

Abstract
The scale of tumor genomic profiling is rapidly outpacing human cognitive capacity to make clinical decisions without the aid of tools. New frameworks are needed to help researchers and clinicians process the information emerging from the explosive growth in both the number of tumor genetic variants routinely tested and the respective knowledge to interpret their clinical significance. We review the current state, limitations, and future trends in methods to support the clinical analysis and interpretation of cancer genomes. This includes the processes of genome-scale variant identification, including tools for sequence alignment, tumor–germline comparison, and molecular annotation of variants. The process of clinical interpretation of tumor variants includes classification of the effect of the variant, reporting the results to clinicians, and enabling the clinician to make a clinical decision based on the genomic information integrated with other clinical features. We describe existing knowledge bases, databases, algorithms, and tools for identification and visualization of tumor variants and their actionable subsets. With the decreasing cost of tumor gene mutation testing and the increasing number of actionable therapeutics, we expect the methods for analysis and interpretation of cancer genomes to continue to evolve to meet the needs of patient-centered clinical decision making. The science of computational cancer medicine is still in its infancy; however, there is a clear need to continue the development of knowledge bases, best practices, tools, and validation experiments for successful clinical implementation in oncology.
INTRODUCTION
As the amount of information generated by an individual patient's tumor genomic profiling increases, clinicians will no longer be able to analyze, interpret, and act on this information without tools that effectively mine the data for clinical use. This remarkable increase in scale is due to the increasing number of altered genes that may portend clinical impact, the larger number of gene variants being routinely tested in some tumor types,1–3 and more generally the explosive growth in clinical knowledge linking somatic or germline variants to predictive or prognostic information. In the future, providers across the health care system—from molecular pathologists and oncologists to genetic counselors—will be challenged to rapidly analyze and interpret the results of emerging genomic technologies for effective patient care.
The clinical workflow for analysis and interpretation of the cancer genome (Fig 1) begins with the genome testing modality, that is, the technology or platform used to generate raw genetic or molecular data (modality options are reviewed in detail elsewhere4). After the assay has been performed, the molecular pathologist identifies alterations in the tumor. Depending on the assay modality, variant identification may be done by visual inspection by a trained specialist (eg, ALK fusion detection assays5) or require the assistance of automated algorithms (eg, assays that assess the entire sequence of one or more genes6). Automated methods for variant identification consist of several processing steps, including algorithms for sequence alignment that take as input a reference sequence, algorithms for comparison of the tumor and normal genome, and molecular annotation of variants. As the scale of data rises from a few well-defined, commonly mutated alterations (hotspots) to individual whole genes to entire genomes, several factors make analysis increasingly difficult. These include artifacts related to sequencing, overall breadth of coverage across one or more genes, differences in depth of coverage at a particular site that affect the power to detect variants, and variability in the reference genome data set as it relates to an individual patient.
Fig 1.

Clinical workflow for tumor genome analysis and interpretation. The workflow begins with the genome-testing modality that may or may not have digital output. Variant identification may be done manually or with the assistance of automated algorithms, depending on the modality. Automated methods for variant identification include several processes shown at the top, including algorithms for sequence alignment that take as input a reference sequence, algorithms for comparison of the tumor and normal genome, and variant annotation. Clinical interpretation of the variant is a process that includes a determination of the size of variant effect and its interaction with other variants as well as an analysis of the strength of the evidence of the effect. These processes require the use of knowledge bases of variant–drug–disease relationships. Actionable results are reported as well as variants of unknown significance (VUS). The process culminates with a clinician using the information to make a clinical decision.
The clinical interpretation of variants identified by genomic profiling is a process that includes classifying the clinical effect of the variant and reporting the results in ways that are meaningful to practicing oncologists. The need for robust clinical interpretation requires considerations that are distinct from those of both the assay testing modality and the number of genes or variants assessed. Unlike many other clinical assays, multiplex or sequencing-based tumor testing may result in many simultaneous and significant outcomes per gene that occur in any given tumor sample. Furthermore, various genetic alterations may predict either sensitivity or resistance (intrinsic or acquired) to a particular drug. For example, in non–small-cell lung cancer, some EGFR mutations are associated with sensitivity to small-molecule epidermal growth factor receptor (EGFR) antagonists (eg, EGFR L858R mutation7), whereas others confer primary or secondary resistance to the same drugs (eg, EGFR T790M8). Furthermore, there are even more variants in EGFR (and most other cancer genes) whose clinical significance is currently unknown; that is, there exist no definitive clinical data to suggest sensitivity or resistance to EGFR-targeted therapy for tumors harboring those particular EGFR alterations.9,10 Similarly, mutations that predict responsiveness to a therapy in some contexts (eg, RAF inhibitors in BRAF[V600]-mutant melanoma) may be associated wtih entirely different clinical interpretations in others (eg, RAF inhibitors in BRAF[V600]-mutant colorectal carcinoma11). In the end, the physician must interpret the utility of tumor genomic results in the context of additional clinical, histologic, and molecular information to make an informed clinical decision.
With hundreds to thousands of tumor variants observed in the coding region of an individual's genome and thousands to millions of variants across an individual's cancer genome,12 it will not be possible for molecular pathologists and oncologists to identify and appropriately annotate the clinical significance of each variant by manually assigning an individual interpretation to the variants found in every patient. Although the computational algorithms and knowledge resources available today to assist this process are still in their infancy, they demonstrate great potential for realizing this aim and thus paving the way for widespread applications of genomic profiling in clinical oncology.
THE PROCESS OF GENOME-SCALE VARIANT IDENTIFICATION
Current clinical genomic testing typically focuses on one or two well-defined, targetable alterations (hotspots) in a small number of genes, and the binary presence or absence of single mutational events is often sufficient to guide clinical decision making. Existing research-grade algorithms for revealing biologically significant alterations within the high volume of genomics data, although powerful for discovery purposes, are not sufficient for clinical deployment. To bring genome-scale data to the clinic, the computational process for variant identification (variant calling) requires adaptations of existing tools and the development of new annotation methods to allow for effective downstream interpretation of these variants.
Sequence Alignment
Large-scale genomics platforms (eg, those that use massively parallel sequencing) produce data reflecting the raw sequence reads that emerge from the sequencing instrument used by the clinical platform. Regardless of whether that output consists of the coding regions of a subset of genes (targeted sequencing), the entire coding regions of the genome (exome), the entire genome, or the spectrum of RNA transcription across the genome (transcriptome), the first step in computational analysis is aligning the raw data to the reference human genome to better understand variations (Fig 2). Multiple tools exist for this process13–21 and can be adapted for clinical use. Critically, depending on the genomic territory probed with the sequencing modality, sequence alignment can still be a computationally intensive and therefore time-consuming process that may challenge existing clinical informatics infrastructures to ensure clinically useful data turnaround. In addition, although cross-comparative analysis of multiple aligners for research purposes has been initiated,22 determination of the most effective clinical alignment algorithm has not yet been established. Because alignment to the reference genome is the foundation for all downstream analysis, clinically focused cross-comparator studies are needed to determine the optimal alignment approach, as these may inform understanding of the sensitivity and specificity of such approaches.
Fig 2.
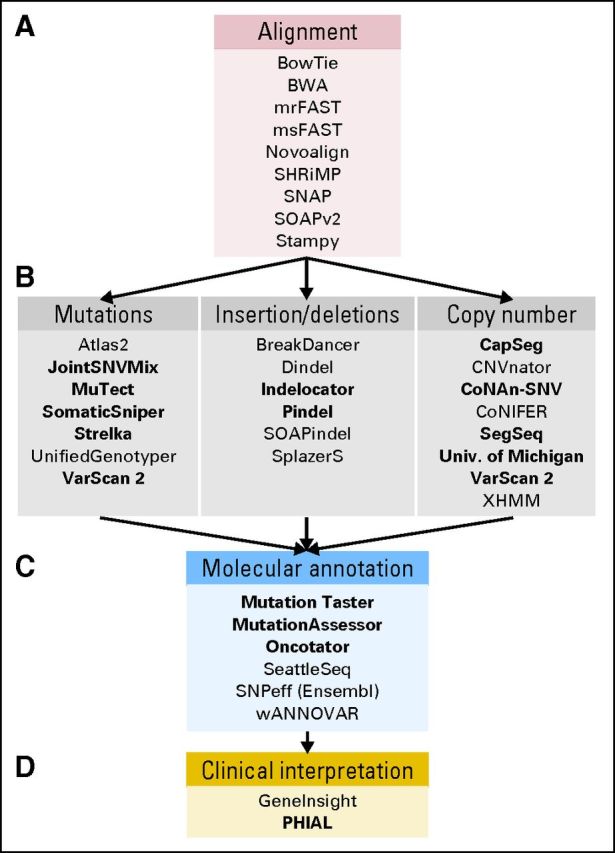
A representative set of tools for the analysis and interpretation of genome sequencing data. These include (A) a listing of representative algorithms for sequencing alignment, (B) variant identification, (C) variant annotation, and (D) clinical interpretation. Boldfaced entries are those specifically geared toward tumor versus normal analysis.
Variant Identification and Tumor/Germline Comparison
After sequence alignment, several analytic tools are available to identify variants in the tumor genome. Targeted gene panels that focus on a subset of highly characterized tumor genes may proceed directly to variant identification without analysis relative to the patient's germline data because well-defined germline events may be filtered out.6,23 However, such avenues may have difficulty in distinguishing some private or low-frequency germline alterations from bona fide somatic alterations in tumors; parallel analysis of paired germline DNA is needed to enable the maximal power to distinguish true somatic events. The variant identification algorithms developed for exome or genome-wide analyses to nominate putative mutations,24–31 small insertions/deletions (“indels”),30,32–37 and copy number alterations29,38–44) can be adapted for targeted panels (Fig 2).
As noted previously, effective variant identification for an entire tumor exome or genome always requires direct comparison of the tumor genome to the patient's germline to identify somatic variants. Many variant identification software packages for exome or genome-level data are specifically primed for use in clinical cancer care (Fig 2). However, clinical-grade sequencing may require additional quality-assurance methods to ensure that these variant identification algorithms do not accidentally filter out known, clinically significant variants. Furthermore, identifying the high-confidence absence of expected or likely tumor variants that have clinical implications (pertinent negatives) will be equally relevant for subsequent clinical interpretation and represents an essential component of clinical variant identification tasks.
Reliable detection of clinically significant rearrangements (eg, ALK fusions) can be difficult without genome or transcriptome-level data, because targeted panels or exome sequencing are unlikely to capture all intronic regions where rearrangements may occur. In the future, potential solutions may include concomitant transcriptome sequencing to capture novel fusion transcripts45,46 or intentional tiling of baits across high-yield intronic regions as part of a targeted gene panel.23 Analysis tools to reliably identify fusions in such regions exist and may be deployed for clinical use once the appropriate platform is chosen.32,47,48
Molecular Annotation of Variants
The somatic events observed with analysis tools are typically represented in computer files as genomic coordinates with allele changes or segments of copy number gain or loss. To proceed with effective clinical interpretation of these events, translation of these data for human use with effective molecular annotation is necessary. Publically available annotation tools exist to convert these data into formats that use gene names and protein changes based on established public resources.49–54 Salient sequencing data metrics may include alternative transcripts expressed from query loci, locus-specific coverage and the variant allelic fraction (defined as the number of alternate reads at the site divided by the total number of reads at that site). Additional resources may link genetic alterations to other databases that can aid downstream clinical interpretation, including the predicted effect of the variant on the protein52,55,56 or the frequency of this event in published cancer genomics research studies.57 At the present time, these annotations are typically focused on research-oriented pursuits, and new databases will be needed to frame clinically oriented molecular annotation.
THE PROCESS OF CLINICAL INTERPRETATION OF TUMOR VARIANTS
Once all tumor variants in a patient's genome have been identified, clinical interpretation of each variant is needed to identify the subset that may affect medical decision making. The process of clinical interpretation includes classification of the effect of the variant, reporting the results to clinicians, and enabling the physician to make a management decision based on the genomic information integrated with other clinical features.
Classifying the Clinical Effects of Genomic Variants
Interpretation of the clinical effect of a tumor variant includes classifying (1) the type of clinical effect, (2) the strength of evidence of the effect, and (3) the size of the effect. Framing observed variants in this manner facilitates their clinical interpretation by focusing attention on variants of different types with the most size and strength of effect for an individual patient (Fig 3).
Fig 3.

Classification of the clinical effect of the variant taking into account the type of effect, strength of the evidence, and the size of the effect. Variants in the top right quadrant should have the highest priority with respect to actionable clinical decisions.
Classification of the type of clinical effect.
As with other clinical biomarkers, cancer-related gene variants can be classified by their type of clinical effect as biomarkers that predict risk of disease, confirm a diagnosis, predict prognosis, predict response or resistance to treatment, or measure response to treatment.58 For example, deleterious germline BRCA1 mutations confer increased risk of breast cancer,59 IDH1 mutations may predict favorable prognosis in gliomas,60 BRAF mutations may predict sensitivity to BRAF inhibitors in melanoma,61 KRAS mutations may predict resistance to anti-EGFR antibody treatment,62 and NPM1 is used to measure minimal residual disease in acute myeloid leukemia.63 Some tumor variants serve multiple clinical utilities. For example, BCR-ABL translocations are diagnostic, prognostic, and predictive of response to treatment with imatinib,64 defining a distinct clinical subtype of chronic myelogenous leukemia.
Likewise, the clinical effect may be influenced by multiple variants that may be operant simultaneously. In acute myeloid leukemia, for example, the combination of certain NPM1 mutations without concurrent FLT3 mutations has been associated with complete remission and favorable outcomes.65 Similarly, multiple EGFR mutations may occur simultaneously in EGFR mutant lung adenocarcinoma that confer opposite sensitivities to EGFR tyrosine kinase inhibitors.66 Initial efforts to computationally represent these complex relationships are underway, including one approach to systematically annotate genomic relationships linked to clinical actions and build a series of heuristics to score the events relative to one another.67
Classification of the strength of evidence.
Large-scale profiles of tumor gene alterations present several challenges with respect to classifying and communicating the strength of the evidence underlying a given clinical effect. Strength of evidence is based on the maturity, quality, and type of studies that inform the potential clinical utility of a genomic alteration.68 When reporting clinical significance, it is not sufficient to simply describe the clinical effect with respect to disease and treatment. More detailed information on the evidence for those relationships is typically required for clinical decision making.
Evidence-based approaches to biomarker testing for routine clinical care limit testing to biomarkers with well-established clinical utility. Multiple frameworks have been proposed that classify the evidence related to the clinical utility of tumor biomarkers, including: (1) the Tumor Marker Utility Grading Systems,68 (2) levels of evidence for using archived tissue to determine the clinical validity of tumor markers,69 (3) levels of evidence combined with tiers to represent different clinical scenarios for a genomic event (eg, sensitivity, resistance, or prognostic/diagnostic effect),6 and (4) the National Comprehensive Cancer Network categories of evidence and consensus.70 These frameworks provide a consistent language for clinical practice guidelines to make recommendations on the use of a biomarker or biomarker-driven therapy in clinical practice. At the present time, however, relatively few tumor genes meet criteria for routine testing.70
As the cost of deep sequencing drops and the clinical utility of multiple tumor biomarkers is established, it should become increasingly cost effective to perform large-scale cancer gene mutation profiling routinely. The resulting proliferation of cancer genomic data may present specific challenges with respect to communication of the strength of evidence for the clinical effects to physicians and patients. The preceding frameworks rely on clinical evidence from relatively large populations of patients. Such recommendations are feasible with variants of relatively high prevalence; however, many tumor variants occur with a frequency of less than 1% in a particular subpopulation. For instance, more than 100 different primary and secondary EGFR mutations have been described in literature case reports for patients with non–small-cell lung cancer.57 Many of these variants occur with extremely low frequency and are of unknown clinical significance. Routine testing of the full gene sequence as opposed to testing strategies that only assess for high-prevalence variants are likely to identify more of these rare variants of unknown significance in tumor genes that have well-established clinical utility.
Thus, as genomic testing expands to large numbers of genes, there will often be no prospective clinical trials with sufficient power to determine the clinical significance of rare variants. Yet clinicians will still need assistance to interpret the clinical significance of these variants. One group describes the evolving evidence perspective leading toward patient-centered outcomes research.71 This framework describes levels of evidence ranging from expert opinion (N of 1), evidence-based medicine (N of many), comparative effectiveness research (N of many), and patient-centered outcomes research (many N′s of 1). The idea is that for variants of unknown significance, providers will make recommendations to individual patients based on as many similar patients that can be found. When such clinical data are not available, predictive modeling or preclinical data may be the only evidence of possible clinical effect. Such information may be useful to guide clinical trial selection in certain research settings, but as yet there exists no validated framework to incorporate preclinical evidence into clinical decision making regarding the use of tumor genomic data. Going forward, a deeper biologic understanding of targetable pathways in the appropriate cell or lineage contexts will likely also play a key role in identifying clinically relevant variants.
Classification of the size of the effect.
The size of the effect relates to the degree of benefit or harm a biomarker predicts.68 The clinical vernacular for classifying tumor variants based on the type and size of clinical effect is rapidly evolving. For example, “EGFR mutations sensitive to EGFR tyrosine kinase inhibitors” has become a common description in clinical trial eligibility criteria. It refers to the known subgroup of EGFR mutations that are sensitive to drugs such as erlotinib and gefitinib. Binary classifications such as “sensitive” and “resistant” are becoming more common in describing biomarker-related drug effects. Other frameworks describe the size of the clinical effect on a continuum from likely benefit versus possible harm.57 Such frameworks use phrases on a continuum of size of effect such as “treatment should be administered,” “it is reasonable to consider treatment,” “treatment may be considered,” and “treatment should not be administered.” Others have adopted “+” versus “–” classification of the size of effects such as the framework used in the well-established Sanford Guide for Antimicrobials,72 which uses “+++” to describe the relative sensitivity of an antimicrobial to treat a particular microorganism. The clinical vernacular for communicating the effect size of a tumor variant specifically with respect to treatment choice is still evolving, but it is expected that binary classifications will not capture the nuances of effect size as more data about variants within a patient's genome become available in preclinical and clinical studies.
Reporting Results
Whether testing a single gene variant or the entire cancer genome, multiple challenges remain in discerning the optimal means to report results in ways that are clinically useful for practicing oncologists. Open-ended issues for the clinical audience include (1) what to report, (2) how to format the report, and (3) if, when, and how to notify providers when new clinical evidence emerges.
Choosing what to report.
After observed variants have been comprehensively classified with clinical applicability in mind, they must be reported to the clinician in a manner that enables facile decision support. Multiple approaches to reporting exist, reflecting a spectrum of data access or restriction. Approaches favoring full access report all variants and allow clinicians complete control over what actions to take. Approaches favoring restricted access only report variants with well-established size and strength of effects. Overall, the challenge with reporting is to balance the need for deep exploration of a tumor genome while simultaneously being user-friendly to allow for efficient assessment of clinically pertinent results. It should be noted that the optimal return of genetic results to clinicians and patients also remains an area of active investigation from an ethical, legal, and social perspective.73–75
Formatting the report for a clinical audience.
Because tumor variants may be classified in multiple ways, it is important to organize the content of the reports in multiple ways to facilitate clinical decisions of different types. One common framework is a gene-oriented approach in which genomic results are organized by their type of clinical effect (see Classification of the Type of Clinical Effect, earlier). When large numbers of genes are assessed simultaneously, clinical decision making may be aided by segregating the results based on the type of clinical action that may follow. For instance, reports of assays that assess both tumor and germline variants should draw attention to variants that may reveal a hereditary cancer syndrome in a separate section from those that confer sensitivity or resistance to a particular therapy. Germline variants unrelated to cancer that have clear medical implications for either the patient or her family should also be noted.
Likewise, it is useful to organize treatment-related results by the drugs they may affect. This is especially important in the context of genomic aberrations that confer resistance to drugs, because they can co-occur with aberrations that confer sensitivity to the same drug (eg, EGFR T790M).8 A drug-oriented view of the results can thus take into account multiple findings simultaneously. Such views are often grouped based on those drugs that are likely to be effective and those that are not likely to be effective. This simplifying approach can be very helpful for clinical decision making; however, care must be taken to ensure that off-label drug associations are clearly stated.
From this perspective, reports of tumor genomic profiles should include an assessment of the level of evidence and strength of the effect with reference to the primary literature. Ideally, content should likewise be formatted in such a way as to draw the clinician's attention to those results with the highest level of evidence and strength of effect (Fig 3).
Several groups that perform tumor mutation analysis are convening molecular tumor boards before finalizing the contents of the interpretive report (eg, Roychowdhury et al3 and Tran et al 76). Other groups that perform tumor genomic analysis on a fixed number of cancer genes have developed electronic versions of their reports that link to outside knowledge resources.2,77 This interactive reporting strategy has the advantage of reporting the analytic validity of an assay at a moment in time, while allowing the clinical utility of the results to continuously be updated by outside and centrally maintained sources. Several tools in use78 or under development67 enable molecular pathologists to more easily create interpretive reports with the frameworks described previously. They are being designed to leverage the information from multiple publically available web-based resources, such that as much information as desired can be obtained with ease and within the framework of an electronic medical record.
The knowledge resources available today vary in their degree of clinical applicability, as many were originally created to facilitate cancer research. They also vary in their scope and degree of coverage of the cancer domain. Some only cover information at the gene level, whereas others cover information at the gene variant level. Some focus on gene–disease relationships and others on gene–drug relationships, which correspond to the types of clinic effects (eg, diagnosis v predict response to treatment) that can be inferred from the knowledge. Finally, these resources vary in the levels of evidence that they cover and their public accessibility. Table 1 describes the cancer-specific databases and knowledge bases for tumor variant information that are potentially clinically applicable. Table 2 describes general gene–drug databases that also may provide some utility for the interpretation of the clinical significance of tumor variants.
Table 1.
Cancer-Specific Knowledge Bases to Assist in Clinical Interpretation of Tumor Variants
| Knowledge Base or Database | Description | Types of Relationships | Clinical Domains | Size of Database (2012) |
|---|---|---|---|---|
| Catalogue of Somatic Mutations in Cancer (COSMIC)57 | COSMIC is a database of somatic mutations in cancers derived from curation of case reports in the literature | Tumor variant–disease | All cancer diagnoses | > 700,000 tumors cases with > 280,000 mutations |
| Genomics of Drug Sensitivity in Cancer79 | The Genomics of Drug Sensitivity in Cancer database contains information on the drug sensitivity of cancer cell lines | Tumor gene–drug | Cancer cell lines | Screen of several hundred cancer cell lines with 130 cancer drugs |
| The Cancer Genome Atlas (TCGA) data portal80 | The TCGA data portal contains cases of tumor with extensive molecular annotations and more limited clinical annotations | Tumor variant–disease–drug–outcome | 21 cancer types | 21 cancer diagnoses with > 6,000 tumor samples with genome and clinical annotations |
| DNA-Mutation Inventory to Refine and Enhance Cancer Treatment (DIRECT)81 | DIRECT is a database of cases of non–small-cell lung cancer reported in the literature with tumor variant(s), drug exposure, and clinical outcome annotations25 | Tumor variant-disease-drug-outcome | Non–small-cell lung cancer | > 1,800 patient cases, 150 EGFR variants |
| My Cancer Genome82 | My Cancer Genome is a knowledge base describing the clinical significance of common tumor gene mutations and their relationships to disease and drugs. It includes annotations of gene directed cancer clinical trials27 | Tumor variant-disease-drug-outcome | All cancers | > 10,000 active cancer trials annotated with over 400 cancer genes. Detailed reviews for 17 cancer types, 25 cancer genes, over 280 tumor variant-disease relationships |
| Atlas Genetics Oncology83 | Atlas Genetics Oncology is a peer reviewed on-line journal and database in free access on internet devoted to genes, cytogenetics, and clinical entities in cancer, and cancer-prone diseases. It presents concise and updated reviews (cards) or longer texts (deep insights)29 | Tumor variant–disease relationships | All cancer diagnoses with a focus on hematologic malignancies | Tumor cytogenetic aberrations in all 24 chromosomes. Not currently up to date in all topics |
| cBio Cancer Genomics Portal84 | This portal contains data from multiple large-scale cancer genomic data sets and allows for explorative analysis | Tumor variant–pathway–disease relationships | 19 cancer types | The portal data includes 7,848 tumor samples from 26 cancer studies |
Table 2.
Gene–Drug Knowledge Bases to Assist in Clinical Interpretation of Tumor Variants
| Knowledge Base | Description | Types of Relationships | Clinical Domains |
|---|---|---|---|
| Pharmacogenomics Knowledge Base (PharmGKB)85 | Contains annotations on genetic variants and gene–drug–disease relationships via literature reviews | Gene–drug, germline variant–drug–host response | Somatic and germline pharmacogenomics |
| DrugBank72 | A compilation of detailed drug data with comprehensive drug target information | Gene–drug, drug–pathway | Somatic and germline pharmacogenomics |
| STITCH86 | An interaction network of genes, drugs, and proteins | Gene–drug, gene–chemical | Somatic and germline pharmacogenomics |
| Therapeutic Target Database (TTD)87 | A database of known and explored therapeutic drug targets, the targeted disease, pathway information, and the associated drugs | Gene–drug, drug–pathway, drug–target | Somatic and germline pharmacogenomics |
Challenges with respect to evolving clinical knowledge.
An active area of study concerns the questions of if, when, and how to notify providers when new clinical evidence emerges regarding clinically actionable genomic alterations in tumor specimens previously reported. The current standard of care in laboratory medicine is to report the most up-to-date knowledge regarding the clinical significance of the test result at the time the initial report is created. There are no standards for reanalyzing the data or contacting providers when new knowledge or analysis algorithms become available. Ethical guidelines on this matter are being considered for germline studies,88 but are lacking for somatic studies. These issues are especially important when new therapies become available (as standard of care or clinical trials) or when new knowledge emerges regarding variant-specific drug sensitivity or resistance. The dynamic reports described in Formatting the Report for a Clinical Audience may be one way to address this issue but require the clinician to refresh the report at the time of transitions in treatment. The next section describes other opportunities for more active forms of clinical decision support to potentially address this issue. However, guidelines and realistic workflow strategies are still needed.
Clinical Decision Support Tools
Reporting results comprises just one way to provide clinicians with decision support regarding potentially clinically actionable results. This type of “passive” clinical decision support requires the clinician to review the report and supporting evidence at the point of making a clinical decision. In contrast, “active” forms of clinical decision support, such as alerts and notifications, are interruptive at the point of finalizing clinical decisions. Active decision support includes alerts for possible contraindications to a selected therapy at the point of provider order entry. For example, if the clinician is attempting to prescribe erlotinib for a patient with EGFR-mutant lung cancer, a decision support system could alert the clinician if the tumor has one of the variants that is known to confer primary or secondary resistance to erlotinib, thereby sparing the patient from a potentially ineffective therapy. The advantage of this approach is the ability to update clinical evidence at the point of treatment action, a major benefit over static reporting strategies. Other opportunities exist to filter and prioritize the list of potential therapies within a chemotherapy order management system at the point of plan selection, taking into account multiple clinical features including the tumor genome. However, such tools are under development for only a small subset of genomic biomarkers.
In conclusion, this science of computational cancer medicine is still in its infancy. With the decreasing cost of tumor gene alteration testing and the increasing number of targeted therapeutics, we expect the methods for analysis and interpretation of cancer genomes to continue to evolve to meet the needs of patient-centered clinical decision making. By successfully bridging research-oriented genomics analysis tools with clinically focused interpretation methods, oncologists may be able to incorporate vast amounts of genomic information into individual patient care. Although it is as yet unknown if this amount of data will result in improvements in clinical outcomes, it is now conceivable to harness the weight of these data and rationally design clinical trials to answer this question across cancer types.
There remains a pressing need to continue to develop knowledge bases, best practices, tools, and validation experiments for the clinical analysis and interpretation of the cancer genome. This effort will require significant and longitudinal collaborations between medical oncologists, molecular pathologists, computational biologists, and biomedical informaticians working together to implement prospective large-scale sequencing. As these tools are developed in conjunction with prospective assessments of the impact cancer genomics may have on patient care, robust analytic and interpretive frameworks should emerge to facilitate clinical cancer genomics throughout the field and across care centers.
In addition, as such analytic and interpretation platforms become more refined, it will be imperative to ensure that these tools and knowledge bases are open source and web based so that they may be accessed by physicians and patients regardless of the care center context. This may require increased regulation of these tools by the US Food and Drug Administration, especially for interpretation algorithms, so that genomic data are not used irresponsibly. Finally, the ethical ramifications of the large amount of data being revealed by these analytic and interpretation algorithms cannot be overlooked; collaborations between ethicists and oncologists armed with this data are necessary and reinforce the need for national guidelines that help guide clinicians on how to safely use information that may have an impact on the patient and his or her family.
GLOSSARY TERMS
- Breadth of coverage:
The amount of genomic territory sequenced in a given testing modality.
- Copy number alteration:
A structural variation in the genome with an increased (amplification) or decreased (deletion) number of copies of a gene or region.
- Depth of coverage:
The number of times a particular place in the genome (eg, base, exon, or region) has been sequenced, often to ensure data accuracy and sensitivity.
- Exome:
Part of the genome formed by genes that code for proteins and other functional gene products (known as exons).
- Genome:
The complete set of genetic material.
- Genome testing modality:
The technology or platform that is used to translate biologic tissue into genetic or molecular data, including Sanger sequencing, mass spectrometric genotyping, allele-specific polymerase chain reaction–based technologies, and massively parallel sequencing, among other options.
- Germline alterations:
Alterations in a gene present at conception that are incorporated into every cell of an individual.
- Hotspots:
Sites in a specific gene known to harbor recurrent alterations that have well-defined biologic and/or clinical significance.
- Insertion/deletion (“indel”):
A local net gain or loss of nucleotides (generally, between one and 50 bases) that results in a frameshift event.
- Massively parallel sequencing:
A high-throughput approach to sequencing DNA or RNA, also known as next-generation or second-generation sequencing.
- Mutation:
A change of one base in a nucleotide sequence that may result in a change in the amino acid sequence.
- Rearrangement:
A genomic alteration resulting from a chromosomal breakpoint that leads to a large structural change in or between chromosomes.
- Somatic alterations:
Alterations in DNA that occur in an individual after conception; here used to denote alterations that occur uniquely in a tumor cell.
- Targeted sequencing:
Sequencing the coding regions or other selected regions from a relatively small subset of genes.
- Transcriptome:
The complete expressed product of the entire genome in a particular cell, tissue, or biofluid at a specific point in time.
- Variant:
A change in the DNA sequence from the reference genome that may or may not have functional consequences.
- Variant calling:
The method for identifying variants in a tumor sample.
Footnotes
Supported by Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation and the Dana-Farber Leadership Council.
Terms in blue are defined in the glossary, found at the end of this article and online at www.jco.org.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships markedwith a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors
Employment or Leadership Position: None Consultant or Advisory Role: Nikhil Wagle, Foundation Medicine (C) Stock Ownership: Nikhil Wagle, Foundation Medicine Honoraria: None Research Funding: None Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Su Z, Dias-Santagata D, Duke M, et al. A platform for rapid detection of multiple oncogenic mutations with relevance to targeted therapy in non-small-cell lung cancer. J Mol Diagn. 2011;13:74–84. doi: 10.1016/j.jmoldx.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacConaill LE, Campbell CD, Kehoe SM, et al. Profiling critical cancer gene mutations in clinical tumor samples. PloS One. 2009;4:e7887. doi: 10.1371/journal.pone.0007887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roychowdhury S, Iyer MK, Robinson DR, et al. Personalized oncology through integrative high-throughput sequencing: A pilot study. Sci Transl Med. 2011;3:111ra21. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macconaill LE, Garraway LA. Clinical implications of the cancer genome. J Clin Oncol. 2010;28:5219–5228. doi: 10.1200/JCO.2009.27.4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mino-Kenudson M, Chirieac LR, Law K, et al. A novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistry. Clin Cancer Res. 2010;16:1561–1571. doi: 10.1158/1078-0432.CCR-09-2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagle N, Berger MF, Davis MJ, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012;2:82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;3:239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 8.Su KY, Chen HY, Li KC, et al. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol. 2012;30:433–440. doi: 10.1200/JCO.2011.38.3224. [DOI] [PubMed] [Google Scholar]
- 9.Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Govindan R, Ding L, Griffith M, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 12.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11:685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 13.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu CM, Wong T, Wu E, et al. SOAP3: Ultra-fast GPU-based parallel alignment tool for short reads. Bioinformatics. 2012;28:878–879. doi: 10.1093/bioinformatics/bts061. [DOI] [PubMed] [Google Scholar]
- 15.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alkan C, Kidd JM, Marques-Bonet T, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hach F, Hormozdiari F, Alkan C, et al. MrsFAST: A cache-oblivious algorithm for short-read mapping. Nat Methods. 2010;7:576–577. doi: 10.1038/nmeth0810-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Homer N. A survey of sequence alignment algorithms for next-generation sequencing. Brief Bioinform. 2010;11:473–483. doi: 10.1093/bib/bbq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lunter G, Goodson M. Stampy: A statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011;21:936–939. doi: 10.1101/gr.111120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rumble SM, Lacroute P, Dalca AV, et al. SHRiMP: Accurate mapping of short color-space reads. PLoS Comput Biol. 2009;5:e1000386. doi: 10.1371/journal.pcbi.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zaharia M, Bolosky WJ, Curtis K, et al. Faster and more accurate sequence alignment with SNAP 2011. http://arxiv.org/abs/1111.5572.
- 22.Ruffalo M, LaFramboise T, Koyutürk M. Comparative analysis of algorithms for next-generation sequencing read alignment. Bioinformatics. 2011;27:2790–2796. doi: 10.1093/bioinformatics/btr477. [DOI] [PubMed] [Google Scholar]
- 23.Lipson D, Capelletti M, Yelensky R, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382–384. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cibulskis K. MuTect 2011. http://www.broadinstitute.org/cancer/cga/mutect.
- 26.Larson DE, Harris CC, Chen K, et al. SomaticSniper: Identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28:311–317. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roth A, Ding J, Morin R, et al. JointSNVMix: A probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics. 2012;28:907–913. doi: 10.1093/bioinformatics/bts053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saunders CT, Wong WS, Swamy S, et al. Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 29.Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Challis D, Yu J, Evani US, et al. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinformatics. 2012;13:8. doi: 10.1186/1471-2105-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen K, Wallis JW, McLellan MD, et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emde AK, Schulz MH, Weese D, et al. Detecting genomic indel variants with exact breakpoints in single- and paired-end sequencing data using SplazerS. Bioinformatics. 2012;28:619–627. doi: 10.1093/bioinformatics/bts019. [DOI] [PubMed] [Google Scholar]
- 34.Broad Institute. Cancer Genome Analysis: Indelocator. http://www.broadinstitute.org/cancer/cga/indelocator.
- 35.Ye K, Schulz MH, Long Q, et al. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li S, Li R, Li H, et al. SOAPindel: Efficient identification of indels from short paired reads. Genome Res. 2013;23:195–200. doi: 10.1101/gr.132480.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Albers CA, Lunter G, MacArthur DG, et al. Dindel: Accurate indel calls from short-read data. Genome Res. 2011;21:961–973. doi: 10.1101/gr.112326.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crisan A, Goya R, Ha G, et al. Mutation discovery in regions of segmental cancer genome amplifications with CoNAn-SNV: A mixture model for next generation sequencing of tumors. PloS One. 2012;7:e41551. doi: 10.1371/journal.pone.0041551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lonigro RJ, Grasso CS, Robinson DR, et al. Detection of somatic copy number alterations in cancer using targeted exome capture sequencing. Neoplasia. 2011;13:1019–1025. doi: 10.1593/neo.111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krumm N, Sudmant PH, Ko A, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22:1525–1532. doi: 10.1101/gr.138115.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abyzov A, Urban AE, Snyder M, et al. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–984. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiang DY, Getz G, Jaffe DB, et al. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods. 2009;6:99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carter SL. CapSeg 2013. http://www.broadinstitute.org/cancer/cga/capseg.
- 44.Fromer M, Moran JL, Chambert K, et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am J Hum Genet. 2012;91:597–607. doi: 10.1016/j.ajhg.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson DR, Wu YM, Kalyana-Sundaram S, et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet. 2013;45:180–185. doi: 10.1038/ng.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maher CA, Kumar-Sinha C, Cao X, et al. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim D, Salzberg SL. TopHat-Fusion: An algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drier Y, Lawrence MS, Carter SL, et al. Somatic rearrangements across cancer reveal classes of samples with distinct patterns of DNA breakage and rearrangement-induced hypermutability. Genome Res. 2013;23:228–235. doi: 10.1101/gr.141382.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang X, Wang K. wANNOVAR: Annotating genetic variants for personal genomes via the web. J Med Genet. 2012;49:433–436. doi: 10.1136/jmedgenet-2012-100918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.SeattleSeq. Annotation 137. 2013. http://snp.gs.washington.edu/SeattleSeqAnnotation137/
- 51.Schwarz JM, Rödelsperger C, Schuelke M, et al. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 52.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cingolani P, Platts A, Wang le L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramos A. Oncotator 2013. 2013. http://www.broadinstitute.org/oncotator/
- 55.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 56.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Forbes SA, Bindal N, Bamford S, et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henry NL, Hayes DF. Cancer biomarkers. Mol Oncol. 2012;6:140–146. doi: 10.1016/j.molonc.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hall JM, Lee MK, Newman B, et al. Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990;250:1684–1689. doi: 10.1126/science.2270482. [DOI] [PubMed] [Google Scholar]
- 60.Houillier C, Wang X, Kaloshi G, et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology. 2010;75:1560–1566. doi: 10.1212/WNL.0b013e3181f96282. [DOI] [PubMed] [Google Scholar]
- 61.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Allegra CJ, Jessup JM, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: Testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091–2096. doi: 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- 63.Krönke J, Schlenk RF, Jensen KO, et al. Monitoring of minimal residual disease in NPM1-mutated acute myeloid leukemia: A study from the German-Austrian acute myeloid leukemia study group. J Clin Oncol. 2011;29:2709–2716. doi: 10.1200/JCO.2011.35.0371. [DOI] [PubMed] [Google Scholar]
- 64.O'Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 65.Schlenk RF, Döhner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 66.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van Allen E, Wagle N, Kryukov G, et al. A heuristic platform for clinical interpretation of cancer genome sequencing data. J Clin Oncol. 2012;30(suppl; abstr 10502):656s. [Google Scholar]
- 68.Hayes DF, Bast RC, Desch CE, et al. Tumor marker utility grading system: A framework to evaluate clinical utility of tumor markers. J Natl Cancer Inst. 1996;88:1456–1466. doi: 10.1093/jnci/88.20.1456. [DOI] [PubMed] [Google Scholar]
- 69.Simon RM, Paik S, Hayes DF. Use of archived specimens in evaluation of prognostic and predictive biomarkers. J Natl Cancer Inst. 2009;101:1446–1452. doi: 10.1093/jnci/djp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Febbo PG, Ladanyi M, Aldape KD, et al. NCCN Task Force report: Evaluating the clinical utility of tumor markers in oncology. J Natl Compr Canc Netw. 2011;9(suppl 5):S1–32. doi: 10.6004/jnccn.2011.0137. [DOI] [PubMed] [Google Scholar]
- 71.Wallace P. Informatics needs and challenges in cancer research: A workshop. 2012. http://www.iom.edu/Activities/Disease/NCPF/2012-FEB-27.aspx. [PubMed]
- 72.Knox C, Law V, Jewison T, et al. DrugBank 3.0: A comprehensive resource for ‘omics' research on drugs. Nucleic Acids Res. 2011;39:D1035–D1041. doi: 10.1093/nar/gkq1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wolf SM, Crock BN, Van Ness B, et al. Managing incidental findings and research results in genomic research involving biobanks and archived data sets. Genet Med. 2012;14:361–384. doi: 10.1038/gim.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Simon C, Shinkunas LA, Brandt D, et al. Individual genetic and genomic research results and the tradition of informed consent: Exploring US review board guidance. J Med Ethics. 2012;38:417–422. doi: 10.1136/medethics-2011-100273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Clayton EW, McGuire AL. The legal risks of returning results of genomics research. Genet Med. 2012;14:473–477. doi: 10.1038/gim.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tran B, Brown AM, Bedard PL, et al. Feasibility of real time next generation sequencing of cancer genes linked to drug response: Results from a clinical trial. Int J Cancer. 2012;132:1547–1555. doi: 10.1002/ijc.27817. [DOI] [PubMed] [Google Scholar]
- 77.Levy MA, Lovly CM, Pao W. Translating genomic information into clinical medicine: Lung cancer as a paradigm. Genome Res. 2012;22:2101–32108. doi: 10.1101/gr.131128.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aronson SJ, Clark EH, Babb LJ, et al. The GeneInsight Suite: A platform to support laboratory and provider use of DNA-based genetic testing. Hum Mutat. 2011;32:532–536. doi: 10.1002/humu.21470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanger Institute. Genomics of drug sensitivity in cancer 2013. http://www.cancerrxgene.org.
- 80.National Cancer Institute. National Human Genome Research Institute: The Cancer Genome Atlas data portal 2013. https://tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp.
- 81.My Cancer Genome: DNA-mutation Inventory to Refine and Enhance Cancer Treatment (DIRECT) 2013. http://www.mycancergenome.org/about/direct. [DOI] [PMC free article] [PubMed]
- 82.My Cancer Genome 2013. http://www.mycancergenome.org.
- 83.Atlas of Genetics and Cytogenetics in Oncology and Haematology 2013. http://AtlasGeneticsOncology.org.
- 84.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McDonagh EM, Whirl-Carrillo M, Garten Y, et al. From pharmacogenomic knowledge acquisition to clinical applications: The PharmGKB as a clinical pharmacogenomic biomarker resource. Biomarkers Med. 2011;5:795–806. doi: 10.2217/bmm.11.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kuhn M, Szklarczyk D, Franceschini A, et al. STITCH 3: Zooming in on protein-chemical interactions. Nucleic Acids Res. 2012;40:D876–D880. doi: 10.1093/nar/gkr1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhu F, Shi Z, Qin C, et al. Therapeutic target database update 2012: A resource for facilitating target-oriented drug discovery. Nucleic Acids Res. 2012;40:D1128–D1136. doi: 10.1093/nar/gkr797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shirts BH, Parker LS. Changing interpretations, stable genes: Responsibilities of patients, professionals, and policy makers in the clinical interpretation of complex genetic information. Genet Med. 2008;10:778–783. doi: 10.1097/GIM.0b013e31818bb38f. [DOI] [PubMed] [Google Scholar]