be expected. However, we find that ER binds with essentially the same affinity to these well-separated locations in the HMGB1-restructured nucleosome. This indicates that HMGB1 may efficiently facilitate global instability of the forces in the nucleosome. In this case, ER binding occurs independent of an enhancement from a “site exposure” unraveling of the DNA from the ends of the nucleosome.
The stabilities of the restructured nucleosomes (N’, N’’) are distinctly different than the canonical nucleosome (N). The canonical nucleosome, N, is unaffected by temperatures up to 37 °C and by the NaCl and DNA levels that we investigated. On the other hand, N’ and N’’ are stable at 4 °C, but at 37 °C, convert to N in about 1 h. This would suggest that the HMGB1 interactions, perhaps their lifetime bound to the nucleosome, is decreased with increasing temperature and therefore its effect is eliminated. Increasing levels of NaCl differentially convert N’ and N’’ (the more unstable form) into the canonical form, with complete conversion of both states at about 200 mmol/L NaCl. This supports the view that the electrostatic forces in all three nucleosome states are different. This stability characteristic of the HMGB1-restructured nucleosomes, N’/N’’, is similar to that observed for the altered nucleosome produced by the CRC, RSC, which also decreases with increasing NaCl levels[109]. Interestingly, NMR evidence suggests that the N-terminal domains of H3 and H4 appear to remain tightly bound to the DNA in the core particle up to about 0.3 mol/L NaCl[110]. This is about a NaCl level that is about 50% greater than that which converts the N’, N’’ states to the canonical nucleosome. This is in line with the notion that the binding interactions of the histone tails of H3 and H4 with DNA in nucleosomes is important for its integrity and that these interactions may be weaker in the restructured nucleosome states that in the canonical nucleosome. The increasing levels of DNA would compete for the HMGB1 and eliminate its effect on N’, N’’, consequently leading to the conversion to the canonical form. Even though the restructured nucleosomes are unstable to challenges by increasing temperature, NaCl and DNA, while the canonical nucleosomes are not, the restructured nucleosomes are stable at -20 °C for extended periods of time - being several months - in our hands.
ER BINDING TO TAILLESS CANONICAL NUCLEOSOMES WITHOUT THE AID OF HMGB1
Removal of the core histone tails from the canonical nucleosome eliminates about 30% of the histone proteins, many of the positive charged residues and virtually all of the tail segments that extend out from the nucleosome and can interact with the DNA backbone and facilitate higher-order chromatin structure. The finding that the Kd value for ER binding was greatly reduced clearly indicates that the presence of the positively charged tails in the canonical nucleosomes does inhibit ER access to the cERE, presumably by either their preferred or stochastic interaction with phosphates in the DNA backbone. The finding that the Kd value was virtually the same as observed on the HMGB1-restructured nucleosomes is consistent with an HMGB1 effect that is derived, in large part, from continuously and transiently disrupting the DNA-tail interactions to increase access to the cERE.
ENERGY LANDSCAPE MODEL: EFFECT OF HMGB1 ON DYNAMICS OF NUCLEOSOME STRUCTURE
The interaction of HMGB1 protein with canonical nucleosomes (N) alters or destabilizes their structural characteristics and facilitates ER access to its ERE in the restructured conformers (N’ and N’’). Challenges to these nucleosome conformational isomers by increasing NaCl or DNA levels reveal all three conformers (N, N’ and N’’) are observed simultaneously in a dynamic equilibrium. A model proposed to explain the conformational changes in protein structure as a result of ligand binding - the preequilibrium/conformational selection model - can be extended to the nucleosome findings[111,112]. In this paradigm, multiple states of the nucleosomes can be considered to preexist in a statistical ensemble of conformers on the nucleosome energy landscape, with the population of the three states sensitive to the immediate microenvironment. The interaction of HMGB1 with the N conformation can be regarded as a “ligand driven” conformational selection of states that drives a population shift in the equilibrium ensemble. In the case of interest, ER binding can further influence the direction and magnitude of the population shift.
Figure 4A displays a limited representation of just two hypothetical three-dimensional energy landscapes for the equilibrium between the three conformational states - the canonical state, N, and the HMGB1-restructured states, N’ and N’’. Using the conventional preparation/isolation conditions for nucleosomes, the canonical nucleosome, N, is the predominant species (energy landscape I) with N’ and N’’ kinetically trapped and at levels that are undetectable by EMSA. The presence and interaction of HMGB1 with the nucleosome drives a population shift as the interaction perturbs the distribution of states, and converts energy landscape I into landscape II. The new landscape now shows the population shift in which HMGB1 interaction selectively stabilizes the N’ and N’’ states, leading to a significant increase in their population, with a concomitant decrease in the population of the canonical nucleosome. Once isolated, the canonical and the restructured nucleosomes are stable at low temperatures, but the N’ and N’’ states can be readily converted to the canonical state by “stressful” conditions, such as increased temperature or increased levels of either NaCl or DNA. Figure 4B provides an overall picture for the reaction of ER with the various nucleosome states. Pathway 1 shows that ER does not bind to the canonical nucleosome. Pathway 2 shows that reaction of 400 nmol/L HMGB1 with the nucleosomes leads to a population shift, which effectively is a restructuring of the nucleosome that facilitates strong ER binding. Pathway 3 shows the route to isolation of the N’ and N’’ states. The nucleosomes were reacted with 1600 nmol/L HMGB1 and then subjected to a sucrose gradient. The gradient fraction also contained 10 nmol/L HMGB1, which corresponds to about 2 HMGB1/nucleosome. ER binds strongly to these well-defined nucleosome states.
Figure 4.
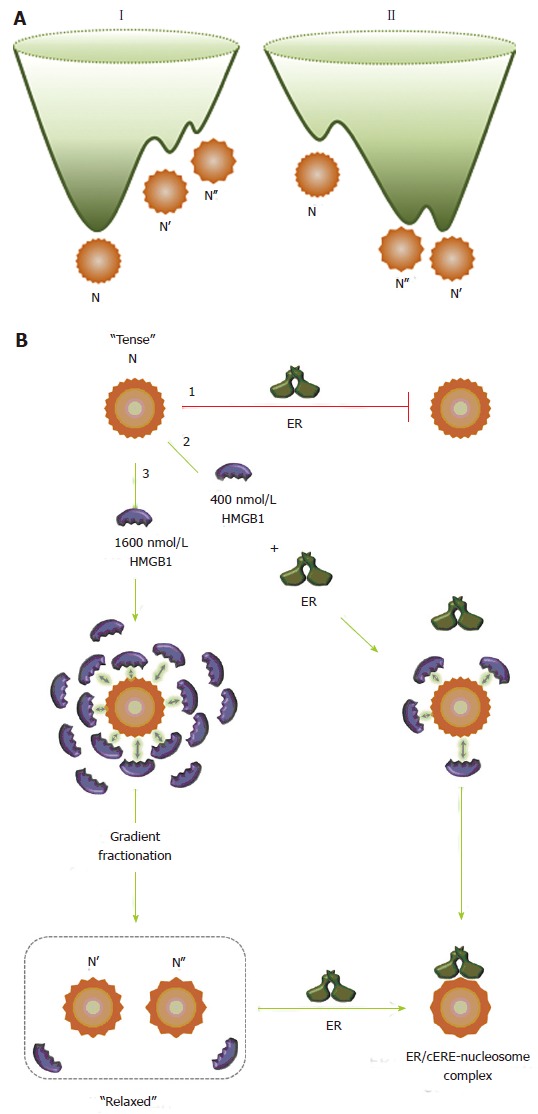
High mobility group protein 1 relaxes the canonical nucleosome structure and facilitates estrogen receptor binding. A: Energy landscapes for canonical (I) and HMGB1-restructured nucleosomes (II). A hypothetical representation for the energy landscape of the canonical nucleosome, N, and the HMGB1-restructured nucleosomes, N’ and N’’. Using conventional isolation protocols, the canonical nucleosome, N, is the predominant and thermodynamically most stable conformation. N’ and N’’ are in low abundance, higher energy conformational isomers that are kinetically trapped near the bottom of energy landscape I. HMGB1 interaction with N reduces intranucleosomal constraints, which resets the energy landscape (II), resulting in a population shift in which the N population significantly decreases and the population of the more “relaxed” and accessible N’ and N’’ states increases. The more unstable form, N’’, sets in a shallower potential well than that for N’. Although interactions with HMGB1 provide the driving force to restructure N into these states, these forms remain stable and although in equilibrium with the canonical state under many solution conditions, can revert to the canonical nucleosome on challenge with increasing concentrations of NaCl and DNA; B: Interaction of the nucleosome with HMGB1 and ER. The canonical nucleosome (N) represents a “tense” and relatively inaccessible conformational isomer (pathway 1) and ER does not bind to the canonical nucleosome state. In the presence of 400 nmol/L HMGB1 (pathway 2), due to the transient and dynamic “hit and run” interaction of HMGB1 with the nucleosome, represented by arrows, the intranucleosomal constraints are relaxed, which facilitates ER binding. ER binds to the nucleosome to form ER/cERE nucleosome complex. In the presence of 1600 nmol/L (pathway 3), the intranucleosomal constraints are relaxed due to increased “hit and run” interaction of HMGB1. After gradient fractionation, the restructured nucleosomes (N’ and N’’) are isolated and contain only low nmol/L levels of HMGB1, which maintain the more accessible and “relaxed” conformational isomers (N’ and N’’) that permit ER binding. HMGB1: High mobility group protein 1; ER: Estrogen receptor.
The interaction of HMGB1 with nucleosomes may be generally viewed in an analogous fashion to the scheme presented by Perutz et al[113] for small molecule interactions with hemoglobin. The interaction of HMGB1 with the nucleosome alters the forces within the nucleosome and drives the population shift to the nucleosome state in which the DNA sites are accessible to ER binding. The canonical nucleosome can be viewed as a highly constrained “tense” state of the nucleosome. As a result, its binding sites are inaccessible to TFs and this nucleosome state is effectively a functionally inactive state. On the other hand, HMGB1 interactions disrupt a level of constraints to restructure the nucleosome to a less constrained, “relaxed” state that is more accessible and permissive to TF binding.
The energy landscape presented here can be considered as only the first level in the ensemble of dynamic nucleosome states. In the most general paradigm, the impact of HMGB1 can be expected to be only one of a multitude of influences on the nucleosome state - including DNA methylation, post-translational modifications of the core histone tails, alternative forms of histone proteins and other factors that control the level of chromatin compaction. Each influence may be expected to exert a different effect, dependent on the collective (or pool) of factors that impinge on the ensemble of states. This collection of factors will expand the population of energy states within the ensemble and will further fine-tune chromatin structure and its functional capabilities. This suggests the presence of a very heterogeneous and dynamic nucleosome population within chromatin, with a varying, yet enormous capacity to respond to changes in environmental ques. These dynamic changes in nucleosome restructuring, driven by a variety of (external) forces, is reminiscent of the simple manipulation and controlled reshaping of a toy “stress ball” (by the pressure from your fingers). As a result, we refer to this dynamic nucleosome model, with its diverse forces and binding interactions affecting the changing aspects of (structural and functional) the nucleosome states, as the “stress ball” model.
THE COOPERATIVE OR CONFLICTING ROLES OF HMGB1 AND HISTONE H1 PROTEINS IN TRANSCRIPTIONAL REGULATION
It is generally thought that HMGB1 and histone H1 compete for binding in, or about, the linker region of the nucleosome and that HMGB1 provides a positive influence on transcription, while histone H1 acts counter to that. Although most in vitro studies agree with both the binding competition and transcriptional regulation, this may be too rigid a picture for in vivo findings[80,87].
Ju et al[88] find HMGB1 in required for transcriptional activation at the pS2 promoter. They find that as HMGB1 enters the complex, it displaces linker histone H1, which is consistent with HMGB1 and histone H1 competing for the same or an overlapping site - presumably on the linker DNA within the nucleosome[88,89].
On the other hand, El Gazzar et al[90] present the unique finding both HMGB1 and H1 are found simultaneously in the TNF-α promoter and are required for endotoxin-mediated silencing of TNF-α promoter in THP-1 human promonocytes.
These studies bring out a challenge to our current thinking about the extent to which in vitro findings can be translated to the in vivo state, in at least two respects. First, both these studies and the report by Poser et al[54], used chromatin immunoprecipitation (ChIP) to reveal that HMGB1 was a stably bound component of the transcriptional machinery. This is unlike the transient binding characteristically found in vitro for HMGB1 interactions with one or two isolated components, such as DNA or nucleosomes. The in vivo studies suggest that HMGB1 makes multiple interactions in these larger complexes that increases their residence times so that they can be revealed by a ChIP analysis.
Second, El Gazzar et al[90] find that both HMGB1 and histone H1 reside together simultaneously at the TNF-α promoter. This indicates that HMGB1 and histone H1 are not competing for the same singular site (linker DNA) on the nucleosome and that, again, in such large promoter complexes, more extensive and different interactions can be expected.
COMPARISON OF ER, PROGESTERONE RECEPTOR AND GR BINDING TO THEIR RESPONSE ELEMENTS IN NUCLEOSOMAL DNA
ER, progesterone receptor (PR) and GR exhibit a similar modular structure, bind to palindromic response elements and bind to these response elements as homodimers. As a result, they are designated as Class I steroid hormone receptors. However, the ER findings, together with previous data on GR and (unpublished) data for PR from our lab, present a credible challenge to the generally held view that members of the steroid receptor family exhibit similar binding characteristics and bind strongly to their response elements within the nucleosomal DNA[50,114]. This led us to compare the effect of HMGB1 on their binding characteristics (Table 2). For both the GR and PR studies, the same DNA that was used in the ER studies was employed, except that the response element was changed to GRE, which both PR and GR bind to. Although PR binds strongly to the GRE in DNA, its binding profile was similar to that for ER in that it does not bind to its response element (GRE) when located at the dyad in the nucleosomal DNA. However, quite unlike the robust effect on ER binding, the presence of HMGB1 did not affect the PR binding affinity. Previous studies indicated that PR binds to the sequence in the mouse mammary tumor virus (MMTV) in a nucleosome, with its binding affinity on DNA being only about a factor of 3-5 stronger than in a nucleosome. This result was quite different than in our comparative findings and may be due to the different response elements used[51]. Again using this same DNA, it was previously shown that GR binds strongly to GRE in DNA and the binding on nucleosomal DNA was reduced by only a factor of about 3, suggesting that nucleosome only marginally inhibits GR binding and suggesting that HMGB1 would probably have only a small, if any, effect on GR binding affinity[50]. This comparative data indicate that HMGB1 exerts a very different effect on the binding affinity for these three steroid hormone receptors at the nucleosomal level. HMGB1 enhances only the binding affinity for ER. This is consistent with recent findings and the proposal that HMGB1 facilitates ER to bind by a variety of modes, which is quite unlike that of other steroid hormone receptors[46-48,93]. This is also consistent with the finding that although HMGB1 enhances GR binding to the GRE in MMTV DNA, the influence of HMGB1 on GR binding to MMTV chromatin was minimal[115].
Table 2.
The effect of high mobility group protein 1 on the binding of steroid hormone receptors to their response elements
| Kd (nmol/L) values | |||
|
Receptor |
DNA |
Nucleosome |
(Nucleosome/HMGB1) |
| ER | 10 | Approximately 300 | Approximately 50 |
| PR | Approximately 2 | Approximately 300 | Approximately 300 |
| GR | Comparable (no specific Kd values) | No data | |
HMGB1: High mobility group protein 1; ER: Estrogen receptor; PR: Progesterone receptor; GR: Glucocorticoid receptor.
Interestingly, these results also in accord with the findings that HMGB1 is an active participant in the activation of the estrogen responsive pS2 gene by ER, in addition to that for the AR, RAR and the T3R[88].
THE ABCS OF A PROPOSED MECHANISM OF ACTION FOR HMGB1 IN RESTRUCTURING THE CANONICAL NUCLEOSOME
HMGB1 has three major modular domains. The A- and the B-boxes are basic and will electrostatically bind to the highly bent nucleosomal DNA. The C-terminal domain is completely acidic and can target positively charged residues or regions in the histone proteins. We propose a model in which the A- and/or B-boxes in HMGB1 can interact in the minor groove of DNA to bend or provide a dynamic flexure in the DNA, disrupt electrostatic interactions and reduce the affinity and life-time of the core histone-DNA constraints. In contrast to the effect of proteins binding in the major groove, minor groove binders, such as HMGB1, can produce these bends and reduce the rigidity of DNA by asymmetric phosphate neutralization, insertion of protein side-chains in between the base pairs and release bound waters and counter ions. These interactions will clearly have similarities to those also reported for the action of the FACT complex on nucleosomes[40,116].
The acidic C-terminus of HMGB1 can play a role also as it can interact with the positively charged core histone tails to reduce the residence time of the tail interactions with DNA and thereby widen the window of opportunity for greater ER access to the cERE. This is in line with the finding that the removal of the core histone tails, or alternatively, the acetylation of lysine residues in the tails did not alter the DNase I 10 bp pattern and did increase the accessibility of transcription factors for their target sequences[100,117-120]. In addition, the C-terminal of HMGB1 was shown to play an essential role in stimulating transcription and that it interacted with the N-terminus of histone H3, which may specifically alter H3 interactions with DNA[82,84,85,121]. The tails are also implicated in interacting with other nucleosomes to build higher-order chromatin structure. Whether HMGB1 disruption of the histone tail contacts with DNA will have any effect on local higher-order structure will be interesting to explore.
The binding of regulatory factors, such as ER, to their response element, act to promote the recruitment of coregulatory factors that serve as “operations managers” on post-translational modification of the tails in the core histones. This permits them to communicate with the transcriptional machinery at the promoter to regulate the transcriptome for estrogen-responsive genes. However, this simple scenario leads us to confront a causality dilemma that asks that if the restructuring or remodeling factors first bind nonspecifically to permit the transcription factor access, what is the mechanism by which the factor targets the site of interest On the other hand, if the transcription factor targets an inaccessible site, how can it gain access and bind to it without the help of a remodeling factor HMGB1 protein is an unusual protein in that it is estimated that there are as many as a million copies in the cell. This being the case, it may, simply by a stochastic nature, be somewhat effective in opening up some regions of chromatin. However, it was shown that ER and HMGB1 form a complex in solution, with HMGB1 binding to the carboxyl terminal end (CTE) of ER, which is immediately adjacent to the DBD domain[44]. This is a situation in which the regulatory factor (ER) chaperones a restructuring factor (HMGB1) to the response element target to provide an effective mechanism to restructure the nucleosome for ER to bind to a more accessible site. This novel avenue would increase the level of HMGB1 at the ERE target site to initiate a restructuring of the nucleosome, while ER then binds to the more accessible site in a step-wise or concerted fashion.
It has been shown that the activity of multiple CRCs is often required to make a site accessible to a regulatory factor. In addition, different CRCs exhibit different capabilities and with the expected redundancies in this spectrum of factors, one can expect that HMGB1 may collaborate with some of these coregulatory factors at this early stage of transcriptional activation. One might imagine that many of these enormous remodeling/coregulatory complexes are really “amalgams” of essential, collaborative factors, with a composition that may vary depending on the temporal state of the cell, the availability of factors and the variety of signals that are occurring at that moment in time[33,35].
The pS2 promoter has the cERE and the TATA box on adjacent nucleosomes, T and E, respectively. This type of ER chaperoning in HMGB1 to the ERE may have consequences in its essential role in transcriptional activation at the pS2 promoter in that it may provide a driving force for ER and HMGB1 binding simultaneously and perhaps an obvious avenue to compete with and expel H1 from the nucleosome early in the activation process[52,88]. In this, perhaps, unusual alignment of the regulatory response element and the TATA box on adjacent nucleosomes in the pS2 promoter, the acidic C-terminus of HMGB1 may have competitive demands on it. It has been shown to interact strongly with the TBP, with evidence that this involves a hydrogen bonding interaction between the acidic residues in HMGB1 and the glutamine (Q) tract in TBP[71].
The binding of ER to cERE was previously shown to bend the cERE toward ER[122,123]. However, when DNA is bound in the nucleosome, the cERE is physically bent in the opposite direction, with the bend toward the histone octamer. The flexure in the DNA that is introduced by the HMGB1 interaction will increase the probability that the DNA and the cERE sequence will have a greater lifetime at or near a bending angle that would be more favorable or amenable to ER binding in its preferred direction.
CONCLUSION
HMGB1 appears to play a role in a multitude of transactions that take place on DNA within chromatin. Its divergent structural domains provide it with the opportunity to interact with DNA and a host of proteins to help mold nucleoprotein complexes into optimum structures to perform their functions. An optimally oriented cERE in a nucleosome proves to be inaccessible to ER. HMGB1 can act on the canonical nucleosome to alter the internal forces between the DNA and the core histones in a nonenzymatic manner to reveal two stable, physically distinct nucleosome conformers. This restructures the nucleosome in a way that facilitates ER access to the cERE and results in a strong binding affinity. The initial stage of characterizing the HMGB1-restructured nucleosome has led to a proposal for HMGB1 acting on an ensemble of nucleosome conformers, in which HMGB1 alters the population of nucleosome states by a “ligand driven” conformational selection process. The current level of characterization also permits us to propose mechanisms by which HMGB1 may act to restructure the nucleosome and lead to the activation of transcription.
ACKNOWLEDGMENTS
I thank Wrange O for the pGEM-Q2 plasmid, Lu P for assistance with the AFM experiment and my coworkers, Joshi SR, Sarpong YC and Peterson RC who made this work a reality.
Footnotes
Supported by The National Institutes of Health [GM054357-04].
Conflict-of-interest statement: There is no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: June 4, 2015
First decision: July 6, 2015
Article in press: March 14, 2016
P- Reviewer: Choe W, Yeligar SM S- Editor: Gong XM L- Editor: A E- Editor: Wu HL
References
- 1.Schlichting CD, Pigliucci M. Control of phenotypic plasticity via regulatory genes. Am Nat. 1993;142:366–370. doi: 10.1086/285543. [DOI] [PubMed] [Google Scholar]
- 2.Pigllucci M. How organisms respond to environmental changes: from phenotypes to molecules (and vice versa) Trends Ecol Evol. 1996;11:168–173. doi: 10.1016/0169-5347(96)10008-2. [DOI] [PubMed] [Google Scholar]
- 3.Schlichting CD, Smith H. Phenotypic plasticity: Linking molecular mechanisms with evolutionary outcomes. Evol Ecol. 2002;16:189–201. [Google Scholar]
- 4.Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502:489–498. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
- 5.Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. 2015;16:258–264. doi: 10.1038/nrm3931. [DOI] [PubMed] [Google Scholar]
- 6.Ribeiro RC, Kushner PJ, Baxter JD. The nuclear hormone receptor gene superfamily. Annu Rev Med. 1995;46:443–453. doi: 10.1146/annurev.med.46.1.443. [DOI] [PubMed] [Google Scholar]
- 7.Chen G, Editor . Estrogen receptors: Mechanisms, structure and role in disease. New York: Nova Science Publishers; 2002. pp. 1–59. [Google Scholar]
- 8.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Ström A, Treuter E, Warner M, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 9.Sanchez R, Nguyen D, Rocha W, White JH, Mader S. Diversity in the mechanisms of gene regulation by estrogen receptors. Bioessays. 2002;24:244–254. doi: 10.1002/bies.10066. [DOI] [PubMed] [Google Scholar]
- 10.Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006;41:105–178. doi: 10.1080/10409230600648736. [DOI] [PubMed] [Google Scholar]
- 11.Poss ZC, Ebmeier CC, Taatjes DJ. The Mediator complex and transcription regulation. Crit Rev Biochem Mol Biol. 2013;48:575–608. doi: 10.3109/10409238.2013.840259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malik S, Roeder RG. The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat Rev Genet. 2010;11:761–772. doi: 10.1038/nrg2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Arensbergen J, van Steensel B, Bussemaker HJ. In search of the determinants of enhancer-promoter interaction specificity. Trends Cell Biol. 2014;24:695–702. doi: 10.1016/j.tcb.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 15.Luger K, Richmond TJ. DNA binding within the nucleosome core. Curr Opin Struct Biol. 1998;8:33–40. doi: 10.1016/s0959-440x(98)80007-9. [DOI] [PubMed] [Google Scholar]
- 16.Beato M, Eisfeld K. Transcription factor access to chromatin. Nucleic Acids Res. 1997;25:3559–3563. doi: 10.1093/nar/25.18.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imbalzano AN, Kwon H, Green MR, Kingston RE. Facilitated binding of TATA-binding protein to nucleosomal DNA. Nature. 1994;370:481–485. doi: 10.1038/370481a0. [DOI] [PubMed] [Google Scholar]
- 18.Wechsler DS, Papoulas O, Dang CV, Kingston RE. Differential binding of c-Myc and Max to nucleosomal DNA. Mol Cell Biol. 1994;14:4097–4107. doi: 10.1128/mcb.14.6.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyes J, Omichinski J, Clark D, Pikaart M, Felsenfeld G. Perturbation of nucleosome structure by the erythroid transcription factor GATA-1. J Mol Biol. 1998;279:529–544. doi: 10.1006/jmbi.1998.1783. [DOI] [PubMed] [Google Scholar]
- 20.Hayes JJ, Clark DJ, Wolffe AP. Histone contributions to the structure of DNA in the nucleosome. Proc Natl Acad Sci USA. 1991;88:6829–6833. doi: 10.1073/pnas.88.15.6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng C, Hayes JJ. Structures and interactions of the core histone tail domains. Biopolymers. 2003;68:539–546. doi: 10.1002/bip.10303. [DOI] [PubMed] [Google Scholar]
- 22.Pruss D, Hayes JJ, Wolffe AP. Nucleosomal anatomy--where are the histones. Bioessays. 1995;17:161–170. doi: 10.1002/bies.950170211. [DOI] [PubMed] [Google Scholar]
- 23.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 24.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 25.Carruthers LM, Hansen JC. The core histone N termini function independently of linker histones during chromatin condensation. J Biol Chem. 2000;275:37285–37290. doi: 10.1074/jbc.M006801200. [DOI] [PubMed] [Google Scholar]
- 26.Hayes JJ, Hansen JC. Nucleosomes and the chromatin fiber. Curr Opin Genet Dev. 2001;11:124–129. doi: 10.1016/s0959-437x(00)00168-4. [DOI] [PubMed] [Google Scholar]
- 27.Horn PJ, Peterson CL. Molecular biology. Chromatin higher order folding--wrapping up transcription. Science. 2002;297:1824–1827. doi: 10.1126/science.1074200. [DOI] [PubMed] [Google Scholar]
- 28.Cairns BR. The logic of chromatin architecture and remodelling at promoters. Nature. 2009;461:193–198. doi: 10.1038/nature08450. [DOI] [PubMed] [Google Scholar]
- 29.Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Curr Opin Cell Biol. 2003;15:172–183. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 30.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 31.Berger SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev. 2002;12:142–148. doi: 10.1016/s0959-437x(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 32.Zlatanova J, Bishop TC, Victor JM, Jackson V, van Holde K. The nucleosome family: dynamic and growing. Structure. 2009;17:160–171. doi: 10.1016/j.str.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 33.Morris SA, Baek S, Sung MH, John S, Wiench M, Johnson TA, Schiltz RL, Hager GL. Overlapping chromatin-remodeling systems collaborate genome wide at dynamic chromatin transitions. Nat Struct Mol Biol. 2014;21:73–81. doi: 10.1038/nsmb.2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J, Kinyamu HK, Archer TK. Changes in attitude, changes in latitude: nuclear receptors remodeling chromatin to regulate transcription. Mol Endocrinol. 2006;20:1–13. doi: 10.1210/me.2005-0192. [DOI] [PubMed] [Google Scholar]
- 35.Aalfs JD, Kingston RE. What does ‘chromatin remodeling’ mean. Trends Biochem Sci. 2000;25:548–555. doi: 10.1016/s0968-0004(00)01689-3. [DOI] [PubMed] [Google Scholar]
- 36.Fan HY, Narlikar GJ, Kingston RE. Noncovalent modification of chromatin: different remodeled products with different ATPase domains. Cold Spring Harb Symp Quant Biol. 2004;69:183–192. doi: 10.1101/sqb.2004.69.183. [DOI] [PubMed] [Google Scholar]
- 37.Luger K. Dynamic nucleosomes. Chromosome Res. 2006;14:5–16. doi: 10.1007/s10577-005-1026-1. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, Smith CL, Saha A, Grill SW, Mihardja S, Smith SB, Cairns BR, Peterson CL, Bustamante C. DNA translocation and loop formation mechanism of chromatin remodeling by SWI/SNF and RSC. Mol Cell. 2006;24:559–568. doi: 10.1016/j.molcel.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reinberg D, Sims RJ. de FACTo nucleosome dynamics. J Biol Chem. 2006;281:23297–23301. doi: 10.1074/jbc.R600007200. [DOI] [PubMed] [Google Scholar]
- 40.Formosa T. The role of FACT in making and breaking nucleosomes. Biochim Biophys Acta. 2012;1819:247–255. doi: 10.1016/j.bbagrm.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xin H, Takahata S, Blanksma M, McCullough L, Stillman DJ, Formosa T. yFACT induces global accessibility of nucleosomal DNA without H2A-H2B displacement. Mol Cell. 2009;35:365–376. doi: 10.1016/j.molcel.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rhoades AR, Ruone S, Formosa T. Structural features of nucleosomes reorganized by yeast FACT and its HMG box component, Nhp6. Mol Cell Biol. 2004;24:3907–3917. doi: 10.1128/MCB.24.9.3907-3917.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell. 2014;157:255–266. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Melvin VS, Roemer SC, Churchill ME, Edwards DP. The C-terminal extension (CTE) of the nuclear hormone receptor DNA binding domain determines interactions and functional response to the HMGB-1/-2 co-regulatory proteins. J Biol Chem. 2002;277:25115–25124. doi: 10.1074/jbc.M110400200. [DOI] [PubMed] [Google Scholar]
- 45.Melvin VS, Harrell C, Adelman JS, Kraus WL, Churchill M, Edwards DP. The role of the C-terminal extension (CTE) of the estrogen receptor alpha and beta DNA binding domain in DNA binding and interaction with HMGB. J Biol Chem. 2004;279:14763–14771. doi: 10.1074/jbc.M313335200. [DOI] [PubMed] [Google Scholar]
- 46.Das D, Peterson RC, Scovell WM. High mobility group B proteins facilitate strong estrogen receptor binding to classical and half-site estrogen response elements and relax binding selectivity. Mol Endocrinol. 2004;18:2616–2632. doi: 10.1210/me.2004-0125. [DOI] [PubMed] [Google Scholar]
- 47.El Marzouk S, Gahattamaneni R, Joshi SR, Scovell WM. The plasticity of estrogen receptor-DNA complexes: binding affinity and specificity of estrogen receptors to estrogen response element half-sites separated by variant spacers. J Steroid Biochem Mol Biol. 2008;110:186–195. doi: 10.1016/j.jsbmb.2008.03.034. [DOI] [PubMed] [Google Scholar]
- 48.Joshi SR, Ghattamaneni RB, Scovell WM. Expanding the paradigm for estrogen receptor binding and transcriptional activation. Mol Endocrinol. 2011;25:980–994. doi: 10.1210/me.2010-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Q, Wrange O. Accessibility of a glucocorticoid response element in a nucleosome depends on its rotational positioning. Mol Cell Biol. 1995;15:4375–4384. doi: 10.1128/mcb.15.8.4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Q, Wrange O. Translational positioning of a nucleosomal glucocorticoid response element modulates glucocorticoid receptor affinity. Genes Dev. 1993;7:2471–2482. doi: 10.1101/gad.7.12a.2471. [DOI] [PubMed] [Google Scholar]
- 51.Piña B, Brüggemeier U, Beato M. Nucleosome positioning modulates accessibility of regulatory proteins to the mouse mammary tumor virus promoter. Cell. 1990;60:719–731. doi: 10.1016/0092-8674(90)90087-u. [DOI] [PubMed] [Google Scholar]
- 52.Sewack GF, Hansen U. Nucleosome positioning and transcription-associated chromatin alterations on the human estrogen-responsive pS2 promoter. J Biol Chem. 1997;272:31118–31129. doi: 10.1074/jbc.272.49.31118. [DOI] [PubMed] [Google Scholar]
- 53.Parkkinen J, Raulo E, Merenmies J, Nolo R, Kajander EO, Baumann M, Rauvala H. Amphoterin, the 30-kDa protein in a family of HMG1-type polypeptides. Enhanced expression in transformed cells, leading edge localization, and interactions with plasminogen activation. J Biol Chem. 1993;268:19726–19738. [PubMed] [Google Scholar]
- 54.Poser I, Golob M, Buettner R, Bosserhoff AK. Upregulation of HMG1 leads to melanoma inhibitory activity expression in malignant melanoma cells and contributes to their malignancy phenotype. Mol Cell Biol. 2003;23:2991–2998. doi: 10.1128/MCB.23.8.2991-2998.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moisy D, Avilov SV, Jacob Y, Laoide BM, Ge X, Baudin F, Naffakh N, Jestin JL. HMGB1 protein binds to influenza virus nucleoprotein and promotes viral replication. J Virol. 2012;86:9122–9133. doi: 10.1128/JVI.00789-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nowak P, Barqasho B, Treutiger CJ, Harris HE, Tracey KJ, Andersson J, Sönnerborg A. HMGB1 activates replication of latent HIV-1 in a monocytic cell-line, but inhibits HIV-1 replication in primary macrophages. Cytokine. 2006;34:17–23. doi: 10.1016/j.cyto.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 57.Lange SS, Vasquez KM. HMGB1: the jack-of-all-trades protein is a master DNA repair mechanic. Mol Carcinog. 2009;48:571–580. doi: 10.1002/mc.20544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Y, Prasad R, Wilson SH. HMGB1: roles in base excision repair and related function. Biochim Biophys Acta. 2010;1799:119–130. doi: 10.1016/j.bbagrm.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Little AJ, Corbett E, Ortega F, Schatz DG. Cooperative recruitment of HMGB1 during V(D)J recombination through interactions with RAG1 and DNA. Nucleic Acids Res. 2013;41:3289–3301. doi: 10.1093/nar/gks1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ciubotaru M, Trexler AJ, Spiridon LN, Surleac MD, Rhoades E, Petrescu AJ, Schatz DG. RAG and HMGB1 create a large bend in the 23RSS in the V(D)J recombination synaptic complexes. Nucleic Acids Res. 2013;41:2437–2454. doi: 10.1093/nar/gks1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dai Y, Wong B, Yen YM, Oettinger MA, Kwon J, Johnson RC. Determinants of HMGB proteins required to promote RAG1/2-recombination signal sequence complex assembly and catalysis during V(D)J recombination. Mol Cell Biol. 2005;25:4413–4425. doi: 10.1128/MCB.25.11.4413-4425.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Andersson U, Erlandsson-Harris H, Yang H, Tracey KJ. HMGB1 as a DNA-binding cytokine. J Leukoc Biol. 2002;72:1084–1091. [PubMed] [Google Scholar]
- 63.Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, Huang J, Yu Y, Fan XG, Yan Z, et al. HMGB1 in health and disease. Mol Aspects Med. 2014;40:1–116. doi: 10.1016/j.mam.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ellerman JE, Brown CK, de Vera M, Zeh HJ, Billiar T, Rubartelli A, Lotze MT. Masquerader: high mobility group box-1 and cancer. Clin Cancer Res. 2007;13:2836–2848. doi: 10.1158/1078-0432.CCR-06-1953. [DOI] [PubMed] [Google Scholar]
- 65.Kang R, Zhang Q, Zeh HJ, Lotze MT, Tang D. HMGB1 in cancer: good, bad, or both. Clin Cancer Res. 2013;19:4046–4057. doi: 10.1158/1078-0432.CCR-13-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang H, Tracey KJ. Targeting HMGB1 in inflammation. Biochim Biophys Acta. 2010;1799:149–156. doi: 10.1016/j.bbagrm.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thomas JO, Travers AA. HMG1 and 2, and related ‘architectural’ DNA-binding proteins. Trends Biochem Sci. 2001;26:167–174. doi: 10.1016/s0968-0004(01)01801-1. [DOI] [PubMed] [Google Scholar]
- 68.Ross ED, Hardwidge PR, Maher LJ. HMG proteins and DNA flexibility in transcription activation. Mol Cell Biol. 2001;21:6598–6605. doi: 10.1128/MCB.21.19.6598-6605.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bustin M. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol Cell Biol. 1999;19:5237–5246. doi: 10.1128/mcb.19.8.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta. 2010;1799:101–113. doi: 10.1016/j.bbagrm.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 71.Das D, Scovell WM. The binding interaction of HMG-1 with the TATA-binding protein/TATA complex. J Biol Chem. 2001;276:32597–32605. doi: 10.1074/jbc.M011792200. [DOI] [PubMed] [Google Scholar]
- 72.Butteroni C, De Felici M, Schöler HR, Pesce M. Phage display screening reveals an association between germline-specific transcription factor Oct-4 and multiple cellular proteins. J Mol Biol. 2000;304:529–540. doi: 10.1006/jmbi.2000.4238. [DOI] [PubMed] [Google Scholar]
- 73.Jayaraman L, Moorthy NC, Murthy KG, Manley JL, Bustin M, Prives C. High mobility group protein-1 (HMG-1) is a unique activator of p53. Genes Dev. 1998;12:462–472. doi: 10.1101/gad.12.4.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Melvin VS, Edwards DP. Coregulatory proteins in steroid hormone receptor action: the role of chromatin high mobility group proteins HMG-1 and -2. Steroids. 1999;64:576–586. doi: 10.1016/s0039-128x(99)00036-7. [DOI] [PubMed] [Google Scholar]
- 75.Najima Y, Yahagi N, Takeuchi Y, Matsuzaka T, Sekiya M, Nakagawa Y, Amemiya-Kudo M, Okazaki H, Okazaki S, Tamura Y, et al. High mobility group protein-B1 interacts with sterol regulatory element-binding proteins to enhance their DNA binding. J Biol Chem. 2005;280:27523–27532. doi: 10.1074/jbc.m414549200. [DOI] [PubMed] [Google Scholar]
- 76.Oñate SA, Prendergast P, Wagner JP, Nissen M, Reeves R, Pettijohn DE, Edwards DP. The DNA-bending protein HMG-1 enhances progesterone receptor binding to its target DNA sequences. Mol Cell Biol. 1994;14:3376–3391. doi: 10.1128/mcb.14.5.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zappavigna V, Falciola L, Helmer-Citterich M, Mavilio F, Bianchi ME. HMG1 interacts with HOX proteins and enhances their DNA binding and transcriptional activation. EMBO J. 1996;15:4981–4991. [PMC free article] [PubMed] [Google Scholar]
- 78.Agresti A, Lupo R, Bianchi ME, Müller S. HMGB1 interacts differentially with members of the Rel family of transcription factors. Biochem Biophys Res Commun. 2003;302:421–426. doi: 10.1016/s0006-291x(03)00184-0. [DOI] [PubMed] [Google Scholar]
- 79.Yu M, Wang J, Li W, Yuan YZ, Li CY, Qian XH, Xu WX, Zhan YQ, Yang XM. Proteomic screen defines the hepatocyte nuclear factor 1alpha-binding partners and identifies HMGB1 as a new cofactor of HNF1alpha. Nucleic Acids Res. 2008;36:1209–1219. doi: 10.1093/nar/gkm1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Catez F, Yang H, Tracey KJ, Reeves R, Misteli T, Bustin M. Network of dynamic interactions between histone H1 and high-mobility-group proteins in chromatin. Mol Cell Biol. 2004;24:4321–4328. doi: 10.1128/MCB.24.10.4321-4328.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee KB, Thomas JO. The effect of the acidic tail on the DNA-binding properties of the HMG1,2 class of proteins: insights from tail switching and tail removal. J Mol Biol. 2000;304:135–149. doi: 10.1006/jmbi.2000.4206. [DOI] [PubMed] [Google Scholar]
- 82.Ueda T, Chou H, Kawase T, Shirakawa H, Yoshida M. Acidic C-tail of HMGB1 is required for its target binding to nucleosome linker DNA and transcription stimulation. Biochemistry. 2004;43:9901–9908. doi: 10.1021/bi035975l. [DOI] [PubMed] [Google Scholar]
- 83.Kawase T, Sato K, Ueda T, Yoshida M. Distinct domains in HMGB1 are involved in specific intramolecular and nucleosomal interactions. Biochemistry. 2008;47:13991–13996. doi: 10.1021/bi8013449. [DOI] [PubMed] [Google Scholar]
- 84.Heo K, Kim B, Kim K, Choi J, Kim H, Zhan Y, Ranish JA, An W. Isolation and characterization of proteins associated with histone H3 tails in vivo. J Biol Chem. 2007;282:15476–15483. doi: 10.1074/jbc.M610270200. [DOI] [PubMed] [Google Scholar]
- 85.Watson M, Stott K, Fischl H, Cato L, Thomas JO. Characterization of the interaction between HMGB1 and H3-a possible means of positioning HMGB1 in chromatin. Nucleic Acids Res. 2014;42:848–859. doi: 10.1093/nar/gkt950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bonaldi T, Längst G, Strohner R, Becker PB, Bianchi ME. The DNA chaperone HMGB1 facilitates ACF/CHRAC-dependent nucleosome sliding. EMBO J. 2002;21:6865–6873. doi: 10.1093/emboj/cdf692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ragab A, Travers A. HMG-D and histone H1 alter the local accessibility of nucleosomal DNA. Nucleic Acids Res. 2003;31:7083–7089. doi: 10.1093/nar/gkg923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–1802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- 89.Haince JF, Rouleau M, Poirier GG. Transcription. Gene expression needs a break to unwind before carrying on. Science. 2006;312:1752–1753. doi: 10.1126/science.1129808. [DOI] [PubMed] [Google Scholar]
- 90.El Gazzar M, Yoza BK, Chen X, Garcia BA, Young NL, McCall CE. Chromatin-specific remodeling by HMGB1 and linker histone H1 silences proinflammatory genes during endotoxin tolerance. Mol Cell Biol. 2009;29:1959–1971. doi: 10.1128/MCB.01862-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lu W, Peterson R, Dasgupta A, Scovell WM. Influence of HMG-1 and adenovirus oncoprotein E1A on early stages of transcriptional preinitiation complex assembly. J Biol Chem. 2000;275:35006–35012. doi: 10.1074/jbc.M004735200. [DOI] [PubMed] [Google Scholar]
- 92.Dasgupta A, Scovell WM. TFIIA abrogates the effects of inhibition by HMGB1 but not E1A during the early stages of assembly of the transcriptional preinitiation complex. Biochim Biophys Acta. 2003;1627:101–110. doi: 10.1016/s0167-4781(03)00080-0. [DOI] [PubMed] [Google Scholar]
- 93.Scovell WM, Joshi SR. The changing paradigm: Estrogen receptor recognition on DNA and within the dynamic nature of nucleosomes. AIMS Mol Sci. 2015;2:48–63. [Google Scholar]
- 94.Joshi SR, Sarpong YC, Peterson RC, Scovell WM. Nucleosome dynamics: HMGB1 relaxes canonical nucleosome structure to facilitate estrogen receptor binding. Nucleic Acids Res. 2012;40:10161–10171. doi: 10.1093/nar/gks815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Narlikar GJ, Phelan ML, Kingston RE. Generation and interconversion of multiple distinct nucleosomal states as a mechanism for catalyzing chromatin fluidity. Mol Cell. 2001;8:1219–1230. doi: 10.1016/s1097-2765(01)00412-9. [DOI] [PubMed] [Google Scholar]
- 96.Schnitzler GR, Cheung CL, Hafner JH, Saurin AJ, Kingston RE, Lieber CM. Direct imaging of human SWI/SNF-remodeled mono- and polynucleosomes by atomic force microscopy employing carbon nanotube tips. Mol Cell Biol. 2001;21:8504–8511. doi: 10.1128/MCB.21.24.8504-8511.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Guyon JR, Narlikar GJ, Sif S, Kingston RE. Stable remodeling of tailless nucleosomes by the human SWI-SNF complex. Mol Cell Biol. 1999;19:2088–2097. doi: 10.1128/mcb.19.3.2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Imbalzano AN, Schnitzler GR, Kingston RE. Nucleosome disruption by human SWI/SNF is maintained in the absence of continued ATP hydrolysis. J Biol Chem. 1996;271:20726–20733. doi: 10.1074/jbc.271.34.20726. [DOI] [PubMed] [Google Scholar]
- 99.Côté J, Peterson CL, Workman JL. Perturbation of nucleosome core structure by the SWI/SNF complex persists after its detachment, enhancing subsequent transcription factor binding. Proc Natl Acad Sci USA. 1998;95:4947–4952. doi: 10.1073/pnas.95.9.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang Z, Zheng C, Thiriet C, Hayes JJ. The core histone N-terminal tail domains negatively regulate binding of transcription factor IIIA to a nucleosome containing a 5S RNA gene via a novel mechanism. Mol Cell Biol. 2005;25:241–249. doi: 10.1128/MCB.25.1.241-249.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ausio J, Dong F, van Holde KE. Use of selectively trypsinized nucleosome core particles to analyze the role of the histone “tails” in the stabilization of the nucleosome. J Mol Biol. 1989;206:451–463. doi: 10.1016/0022-2836(89)90493-2. [DOI] [PubMed] [Google Scholar]
- 102.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 103.Yang XJ. Lysine acetylation and the bromodomain: a new partnership for signaling. Bioessays. 2004;26:1076–1087. doi: 10.1002/bies.20104. [DOI] [PubMed] [Google Scholar]
- 104.Hamiche A, Kang JG, Dennis C, Xiao H, Wu C. Histone tails modulate nucleosome mobility and regulate ATP-dependent nucleosome sliding by NURF. Proc Natl Acad Sci USA. 2001;98:14316–14321. doi: 10.1073/pnas.251421398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cairns BR. Chromatin remodeling: insights and intrigue from single-molecule studies. Nat Struct Mol Biol. 2007;14:989–996. doi: 10.1038/nsmb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Anderson JD, Widom J. Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites. J Mol Biol. 2000;296:979–987. doi: 10.1006/jmbi.2000.3531. [DOI] [PubMed] [Google Scholar]
- 107.Anderson JD, Thåström A, Widom J. Spontaneous access of proteins to buried nucleosomal DNA target sites occurs via a mechanism that is distinct from nucleosome translocation. Mol Cell Biol. 2002;22:7147–7157. doi: 10.1128/MCB.22.20.7147-7157.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li G, Widom J. Nucleosomes facilitate their own invasion. Nat Struct Mol Biol. 2004;11:763–769. doi: 10.1038/nsmb801. [DOI] [PubMed] [Google Scholar]
- 109.Lorch Y, Cairns BR, Zhang M, Kornberg RD. Activated RSC-nucleosome complex and persistently altered form of the nucleosome. Cell. 1998;94:29–34. doi: 10.1016/s0092-8674(00)81218-0. [DOI] [PubMed] [Google Scholar]
- 110.Cary PD, Moss T, Bradbury EM. High-resolution proton-magnetic-resonance studies of chromatin core particles. Eur J Biochem. 1978;89:475–482. doi: 10.1111/j.1432-1033.1978.tb12551.x. [DOI] [PubMed] [Google Scholar]
- 111.Boehr DD, Nussinov R, Wright PE. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Csermely P, Palotai R, Nussinov R. Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem Sci. 2010;35:539–546. doi: 10.1016/j.tibs.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Perutz MF. Hemoglobin structure and respiratory transport. Sci Am. 1978;239:92–125. doi: 10.1038/scientificamerican1278-92. [DOI] [PubMed] [Google Scholar]
- 114.Pham TA, McDonnell DP, Tsai MJ, O’Malley BW. Modulation of progesterone receptor binding to progesterone response elements by positioned nucleosomes. Biochemistry. 1992;31:1570–1578. doi: 10.1021/bi00120a039. [DOI] [PubMed] [Google Scholar]
- 115.Fletcher TM, Xiao N, Mautino G, Baumann CT, Wolford R, Warren BS, Hager GL. ATP-dependent mobilization of the glucocorticoid receptor during chromatin remodeling. Mol Cell Biol. 2002;22:3255–3263. doi: 10.1128/MCB.22.10.3255-3263.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Privalov PL, Dragan AI, Crane-Robinson C. The cost of DNA bending. Trends Biochem Sci. 2009;34:464–470. doi: 10.1016/j.tibs.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 117.Vignali M, Steger DJ, Neely KE, Workman JL. Distribution of acetylated histones resulting from Gal4-VP16 recruitment of SAGA and NuA4 complexes. EMBO J. 2000;19:2629–2640. doi: 10.1093/emboj/19.11.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Polach KJ, Lowary PT, Widom J. Effects of core histone tail domains on the equilibrium constants for dynamic DNA site accessibility in nucleosomes. J Mol Biol. 2000;298:211–223. doi: 10.1006/jmbi.2000.3644. [DOI] [PubMed] [Google Scholar]
- 119.Vettese-Dadey M, Walter P, Chen H, Juan LJ, Workman JL. Role of the histone amino termini in facilitated binding of a transcription factor, GAL4-AH, to nucleosome cores. Mol Cell Biol. 1994;14:970–981. doi: 10.1128/mcb.14.2.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brower-Toland B, Wacker DA, Fulbright RM, Lis JT, Kraus WL, Wang MD. Specific contributions of histone tails and their acetylation to the mechanical stability of nucleosomes. J Mol Biol. 2005;346:135–146. doi: 10.1016/j.jmb.2004.11.056. [DOI] [PubMed] [Google Scholar]
- 121.Stros M. Binding of non-histone chromosomal protein HMG1 to histone H3 in nucleosomes detected by photochemical cross-linking. Biochem Biophys Res Commun. 1987;147:301–308. doi: 10.1016/s0006-291x(87)80121-3. [DOI] [PubMed] [Google Scholar]
- 122.Nardulli AM, Shapiro DJ. Binding of the estrogen receptor DNA-binding domain to the estrogen response element induces DNA bending. Mol Cell Biol. 1992;12:2037–2042. doi: 10.1128/mcb.12.5.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nardulli AM, Grobner C, Cotter D. Estrogen receptor-induced DNA bending: orientation of the bend and replacement of an estrogen response element with an intrinsic DNA bending sequence. Mol Endocrinol. 1995;9:1064–1076. doi: 10.1210/mend.9.8.7476980. [DOI] [PubMed] [Google Scholar]