

Abstract
RPE65, an abundant membrane-associate protein in the retinal pigment epithelium (RPE), is a key retinoid isomerase of the visual cycle necessary for generating 11-cis-retinal that functions not only as a molecular switch for activating cone and rod visual pigments in response to light stimulation, but also as a chaperone for normal trafficking of cone opsins to the outer segments. Many mutations in RPE65 are associated with Leber congenital amaurosis (LCA). A R91W substitution, the most frequent LCA-associated mutation, results in a severe decrease in protein level and enzymatic activity of RPE65, causing cone opsin mislocalization and early cone degeneration in the mutation knock-in mouse model of LCA. Here we show that R91W RPE65 undergoes ubiquitination-dependent proteasomal degradation in the knock-in mouse RPE due to misfolding. The 26S proteasome non-ATPase regulatory subunit 13 mediated degradation specifically of misfolded R91W RPE65. The mutation disrupted membrane-association and colocalization of RPE65 with lecithin:retinol acyltransferase (LRAT) that provides the hydrophobic substrate for RPE65. Systemic administration of sodium 4-phenylbutyrate (PBA), a chemical chaperone, increased protein stability, enzymatic activity, membrane-association, and colocalization of R91W RPE65 with LRAT. This rescue effect increased synthesis of 11-cis-retinal and 9-cis-retinal, a functional iso-chromophore of the visual pigments, led to alleviation of S-opsin mislocalization and cone degeneration in the knock-in mice. Importantly, PBA-treatment also improved cone-mediated vision in the mutant mice. These results indicate that PBA, a U.S. Food and Drug Administration-approved safe oral medication, may provide a noninvasive therapeutic intervention that delays daylight vision loss in patients with RPE65 mutations.
SIGNIFICANCE STATEMENT LCA is a severe early onset retinal dystrophy. Recent clinical trials of gene therapy have implicated the need of an alternative or combination therapy to improve cone survival and function in patients with LCA caused by RPE65 mutations. Using a mouse model carrying the most frequent LCA-associated mutation (R91W), we found that the mutant RPE65 underwent ubiquitination-dependent proteasomal degradation due to misfolding. Treatment of the mice with a chemical chaperone partially corrected stability, enzymatic activity, and subcellular localization of R91W RPE65, which was also accompanied by improvement of cone survival and vision. These findings identify an in vivo molecular pathogenic mechanism for R91W mutation and provide a feasible pharmacological approach that can delay vision loss in patients with RPE65 mutations.
Keywords: chemical chaperone, cone photoreceptor, Leber congenital amaurosis, retina, retinoid visual cycle, Rpe65
Introduction
Visual signal starts with photoisomerization of 11-cis-retinal to all-trans-retinal in the visual pigments of cone and rod photoreceptors. To sustain vision, 11-cis-retinal must be regenerated. RPE65, a retinal pigment epithelium (RPE)-specific microsomal protein (Hamel et al., 1993), is a key retinoid isomerase in the visual cycle necessary for regenerating 11-cis-retinal (Jin et al., 2005; Moiseyev et al., 2005; Redmond et al., 2005). RPE65 uses all-trans-retinyl esters synthesized by LRAT (Ruiz et al., 1999) and other enzymes (Kaschula et al., 2006; Kaylor et al., 2015) as its substrate (Moiseyev et al., 2003). Mice lacking RPE65 cannot synthesize 11-cis-retinoids, thereby their photoreceptors almost completely lose light-sensitivity (Redmond et al., 1998; Pang et al., 2005). Rpe65−/− mice also exhibit cone opsin mislocalization and early cone degeneration (Rohrer et al., 2005; Znoiko et al., 2005; Fan et al., 2008).
The significance of RPE65 function in vision and retinal health is also reflected by the facts that many mutations in RPE65 cause Leber congenital amaurosis (LCA) or retinitis pigmentosa (RP; Gu et al., 1997; Marlhens et al., 1997; Lotery et al., 2000; Thompson et al., 2000). RPE65 mutations are estimated to account for ∼16% of LCA and 2% of recessive RP (RetNet). Although night blindness is the first significant symptom in most patients with RPE65 mutations (Felius et al., 2002), in vivo microscopy of the fovea demonstrated that many patients exhibited severe cone photoreceptor loss at very early ages (Jacobson et al., 2005, 2009). The importance of RPE65 for the central vision is also supported by its abundant expression and higher retinoid isomerase activity in the macaque central RPE layer localized to the cone-rich area (Jacobson et al., 2007).
Recent clinical trials of gene therapy that express normal RPE65 in patients' RPE have shown some improvement of vision in some patients (Bainbridge et al., 2008; Cideciyan et al., 2008; Hauswirth et al., 2008; Maguire et al., 2008). Subsequent studies, however, showed that gene therapy could not stop the progressive retinal degeneration and vision loss (Cideciyan et al., 2013; Bainbridge et al., 2015). The rate of loss of photoreceptors in the treated retinas was the same as that in the untreated retina (Jacobson et al., 2015). In addition, topographic maps of visual sensitivity in the treated region indicated that the areas of improved vision had progressive diminution (Jacobson et al., 2015). It is therefore urgently needed to develop an alternative therapy that alleviates cone degeneration or enhances the efficacy of gene therapy in patients with RPE65 mutations.
A R91W substitution in RPE65 is one of the most frequent mutations causing LCA (Morimura et al., 1998; Thompson et al., 2000). Although patients with this mutation had useful vision in the first decade of life, optical coherence tomography demonstrated that 3- and 7-year-old patients already had cone degeneration (Jacobson et al., 2007, 2008). Similarly, a mouse model carrying R91W mutation displayed early cone degeneration (Samardzija et al., 2009), due to shortage of 11-cis-retinal supply (Samardzija et al., 2008). RPE65 in the R91W mouse was decreased to ∼5% of wild-type RPE65 level by an unknown mechanism (Samardzija et al., 2008). In vitro and animal studies showed that not only R91W mutation but also many other mutations resulted in a drastic decrease of RPE65 (Chen et al., 2006; Takahashi et al., 2006; Bereta et al., 2008; Philp et al., 2009; Nikolaeva et al., 2010; Wright et al., 2013; S. Li et al., 2014, 2015). In vivo identification of the underlying molecular mechanism of this phenotype is important not only for understanding the disease mechanisms of RPE65 mutations but also for developing a therapeutic intervention that prevents or delays loss of RPE65 function in patients.
In this study, we analyzed the molecular basis and pathway leading to degradation of RPE65 in the R91W knock-in (KI) mouse RPE and tested whether a chemical chaperone-mediated protein repair approach can alleviate cone degeneration and vision loss in the knock-in mouse model of LCA.
Materials and Methods
Animals.
Wild-type 129S2/Sv (Charles River Laboratories), R91W KI (Samardzija et al., 2008) and rd12 (The Jackson Laboratory; Pang et al., 2005) mice were maintained in 12 h cyclic light at ∼30 lux. RPE65 in these mice has a leucine residue at position 450 (Leu450). All animal experiments followed the ARVO statement for the use of animals in ophthalmic and vision research and the protocols approved by the Institutional Animal Care and Use Committee. Except where noted, animals of either sex were daily injected intraperitoneally with 50 mg sodium 4-phenylbutyrate (PBA) in saline per kilogram body weight for 3 or 8 weeks, beginning on postnatal day (P)14. Although PBA is a FDA-approved oral medication, we performed intraperitoneal injection to strictly control the amounts of PBA treated. Mice intraperitoneally injected with the same volume of saline were used as controls.
Eyecup ex vivo experiments.
After removal of the anterior section and neural retina, the RPE in mouse eyecups were maintained in DMEM-F12 medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum, 100 U/ml penicillin G, and 100 μg/ml streptomycin at 37°C or 30°C <5% CO2. Transfection of plasmid DNA (pcDNA, pEGFP, pRK5, and pPSMD13) and siRNA (PSMD13 siRNA and scrambled negative control siRNA from OriGene Technologies) was performed using the PolyJet transfection reagent (SignaGen Laboratories) according to the manufacturer's procedure. Forty hours post-transfection, the eyecups were subjected to immunoblot analysis, immunoprecipitation or microscopic analysis. For proteasome and lysosome inhibitor experiments, mouse eyecups were incubated with increasing concentrations of MG132 (15–25 μm), pepstatin A (10–30 μm) or DMSO for 5 h.
Electroretinography.
Overnight dark-adapted 10-week-old mice treated with PBA or saline were anesthetized with intraperitoneal ketamine and xylazine. The pupils were dilated with 1% tropicamide. Electroretinography (ERG) was recorded from the corneal surface using a silver-silver chloride wire electrode referenced to a subcutaneous electrode in the mouth. A needle electrode in the tail served as the ground. A drop of 2% methylcellulose was placed on the cornea to prevent corneal desiccation. Animals were light adapted for 10 min by exposing to a white 32 cd/m2 light, and photopic ERG responses were obtained with white flashes (−1 to 2.4 log cd · s/m2) on the rod-saturating background (32 cd/m2). Five responses to 10 s interval flashes were averaged for each step. Intensity-response amplitude data were displayed on log-linear coordinates using the SigmaPlot 11 software. For recording S-cone ERG, animals were light adapted for 10 min by exposing to a white 40 cd/m2 light, and S-cone ERG responses were obtained on the 40 cd/m2 background with xenon flashes (0.1–1.4 log cd · s/m2) equipped with a Hoya U-360 filter (360 nm peak, Edmund Optics; Oh et al., 2008).
Immunoblot analysis.
Protein samples in the Laemmli sample buffer containing 50 mm dithiothreitol were heated for 10 min at 70°C, separated by SDS-PAGE in a 10%, 12% or 4–12% gradient polyacrylamide gel, and transferred to an Immobilon-P membrane (EMD Millipore). The membrane was incubated in blocking buffer, primary antibody, and horseradish peroxidase-conjugated anti-rabbit or mouse IgG secondary antibody. Antibodies against RPE65 (Abcam and EMD Millipore; Jin et al., 2007), PSMD13 (Proteintech), PSMD11 (Abnova), LRAT (Batten et al., 2004; Golczak et al., 2005), ubiquitin (Enzo Life Sciences), S-opsin (Santa Cruz Biotechnology), M-opsin (EMD Millipore), cone arrestin (EMD Millipore), cadherin-1 (Santa Cruz Biotechnology), or β-actin (Sigma-Aldrich) were used as the primary antibodies. Immunoblots were visualized with the enhanced ECL-Prime and quantified as described previously (S. Li et al., 2013b).
Immunoprecipitation.
RPE cells in mouse eyecups were lysed in a lysis buffer (S. Li et al., 2013b) containing protease-inhibitor mixture (Roche Applied Science), 10 mm N-ethylmaleimide, and 100 μm PR619. After centrifugation for 5 min at 1000 × g, the supernatants were incubated with an anti-RPE65 monoclonal antibody bound to the GammaBind G Sepharose (GE Healthcare). Because protein levels of R91W RPE65 in the KI mice are <10% of RPE65 levels in WT mice (Samardzija et al., 2008), we used 200 μg of WT and 2 mg of KI mice RPE proteins for immunoprecipitation. The beads were washed three times with the lysis buffer containing the inhibitors of proteases and deubiquitylating enzymes. Precipitated proteins were dissolved in Laemmli buffer and heated at 70°C for 10 min for immunoblot analysis.
Preparation of mouse RPE membrane pellets.
After removal of the anterior section and neural retina, RPE in mouse eyecups were homogenized in ice-cold 10 mm HEPES buffer, pH 7.4, containing 280 mm sucrose, 10 mm MgCl2, and protease-inhibitor mixture. This homogenate was used as total RPE cell lysate. A portion of each homogenate was centrifuged for 10 min at 1000 × g. The supernatants were collected and centrifuged for 1 h at 100,000 × g. The resulting ultra-pellets were rinsed in the same buffer one time. The total cell lysates and ultra-pellets were subjected to immunoblot analysis.
Proteasomal activity measurements.
Chymotrypsin-like and trypsin-like proteolytic activities of proteasomes were measured with the fluorogenic 7-Amino-4-methylcoumarin-conjugated peptide substrates as described by Lobanova et al. (2013).
Retinoid isomerase assay.
Mice eyecups without the neural retina were irradiated for 10 min on ice with 365 nm light from a Spectroline Model EN-140L ultraviolet light source to destroy endogenous retinoids. RPE homogenates in 20 mm HEPES buffer were prepared from the eyecups. Each assay mixture contained 500 μg cell homogenate, 10 μm all-trans-retinol, and 6% bovine serum albumin. After incubating for 2 h in darkness at 37°C, retinoids were extracted with hexane and were analyzed by HPLC, as described below.
Analysis of retinoids.
Retinoids in eyecups of overnight dark-adapted mice were extracted with hexane as described previously and analyzed by normal-phase HPLC (Jin et al., 2009). In brief, retinoids in hexane extractions were evaporated, dissolved in 100 μl of hexane, and separated on a silica column (Zorbax-Sil 5 μm, 250 × 4.6 mm, Agilent Technologies) by gradient (0.2–10% dioxane in hexane at 2.0 ml/min flow rate) or nongradient (10% dioxane in hexane at 1.0 ml/min flow rate) elution of mobile phase on an Agilent 1100 HPLC system equipped with a photo-diode array detector (Agilent Technologies). Spectral data were acquired for all eluted peaks. Quantitation was performed by comparison of peak areas to calibration curves established with authentic retinoid standards.
Immunohistochemistry.
Mouse retinal cryosections were prepared as described previously (Sato et al., 2013). Briefly, mouse eyeballs were fixed overnight with 4% paraformaldehyde in 0.1 m phosphate buffer (PB). After removing cornea and lens, eyecups were immersed in 15% sucrose in 0.1 m PB for 2 h, in 30% sucrose in 0.1 m PB for 2 h, and then in a 1:1 mixture of 30% sucrose and Optimal Cutting Temperature (OCT) medium (Sakura Finetechnical) overnight at 4°C. After embedding eyecups in OCT, 15-μm-thick sections were cut on a Shandon Cryotome SME cryostat (Thermo Scientific). The sections were immunostained with the primary antibodies listed in the method of immunoblot analysis and secondary antibodies, as described previously (S. Li et al., 2013a). Nuclei were labeled with DAPI (Sigma-Aldrich). Images were captured with a Zeiss LSM710 Meta confocal microscope with a 40× oil-immersion objective.
Quantitation of cone arrestin-positive cone and M-cone numbers.
Retinal sections of 10-week-old mice were immunostained for cone arrestin (CAR) and M-opsin, as described above. Numbers of CAR-positive outer segments and middle-wavelength cones (M-cones) in whole retinal sections were counted using an Olympus BX61VS microscope equipped with VS-ASW FL software (Sato et al., 2013).
Quantification of S-opsin mislocalization.
Immunostaining of S-opsin was performed on retinal cryosections of 5-week-old WT and KI mice, as described above. Fluorescence intensities in the outer segments (OS) and outer plexiform layer (OPL) were measured using the Olympus BX61VS microscope. We determined the fraction of mislocalized S-opsin according to the following formula. Mislocalization = [OPL fluorescence/(OS fluorescence + OPL fluorescence)] × 100%.
Statistics.
Data were expressed as the mean ± SD of three or more independent experiments. Differences between test and control groups were determined with an unpaired two-tailed Student's t test, using SigmaPlot v11. P values <0.05 were considered statistically significant.
Results
Ubiquitination-dependent proteasomal degradation of R91W RPE65 in RPE
R91W mutation results in dramatic decrease of RPE65 in the R91W KI mouse RPE (Samardzija et al., 2008). To identify the pathway causing reduction of RPE65, we treated RPE in WT and KI eyecups with different concentrations of MG132 (a proteasome inhibitor) or pepstatin A (a lysosome inhibitor). Immunoblot analysis of the eyecups showed that protein levels of R91W RPE65 were increased at least twofold in MG132-treated, but not pepstatin A-treated, eyecups (Fig. 1A). This result suggests that the proteasome plays a critical role in degrading the mutant RPE65. Because ubiquitination is critical for protein degradation in the proteasomes, we tested whether R91W RPE65 is ubiquitinated in the RPE. Under conditions that inhibit protein deubiquitination, we performed immunoprecipitation of RPE65. We then analyzed the immunoprecipitates by immunoblot analysis using antibodies against ubiquitin or RPE65. As shown in Figure 1B, R91W RPE65 was strongly polyubiquitinated in the KI mouse RPE. MG132-treatment of the KI eyecup further increased the signal intensities of the ubiquitinated R91W RPE65 (Fig. 1B). We observed very weak signals of ubiquitinated wild-type RPE65 under the same experimental conditions (Fig. 1B).
Figure 1.

Ubiquitination-dependent proteasomal degradation of R91W RPE65 in mouse RPE. A, Immunoblot analysis of RPE65 and LRAT in WT and R91W, KI mice eyecups treated with DMSO or the indicated concentrations of MG132 or pepstatin A. Beta-actin served as a loading control. Relative intensities of RPE65 immunoblots were quantified, normalized by the intensities of LRAT, and expressed in the histograms as percentage of RPE65 in DMSO-treated eyecups. Asterisks indicate significant differences between DMSO- and MG132-treated groups (*p < 0.01, **p < 0.005). Error bars show SD (n = 4). B, RPE65 in WT and KI eyecups treated with 20 μm MG132 or DMSO was immunoprecipitated, and the immunoprecipitates were probed with antibodies against ubiquitin (Ub) or RPE65. Polyubiquitinated RPE65 (Ub-RPE65) and monomeric RPE65 (arrowhead) are indicated.
PSMD13 promoted degradation of misfolded RPE65 in RPE
We recently showed that the 26S proteasome non-ATPase regulatory subunit 13 (PSMD13) promotes degradation of several mutant RPE65s in culture cells (S. Li et al., 2014, 2015). To test whether PSMD13 has the same function in vivo, we transfected PSMD13-specific siRNA (100 ∼ 200 nm) or control siRNA (200 nm) into RPE cells in WT and KI eyecups. Immunoblot analysis showed that R91W RPE65 was increased as PSMD13 decrease in eyecups transfected with PSMD13 siRNA (Fig. 2A–C). In agreement with this result, chymotrypsin-like proteolytic activity of the proteasomes was decreased in PSMD13 siRNA-transfected WT and KI eyecups compared with control siRNA-transfected eyecups (Fig. 2E). Transfection efficiency test showed that 40 ∼ 50% RPE cells were positive for EGFP in EGFP-transfected eyecups whereas mock vector (pcDNA)-transfected eyecups had no EGFP-positive cells under the same experimental conditions (Fig. 2D). To confirm these results, we transfected pPSMD13 or pRK5 mock vector into RPE cells in WT and KI mice eyecups. As shown in Figure 2F–H, R91W RPE65 was markedly decreased in RPE overexpressing PSMD13. Both chymotrypsin-like and trypsin-like proteolytic activities were increased 35 ∼ 40% in PSMD13-transfected KI eyecups compared with pRK5-transfected KI eyecups (Fig. 2I).
Figure 2.

PSMD13-mediated ubiquitination-dependent degradation of misfolded R91W RPE65. A, Immunoblot analysis of RPE65 and PSMD13 in RPE of WT and KI eyecups transfected with the indicated amounts of PSMD13 siRNA (siPSMD13) or negative control siRNA (siCont, 200 nm). The 26S proteasome non-ATPase regulatory subunit 11 (PSMD11) and LRAT were detected as internal maker for the 19S cap of the proteasome and RPE, respectively. B, C, Relative immunoblot intensities of RPE65 and PSMD13 normalized to LRAT were expressed as fold of RPE65 (B) or percentage of PSMD13 (C) in eyecups transfected with control siRNA. D, Confocal microscopic images of RPE cells in 129S2/Sv mice eyecups transfected with pcDNA or pcDNA encoding EGFP. Nuclei were stained with DAPI. E, Chymotrypsin-like and trypsin-like activities of the proteasomes in RPE of WT and KI mice eyecups transfected with 200 nm PSMD13 siRNA or control siRNA. F, Immunoblot analysis of RPE65, PSMD13, and the indicated proteins in WT and KI eyecups transfected with pRK5 or pRK5 encoding PSMD13. G, H, Relative immunoblot intensities of RPE65 and PSMD13 normalized to LRAT in F. I, Chymotrypsin-like and trypsin-like activities of the proteasomes in RPE of WT and KI mice eyecups transfected with pRK5 or PSMD13 construct (pPSMD13). J, RPE65 in WT and KI eyecups transfected with siPSMD13 or siCont was immunoprecipitated and probed with an Ub antibody. K, Immunoblot analysis of RPE65 and PSMD13 in RPE of WT and KI eyecups maintained at 37°C or 30°C after transfecting pRK5 or pPSMD13. L, Relative immunoblot intensities of RPE65 and PSMD13 (bottom) in the KI mice eyecups in K. Asterisks indicate statistically significant differences between test and control groups (*p < 0.05, **p < 0.005). Error bars indicate SD (n = 4).
Mutation in the yeast Rpn9, a PSMD13 homolog, results in accumulation of ubiquitinated proteins (Takeuchi et al., 1999). We therefore tested whether PSMD13 promotes degradation of R91W RPE65 via the ubiquitination-dependent pathway. We performed immunoprecipitation of RPE65 in WT and KI eyecups transfected with PSMD13 siRNA or control siRNA. Immunoblot analysis of the immunoprecipitates showed that ubiquitinated R91W RPE65 was increased in PSMD13 siRNA-transfected KI eyecups (Fig. 2J), suggesting that PSMD13 promoted degradation of ubiquitinated mutant RPE65 that might be misfolded.
To test whether PSMD13 promotes degradation of misfolded RPE65 in vivo, we overexpressed PSMD13 in RPE of WT and KI eyecups at 37°C and low temperature, which has been shown to help proper folding of mutant proteins (Denning et al., 1992; Vollrath and Liu, 2006; S. Li et al., 2013b). Immunoblot analysis showed that the full-length R91W RPE65 was further decreased in the PSMD13-overexpressing KI eyecups at 37°C compared with control (pRK5-transfected) KI eyecups (Fig. 2K,L). In contrast, cleaved fragments from the mutant RPE65, but not from wild-type RPE65, were increased in the PSMD13-overexpressing eyecups at 37°C (Fig. 2K). At 30°C, however, the full-length R91W RPE65 was drastically increased in both KI eyecups overexpressing and not overexpressing PSMD13 (Fig. 2K,L). These results are consistent with our previous studies showing that low temperature rescues membrane-association and the isomerase activity of several disease-causing RPE65 mutants, including R91W RPE65, in culture cells (S. Li et al., 2014; 2015; Jin et al., 2016), and suggest that PSMD13 preferentially promoted degradation of misfolded mutant RPE65.
PBA increased synthesis of 11-cis-retinal and 9-cis-retinal in the KI mice
The results described above suggest that misfolding is the molecular basis for the degradation of R91W RPE65. To test this possibility, we treated the KI mice with a series of varying amounts of PBA, which has been shown to help proper folding of some mutant proteins (Rubenstein and Zeitlin, 1998; Bonapace et al., 2004; Zode et al., 2011). Protein levels of R91W RPE65 were increased nearly threefold in 5-week-old KI mice treated with 50 mg PBA/kg body weight for 3 weeks compared with age-matched KI mice treated with saline (Fig. 3A). Consistent with this result, the retinoid isomerase activity in RPE homogenates of KI mice treated with 50 mg PBA/kg were ∼2-fold higher than that in saline-treated KI mice RPE (Fig. 3B). We used 50 mg PBA/kg for all of the following experiments.
Figure 3.

PBA increased the isomerase activity and chromophore synthesis in KI mice. A, Immunoblot analysis of RPE65 in RPE of 5-week-old KI mice injected intraperitoneally with the indicated amounts of PBA or saline for 3 weeks, starting at P14. Histogram shows relative immunoblot intensities of RPE65 in PBA-treated mice versus control mice. B, Representative HPLC chromatograms of retinoid isomerase assays of RPE from WT and KI mice treated with 50 mg PBA or saline. The peaks of 11-cis-retinol (11cROL) are indicated by arrows. Histograms show relative isomerase activities of WT and R91W RPE65 in PBA-treated versus saline-treated mice. C, Amounts of atRE in WT and the indicated mutant mice eyes. D, Amounts of the indicated ocular retinoid (11cRAL, 11-cis-retinal; atRAL, all-trans-retinal; 9cRAL, 9-cis-retinal and atROL, all-trans-retinol) in KI mice treated with saline or PBA. Asterisks indicate statistically significant differences between PBA- and saline-treated KI mice (p < 0.01). Error bars indicate SD (n = 6).
To test the effect of PBA on the visual cycle of the KI mice, we analyzed ocular retinoids in dark adapted 5-week-old WT, rd12 (RPE65-null) and KI mice treated with PBA or saline. In agreement with the previous study on Rpe65−/− and the KI mice (Redmond et al., 1998; Samardzija et al., 2008), rd12 and KI mice treated with saline contained dramatically increased amounts of all-trans-retinyl esters (atRE) in the eyes. WT mice contained 38 ± 5.4 pmol atRE in an eye, whereas rd12 and saline-treated KI mice contained 463 ± 41.2 pmol or 476 ± 35.8 pmol atRE per eye, respectively (Fig. 3C). In PBA-treated KI mice, the amounts of atRE were reduced by ∼15% to 404 ± 33.5 pmol/eye compared with saline-treated KI mice (Fig. 3C). Immunoblot analysis of the lecithin:retinol acyltransferase (LRAT) indicates that this reduction of atRE was not due to decrease in LRAT expression in PBA-treated KI mice (Fig. 4C). Both 11-cis-retinal and 9-cis-retinal were increased at least 70% in PBA-treated KI mice compared with saline-treated KI mice (Fig. 3D). 9-cis-Retinal has been shown to form functional iso-rhodopsin in Rpe65−/− mice (Fan et al., 2003).
Figure 4.

Increase in membrane-association and colocalization of R91W RPE65 with LRAT in KI mice treated with PBA. A, Immunoblot analyses of RPE65 and cadherin-1 (CDH1) in membrane fractions of RPE from 10-week-old WT and KI mice treated with 50 mg PBA or saline for 8 weeks. Histograms show relative immunoblot intensities of RPE65 in the membrane fractions. B, Immunohistochemistry showing increase in colocalization of R91W RPE65 with LRAT in RPE of KI mice treated with PBA. Arrows indicate colocalization areas. Scale bar, 20 μm. C, Representative immunoblot analysis showing increase of R91W RPE65, but not LRAT, in RPE of KI mice treated with PBA for 8 weeks. Histogram shows relative expression levels of RPE65 and LRAT in WT mice and KI mice treated with saline or PBA. Asterisks indicate significant differences between PBA- and saline-treated KI mice (p < 0.005). Error bars indicate SD (n = 4).
PBA increased membrane-association and colocalization of R91W RPE65 with LRAT
Association of RPE65 with membranes is critical for its enzymatic activity (Jin et al., 2007; Nikolaeva et al., 2009; Takahashi et al., 2009; Golczak et al., 2010). We therefore tested whether PBA-treatment enhanced association of R91W RPE65s with membranes. We prepared membrane fractions from RPE of 10-week-old WT and KI mice treated with PBA or saline for 8 weeks. Immunoblot analysis of RPE65 in the membrane fractions showed that PBA-treatment increased membrane-association of R91W RPE65 at least 3.5-fold, but had no influence on wild-type RPE65 (Fig. 4A).
We then tested whether PBA-treatment promoted colocalization of R91W RPE65 with LRAT that synthesizes the hydrophobic substrate of RPE65. As expected, immunohistochemistry showed that the majority of RPE65 and LRAT proteins were colocalized and distributed throughout the RPE cells in WT retinas (Fig. 4B, top). In saline-treated KI mice, R91W RPE65 mainly localized to the basal side of the RPE and formed aggresome-like inclusion bodies in close proximity to the nuclei of RPE cells (Fig. 4B, middle). This distribution pattern was completely different from that of LRAT in the same retinas (Fig. 4B, middle). In PBA-treated KI mice, however, many R91W mutant proteins distributed to cytosol area and colocalized with LRAT (Fig. 4B, bottom). PBA-treatment for 8 weeks increased protein level of R91W RPE65, but not LRAT (Fig. 4C).
PBA reduced mislocalization of S-opsin in the KI mice
Because S-opsin is mislocalized in the KI mice (Samardzija et al., 2009), we tested whether PBA can prevent or reduce mislocalization of S-opsin in 5-week-old KI mice treated with PBA or saline for 3 weeks. Immunohistochemistry showed that S-opsin was mainly confined to the cone OS in WT retina (Fig. 5A). In KI mice treated with saline, a greater fraction of S-opsin immunoreactivity was present in the OPL and some cell bodies of cones (Fig. 5A). In the PBA-treated KI mice, however, S-opsin immunoreactivity was largely reduced in the OPL but increased in the OS (Fig. 5A). By measuring fluorescence intensity in the OS and OPL, we estimated the fraction of S-opsin mislocalization in WT mice, saline-treated, and PBA-treated KI mice. The fractional mislocalization of S-opsin was reduced ∼50% in PBA-treated KI mice compared with saline-treated KI mice (Fig. 5B).
Figure 5.

PBA attenuated S-opsin mislocalization in KI mice. A, Representative S-opsin immunoreactivity in retinal sections of WT and KI mice treated with PBA (50 mg/kg) or saline. ONL, Outer nuclear layer; INL, inner nuclear layer. Scale bar, 20 μm. B, Percentage of S-opsin (S-op) mislocalization estimated by dividing S-opsin immunofluorescence in the OPL by the sum of immunofluorescence in OPL and OSs. Note the decrease in S-opsin mislocalization in KI mice after administering PBA. C, Immunoblot analysis of S-opsin in retinas of WT and KI mice treated with PBA or saline. Histogram shows relative expression levels of S-opsin in PBA- or saline-treated KI mice versus saline-treated WT mice. Asterisks indicate significant differences between PBA- and saline-treated KI mice (p < 0.007). Error bars indicate SD (n = 4).
If S-opsin mislocalization is one of the pathogenic factors leading to S-cone degeneration in the KI mice, the decrease in S-opsin mislocalization should be accompanied by increase of S-opsin in the PBA-treated KI mice. To test this possibility, we performed quantitative immunoblot analysis for S-opsin. As shown in Figure 5C, S-opsin was increased ∼2-fold in PBA-treated KI mice compared with saline-treated KI mice.
PBA reduced cone degeneration in the KI mice
To test whether PBA can protect cones from early degeneration in the KI mice, we performed immunohistochemistry for CAR and M-opsin on retinal sections of 10-week-old WT and KI mice treated with PBA or saline for 8 weeks. We first detected CAR, which expresses in both M- and S-cones. Low-magnification images for whole retinal sections showed that the KI mice exhibited a severe loss of cones in the inferior retina (Fig. 6A), which is consistent with our previous result (Samardzija et al., 2009). This observation was confirmed by high-magnification images for central areas of the superior and inferior retinas (Fig. 6B). We then compared with total numbers of CAR-positive cone OS in the retinas of KI mice treated with PBA or saline. Numbers of CAR-positive cone OS in the inferior and superior retinas of PBA-treated KI mice were increased by 38% and 21%, respectively, compared with those in saline-treated KI mice (Fig. 6C). CAR-positive cone OS in PBA-treated KI mice were notably longer than those in saline-treated KI mice (Fig. 6B). Consistent with these observations, immunoblot analysis showed that protein levels of CAR in retinal homogenates were increased by ∼65% in PBA-treated KI mice compared with saline-treated KI mice (Fig. 6D).
Figure 6.
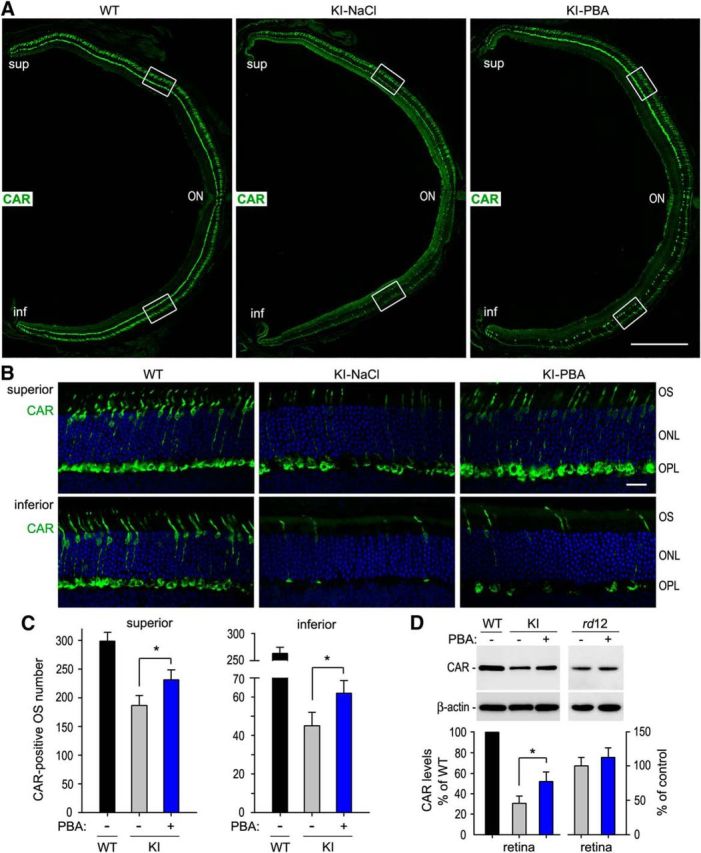
Prolonged cone survival in KI mice treated with PBA. A, Low-magnification images of immunohistochemistry for CAR on retinal sections of 10-week-old WT and KI mice treated with PBA or saline for 8 weeks. ON, Optic nerve. Scale bar, 500 μm. B, High-magnification images showing the superior and inferior central regions highlighted in A. Scale bar, 20 μm. C, Counts of CAR-positive cone OS in the superior and inferior retinas from the indicated mice. D, Quantitative immunoblot analysis of CAR in WT mice, as well as KI and rd12 mice treated with PBA or saline for 8 weeks. Asterisks indicate significant differences between PBA- and saline-treated KI mice (p ≤ 0.01). Note that CAR expression levels in rd12 mice treated with PBA are similar to those in rd12 mice treated with saline. Error bars indicate SD (n = 4).
To know whether PBA improves cone survival in the KI mice by partially rescuing of the mutant RPE65-mediated visual cycle, we treated rd12 (RPE65-null) mice with PBA (50 mg/kg) or saline for 8 weeks, starting at P14. Immunoblot analysis showed that CAR protein levels were not significantly increased in PBA-treated rd12 mice compared with saline-treated rd12 mice (Fig. 6D).
We next counted numbers of M-opsin-positive cone OS in WT and KI mice retinal sections. M-cones were reduced by 45% and 89% in the saline-treated KI mice superior and inferior retinas, respectively, compared with those in WT retinas (Fig. 7A,B). In PBA-treatment KI mice, the numbers of M-cone OS were increased by 28% and 41% in the superior and inferior retinas, respectively, compared with those in saline-treated KI mice (Fig. 7B). In addition, length of M-cone OS was notably longer in PBA-treated KI mice compared with that in saline-treated KI mice (Fig. 7A). Consistent with these observations, quantitative immunoblot analysis showed that M-opsin protein levels were increased by ∼55% in PBA-treated KI retinas compared with saline-treated KI retinas (Fig. 7C).
Figure 7.

PBA reduced M-cone degeneration in KI mice. A, Representative M-opsin immunoreactivity in the superior retinas of 10-week-old WT and KI mice treated with PBA or saline. Scale bar, 20 μm. B, Total numbers of M-cones in the superior and inferior retinal sections of WT and KI mice treated with PBA or saline. C, Immunoblot analysis showing increase of M-opsin in KI mice treated with PBA. Asterisks indicate significant differences between PBA- and saline-treated KI mice (p ≤ 0.01). Error bars indicate SD (n = 4).
PBA improved cone visual function in the KI mice but not in rd12 mice
The partial rescue of RPE65 function (Fig. 3) and cone survival (Figs. 6, 7) prompted us to test whether PBA can improve cone visual function in the KI mice. We measured visual function by ERG on 10-week-old WT and KI mice treated with PBA or saline for 8 weeks. ERG responses were elicited by stimulating mice with a series of achromatic light flashes under a rod-saturating background light (32 cd/m2). Amplitudes of photopic ERG responses to high flash intensities were increased by 30–45% in PBA-treated KI mice compared with saline-treated KI mice (Fig. 8A,B). To confirm whether S-cone vision is improved in PBA-treated KI mice, we recorded ERG responses using 360 nm UV light flashes under a 40 cd/m2 background light. As shown in Figure 8C,D, b-wave amplitudes of S-cone ERG responses to 0.78 and 1.08 log cd · s/m2 UV light flashes were increased ∼90% and ∼52%, respectively, in PBA-treated KI mice compared with those in saline-treated KI mice. In contrast, S-cone ERG responses, elicited with the same UV light flashes, in 10-week-old rd12 mice treatment with PBA for 8 weeks were similar to those in rd12 mice treatment with saline for 8 weeks (Fig. 8C,D).
Figure 8.

PBA improved cone visual function in KI mice but not in rd12 mice. A, Representative photopic ERG in 10-week-old WT and KI mice treated with PBA or saline for 8 weeks. ERG responses were elicited with the indicated flash intensities of white light under a rod-saturating background light. B, Amplitudes of photopic ERG b-waves, evoked with the indicated achromatic light flashes, in WT and KI mice treated with PBA or saline. C, Representative ERG responses of S-cones in 10-week-old WT, KI, and rd12 mice treated with PBA or saline for 8 weeks. Data were acquired using the indicated flash intensities of 360 nm UV light under a rod-saturating background light. D, Amplitudes of S-cone b-waves, evoked with the indicated UV light flashes, in KI and rd12 mice treated with PBA or saline. Asterisks indicate statistically significant differences between PBA- and saline-treated KI mice (p < 0.01). Note the similar b-wave amplitudes in PBA- and saline-treated rd12 mice. Error bars indicate SD (n = 7).
Discussion
This study identified misfolding as the molecular basis for the PSMD13-promoted degradation of ubiquitinated mutant RPE65 in the KI mouse RPE proteasomes. We further demonstrated that PBA, an aromatic short-chain fatty acid functioning as a chemical chaperone, partially rescued R91W RPE65-catalyzed synthesis of the visual chromophores, thereby improved cone survival and vision in the mutant mouse model of LCA.
RPE65 is a highly abundant enzyme (Hamel et al., 1993) with very low catalytic activity (Winston and Rando, 1998; Jin et al., 2005). Therefore, decrease in protein stability will result in a pernicious effect on its function. In this study, we found that the severe decrease of R91W RPE65 in RPE was due to proteasomal degradation (Fig. 1). The copious polyubiquitination and abnormal subcellular localization (Figs. 1B,4B) suggest that R91W RPE65 is misfolded. Our previous studies showed that many other RPE65 mutants were also degraded via the ubiquitin-proteasome pathway (UPP) in culture cells (S. Li et al., 2014, 2015). These results suggest that degradation of RPE65 via the UPP is a common pathogenic mechanism for many missense mutations in RPE65.
The UPP plays a pivotal role in the cellular protein quality control. In this study, we identified PSMD13 as a critical player in eliminating misfolded mutant RPE65 via the UPP in the KI mouse RPE. Knockdown of PSMD13 markedly increased R91W RPE65, including ubiquitinated mutant RPE65, possibly due to reduction of the proteasome activity (Fig. 2A–C,E,J). Conversely, overexpression of PSMD13 reduced full-length mutant RPE65 at 37°C (Fig. 2F–H,K,L), and significantly increased RPE65 fragments cleaved only in the KI, but not in WT, RPE (Fig. 2K). These results are consistent with the previous study in yeast. The yeast PSMD13 homolog (Rpn9) is required for the proteasome activity. It participates in the assembly and/or stability of the 26S proteasome (Takeuchi et al., 1999).
Low temperature has been shown to promote folding of mutant proteins (Denning et al., 1992; Vollrath and Liu, 2006; S. Li et al., 2013b). Consistent with these studies, full-length R91W RPE65 was dramatically increased in RPE at 30°C, even under overexpressing PSMD13 (Fig. 2K,L). Membrane-association and isomerase activity of R91W RPE65 were also increased at 30°C (S. Li et al., 2015). This result and the similar amounts of mutant RPE65 fragments in RPE at 30°C and 37°C suggest that the increase of full-length R91W RPE65 was due to improved folding of R91W RPE65 rather than decrease in the proteasome activity at 30°C. These results also suggest that PSMD13 promotes degradation of misfolded, but not properly folded, RPE65. High expression level of RPE65 and proteasomal overload observed in multiple forms of inherited retinal degeneration (Lobanova et al., 2013) might cause incomplete degradation of misfolded R91W RPE65 (Fig. 2K), which formed aggresome-like structure in RPE (Fig. 4B).
We recently suggested that whether a mutation is mapped on the active or near the active site of RPE65 is a critical parameter in determining whether the isomerase function of the affected RPE65 can be rescued (S. Li et al., 2014, 2015). We found that the isomerase activity of many pathogenic RPE65s with nonactive site mutations, but not with active site mutations, could be rescued at 30°C and/or with chemical chaperones (S. Li et al., 2014, 2015). Importantly, the majority of disease-causing missense mutations are mapped onto the nonactive sites of RPE65. Similar to many disease-causing missense mutations, R91W was mapped onto the nonactive site of the RPE65 crystal structure (Kiser et al., 2012; S. Li et al., 2015), and the activity of R91W RPE65 could be rescued partially by treating cells with low temperature (S. Li et al., 2015). Although most of missense mutations have not been studied in vivo, our in vitro results (S. Li et al., 2014, 2015; Jin et al., 2016) and the results presented in this study suggest that PBA may also rescue the isomerase function in vivo for many disease-associated RPE65s with nonactive site mutations.
Both 11-cis-retinal and 9-cis-retinal function as the light-sensitive chromophore of the visual pigments (Crouch et al., 1975). Administration of QLT091001 (9-cis-retinyl acetate) in patients with RPE65 or LRAT mutation improved visual function (Koenekoop et al., 2014). However, QLT091001 caused some side effects in patients (Koenekoop et al., 2014). In contrast, PBA is a FDA-approved safe medication (Perlmutter, 2002). PBA increased the retinoid isomerase activity and synthesis of 11-cis-retinal and 9-cis-retinal in the KI mice (Fig. 3). Although the isomerase activity in the PBA-treated KI mice RPE was still <5% of WT activity, a recent study on a mouse model with RPE65 P25L knock-in mutation (Y. Li et al., 2015) suggests that the partial rescue of the isomerase function in PBA-treated KI mice is very important. The P25L RPE65 had <10% of WT activity (Lorenz et al., 2008). However, the P25L knock-in mice exhibited almost normal scotopic and photopic ERG responses.
It has been known that association of RPE65 with membranes is critical for its substrate-binding and enzymatic activity (Mata et al., 2004; Jin et al., 2007; Nikolaeva et al., 2009; Takahashi et al., 2009; Golczak et al., 2010). Several missense mutations have been shown to cause aggregation and mislocalization of RPE65 in culture cells (Chen et al., 2006; Takahashi et al., 2006; S. Li et al., 2014, 2015). Similarly, we observed that R91W RPE65 formed aggresome-like inclusion bodies in the RPE (Fig. 4B). This abnormal subcellular distribution might accelerate the decrease of 11-cis-retinol synthesis by dissociating RPE65 from the endoplasmic reticulum where LRAT synthesizes the substrates for RPE65. In addition, the aggregate formation of misfolded RPE65 may be involved in the pathogenic mechanisms for RPE atrophy observed in LCA patients with recessive RPE65 missense mutations (Cideciyan et al., 2015). Importantly, PBA-treatment significantly increased normal subcellular distribution of R91W RPE65 evidenced by the increase in membrane-association and colocalization of the mutant RPE65 with LRAT (Fig. 4A,B). These results and the partial rescue of isomerase activity (Fig. 3B) suggest that PBA enhanced proper folding of R91W RPE65 and reduced degradation of mutant RPE65 by the protein quality control system. Decrease in RPE65 misfolding could reduce the potential cytotoxic effects of RPE65 mutants in patients' RPE.
The most clinically significant result of this study is the improvement of cone survival and vision in the PBA-treated KI mice (Figs. 5–8). Slowing the progression of cone loss could greatly extend the years of useful vision of patients. PBA-mediated alleviation of cone and vision loss may include at least two different mechanisms: (1) PBA helped folding of R91W RPE65, which in turn improved cone survival and function by increasing supply of 11-cis-retinal and 9-cis-retinal; and (2) PBA promoted folding of cone opsins mislocalized due to shortage of chromophore (Samardzija et al., 2009). In Rpe65−/− mice, cone opsins are mislocalized (Rohrer et al., 2005) and M-opsin undergoes proteasomal degradation (Sato et al., 2012). In Lrat−/− retina lacking 11-cis-retinal (Batten et al., 2004), S-opsin is mislocalized (Zhang et al., 2008) and colocalized with ubiquitin (Zhang et al., 2011). These observations suggest that cone opsins are misfolded in the absence or shortage of 11-cis-retinal. As a chemical chaperone, PBA might act on misfolded R91W RPE65 and cone opsins in the KI mice. However, the lack of increase in CAR protein level and S-cone ERG responses in PBA-treated rd12 mice indicates that PBA-mediated partial rescue of R91W RPE65 function contributed to the improvement of the visual cycle, cone survival, and vision in KI mice.
It is remarkable that we detected S-cone function with UV-stimulation even in saline-treated KI mice. Our earlier genetic examination of rod and cone function in the KI mice suggested that rods entrap most of the available 11-cis-retinal leading to undetectable cone function after stimulation with white light (Samardzija et al., 2009). In contrast to white light, however, UV-light of 360 nm activates S-cones with a highly increased efficiency because S-cones have their absorption optimum at ∼355 nm (Jacobs et al., 1991; Lyubarsky et al., 1999). Thus, a small amount of bleachable cone opsin may lead to a recordable response after UV stimulation.
Recently, several proteins, including MYO7A, fatty acid transport proteins (FATPs), and elongation of very long-chain fatty acids-like 1 (ELOVL1), have been identified as regulators of RPE65 (Guignard et al., 2010; Lopes et al., 2011; S. Li et al., 2013a; Eroglu et al., 2016). In addition, lignoceroyl (C24:0)-CoA, a major product of FATP, has also been shown to inhibit synthesis of 11-cis-retinol catalyzed by RPE65 (S. Li et al., 2013a). Because FATP4 inhibits synthesis of 11-cis-retinol by competing with RPE65 for the substrate of RPE65 (S. Li et al., 2013a), it would be interesting to test whether knock-out of FATP4 can increase synthesis of 11-cis-retinol and 11-cis-retinal in the KI mice RPE.
Although PBA can be used as a noninvasive stand-alone treatment, it can also supplement RPE65 gene therapy for patients with RPE65 mutations. A combined application of PBA, AAV-RPE65, and tauroursodeoxycholic acid (Zhang et al., 2012) may be a powerful therapeutic intervention that facilitates long-term improvement of the visual cycle, cone survival and daylight vision in patients with RPE65 mutations.
Footnotes
This work was supported by grants from NIH (EY021208 to M. J. and GM103340 to LSU Neuroscience COBRE facilities), RPB, and Lions Eye Foundation (to LSU Department of Ophthalmology). We thank Drs Marcin Golczak and Krzysztof Palczewski for the antibody against LRAT.
The authors declare no competing financial interests.
References
- Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, Petersen-Jones S, Bhattacharya SS, Thrasher AJ, Fitzke FW, Carter BJ, Rubin GS, Moore AT, Ali RR. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med. 2008;358:2231–2239. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- Bainbridge JW, Mehat MS, Sundaram V, Robbie SJ, Barker SE, Ripamonti C, Georgiadis A, Mowat FM, Beattie SG, Gardner PJ, Feathers KL, Luong VA, Yzer S, Balaggan K, Viswanathan A, de Ravel TJ, Casteels I, Holder GE, Tyler N, Fitzke FW, et al. Long-term effect of gene therapy on Leber's congenital amaurosis. N Engl J Med. 2015;372:1887–1897. doi: 10.1056/NEJMoa1414221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batten ML, Imanishi Y, Maeda T, Tu DC, Moise AR, Bronson D, Possin D, Van Gelder RN, Baehr W, Palczewski K. Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J Biol Chem. 2004;279:10422–10432. doi: 10.1074/jbc.M312410200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereta G, Kiser PD, Golczak M, Sun W, Heon E, Saperstein DA, Palczewski K. Impact of retinal disease-associated RPE65 mutations on retinoid isomerization. Biochemistry. 2008;47:9856–9865. doi: 10.1021/bi800905v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonapace G, Waheed A, Shah GN, Sly WS. Chemical chaperones protect from effects of apoptosis-inducing mutation in carbonic anhydrase IV identified in retinitis pigmentosa 17. Proc Natl Acad Sci U S A. 2004;101:12300–12305. doi: 10.1073/pnas.0404764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Moiseyev G, Takahashi Y, Ma JX. Impacts of two point mutations of RPE65 from Leber's congenital amaurosis on the stability, subcellular localization and isomerohydrolase activity of RPE65. FEBS Lett. 2006;580:4200–4204. doi: 10.1016/j.febslet.2006.06.078. [DOI] [PubMed] [Google Scholar]
- Cideciyan AV, Aleman TS, Boye SL, Schwartz SB, Kaushal S, Roman AJ, Pang JJ, Sumaroka A, Windsor EA, Wilson JM, Flotte TR, Fishman GA, Heon E, Stone EM, Byrne BJ, Jacobson SG, Hauswirth WW. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci U S A. 2008;105:15112–15117. doi: 10.1073/pnas.0807027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Jacobson SG, Beltran WA, Sumaroka A, Swider M, Iwabe S, Roman AJ, Olivares MB, Schwartz SB, Komáromy AM, Hauswirth WW, Aguirre GD. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci U S A. 2013;110:E517–E525. doi: 10.1073/pnas.1218933110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Swider M, Jacobson SG. Autofluorescence imaging with near-infrared excitation: normalization by reflectance to reduce signal from choroidal fluorophores. Invest Ophthalmol Vis Sci. 2015;56:3393–3406. doi: 10.1167/iovs.15-16726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch R, Purvin V, Nakanishi K, Ebrey T. Isorhodopsin II: artificial photosensitive pigment formed from 9,13-dicis retinal. Proc Natl Acad Sci U S A. 1975;72:1538–1542. doi: 10.1073/pnas.72.4.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Eroglu A, Gentleman S, Poliakov E, Redmond TM. Inhibition of RPE65 retinol isomerase activity by inhibitors of lipid metabolism. J Biol Chem. 2016;291:4966–4973. doi: 10.1074/jbc.M115.685651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Rohrer B, Moiseyev G, Ma JX, Crouch RK. Isorhodopsin rather than rhodopsin mediates rod function in RPE65 knock-out mice. Proc Natl Acad Sci U S A. 2003;100:13662–13667. doi: 10.1073/pnas.2234461100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Rohrer B, Frederick JM, Baehr W, Crouch RK. Rpe65−/− and Lrat−/− mice: comparable models of Leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2008;49:2384–2389. doi: 10.1167/iovs.08-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felius J, Thompson DA, Khan NW, Bingham EL, Jamison JA, Kemp JA, Sieving PA. Clinical course and visual function in a family with mutations in the RPE65 gene. Arch Ophthalmol. 2002;120:55–61. doi: 10.1001/archopht.120.1.55. [DOI] [PubMed] [Google Scholar]
- Golczak M, Kuksa V, Maeda T, Moise AR, Palczewski K. Positively charged retinoids are potent and selective inhibitors of the trans-cis isomerization in the retinoid (visual) cycle. Proc Natl Acad Sci U S A. 2005;102:8162–8167. doi: 10.1073/pnas.0503318102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golczak M, Kiser PD, Lodowski DT, Maeda A, Palczewski K. Importance of membrane structural integrity for RPE65 retinoid isomerization activity. J Biol Chem. 2010;285:9667–9682. doi: 10.1074/jbc.M109.063941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton MJ, Gal A. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet. 1997;17:194–197. doi: 10.1038/ng1097-194. [DOI] [PubMed] [Google Scholar]
- Guignard TJ, Jin M, Pequignot MO, Li S, Chassigneux Y, Chekroud K, Guillou L, Richard E, Hamel CP, Brabet P. FATP1 inhibits 11-cis retinol formation via interaction with the visual cycle retinoid isomerase RPE65 and lecithin:retinol acyltransferase. J Biol Chem. 2010;285:18759–18768. doi: 10.1074/jbc.M109.064329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel CP, Tsilou E, Pfeffer BA, Hooks JJ, Detrick B, Redmond TM. Molecular cloning and expression of RPE65, a novel retinal pigment epithelium-specific microsomal protein that is post-transcriptionally regulated in vitro. J Biol Chem. 1993;268:15751–15757. [PubMed] [Google Scholar]
- Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, Wang L, Conlon TJ, Boye SL, Flotte TR, Byrne BJ, Jacobson SG. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19:979–990. doi: 10.1089/hum.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs GH, Neitz J, Deegan JF., 2nd Retinal receptors in rodents maximally sensitive to ultraviolet light. Nature. 1991;353:655–656. doi: 10.1038/353655a0. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Aleman TS, Cideciyan AV, Sumaroka A, Schwartz SB, Windsor EA, Traboulsi EI, Heon E, Pittler SJ, Milam AH, Maguire AM, Palczewski K, Stone EM, Bennett J. Identifying photoreceptors in blind eyes caused by RPE65 mutations: prerequisite for human gene therapy success. Proc Natl Acad Sci U S A. 2005;102:6177–6182. doi: 10.1073/pnas.0500646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Aleman TS, Cideciyan AV, Heon E, Golczak M, Beltran WA, Sumaroka A, Schwartz SB, Roman AJ, Windsor EA, Wilson JM, Aguirre GD, Stone EM, Palczewski K. Human cone photoreceptor dependence on RPE65 isomerase. Proc Natl Acad Sci U S A. 2007;104:15123–15128. doi: 10.1073/pnas.0706367104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Aleman TS, Sumaroka A, Windsor EA, Schwartz SB, Heon E, Stone EM. Photoreceptor layer topography in children with leber congenital amaurosis caused by RPE65 mutations. Invest Ophthalmol Vis Sci. 2008;49:4573–4577. doi: 10.1167/iovs.08-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Aleman TS, Cideciyan AV, Roman AJ, Sumaroka A, Windsor EA, Schwartz SB, Heon E, Stone EM. Defining the residual vision in leber congenital amaurosis caused by RPE65 mutations. Invest Ophthalmol Vis Sci. 2009;50:2368–2375. doi: 10.1167/iovs.08-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Roman AJ, Sumaroka A, Schwartz SB, Heon E, Hauswirth WW. Improvement and decline in vision with gene therapy in childhood blindness. N Engl J Med. 2015;372:1920–1926. doi: 10.1056/NEJMoa1412965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Li S, Moghrabi WN, Sun H, Travis GH. Rpe65 is the retinoid isomerase in bovine retinal pigment epithelium. Cell. 2005;122:449–459. doi: 10.1016/j.cell.2005.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Yuan Q, Li S, Travis GH. Role of LRAT on the retinoid isomerase activity and membrane association of Rpe65. J Biol Chem. 2007;282:20915–20924. doi: 10.1074/jbc.M701432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Li S, Nusinowitz S, Lloyd M, Hu J, Radu RA, Bok D, Travis GH. The role of interphotoreceptor retinoid-binding protein on the translocation of visual retinoids and function of cone photoreceptors. J Neurosci. 2009;29:1486–1495. doi: 10.1523/JNEUROSCI.3882-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Li S, Hu J, Jin HH, Jacobson SG, Bok D. Functional rescue of retinal degeneration-associated mutant RPE65 proteins. Adv Exp Med Biol. 2016;854:525–532. doi: 10.1007/978-3-319-17121-0_70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaschula CH, Jin MH, Desmond-Smith NS, Travis GH. Acyl CoA:retinol acyltransferase (ARAT) activity is present in bovine retinal pigment epithelium. Exp Eye Res. 2006;82:111–121. doi: 10.1016/j.exer.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Kaylor JJ, Radu RA, Bischoff N, Makshanoff J, Hu J, Lloyd M, Eddington S, Bianconi T, Bok D, Travis GH. Diacylglycerol o-acyltransferase type-1 synthesizes retinyl esters in the retina and retinal pigment epithelium. PLoS One. 2015;10:e0125921. doi: 10.1371/journal.pone.0125921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser PD, Farquhar ER, Shi W, Sui X, Chance MR, Palczewski K. Structure of RPE65 isomerase in a lipidic matrix reveals roles for phospholipids and iron in catalysis. Proc Natl Acad Sci U S A. 2012;109:E2747–E2756. doi: 10.1073/pnas.1212025109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenekoop RK, Sui R, Sallum J, van den Born LI, Ajlan R, Khan A, den Hollander AI, Cremers FP, Mendola JD, Bittner AK, Dagnelie G, Schuchard RA, Saperstein DA. Oral 9-cis retinoid for childhood blindness due to Leber congenital amaurosis caused by RPE65 or LRAT mutations: an open-label phase 1b trial. Lancet. 2014;384:1513–1520. doi: 10.1016/S0140-6736(14)60153-7. [DOI] [PubMed] [Google Scholar]
- Li S, Lee J, Zhou Y, Gordon WC, Hill JM, Bazan NG, Miner JH, Jin M. Fatty acid transport protein 4 (FATP4) prevents light-induced degeneration of cone and rod photoreceptors by inhibiting RPE65 isomerase. J Neurosci. 2013a;33:3178–3189. doi: 10.1523/JNEUROSCI.2428-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Yang Z, Hu J, Gordon WC, Bazan NG, Haas AL, Bok D, Jin M. Secretory defect and cytotoxicity: the potential disease mechanisms for the retinitis pigmentosa (RP)-associated interphotoreceptor retinoid-binding protein (IRBP) J Biol Chem. 2013b;288:11395–11406. doi: 10.1074/jbc.M112.418251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Izumi T, Hu J, Jin HH, Siddiqui AA, Jacobson SG, Bok D, Jin M. Rescue of enzymatic function for disease-associated RPE65 proteins containing various missense mutations in non-active sites. J Biol Chem. 2014;289:18943–18956. doi: 10.1074/jbc.M114.552117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hu J, Jin RJ, Aiyar A, Jacobson SG, Bok D, Jin M. Temperature-sensitive retinoid isomerase activity of RPE65 mutants associated with Leber congenital amaurosis. J Biochem. 2015;158:115–125. doi: 10.1093/jb/mvv028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yu S, Duncan T, Li Y, Liu P, Gene E, Cortes-Pena Y, Qian H, Dong L, Redmond TM. Mouse model of human RPE65 P25L hypomorph resembles wild type under normal light rearing but is fully resistant to acute light damage. Hum Mol Genet. 2015;24:4417–4428. doi: 10.1093/hmg/ddv178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanova ES, Finkelstein S, Skiba NP, Arshavsky VY. Proteasome overload is a common stress factor in multiple forms of inherited retinal degeneration. Proc Natl Acad Sci U S A. 2013;110:9986–9991. doi: 10.1073/pnas.1305521110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes VS, Gibbs D, Libby RT, Aleman TS, Welch DL, Lillo C, Jacobson SG, Radu RA, Steel KP, Williams DS. The Usher 1B protein, MYO7A, is required for normal localization and function of the visual retinoid cycle enzyme, RPE65. Hum Mol Genet. 2011;20:2560–2570. doi: 10.1093/hmg/ddr155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz B, Poliakov E, Schambeck M, Friedburg C, Preising MN, Redmond TM. A comprehensive clinical and biochemical functional study of a novel RPE65 hypomorphic mutation. Invest Ophthalmol Vis Sci. 2008;49:5235–5242. doi: 10.1167/iovs.07-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotery AJ, Namperumalsamy P, Jacobson SG, Weleber RG, Fishman GA, Musarella MA, Hoyt CS, Héon E, Levin A, Jan J, Lam B, Carr RE, Franklin A, Radha S, Andorf JL, Sheffield VC, Stone EM. Mutation analysis of 3 genes in patients with Leber congenital amaurosis. Arch Ophthalmol. 2000;118:538–543. doi: 10.1001/archopht.118.4.538. [DOI] [PubMed] [Google Scholar]
- Lyubarsky AL, Falsini B, Pennesi ME, Valentini P, Pugh EN., Jr UV- and midwave-sensitive cone-driven retinal responses of the mouse: a possible phenotype for coexpression of cone photopigments. J Neurosci. 1999;19:442–455. doi: 10.1523/JNEUROSCI.19-01-00442.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire AM, Simonelli F, Pierce EA, Pugh EN, Jr, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, Rossi S, Lyubarsky A, Arruda VR, Konkle B, Stone E, Sun J, Jacobs J, Dell'Osso L, Hertle R, Ma JX, et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlhens F, Bareil C, Griffoin JM, Zrenner E, Amalric P, Eliaou C, Liu SY, Harris E, Redmond TM, Arnaud B, Claustres M, Hamel CP. Mutations in RPE65 cause Leber's congenital amaurosis. Nat Genet. 1997;17:139–141. doi: 10.1038/ng1097-139. [DOI] [PubMed] [Google Scholar]
- Mata NL, Moghrabi WN, Lee JS, Bui TV, Radu RA, Horwitz J, Travis GH. Rpe65 is a retinyl ester binding protein that presents insoluble substrate to the isomerase in retinal pigment epithelial cells. J Biol Chem. 2004;279:635–643. doi: 10.1074/jbc.M310042200. [DOI] [PubMed] [Google Scholar]
- Moiseyev G, Crouch RK, Goletz P, Oatis J, Jr, Redmond TM, Ma JX. Retinyl esters are the substrate for isomerohydrolase. Biochemistry. 2003;42:2229–2238. doi: 10.1021/bi026911y. [DOI] [PubMed] [Google Scholar]
- Moiseyev G, Chen Y, Takahashi Y, Wu BX, Ma JX. RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc Natl Acad Sci U S A. 2005;102:12413–12418. doi: 10.1073/pnas.0503460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, Dryja TP. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc Natl Acad Sci U S A. 1998;95:3088–3093. doi: 10.1073/pnas.95.6.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaeva O, Takahashi Y, Moiseyev G, Ma JX. Purified RPE65 shows isomerohydrolase activity after reassociation with a phospholipid membrane. FEBS J. 2009;276:3020–3030. doi: 10.1111/j.1742-4658.2009.07021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaeva O, Takahashi Y, Moiseyev G, Ma JX. Negative charge of the glutamic acid 417 residue is crucial for isomerohydrolase activity of RPE65. Biochem Biophys Res Commun. 2010;391:1757–1761. doi: 10.1016/j.bbrc.2009.12.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh EC, Cheng H, Hao H, Jia L, Khan NW, Swaroop A. Rod differentiation factor NRL activates the expression of nuclear receptor NR2E3 to suppress the development of cone photoreceptors. Brain Res. 2008;1236:16–29. doi: 10.1016/j.brainres.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang JJ, Chang B, Hawes NL, Hurd RE, Davisson MT, Li J, Noorwez SM, Malhotra R, McDowell JH, Kaushal S, Hauswirth WW, Nusinowitz S, Thompson DA, Heckenlively JR. Retinal degeneration 12 (rd12): a new, spontaneously arising mouse model for human Leber congenital amaurosis (LCA) Mol Vis. 2005;11:152–162. [PubMed] [Google Scholar]
- Perlmutter DH. Chemical chaperones: a pharmacological strategy for disorders of protein folding and trafficking. Pediatr Res. 2002;52:832–836. doi: 10.1203/00006450-200212000-00004. [DOI] [PubMed] [Google Scholar]
- Philp AR, Jin M, Li S, Schindler EI, Iannaccone A, Lam BL, Weleber RG, Fishman GA, Jacobson SG, Mullins RF, Travis GH, Stone EM. Predicting the pathogenicity of RPE65 mutations. Hum Mutat. 2009;30:1183–1188. doi: 10.1002/humu.21033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond TM, Yu S, Lee E, Bok D, Hamasaki D, Chen N, Goletz P, Ma JX, Crouch RK, Pfeifer K. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet. 1998;20:344–351. doi: 10.1038/3813. [DOI] [PubMed] [Google Scholar]
- Redmond TM, Poliakov E, Yu S, Tsai JY, Lu Z, Gentleman S. Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc Natl Acad Sci U S A. 2005;102:13658–13663. doi: 10.1073/pnas.0504167102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer B, Lohr HR, Humphries P, Redmond TM, Seeliger MW, Crouch RK. Cone opsin mislocalization in Rpe65−/− mice: a defect that can be corrected by 11-cis retinal. Invest Ophthalmol Vis Sci. 2005;46:3876–3882. doi: 10.1167/iovs.05-0533. [DOI] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. Use of protein repair therapy in the treatment of cystic fibrosis. Curr Opin Pediatr. 1998;10:250–255. doi: 10.1097/00008480-199806000-00005. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Winston A, Lim YH, Gilbert BA, Rando RR, Bok D. Molecular and biochemical characterization of lecithin retinol acyltransferase. J Biol Chem. 1999;274:3834–3841. doi: 10.1074/jbc.274.6.3834. [DOI] [PubMed] [Google Scholar]
- Samardzija M, von Lintig J, Tanimoto N, Oberhauser V, Thiersch M, Remé CE, Seeliger M, Grimm C, Wenzel A. R91W mutation in Rpe65 leads to milder early-onset retinal dystrophy due to the generation of low levels of 11-cis-retinal. Hum Mol Genet. 2008;17:281–292. doi: 10.1093/hmg/ddm304. [DOI] [PubMed] [Google Scholar]
- Samardzija M, Tanimoto N, Kostic C, Beck S, Oberhauser V, Joly S, Thiersch M, Fahl E, Arsenijevic Y, von Lintig J, Wenzel A, Seeliger MW, Grimm C. In conditions of limited chromophore supply rods entrap 11-cis-retinal leading to loss of cone function and cell death. Hum Mol Genet. 2009;18:1266–1275. doi: 10.1093/hmg/ddp026. [DOI] [PubMed] [Google Scholar]
- Sato K, Ozaki T, Ishiguro S, Nakazawa M. M-opsin protein degradation is inhibited by MG-132 in Rpe65−/− retinal explant culture. Mol Vis. 2012;18:1516–1525. [PMC free article] [PubMed] [Google Scholar]
- Sato K, Li S, Gordon WC, He J, Liou GI, Hill JM, Travis GH, Bazan NG, Jin M. Receptor interacting protein kinase-mediated necrosis contributes to cone and rod photoreceptor degeneration in the retina lacking interphotoreceptor retinoid-binding protein. J Neurosci. 2013;33:17458–17468. doi: 10.1523/JNEUROSCI.1380-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Chen Y, Moiseyev G, Ma JX. Two point mutations of RPE65 from patients with retinal dystrophies decrease the stability of RPE65 protein and abolish its isomerohydrolase activity. J Biol Chem. 2006;281:21820–21826. doi: 10.1074/jbc.M603725200. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Moiseyev G, Ablonczy Z, Chen Y, Crouch RK, Ma JX. Identification of a novel palmitylation site essential for membrane association and isomerohydrolase activity of RPE65. J Biol Chem. 2009;284:3211–3218. doi: 10.1074/jbc.M807248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi J, Fujimuro M, Yokosawa H, Tanaka K, Toh-e A. Rpn9 is required for efficient assembly of the yeast 26S proteasome. Mol Cell Biol. 1999;19:6575–6584. doi: 10.1128/MCB.19.10.6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DA, Gyürüs P, Fleischer LL, Bingham EL, McHenry CL, Apfelstedt-Sylla E, Zrenner E, Lorenz B, Richards JE, Jacobson SG, Sieving PA, Gal A. Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Invest Ophthalmol Vis Sci. 2000;41:4293–4299. [PubMed] [Google Scholar]
- Vollrath D, Liu Y. Temperature sensitive secretion of mutant myocilins. Exp Eye Res. 2006;82:1030–1036. doi: 10.1016/j.exer.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Winston A, Rando RR. Regulation of isomerohydrolase activity in the visual cycle. Biochemistry. 1998;37:2044–2050. doi: 10.1021/bi971908d. [DOI] [PubMed] [Google Scholar]
- Wright CB, Chrenek MA, Foster SL, Duncan T, Redmond TM, Pardue MT, Boatright JH, Nickerson JM. Complementation test of Rpe65 knock-out and tvrm148. Invest Ophthalmol Vis Sci. 2013;54:5111–5122. doi: 10.1167/iovs.13-12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Fan J, Li S, Karan S, Rohrer B, Palczewski K, Frederick JM, Crouch RK, Baehr W. Trafficking of membrane-associated proteins to cone photoreceptor outer segments requires the chromophore 11-cis-retinal. J Neurosci. 2008;28:4008–4014. doi: 10.1523/JNEUROSCI.0317-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Zhang N, Baehr W, Fu Y. Cone opsin determines the time course of cone photoreceptor degeneration in Leber congenital amaurosis. Proc Natl Acad Sci U S A. 2011;108:8879–8884. doi: 10.1073/pnas.1017127108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Baehr W, Fu Y. Chemical chaperone TUDCA preserves cone photoreceptors in a mouse model of Leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2012;53:3349–3356. doi: 10.1167/iovs.12-9851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Znoiko SL, Rohrer B, Lu K, Lohr HR, Crouch RK, Ma JX. Downregulation of cone-specific gene expression and degeneration of cone photoreceptors in the Rpe65−/− mouse at early ages. Invest Ophthalmol Vis Sci. 2005;46:1473–1479. doi: 10.1167/iovs.04-0653. [DOI] [PubMed] [Google Scholar]
- Zode GS, Kuehn MH, Nishimura DY, Searby CC, Mohan K, Grozdanic SD, Bugge K, Anderson MG, Clark AF, Stone EM, Sheffield VC. Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J Clin Invest. 2011;121:3542–3553. doi: 10.1172/JCI58183. [DOI] [PMC free article] [PubMed] [Google Scholar]