

Abstract
The disease mechanisms underlying type 2 diabetes (T2D) remain poorly defined. Here we aimed to explore the pathophysiology of T2D by analyzing gene co-expression networks in human islets. Using partial correlation networks we identified a group of co-expressed genes (‘module’) including F2RL2 that was associated with glycated hemoglobin. F2Rl2 is a G-protein-coupled receptor (GPCR) that encodes protease-activated receptor-3 (PAR3). PAR3 is cleaved by thrombin, which exposes a 6-amino acid sequence that acts as a ‘tethered ligand’ to regulate cellular signaling. We have characterized the effect of PAR3 activation on insulin secretion by static insulin secretion measurements, capacitance measurements, studies of diabetic animal models and patient samples. We demonstrate that thrombin stimulates insulin secretion, an effect that was prevented by an antibody that blocks the thrombin cleavage site of PAR3. Treatment with a peptide corresponding to the PAR3 tethered ligand stimulated islet insulin secretion and single β-cell exocytosis by a mechanism that involves activation of phospholipase C and Ca2+ release from intracellular stores. Moreover, we observed that the expression of tissue factor, which regulates thrombin generation, was increased in human islets from T2D donors and associated with enhanced β-cell exocytosis. Finally, we demonstrate that thrombin generation potential in patients with T2D was associated with increased fasting insulin and insulinogenic index. The findings provide a previously unrecognized link between hypercoagulability and hyperinsulinemia and suggest that reducing thrombin activity or blocking PAR3 cleavage could potentially counteract the exaggerated insulin secretion that drives insulin resistance and β-cell exhaustion in T2D.
Keywords: islets, insulin secretion in vitro, insulin secretion in vivo, pathogenic mechanisms
Introduction
Insulin secretion from the pancreatic β-cells normally maintains glucose levels within a narrow range. Insulin resistance of target cells induces a compensatory upregulation of insulin secretion, and insulin secretion can be 3–4 times higher in obese compared with lean individuals.1 The upregulated insulin output under conditions of insulin resistance has been attributed to both increased β-cell mass and enhanced secretion from the individual β-cells,2-4 but the underlying mechanisms remain poorly understood.3 A vicious cycle has been proposed to precipitate T2D, in which the compensatory hyperinsulinemia exacerbates insulin resistance and contributes to β-cell exhaustion and deteriorating glucose control over time.3,5
Glucose is metabolized in the β-cell to generate cytosolic ATP, which leads to closure of ATP-sensitive K+ channels (KATP-channels) and Ca2+-dependent exocytosis.3 In addition, several mechanisms affect insulin secretion via G-protein-coupled receptors (GPCRs),6 but these pathways are not fully elucidated. GPCRs interact with heterotrimeric G-proteins and integrate a plethora of hormonal signals via second messengers such as cAMP and inositol 1,4,5-trisphosphate (IP3). GPCRs have attracted considerable interest as putative therapeutic targets for type 2 diabetes (T2D) and other metabolic diseases,6 with the glucagon-like peptide-1 receptor (GLP1R) and the free fatty acid receptor 1 (GPR40) as 2 examples.7,8 However, we have still limited knowledge of how GPCRs affect the pathophysiology of T2D.6 Identification of additional GPCRs of relevance to T2D could therefore have substantial impact on our understanding and management of the disease.
Polygenic diseases like T2D typically involve subtle expression changes in a plethora of genes, which may be difficult to identify by differential expression analysis of individual genes.9 It has therefore been suggested that analysis of coordinated changes in gene co-expression networks could be used as an alternative approach to provide pathophysiological insights for complex polygenic diseases.9,10 Previously, we have demonstrated the usefulness of applying network models to global gene expression data from metabolic tissues to identify new disease genes for T2D.11 Here, we use network modeling of gene expression data from human pancreatic islets to find GPCRs of relevance to islet secretory function and T2D and identify F2RL2, which encodes protease-activated receptor-3 (PAR3),12 as a link between hypercoagulability and hyperinsulinemia.
Materials and Methods
Human islets
Experimental procedures were approved by the local ethical committees. Microarray raw data have been deposited in a MIAME database (GEO, accession number: GSE38642).
Bioinformatics
The network analysis was performed using qpgraph in R.13 See also Extended Methods.
Insulin secretion and exocytosis measurements were conducted as previously described.11
[Ca2+]i measurements. [Ca2+]i was estimated by Fluo-5F (Invitrogen, USA).
Thrombin generation in T2D patients
T2D patients from the “Detailed mapping of type 2 diabetes” (DIACT) cohort were analyzed for thrombin generation potential.
Statistical analyses
Student’s t-test was used for comparisons of data from the cellular and animal experiments.
Results
Identification of modules with GPCR-connected genes
To identify GPCRs that regulate β-cell function in T2D we devised a network-based approach using global microarray expression data from human islets donors (n = 43), of which 19 had T2D (Fig. 1 and ESM Table 1). We estimated a gene network from the expression data using limited-order partial correlations to distinguish true gene connections from those that may result from non-biological confounding effects, such as batch variation between microarrays.13 The presence of an ‘edge’ in the network (suggesting a connection between 2 genes) was determined by a cutoff ϵ on the portion of non-rejected tests for partial correlation. We used ϵ = 0.4 as a cutoff, which means that the presence of an edge in the resulting gene network is supported by at least 60% of the partial correlation tests.
Figure 1.
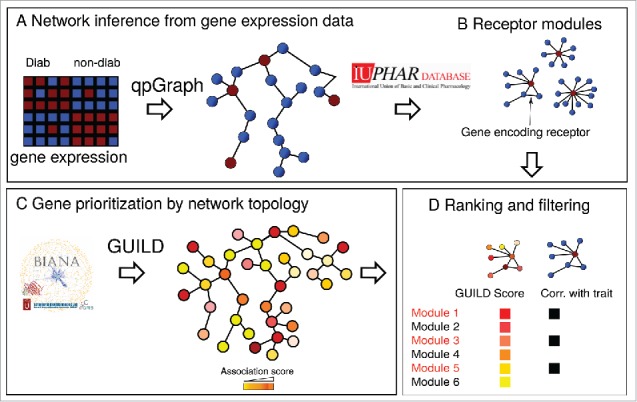
GPCR-containing gene modules. Modules in the regulatory network that contained a GPCR were identified and ranked according to their GUILD score and trait correlation.
We used the gene network to identify GPCRs that are associated with T2D traits. We focused on receptors that have endogenous ligands with known functions to facilitate the coupling of GCPRs to pathophysiological mechanisms. A total of 77 such GPCRs (ESM Table 2) have been reported in the International Union of Pharmacology database.14 We mapped these GPCRs to the network and analyzed their connectedness to other genes. In this manner, we identified 15 gene modules that contained a GPCR that was connected with at least 10 other genes (ESM Table 3).
The identified modules were further characterized using 2 different approaches. The module genes were first mapped to a protein–protein interaction network constructed from publically available data.15 We used the GUILD algorithm to obtain a score for each module based on the number of genes in the module that are suggested to be involved in T2D according to the Online Inheritance in Man database.15 The GUILD algorithm assumes that genes that are connected to disease genes are more likely to be of pathophysiological relevance.15
Rather than analyzing each gene individually, we next computed the singular value decomposition of the gene expression traits of each module. We used the values of the first right-singular vector as summary expression of the module.16 For each module we estimated the correlation between the first right-singular vector and islet insulin secretion or HbA1c of the 43 donors. Four modules in the upper range of the GUILD score rank were nominally associated with HbA1c or glucose-stimulated insulin secretion (ESM Table 3). Two of them contained BDKRB1 and BDKRB2, which encode the bradykinin receptor B1 and B2, respectively. Activation of these receptors has been shown to stimulate insulin secretion both in vitro and in vivo.17-19 One module contained the GLP1R, which mediates the stimulatory effect of glucagon-like peptide-1 on insulin secretion.20 The GPCR contained in the final module, F2RL2, has not previously been implicated in T2D and we therefore investigated its role in insulin secretion in greater detail.
Characterization of F2RL2
F2RL2 encodes PAR3 (also termed coagulation factor II [thrombin] receptor-like-2). Four subtypes of protease-activated receptors (PARs) have been identified (PAR1–4).21 They have a unique mode of activation; cleavage of their N-terminal by thrombin or trypsin exposes a cryptic domain that acts as a ‘tethered ligand’ to trigger receptor auto-activation or activation of other PARs.22,23 The cryptic domain of PAR3 in particular has been shown to activate other PARs.22,23 PARs are present on platelets and mediate coagulation but are also expressed in several other cell types, including human islets.6,21 The receptors have been shown to regulate tumor microenvironment and T-cell activation. PAR3 has not previously been implicated in islet function.
Synthetic peptides 5 to 6 amino acids (aa) long, corresponding to the sequences of the PAR-tethered ligands, can substitute for thrombin in activating PARs and have proved useful for characterizing the cellular effects of PAR activation.21 We treated mouse islets with the 6-aa PAR3-activating peptide (PAR3-AP;SFNGGP) at 20 μmol/l, which is in the range of what has previously been shown to induce cellular effects.22,24 PAR3-AP stimulated insulin secretion by 90% at basal glucose measured during a 1-h static incubation in Krebs-Ringer bicarbonate buffer (KRBB) (p = 0.02). Moreover, PAR3-AP stimulated insulin secretion by 130% at high glucose (p = 0.01;Fig. 2A), which is higher than typically seen in response to GLP1.25 The other PAR-activating peptides (PAR1-AP, PAR2-AP and PAR4-AP) were without effect on insulin secretion.(Fig. 2B)
Figure 2.

Effects of thrombin and PAR3-AP on insulin secretion. (A–B) Insulin secretion in response to 1-h incubations (n = 4–8). C. Accumulated insulin in the incubation medium (n = 5). D–E. Increase in cell capacitance (ΔC), reflecting exocytosis, and integrated Ca2+-current (n = 15–20). F-H. Total capacitance increase in INS-1–832/13-cells (n = 10–20). Means±s .e.m.*P < 0 .05;**P < 0 .01;***P < 0 .001.
Since thrombin is the physiological activator of PAR3 we incubated mouse islets in the standard cell culture medium (at 11 mmol/l glucose) supplemented with 10 nmol/l thrombin (a concentration that has been shown to cleave 80% of PAR3[12]). By immunostaining of rat pancreatic sections we verified that thrombin can normally penetrate into the islets (ESM Fig. 1A). After 2 h incubation, the amount of insulin secreted into the culture medium was 140% higher in the presence of thrombin compared with islets incubated without thrombin (p = 0.03;Fig. 2C), and insulin levels were increased by 87% after 12 h (p = 0.01) and by 29% after 24 h incubation (p = 0.08). Thrombin also tended to increase insulin secretion during 1-h incubations in KRBB with 16.7 mmol/l glucose (ESM Fig. 1B). Long-term incubation with PAR3-AP stimulated insulin release to a similar extent to that of thrombin and increased insulin levels by 173% after 2 h (p = 0.005), by 101% after 12 h (p = 0.01) and by 36% after 24 h compared with islets cultured in the absence of PAR3-AP (p = 0.04;Fig. 2C).
We next used an antibody (H103, generated using aa1–103 of PAR3 as the antigen), which binds to the thrombin cleavage site of PAR3 and blocks thrombin-mediated PAR3 activation.26 Incubation of mouse islets with H103 at 25 μg/ml prevented the stimulatory effect of thrombin on insulin secretion (p = 0.02 at 2 h;p = 0.0002 at 12 h;p = 0.01 at 24 h for comparisons between islets incubated in the presence of thrombin with or without H103;Fig. 2C). Incubation with H103 alone had no significant effect on insulin levels after 2 or 12 h, but tended to reduce the insulin levels by 30% after 24 h (p = 0.08;Fig. 2C). These data suggest that thrombin stimulates insulin secretion by activation of PAR3.
PAR3-AP increases β-cell exocytosis
Insulin is released from the β-cells in a characteristic biphasic manner: a transient first phase (10–15 min) is followed by a sustained second phase.3 We examined insulin exocytosis in single mouse β-cells by measuring cell capacitance. We applied depolarizations to stimulate glucose-induced electrical activity, and monitored the increase in cell area that results when secretory granules fuse with the plasma membrane. The exocytotic response to a train of 10 depolarizations was increased by 70% when 20 μmol/l PAR3-AP was included in the extracellular solution for 10–15 minutes prior to the recording (p = 0.03;Fig. 2D). The integrated Ca2+-current, which reflects the cumulative Ca2+-entry, was also increased in PAR3-AP-treated cells compared with control cells (p = 0.01;Fig. 2E). Both rapid exocytosis (estimated as the response to the first 2 depolarizations) and slow exocytosis (the response to pulses 3–10, proposed to correlate with first- and second-phase insulin secretion3) were increased by PAR3-AP (by 61 and 91%, respectively). These observations were paralleled by insulin secretion experiments of mouse islets at 16.7 mM glucose, showing that PAR3 stimulated insulin secretion by 52% during the first 10 min (p = 0.05) and by 170% (p = 0.07) during 11–60 min of the incubation period (ESM Fig. 1C).
PAR3-AP acts via Gq proteins
PAR3-AP stimulated exocytosis also in clonal insulin-secreting INS-1 832/13 cells (p = 0.01;Fig. 2F), independently of the depolarization pulse length (ESM Fig. 2A). The peptide did not affect apoptosis rate (ESM Fig. 2B). PARs have been suggested to signal via both Gi/Go and Gq proteins.21 In all capacitance recordings, cytosolic cAMP was clamped to the concentrations of the pipette-filling solution (0.1 mmol/l), and altered cAMP production through Gi/Go signaling accordingly cannot account for the effect of PAR3-AP on exocytosis. Gq proteins, on the other hand, activate phospholipase C to generate diacylglycerol and IP3, which results in release of Ca2+ from intracellular stores. We therefore investigated the potential involvement of Gq protein signaling in the effect of PAR3-AP on the exocytotic response.
We first treated INS-1 832/13 cells with the phospholipase C inhibitor U73122 (5 μmol/l). Addition of U73122 alone produced a slight reduction of the exocytotic response compared with the control (30%;p = 0.048;Fig. 2F). Interestingly, the stimulatory effect of PAR3-AP was abolished in the presence of U73122 (p = 2E-5;Fig. 2F). This was paralleled by experiments in which 5 μmol/l xestospongin C, which blocks the receptor for IP3 on the endoplasmic reticulum (ER), was included in the pipette solution that dialyzes the cell interior. Xestospongin C prevented the stimulatory effect of PAR3-AP on exocytosis (p = 0.01;Fig. 2G). We also assessed the involvement of protein kinase C (PKC), which is activated by diacylglycerol, and found that the PKC inhibitor GF109203X (2.4 μmol/l) prevented the effect of PAR3-AP on exocytosis (p = 0.002;Fig. 2H). These findings were paralleled by measurements of insulin secretion in mouse islets, showing that the stimulatory effect of PAR3 on glucose-induced insulin secretion was attenuated by U73122 and GF109203X (ESM Fig. 1D). The data taken together suggest that PAR3-AP stimulates exocytosis via Gq protein signaling.
PAR3-AP increases release of Ca2+ from intracellular stores.
To study the specific effect of PAR3-AP on the intracellular Ca2+ concentration ([Ca2+]i) we conducted confocal imaging of mouse islets incubated with the Ca2+-sensor Fluo-5F. These experiments demonstrated a 2-fold elevation of [Ca2+]i by PAR3-AP at 16.7 mmol/l glucose (Fig. 3A). Elevated [Ca2+]i can result from Ca2+-release from intracellular stores (mediated by IP3) or from Ca2+-influx via voltage-gated Ca2+-channels in the plasma membrane. We examined the effect of PAR3-AP on voltage-gated Ca2+-channels by using the L-type Ca2+-channel blocker isradipine. As expected, isradipine (5 μmol/l) blocked most of the [Ca2+]i oscillations (Fig. 3B). However, even in the presence of isradipine, PAR3-AP was able to significantly increase [Ca2+]i (p = 0.005). These observations were further corroborated by capacitance measurements of INS-1 832/13 cells, which showed that PAR3-AP enhanced exocytosis in the presence of isradipine (5 μmol/l) and SNX-482 (100 nmol/l), applied to block L-type and R-type voltage-gated Ca2+-channels, respectively (Fig. 3C). We then examined the involvement of intracellular Ca2+-stores in the PAR3-AP-mediated increase of [Ca2+]i by using 30 μmol/l cyclopiazonic acid (CPA) that inhibits the Ca2+-ATPase in the endoplasmic reticulum and thus empties the intracellular Ca2+-stores. In contrast to the experiments using isradipine, the stimulatory effect of PAR3-AP on [Ca2+]i was completely abolished by CPA (Fig. 3D). These findings taken together demonstrate that PAR3-AP stimulates β-cell exocytosis by activation of phospholipase C leading to release of Ca2+ from intracellular stores.
Figure 3.
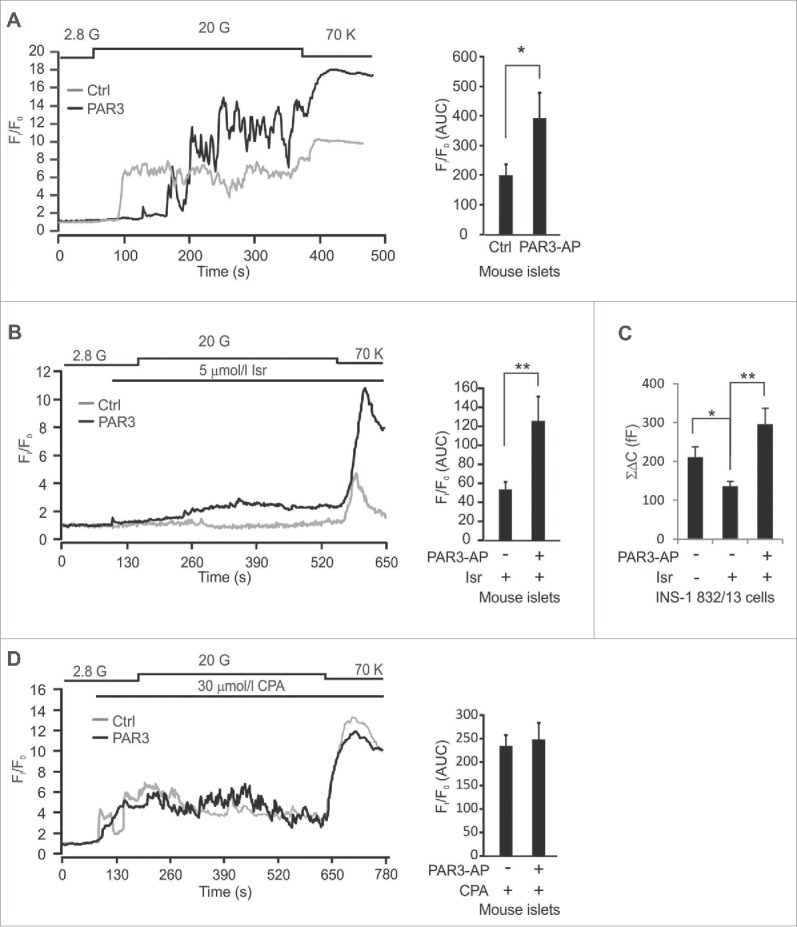
Intracellular Ca2+ recordings. (A, B, D) Fluo-5F-fluorescence in mouse β-cells and the area under the curve at 20 mmol/l glucose (n = 12–18). C. Total capacitance increase (ΣΔC) in INS-1–832/13-cells (n = 15–20).
PAR3-AP stimulates insulin secretion and exocytosis in human β-cells
PAR3 mRNA was detected in human islets (in the same range as that of PDX1) by RT-qPCR (ESM Fig.3). However, we failed to unequivocally demonstrate PAR3 protein expression because of unspecific antibody signals using Western blot (ESM Fig.3). There are several extra-islet sources of activated PAR3, in particular thrombocytes. By interacting with other GPCRs including other PARs, as has previously been shown,22,23 activated PAR3 could therefore have a pathophysiological effect even if PAR3 itself is not expressed in β-cells.
Thus, we next investigated whether PAR3-AP stimulates insulin secretion in human islets. Islets from 3 (non-diabetic) donors were incubated for 1 h at 2.8 or 16.7 mmol/l glucose with or without the PAR3-AP corresponding to the human PAR3 tethered ligand (TFRGAP). PAR3-AP evoked a 105% stimulation of insulin secretion at high glucose (p = 0.0008;Fig. 4A). Single-cell capacitance recordings of human β-cells showed that PAR3-AP induced a 140% potentiation of exocytosis compared with non-treated control cells (p = 0.04;Fig. 4B). These findings show that PAR3-AP stimulates insulin exocytosis in human β-cells with a similar magnitude to that seen in rodent cells.
Figure 4.
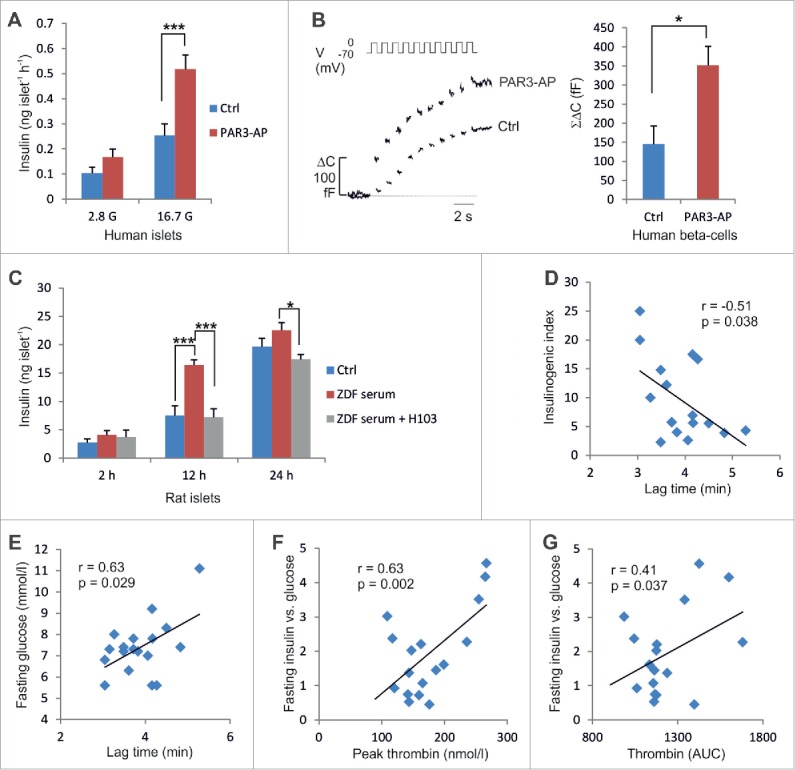
PAR3-AP effects in human islets and thrombin generation in T2D patients. (A) Insulin secretion in human islets (n=4 −8). (B) Increase in cell capacitance (n = 11–12). (C) Accumulated insulin levels following 2, 12 or 24-h incubations of islets from non-diabetic Wistar rats in medium supplemented with plasma from Wistar rats (Ctrl) or ZDF rats with or without 25 μg/ml H103 (n = 5–10). (D–E) The lag time of thrombin activation plotted against insulinogenic index and fasting glucose. (F-G) The peak concentration of thrombin and endogenous thrombin potential (AUC), respectively, plotted against fasting insulin corrected for glucose (n = 17).
Increased thrombin generation is associated with enhanced insulin secretion in T2D
The PAR3 protease thrombin is generated by tissue factor (TF). TF activates factor VII (F7), which in turn increases active factor X (F10) and leads to cleavage of prothrombin to thrombin. Circulating TF and other markers of coagulation activity are elevated in T2D patients.27-29 The mechanisms underlying the increased coagulability in T2D are not fully understood but have been attributed to hyperglycemia and dyslipidemia as well as inflammation.30-31 The effect of hypercoagulability in vivo on insulin secretion has not been investigated.
Increased coagulation activity has also been reported also in animal models of diabetes, including the Zucker Diabetic Fatty (ZDF) rat.32-33 We observed a mild hyperglycemia in 6-week old ZDF rats (ESM Fig. 2C), which was paralleled by a pronounced upregulation of glucose-stimulated insulin secretion in vitro (p = 3E-6;ESM Fig. 2D). By contrast, islets from 12 week old ZDF rats rather displayed a reduction of glucose-stimulated insulin secretion compared to control islets (p = 0.006) and severe hyperglycemia in vivo. These data are in line with previous observations of ZDF rats, which suggest an initial compensatory upregulation of insulin secretion that contributes to β-cell failure.32-33
To investigate whether increased thrombin activity in the plasma of ZDF rats affects insulin secretion capacity we incubated isolated islets from non-diabetic Wistar rats in medium supplemented with plasma from 12-week old ZDF rats in the absence or presence of the H103 antibody. Islets incubated with ZDF plasma secreted 120% more insulin compared to islets incubated with plasma from non-diabetic control rats (p = 0.0002 at 12 h;Fig. 4C). Interestingly, the addition of the H103 antibody to the cell culture medium blocked the stimulatory effect of ZDF plasma on insulin release (p = 8E-5 at 12 h). These findings suggest that PAR3 activation is one factor that contributes to the upregulated insulin secretion in ZDF rats, in addition to elevated plasma levels of lipids and other insulin secretagogues.
The relationship between hypercoagulability and insulin secretion in man has not previously been investigated. The expression of TF was significantly higher in human islets from T2D donors compared with non-diabetic donors (p = 0.011;ESM Fig. 2E), while there were no differences for F7, F10, thrombin and PAR3. The increased expression of TF may be derived either from the islet microvasculature or the endocrine islet cells. There was an association between islet TF expression and donor HbA1c (p = 0.048;β = 0.28), while TF expression was not associated with BMI (p = 0.4) or age (p = 0.4). We next performed capacitance recordings of human β-cells and found an association between increased exocytosis in response to a depolarization train and expression of TF in T2D islets (p = 0.0006;β = 0.64;n = 6) and a weaker association in ND islets (p = 0.03;β = 0.165; n = 18; one-sided comparisons using linear regression).
Based on the present data on TF expression in T2D islets, we postulated that the activity of the extrinsic coagulation cascade (rather than the absolute expression levels of the downstream coagulation factors) is associated with islet function in T2D. To directly test whether there was an association between thrombin generation and insulin secretion in vivo in T2D, we recruited 17 patients with T2D (ESM Table 4) and analyzed the thrombin generation potential in platelet-poor plasma from these patients. Coagulation of the blood samples was initiated in vitro by TF and phospholipids. The time to thrombin generation (lag time), the peak thrombin concentration and the area under the thrombin generation curve (the endogenous thrombin potential, ETP) were determined for each sample. On the same day, the patients also underwent an oral glucose tolerance test (OGTT). We found an association between reduced lag time (i.e. accelerated thrombin generation) and increased insulinogenic index (measuring insulin secretion during the first 30 min of the OGTT) (p = 0.038;r = −0.51; Fig. 4D). There was also an association between reduced lag time and lower fasting glucose (p = 0.029;r = 0.63; Fig. 4E). Moreover, fasting insulin corrected for glucose levels was associated with peak levels of thrombin (p = 0.002;r = 0.63; Fig. 4F) and ETP (p = 0.037;r = 0.41; Fig. 4G). These data taken together demonstrate that increased thrombin generation is associated with exaggerated insulin secretion in T2D patients.
Discussion
The present study demonstrates a previously unrecognized link between hypercoagulability and hyperinsulinemia in T2D. The stimulatory effect of thrombin on islet insulin secretion was prevented by blocking the thrombin cleavage site that exposes the PAR3 tethered ligand. We show that PAR3-AP, which corresponds to the tethered ligand, activates PLC and elicit Ca2+-release from intracellular stores, culminating in increased insulin exocytosis. Activated PAR3 has been suggested to interact with PAR4 and activate both PAR1 and PAR2, and the PLC activation induced by PAR3-AP may therefore involve several interconnected PARs.22 PAR3-AP enhanced exocytosis also in the presence of inhibitors of voltage-gated Ca2+-channels, which argues against a major role of increased voltage-gated Ca2+-influx in the stimulatory effect of PAR3-AP on exocytosis. The data rather support the involvement of Ca2+-release from intracellular stores, since blocking the IP3 receptor abolished the effect of PAR3-AP on exocytosis. Moreover, PAR3-AP-mediated increase of [Ca2+]i was prevented by emptying the intracellular Ca2+-stores by CPA. Previous observations of Ca2+-release from intracellular stores suggest that the ER is located close to the β-cell plasma membrane,33 and Ca2+-release from intracellular stores in the vicinity of the insulin granules will therefore immediately potentiate Ca2+-dependent exocytosis.
We propose a disease model for T2D in which increased thrombin generation activates PAR3, resulting in PLC activation, Ca2+-release from intracellular stores and potentiation of insulin secretion. PAR3-AP enhanced insulin secretion and β-cell exocytosis also in human β-cells. Our findings also demonstrate that enhanced thrombin activity is associated with increased insulin secretion in T2D patients.
What are the pathophysiological consequences of thrombin-mediated stimulation of insulin secretion? Both hyperlipidemia and hyperglycemia have been suggested to increase the coagulation activity, and circulating coagulation markers are elevated in obese individuals who are pre-diabetic.28,30 Moreover, hyperinsulinemia has been shown to enhance the coagulation activity,28,31 and thrombin-stimulated insulin secretion may therefore further aggravate the hypercoagulability. In the short term, upregulated insulin secretion helps to maintain normoglycemia. However, hyperinsulinemia has detrimental long-term consequences involving increased insulin resistance and exhaustion of β-cells. Studies of Pima Indians have shown that fasting hyperinsulinemia predicts T2D.34 Furthermore, individuals with persistent hyperinsulinemic hypoglycemia of infancy have been reported to develop diabetes later in life despite normal insulin sensitivity.35
The experiments in ZDF rats demonstrate that the initial upregulation of insulin secretion at early stages of glucose intolerance (at 6 weeks of age) was followed by a pronounced reduction of glucose-stimulated insulin secretion and severe hyperglycemia at 12 weeks. Several mechanisms, including toxic effects of high lipid levels, have been suggested to cause islet failure in ZDF rats.33 Our observations that plasma from ZDF rats stimulated insulin secretion in isolated islets from non-diabetic rats, an effect that was prevented by blocking PAR3 by H103 antibodies, suggest that increased thrombin generation may contribute to the pathophysiology by inducing a vicious cycle that is driven by exaggerated hypercoagulability and insulin secretion. This is corroborated by recent findings of enhanced thrombin generation in ZDF rats.32 In vivo studies using PAR3 antibodies in diabetic animal models will be of interest to further explore the pathophysiology.
Although we did not observe a direct effect on apoptosis in vitro by PAR3-AP (after 48 h incubation), chronic overstimulation of insulin secretion has been proposed to induce ER stress and oxidative stress and result in β-cell dysfunction.5 Both Ca2+-concentration and PLC-activity in β-cell are normally tightly controlled.36,37 The physiological glucose-regulated activation of voltage-gated Ca2+-channels results in minute bursts of Ca2+-influx sufficient to trigger exocytosis of insulin granules in the vicinity of the channels. By contrast, PAR3-mediated release of Ca2+ from intracellular stores gives a considerably higher Ca2+-load (Fig. 3A). Chronically elevated Ca2+-levels have been shown to impair β-cell function5,35 and there is evidence that low-grade inflammation alters Ca2+-handling resulting in β-cell dysfunction.38 Thrombin-stimulated insulin secretion via increased PAR3 activity could be one mechanism leading to β-cell exhaustion through changes in Ca2+-homeostasis (Ca2+-toxicity).
PAR3 expression was similar in islets from T2D and non-diabetic donors, but the summary expression of the module of genes connected with PAR3 was altered in T2D islets. This demonstrates the potential of the network approach to find disease genes that would not be identified by differential expression analysis of individual genes. The approach could have broad applicability to identify disease-relevant GPCRs for a range of disorders using global gene expression data.
In conclusion, we identify PAR3 as a novel link between thrombin activity and increased insulin secretion. The data suggest a previously unrecognized role of hypercoagulability in the pathophysiology of T2D via thrombin-mediated exaggeration of insulin secretion. Moreover, our findings raise the possibility that antibodies that block PAR3 activation or commonly used drugs that decrease coagulation activity could be a potential therapeutic avenue to counteract the detrimental effects of hyperinsulinemia and excessive Ca2+-load in the β-cells.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank B-M Nilsson and M Ljungkvist for expert technical assistance.
Author Contributions
SH, EG, IGC, RC and AHR performed and interpreted bioinformatics analyses. JW, YT, EZ, ASA, HN, ASS performed experiments. EZ and EB designed and interpreted the data on thrombin generation potential. SH, JW, CBW, IGC, RC and AHR designed the study and wrote the paper. All authors commented on the manuscript.
Funding
Supported by the NovoNordisk foundation, the Hjelt foundation and the Swedish Research Council. S.H. and R.C. acknowledge support from a Spanish MINECO grant (ref. TIN2011-22826) and S.H. and I.C. acknowledge support from the Interdisciplinary Center for Clinical Research within the faculty of Medicine at the RWTH Aachen University.
Supplemental Material
Supplemental Material may be downloaded here: publisher's website
References
- 1.Polonsky KS, Given BD, Hirsch L, Shapiro ET, Tillil H, Beebe C, Galloway JA, Frank BH, Karrison T, Van Cauter E. Quantitative study of insulin secretion and clearance in normal and obese subjects. J Clin Invest 1988; 81(2):435-41; PMID:3276729; http://dx.doi.org/ 10.1172/JCI113338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bergman RN. Minimal model: perspective from 2005. Horm Res 2005; 64 Suppl 3:8-15; PMID:16439839; http://dx.doi.org/ 10.1159/000089312 [DOI] [PubMed] [Google Scholar]
- 3.Ashcroft FM, Rorsman P. Diabetes mellitus and the β cell: the last ten years. Cell 2012; 148(6):1160-71; PMID:22424227; http://dx.doi.org/ 10.1016/j.cell.2012.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weir GC, Bonner-Weir S. Five stages of evolving β-cell dysfunction during progression to diabetes. Diabetes 2004; 53 Suppl 3:S16-21; PMID:15561905; http://dx.doi.org/ 10.2337/diabetes.53.suppl_3.S16 [DOI] [PubMed] [Google Scholar]
- 5.Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009; 58(4):773-95; PMID:19336687; http://dx.doi.org/ 10.2337/db09-9028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amisten S, Salehi A, Rorsman P, Jones PM, Persaud SJ. An atlas and functional analysis of G-protein coupled receptors in human islets of Langerhans. Pharmacol Ther 2013; 139(3):359-91; PMID:23694765; http://dx.doi.org/ 10.1016/j.pharmthera.2013.05.004 [DOI] [PubMed] [Google Scholar]
- 7.Russell S. Incretin-based therapies for type 2 diabetes mellitus: a review of direct comparisons of efficacy, safety and patient satisfaction. Int J Clin Pharm 2013; 35(2):159-72; PMID:23263796; http://dx.doi.org/ 10.1007/s11096-012-9729-9 [DOI] [PubMed] [Google Scholar]
- 8.Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, et al. Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature 2003; 422(6928):173-6; PMID:12629551; http://dx.doi.org/ 10.1038/nature01478 [DOI] [PubMed] [Google Scholar]
- 9.Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabasi AL. Hierarchical organization of modularity in metabolic networks. Science 2002; 297(5586):1551-5; PMID:12202830; http://dx.doi.org/ 10.1126/science.1073374 [DOI] [PubMed] [Google Scholar]
- 10.Schadt EE, Friend SH, Shaywitz DA. A network view of disease and compound screening. Nat Rev Drug Discov 2009; 8(4):286-95; PMID:19337271; http://dx.doi.org/ 10.1038/nrd2826 [DOI] [PubMed] [Google Scholar]
- 11.Mahdi T, Hänzelmann S, Salehi A, Muhammed SJ, Reinbothe TM, Tang Y, Axelsson AS, Zhou Y, Jing X, Almgren P, et al. Secreted frizzled-related protein 4 reduces insulin secretion and is overexpressed in type 2 diabetes. Cell Metab 2012; 16(5):625-33; PMID:23140642; http://dx.doi.org/ 10.1016/j.cmet.2012.10.009 [DOI] [PubMed] [Google Scholar]
- 12.Ishihara H, Connolly AJ, Zeng D, Kahn ML, Zheng YW, Timmons C, Tram T, Coughlin SR. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature 1997; 386(6624):502-6; PMID:9087410; http://dx.doi.org/ 10.1038/386502a0 [DOI] [PubMed] [Google Scholar]
- 13.Castelo R, Roverato A. Reverse engineering molecular regulatory networks from microarray data with qp-graphs. J Comput Biol 2009; 16(2):213-27; PMID:19178140; http://dx.doi.org/ 10.1089/cmb.2008.08TT [DOI] [PubMed] [Google Scholar]
- 14.Sharman JL, Benson HE, Pawson AJ, Lukito V, Mpamhanga CP, Bombail V, Davenport AP, Peters JA, Spedding M, Harmar AJ. IUPHAR-DB: updated database content and new features. Nucleic Acids Res 2013; 41(Database issue):D1083-1088; PMID:23087376; http://dx.doi.org/ 10.1093/nar/gks960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guney E, Oliva B. Exploiting protein-protein interaction networks for genome-wide disease-gene prioritization. PLoS One 2012; 7(9):e43557; PMID:23028459; http://dx.doi.org/ 10.1371/journal.pone.0043557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomfohr J, Lu J, Kepler TB. Pathway level analysis of gene expression using singular value decomposition. BMC Bioinformatics 2005; 6:225; PMID:16156896; http://dx.doi.org/ 10.1186/1471-2105-6-225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang C, Hsu WH. Stimulatory effect of bradykinin on insulin release from the perfused rat pancreas. Am J Physiol 1995; 268(5 Pt 1):E1027-1030; PMID:7762629 [DOI] [PubMed] [Google Scholar]
- 18.Yang C, Lee B, Chen TH, Hsu WH. Mechanisms of bradykinin-induced insulin secretion in clonal β cell line RINm5F. J Pharmacol Exp Ther 1997; 282(3):1247-52; PMID:9316832 [PubMed] [Google Scholar]
- 19.Damas J, Hallet C, Lefebvre PJ. Changes in blood glucose and plasma insulin levels induced by bradykinin in anaesthetized rats. Br J Pharmacol 2001; 134(6):1312-8; PMID:11704652; http://dx.doi.org/ 10.1038/sj.bjp.0704374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahren B. Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov 2009; 8(5):369-85; PMID:19365392; http://dx.doi.org/ 10.1038/nrd2782 [DOI] [PubMed] [Google Scholar]
- 21.Kaufmann R, Hollenberg MD. Proteinase-activated receptors (PARs) and calcium signaling in cancer. Adv Exp Med Biol 2012; 740:979-1000; PMID:22453980; http://dx.doi.org/ 10.1007/978-94-007-2888-2_45 [DOI] [PubMed] [Google Scholar]
- 22.Hansen KK, Saifeddine M, Hollenberg MD. Tethered ligand-derived peptides of proteinase-activated receptor 3 (PAR3) activate PAR1 and PAR2 in Jurkat T cells. Immunology 2004; 112(2):183-90; PMID:15147561; http://dx.doi.org/ 10.1111/j.1365-2567.2004.01870.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trejo J, Altschuler Y, Fu HW, Mostov KE, Coughlin SR. Protease-activated receptor-1 down-regulation: a mutant HeLa cell line suggests novel requirements for PAR1 phosphorylation and recruitment to clathrin-coated pits. J Biol Chem 2000; 275:31255; PMID:10893235; http://dx.doi.org/ 10.1074/jbc.M003770200 [DOI] [PubMed] [Google Scholar]
- 24.Vidwan P, Pathak A, Sheth S, Huang J, Monroe DM, Stouffer GA. Activation of protease-activated receptors 3 and 4 accelerates tissue factor-induced thrombin generation on the surface of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2010; 30(12):2587-96; PMID:20930172; http://dx.doi.org/ 10.1161/ATVBAHA.110.211177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jimenez-Feltstrom J, Lundquist I, Salehi A. Glucose stimulates the expression and activities of nitric oxide synthases in incubated rat islets: an effect counteracted by GLP-1 through the cyclic AMP/PKA pathway. Cell Tissue Res 2005; 319(2):221-30; PMID:15558323; http://dx.doi.org/ 10.1007/s00441-004-1013-4 [DOI] [PubMed] [Google Scholar]
- 26.Burnier L, Mosnier LO. Novel mechanisms for activated protein C cytoprotective activities involving noncanonical activation of protease-activated receptor 3. Blood 2013; 122(5):807-16; PMID:23788139; http://dx.doi.org/ 10.1182/blood-2013-03-488957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sudic D, Razmara M, Forslund M, Ji Q, Hjemdahl P, Li N. High glucose levels enhance platelet activation: involvement of multiple mechanisms. Br J Haematol 2006; 133(3):315-22; PMID:16643434; http://dx.doi.org/ 10.1111/j.1365-2141.2006.06012.x [DOI] [PubMed] [Google Scholar]
- 28.Alessi MC, Juhan-Vague I. Metabolic syndrome, haemostasis and thrombosis. Thromb Haemost 2008; 99(6):995-1000; PMID:18521499 [DOI] [PubMed] [Google Scholar]
- 29.Diamant M, Nieuwland R, Pablo RF, Sturk A, Smit JW, Radder JK. Elevated numbers of tissue-factor exposing microparticles correlate with components of the metabolic syndrome in uncomplicated type 2 diabetes mellitus. Circulation 2002; 106(19):2442-7; PMID:12417540; http://dx.doi.org/ 10.1161/01.CIR.0000036596.59665.C6 [DOI] [PubMed] [Google Scholar]
- 30.Boden G, Rao AK. Effects of hyperglycemia and hyperinsulinemia on the tissue factor pathway of blood coagulation. Curr Diab Rep 2007; 7(3):223-7; PMID:17547839; http://dx.doi.org/ 10.1007/s11892-007-0035-1 [DOI] [PubMed] [Google Scholar]
- 31.Vaidyula VR, Rao AK, Mozzoli M, Homko C, Cheung P, Boden G. Effects of hyperglycemia and hyperinsulinemia on circulating tissue factor procoagulant activity and platelet CD40 ligand. Diabetes 2006; 55(1):202-8; PMID:16380494; http://dx.doi.org/ 10.2337/diabetes.55.01.06.db05-1026 [DOI] [PubMed] [Google Scholar]
- 32.Shang J, Chen Z, Wang M, Li Q, Feng W, Wu Y, Wu W, Graziano MP, Chintala M. Zucker Diabetic Fatty rats exhibit hypercoagulability and accelerated thrombus formation in the Arterio-Venous shunt model of thrombosis. Thromb Res 2014; 134(2):433-9; PMID:24796819 [DOI] [PubMed] [Google Scholar]
- 33.Unger RH. How obesity causes diabetes in Zucker diabetic fatty rats. Trends Endocrinol Metab 1997; 8(7):276-82; PMID:18406816; http://dx.doi.org/ 10.1016/S1043-2760(97)00094-5 [DOI] [PubMed] [Google Scholar]
- 34.Weyer C, Hanson RL, Tataranni PA, Bogardus C, Pratley RE. A high fasting plasma insulin concentration predicts type 2 diabetes independent of insulin resistance: evidence for a pathogenic role of relative hyperinsulinemia. Diabetes 2000; 49(12):2094-101; PMID:11118012; http://dx.doi.org/ 10.2337/diabetes.49.12.2094 [DOI] [PubMed] [Google Scholar]
- 35.Huopio H, Otonkoski T, Vauhkonen I, Reimann F, Ashcroft FM, Laakso M. A new subtype of autosomal dominant diabetes attributable to a mutation in the gene for sulfonylurea receptor 1. Lancet 2003; 361(9354):301-7; PMID:12559865; http://dx.doi.org/ 10.1016/S0140-6736(03)12325-2 [DOI] [PubMed] [Google Scholar]
- 36.Wiederkehr A, Wollheim CB. Mitochondrial signals drive insulin secretion in the pancreatic β-cell. Mol Cell Endocrinol 2012; 353(1-2):128-37; PMID:21784130; http://dx.doi.org/ 10.1016/j.mce.2011.07.016 [DOI] [PubMed] [Google Scholar]
- 37.Thore S, Dyachok O, Tengholm A. Oscillations of phospholipase C activity triggered by depolarization and Ca2+ influx in insulin-secreting cells. J Biol Chem 2004; 279(19):19396-400; PMID:15044448; http://dx.doi.org/ 10.1074/jbc.C400088200 [DOI] [PubMed] [Google Scholar]
- 38.Dula SB, Jecmenica M, Wu R, Jahanshahi P, Verrilli GM, Carter JD, Brayman KL, Nunemaker CS. Evidence that low-grade systemic inflammation can induce islet dysfunction as measured by impaired calcium handling. Cell Calcium 2010; 48(2-3):133-42; PMID:20800281; http://dx.doi.org/ 10.1016/j.ceca.2010.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.