

Abstract
Anti‐transferrin receptor (TfR)‐based bispecific antibodies have shown promise for boosting antibody uptake in the brain. Nevertheless, there are limited data on the molecular properties, including affinity required for successful development of TfR‐based therapeutics. A complex nonmonotonic relationship exists between affinity of the anti‐TfR arm and brain uptake at therapeutically relevant doses. However, the quantitative nature of this relationship and its translatability to humans is heretofore unexplored. Therefore, we developed a mechanistic pharmacokinetic‐pharmacodynamic (PK‐PD) model for bispecific anti‐TfR/BACE1 antibodies that accounts for antibody‐TfR interactions at the blood‐brain barrier (BBB) as well as the pharmacodynamic (PD) effect of anti‐BACE1 arm. The calibrated model correctly predicted the optimal anti‐TfR affinity required to maximize brain exposure of therapeutic antibodies in the cynomolgus monkey and was scaled to predict the optimal affinity of anti‐TfR bispecifics in humans. Thus, this model provides a framework for testing critical translational predictions for anti‐TfR bispecific antibodies, including choice of candidate molecule for clinical development.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? ☑ Intermediate affinity antibodies against TfR have been demonstrated to cross BBB at pharmacologically relevant levels in the mouse model. Two antibodies against primate‐TfR have also been shown to cross the BBB but the properties of an optimal antibody are unexplored. ☑ WHAT QUESTION DID THIS STUDY ADDRESS? ☑ How can preclinical data be utilized to predict the optimal anti‐TfR affinity for human‐brain penetration and expected clinical efficacy of anti‐TfR bispecific compared to corresponding bivalent antibody for a range of targets. ☑ WHAT THIS STUDY ADDS TO OUR KNOWLEDGE ☑ The modeling framework is capable of predicting antibody PK and CSF PD for a wide range of brain‐targeted antibody characteristics in nonhuman primates. The workflow allows predictions for expected human response to anti‐TfR bispecifics targeting brain‐targets at varied concentrations and turnover rates. ☑ HOW THIS MIGHT CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS ☑ The model prospectively specifies criteria for optimal antibody design and translation to clinical setting. It provides clearly defined clinically testable predictions for expected human response to anti‐TfR platform and thus helps validate its clinical utility.
The biology of the blood‐brain barrier (BBB) and the mechanisms by which it regulates the passage of molecules from the vascular space to the brain has been an important subject of research in the last few decades.1, 2 The presence of tight junctions between endothelial cells in brain capillaries impedes the passage of large molecules, including antibodies across the endothelial barrier. An important mechanism that allows the transport of large molecules across the BBB is specific binding to receptors that internalize and release the ligand across the capillary endothelium (e.g., the transferrin receptor [TfR]‐transferrin pair).3, 4 The TfR present on capillary endothelium binds and internalizes the ligand, subsequently releasing it on the luminal side of the endothelial cell into the brain tissue. Multiple attempts have been made in the past to utilize this system for delivery of therapeutic antibodies across the BBB.5, 6, 7, 8, 9, 10 In previous studies, micro doses of radiolabeled antibody with a high affinity against TfR successfully crossed the vascular wall.10 Yet, whether the antibodies penetrated into the brain at pharmacologically relevant doses remained to be tested.
Previous works have clearly demonstrated the existence of a nonmonotonic relationship between brain uptake and affinity of anti‐TfR antibodies in the mouse.11, 12 These studies utilized anti‐TfR as the brain targeting arm (affinity ranging from 10s–1000s of nM), and anti‐BACE1 (β‐amyloid precursor protein cleavage enzyme) as the therapeutic arm. BACE1 is an enzyme that cleaves membrane amyloid precursor protein and releases soluble Aβ into the brain interstitium. Inhibition of BACE1 activity leads to reduction in soluble‐Aβ levels in the brain, which serves as an easily measured preclinical brain‐pharmacodynamic (PD) readout.
The authors showed that high affinity anti‐TfR antibodies bound TfR tightly and were subsequently internalized but degraded in lysosomes. Therefore, they were less likely to be released from the TfR and penetrate into the brain tissue.13 On the other hand, very low affinity anti‐TfR antibodies were not efficiently transported across the BBB because of low binding to TfR.11, 12 Antibodies with intermediate affinity to TfR yielded the best delivery by balancing binding of TfR on the luminal side and efficient release to the brain tissue. Subsequently, antibodies were generated against cynomolgus monkey TfR that demonstrated BBB penetration and delivery into the brain tissue.14 In this cynomolgus monkey study, cerebrospinal fluid (CSF) Aβ was used as a biomarker for brain Aβ. Previous studies have demonstrated that Aβ lowering in the CSF was predictive of the Aβ lowering in the brains in mice, rats, and guinea pigs for various enzymes involved in cleavage of amyloid precursor protein.15 Accordingly, previous pharmacokinetic‐pharmacodynamic (PK‐PD) models for small molecule inhibitors of amyloid precursor protein cleavage have successfully utilized CSF Aβ level as readout for the PD effect of the drug in the brain.16
Interestingly, the optimal anti‐TfR affinity required for maximal brain exposure was different in mice when compared to cynomolgus monkeys.12, 14 In mice, lower affinity anti‐TfR bispecifics (with equilibrium dissociation constant, KD ∼ 600 nM) penetrated the brain better than higher anti‐TfR affinity bispecifics (KD ∼ 32 nM). In contrast, the trend seemed to be shifted in cynomolgus monkeys, with higher affinity antibodies (KD ∼ 37 nM) penetrating better than the lower affinity antibodies (KD ∼ 1650 nM). To identify the antibody characteristics that would result in maximal brain exposure, it was important to quantify the relationship between brain exposure and affinity of antibodies for TfR in a primate model.
Previously, Gadkar et al.17 developed a PK‐PD model for anti‐TfR/BACE1 antibodies in mice. The model accounts for target‐mediated clearance of anti‐TfR bispecific antibodies in plasma using a Michaelis‐Menten approximation and related TfR‐affinity to antibody uptake into the brain tissue using an empirical function fitted to data from three different anti‐TfR/BACE1 bispecifics of varying TfR affinity and one non‐TfR binding antibody. The model successfully described the nonmonotonic TfR affinity‐dependence for brain uptake of antibody and thus Aβ reduction in the brain. However, data from multiple anti‐TfR/BACE1 antibodies are needed to parametrize the relationships between binding affinity and the target‐mediated processes of clearance and brain uptake. Furthermore, the form of the empirical function for brain uptake of antibody was chosen ad hoc based on data from numerous anti‐TfR/BACE1 bispecifics. The translational relevance of this empirical model is not known, nor is its applicability to other potential shuttling mechanisms that may be of interest in the future.
This article presents a novel PK‐PD model with mechanistic detail for systemic target‐mediated clearance and the affinity‐dependent brain uptake that have been calibrated and validated for data from the cynomolgus monkey and utilized for predictions of potential human studies. The model accounts for systemic‐clearance and brain‐uptake by representing anti‐TfR interactions in systemic compartment and at the BBB, respectively. Finally, the model represents the Aβ production and clearance in the brain, and the PD readout of brain Aβ reduction in response to inhibition of the BACE1 enzyme. This model was first calibrated with previously published cynomolgus monkey PK‐PD data obtained using two anti‐TfR/BACE1 bispecific antibodies. PK‐PD data from three additional bispecific antibodies with different affinities were reserved for model validation. We show that the model quantitatively predicts the systemic pharmacokinetic (PK) and CSF Aβ reduction for these three additional antibodies without requiring additional tuning of parameters or model structure. The model was then applied to predict the optimal affinity in the cynomolgus monkeys and translational predictions for ideal anti‐TfR bispecific antibodies in humans. This work allows for greater insight into the multiple mechanisms, including steps in the transcytosis process, by which anti‐TfR affinity influences brain uptake. Thus, it will aid in the design of anti‐TfR antibodies with optimal affinity and subsequent clinical studies to maximize the therapeutic potential for this brain‐targeting platform. Furthermore, using this framework, we compare expected target engagement for anti‐TfR bispecific antibodies and its corresponding bivalent antibodies. We demonstrate that the most suitable choice depends on the target level, target turnover rate, and dosing regimen, and therefore the anti‐TfR bispecific platform should not be treated as a “one size fits all” solution to targeting in the brain.
MATERIALS AND METHODS
Structural model development
The PK‐PD model includes two modules: systemic PK, which interacts with brain‐uptake that ultimately drives the downstream PD. The systemic PK is represented with a two‐compartment model along with a mechanistic representation of target‐mediated drug disposition whereby the antibody clears from the body because of TfR‐binding and degradation (Figure 1 a). The novel representation of brain‐uptake depicted in Figure 1 b, is based on two mechanisms of transcytosis. The first mechanism is TfR‐mediated transcytosis. The antibody in the capillary reversibly binds to TfR at the BBB and the antibody‐TfR complex irreversibly internalizes into the capillary endothelium. Once internalized, the antibody‐TfR complex either dissociates or is degraded.13 The dissociated antibody can then be released into the brain. The second mechanism is the non‐TfR‐mediated uptake of antibodies into the brain via nonspecific mechanisms. It is assumed that all antibodies are passively cleared from the brain at the same rate. Active TfR‐mediated clearance from the brain is considered to be subsumed within the passive clearance term because the measurements allow us to estimate the net flux of antibody from the central to brain compartment but not the individual influx and outflux terms.
Figure 1.
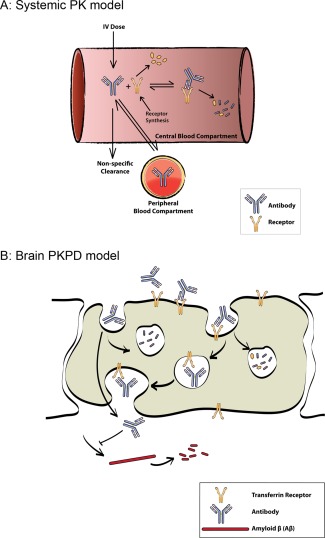
Schematic of pharmacokinetic‐pharmacodynamic (PK‐PD) model. (a) Mechanistic target‐mediated drug disposition PK model that accounts for explicit binding of antibody to systemic transferrin receptor (TfR) and subsequent target‐mediated degradation. (b) Mechanism‐based model for binding of antibodies to TfR on the surface of vascular endothelium, internalization of complex, antibody‐receptor dissociation in endosomes, and transcytosis of antibodies to the brain tissue. Finally, the antibody clears from the brain. The PD model accounts for Aβ production and clearance from the brain tissue.
The PD module is based on continuous production and clearance of Aβ (Figure 1 b). Engagement of BACE1 by the antibody reduces the production rate of Aβ. This inhibition of the production rate of Aβ is represented by a Hill function term within the production rate.16, 17
The detailed structure and the ordinary differential equations that describe the mechanistic model are provided in the Supplementary Material.
Calibration and validation methodology
Model parameters were optimized using particle swarm optimization. The application of particle swarm optimization to parameterizing a model that mechanistically represents complex biological processes has been described previously.18, 19 The objective function to be minimized was defined as the difference between model simulations and experimental measurements normalized by the SD of experimental measurement. Because particle swarm optimization is a heuristic method, it does not provide the precision for the parameter estimates. Rather, confidence in the model's predictive capability is established by showing successful prediction of additional data that were not used for parameter estimation. This approach of focusing on model predictions is utilized by “quantitative systems pharmacology” models because these models are typically underconstrained but contain several “sloppy parameters” whose precise values do not influence the predictions of interest.20, 21 Alternate parameterizations are considered in determining the confidence interval of predictions. Additional details for objective function evaluation and estimation of confidence intervals are provided in the Supplementary Material.
Translational scaling for predictions
The cynomolgus monkey PK‐PD model was scaled to humans, as described earlier.22 Specifically, the clearance parameter was translated using allometric scaling with an exponent of 0.8 and the rest of the parameters were scaled 1:1 except for the Aβ turnover rate in the brain. Based on measurements by Randall Bateman's laboratory, Aβ turnover in humans was assumed to be 20% slower than that in cynomolgus monkeys.23, 24
RESULTS
Model calibration and validation
Calibration was performed in two steps. First, the PK module was fitted to PK data for two anti‐TfR antibodies (anti‐TfR1/BACE1 and anti‐TfR2/BACE1) and a nonspecific anti‐gD antibody.14 This fitting provided values for the two‐compartment PK parameters (compartment volumes, distributional clearance, and nonspecific elimination), as well as those associated with TfR‐mediated clearance (TfR turnover in the central compartment and degradation rates of TfR‐antibody complex; Table 1, 2). The on rate and KD for antibody‐TfR binding were set directly to values measured in vitro, without additional fitting (Table 3). This approach decreases the number of degrees of freedom in the model and allows for a more robust estimation of the free parameters. Furthermore, it would enable direct use of in vitro measurements for in vivo predictions for future molecules. Figure 2 a shows that the model is able to accurately describe antibody PK using the in vitro binding kinetics for binding to and unbinding from the receptor.
Table 1.
List of model species
| Name | Description | Initial Condition (nM) | |
|---|---|---|---|
| Ab C/P | Concentration of antibody in central/peripheral compartment | 0 | |
| RC/b | Concentration of TfR in central/brain compartment |
|
|
| [Ab: R]C/b | Concentration of antibody‐TfR complex in central/brain compartment | 0 | |
| [Ab: R]bi | Concentration of internalized antibody‐TfR complex in brain compartment | 0 | |
| Abbi/bc | Concentration of internalized/transcytosed antibody in brain compartment | 0 | |
| T/Aβ | Concentration of generic‐target T or Aβ in the brain cortex |
|
|
| [Ab:T]bc | Concentration of antibody‐target complex in brain cortex | 0 |
TfR, transferrin receptor.
Table 2.
List of model parameters (kon and KD are the only parameters that were fixed based on in vitro measurements and the rest were estimated in the model)
| Name | Description | Value (Units) | |
|---|---|---|---|
| CL | Non‐specific elimination clearance of antibody | 0.0039 (L/day/kg) | |
| CLd | Distribution clearance of antibody | 0.0188 (L/day/kg) | |
| kon | Association rate of antibody to TfR (fixed) | 17.9 (1/nM/day) | |
| KD | Dissociation constant for antibody binding to TfR (fixed) | See Table 3 (ug/ml) | |
| Vi | Volume of central/peripheral compartment | 0.0322/0.0289 (L/kg) | |
|
|
Rate of TfR synthesis in central/brain compartment | 886/341 (nM/day) | |
|
|
Rate of TfR degradation in central/brain compartment | 2‐1 / 7 (1/day) | |
|
|
Rate of antibody‐TfR complex degradation in central compartment | 0.305 (1/day) | |
| kint | Rate of antibody‐TfR complex internalization into brain | 5.72 (1/day) | |
| kbuns | Rate of non‐specific uptake of antibody into the brain | 0.016 (1/day) | |
| ktrans | Rate of antibody transcytosis into brain cortex | 200 (1/day) | |
|
|
Rate of antibody clearance from brain cortex | 291 (1/day) | |
| SynA β | Rate of Aβ synthesis in the brain cortex | 100 (nM/day) | |
|
|
Rate of Aβ degradation in brain cortex | 2.5 (1/day) | |
|
|
Maximum effect of BACE inhibition by antibody on AP production (fixed) | 0.75 (dimensionless) | |
|
|
Antibody concentration at which its effect is half the maximum effect | 6.67 (nM) |
TfR, transferrin receptor.
Table 3.
Affinity of anti‐TfR arms described in this study
| Anti‐TfR arm | KD (nM) |
|---|---|
| TfR1 | 37 |
| TfR2 | 1650 |
| TfR52 | 343 |
| TfR53 | 143 |
TfR, transferrin receptor.
Figure 2.
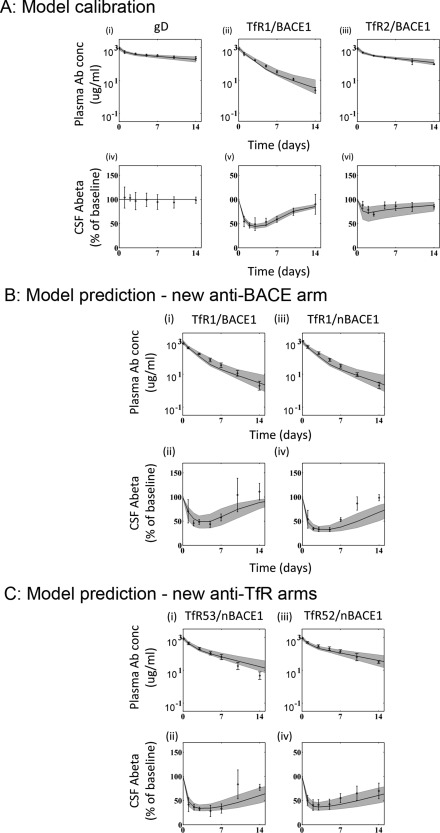
Model calibration and validation. (a) Calibration of model to published pharmacokinetic‐pharmacodynamic (PK‐PD) results: top plots demonstrate model fits (black curve) along with 90% confidence interval (shaded region) to serum PK data for anti‐gD (i), anti‐transferrin receptor (TfR)1/BACE1 (ii), and anti‐TfR2/BACE1 (iii). Bottom plots show model fits (black curve) with 90% confidence intervals (shaded region) to cerebrospinal fluid (CSF) Aβ inhibition profile for anti‐gD (iv), anti‐TfR1/BACE1 (v), and anti‐TfR2/BACE1 (vi). Experimental data is available from five animals in each arm. (b) The PD model validation using a more potent anti‐BACE1 arm (anti‐TfR1/nBACE1): top plots demonstrate model fits (black curve) along with 90% confidence intervals (shaded region) to serum PK data for anti‐TfR1/BACE1 (i) and anti‐TfR1/nBACE1 (ii). Bottom plots show model fits (black curve) with 90% confidence intervals (shaded region) to CSF Aβ inhibition profile for anti‐TfR1/BACE1 (iii) and anti‐TfR1/nBACE1 (iv). Experimental data is available from five animals in each arm. (c) The PK‐PD model validation using anti‐TfR arms with intermediate affinities (anti‐TfR52 or 53/nBACE1): top plots demonstrate model fits (black curve) along with 90% confidence intervals (shaded region) to serum PK data for anti‐TfR53/nBACE1 (i) and anti‐TfR52/nBACE1 (ii). Bottom plots show model fits (black curve) with 90% confidence intervals (shaded region) to CSF Aβ inhibition profile for anti‐TfR53/nBACE1 (iii) and anti‐TfR52/nBACE1 (iv). Experimental data is available from five animals in each arm.
Next, the brain uptake and PD modules were fit simultaneously based on the observed Aβ‐inhibition over 2 weeks after administration of the anti‐TfR1 and anti‐TfR2 antibodies. The brain‐uptake module was also constrained by the levels of antibodies measured in the brain at 24 hours postdose.14 The estimated parameters for the brain‐uptake module were those related to TfR turnover in brain capillary endothelium, internalization of TfR‐antibody complex, nonspecific antibody internalization, and antibody release into and clearance from the brain. The PD module parameters were those related to the Hill equation parameters for inhibition of Aβ production. Similar to the PK module, the brain‐uptake module also utilized the in vitro measurements for binding and dissociation of the antibody to TfR, both at the BBB and inside the capillary endothelium. Figure 2 a shows that the model is able to describe the Aβ inhibition profile for both anti‐TfR1 and anti‐TfR2 antibodies well.
Even after fixing the parameters for antibody‐TfR binding‐unbinding kinetics based on in vitro data, the model is not identifiable such that multiple distinct parameter sets reasonably fit the available PK and PD data for anti‐TfR1, anti‐TfR2, and anti‐gD antibodies. Therefore, to assure that the model adequately captures transcytosis phenomenon in the brain, model predictions were validated using additional experimental data. Note that because the model parameters are set before this validation step, in silico predictions in line with experimental results provide confidence that the model captures the relevant biology and can then be used to investigate additional research questions.
To robustly evaluate whether the model is able to capture the behavior of both arms of the antibody, model predictions were tested using data reserved for model validation corresponding to three additional antibodies: (1) anti‐TfR1/nBACE1, antibody with the same high affinity anti‐TfR arm and a higher‐affinity anti‐BACE1; (2) anti‐TfR52/nBACE1, antibody with a low‐intermediate affinity anti‐TfR arm and the higher affinity anti‐BACE1 arm; and (3) anti‐TfR53/nBACE1, antibody with a high‐intermediate affinity anti‐TfR arm and the higher affinity anti‐BACE1 arm.
The model correctly predicts PK and PD for antibodies with increased potency against BACE1
A new bispecific antibody with the same anti‐TfR1 arm and a higher affinity anti‐BACE1 arm (anti‐nBACE1) with four‐times greater potency against BACE1 than the original anti‐TfR/BACE1 was generated.14, 15, 16, 17, 18, 19, 25, 26 The two anti‐TfR1 antibodies exhibited the same plasma PK, supporting the assumption that binding to TfR is the major driver for target‐mediated clearance and that binding to BACE1 does not have a measurable impact. Because anti‐TfR1/BACE1 antibody was one of the antibodies used for model calibration, it was not surprising that the model fits the PK data for both antibodies with this same TfR1 arm well (Figure 2 b[i]). In order to predict the PD data for the anti‐TfR1/nBACE1 antibody, the parameter was reduced by fourfold, the factor by which anti‐BACE1 and anti‐nBACE1 differ in their potency against BACE1. As presented in Figure 2 b(ii), the model predicted the PD response to anti‐TfR1/nBACE1 without any additional model‐fitting (simulation vs. experiment in Supplementary Figure S1). Importantly, this validation provides confidence in the PD module and demonstrates that in vitro measurement for activity of antibody against BACE1 was sufficient to explain the observed change in PD response in vivo. Thus, the model can be used with increased confidence to explore the expected modulations in PD response with new anti‐BACE1 arms.
The model correctly predicts PK and PD for antibodies with intermediate affinities against TfR
To capture the PK‐PD relationship quantitatively in a species‐dependent manner, the mechanistic model accounts for TfR level and turnover in primates, estimates for which were obtained during model calibration. Because these parameters cannot be tested directly, the brain‐uptake module was validated by comparing model predictions with experimental data for two new antibodies against cynomolgus monkey TfR: anti‐TfR53 (KD−143nM) and anti‐TfR52 (KD−343nM).
Figure 2 c demonstrates that the model developed based on the previous study with anti‐TfR1 and anti‐TfR2 antibodies predicts the PK and PD for both anti‐TfR53 and anti‐TfR52 well without any additional fitting (simulation vs. experiment in Supplementary Figure S1). The only model parameters altered were the kinetics for binding to TfR that were specified for each antibody based on results from a BiaCore binding assay. Thus, the concordant results establish confidence in the ability of the model to mechanistically and quantitatively capture the process of antibody transcytosis across the BBB and support the use of this semiquantitative model to predict PK‐PD of anti‐TfR bispecific antibodies in humans.
Predicted optimal anti‐TfR affinity for humans
The cynomolgus monkey PK‐PD model was extended to predict expected human serum PK and brain Aβ kinetics (a measure of anti‐BACE1 activity) for anti‐TfR antibodies. Figure 3 a(i–iv) shows the predicted human Aβ inhibition profile by anti‐TfR/BACE1 bispecific antibody for four different affinities against TfR, again indicating a biphasic relationship between affinity and Aβ inhibition. For KD values of 2,000 to 20 nM, Aβ inhibition improves with increasing affinity (decreasing KD). However, at higher affinity (KD = 2nM), the inhibition of Aβ is reduced. The relationship between peak and average Aβ inhibition by anti‐TfR/BACE1 bispecific antibodies of different affinity toward TfR is shown in Figure 3 b. The model predicts that an anti‐TfR affinity in the range of 100 to 300 nM would be optimal for achieving maximal average as well as maximal peak Aβ inhibition.
Figure 3.
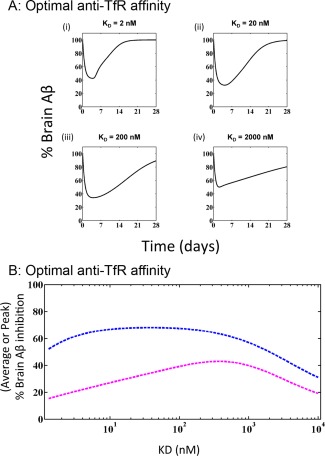
Optimal anti‐transferrin receptor (TfR) affinity in humans. (a) Expected Aβ inhibition profiles in human brain tissue after a single 30 mg/kg i.v. dose of anti‐TfR/nBACE1 antibody with the same anti‐BACE1 arm and different affinities against TfR receptor: 2 nM (i), 20 nM (ii), 200 nM (iii), and 2,000 nM (iv). (b) Expected average (magenta) and maximal (blue) human brain Aβ inhibition expected after a single 30 mg/kg i.v. dose of anti‐TfR/nBACE1 antibody with the same anti‐BACE1 arm and different affinities against TfR receptor ranging from 1 nM to 10 µM. Maximal Aβ inhibition peaks for anti‐TfR antibodies in the range of 10–500 nM, whereas average Aβ inhibition peaks in the range of 100–300 nM.
Comparative predictions for bivalent vs. anti‐TfR bispecific antibodies against generic target
To date, characterization of the anti‐TfR bispecific antibody platform and its relative ability to improve brain uptake have been conducted using anti‐BACE1 as the potential therapeutic target. BACE1 is not only an important target for Alzheimer's Disease, but also provides a robust PD readout that has been indirectly used to quantify improvements in brain uptake gained by using anti‐TfR. Although anti‐BACE1 has been used to support proof‐of‐concept for the anti‐TfR bispecific platform, other targets implicated in neurological disorders are also of interest. Furthermore, many proteins of interest in neurodegenerative diseases have been targeted by direct binding and inhibition.27, 28 Accordingly, we tested the application of an anti‐TfR bispecific approach for improved delivery of antibodies that directly bind other brain targets. However, some targets may benefit more from increasing brain delivery, whereas others may benefit more from having two arms to bind the target. In addition, anti‐TfR bispecific antibodies clear from the body much faster than typical bivalent antibodies because of TfR‐mediated clearance, thus reducing exposure of anti‐TfR bispecific relative to bivalent in the periphery. Therefore, to explore the potential impact of target characteristics on the relative advantages/disadvantages of using an anti‐TfR bispecific format, we extended the model to account for a generic target “T” that is bound and neutralized by either an anti‐TfR/anti‐T bispecific antibody or an anti‐T bivalent antibody. The model was then used to compare the level of target engagement achieved by either antibody format as compared to the baseline level of total target present in the brain.
Figure 4 a(i) demonstrates the percent target engagement by an anti‐TfR bispecific antibody with KD of 100 nM against TfR and 1 nM against T, administered at 30 mg/kg i.v. every 4 weeks. Figure 4 a(ii) demonstrates the percent target engagement by the bivalent antibody with two binding sites against that target with an affinity of 1 nM, administered in the same manner as anti‐TfR bispecific. As expected, both antibodies are more effective against targets with lower steady‐state concentration and longer elimination half‐lives. On the other hand, if the steady‐state concentration of target is too high and/or elimination half‐life is too low, then both antibodies are relatively ineffective (<20%) at the specified dose level, despite marginally better neutralization using the bispecific antibody. The most interesting prediction is related to conditions wherein either the bispecific or the bivalent antibody provides a relative advantage, supporting that rational selection of the most appropriate antibody platform should take key target characteristics into account (Figure 4 a[iii]). For this dosing regimen (30 mg/kg i.v. Q4W), there exists an intermediate range for targets with intermediate concentrations and elimination half‐lives for which the anti‐TfR bispecific platform offers a significant advantage (Figure 4 a[iii]).
Figure 4.
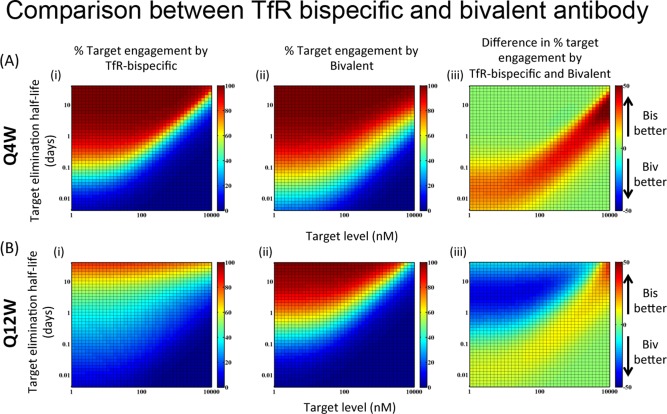
Target engagement by bivalent (anti‐Target KD = 1 nM, two binding sites) vs. bispecific (anti‐transferrin receptor [TfR] KD = 100 nM, anti‐Target KD = 1 nM) antibody for targets with varying expression levels and turnover rates. On the color bar, bispecific and bivalent are abbreviated as “Bis” and “Biv,” respectively. (a) If the antibody is given i.v. at 30 mg/kg every 4 weeks, then at intermediate target expression levels and turnover rates, bispecific antibody provides a significant advantage. (b) If the antibody is given i.v. at 30 mg/kg every 12 weeks, then the advantage of bispecific disappears while the bivalent antibody provides a significant advantage for slow turnover low expression level targets.
In contrast, because of the target‐mediated clearance associated with anti‐TfR antibodies, if the dosing interval is extended (e.g., Q12W instead of Q4W) and the same comparison between bivalent and bispecific antibody is conducted, the advantage of anti‐TfR bispecific platforms diminishes (Figure 4 b). Both antibodies are equally ineffective for targets with high expression level and fast turnover, but the bivalent antibody is actually more effective for targets with low level and low turnover rate (Figure 4 b[iii]).
Thus, the relative efficiency of the bivalent vs. anti‐TfR bispecific antibody format for binding a given central nervous system target is determined by target concentration, target turnover rates, and clinical dosing regimen. For each target and specific clinical requirements, the PK‐PD framework from this model can be used to inform decision‐making around the relative value of using the anti‐TfR bispecific antibody platform over a standard antibody approach.
DISCUSSION
The BBB limits the delivery of large molecules, including antibodies, into the brain. Consequently, achieving sufficient brain exposure to maintain therapeutically effective concentrations of drug poses a significant challenge to treatment of brain diseases with antibody therapeutics. Research into the biology of the BBB is ongoing and has already provided insight into mechanisms by which it regulates the passage of molecules from the vasculature into the brain. Previous works in mice and cynomolgus monkeys have studied and leveraged TfR‐mediated transcytosis by using anti‐TfR bispecific antibodies to improve transport into the brain.14 However, the complex relationship between the antibody affinity for TfR and the resulting brain exposure is not well understood.
Gadkar et al.17 previously presented a PK‐PD model for the behavior of murine anti‐TfR bispecifics in mice to address the complex systemic and brain PK‐PD relationships observed. However, the empirical representation of antibody‐TfR interaction restricts the ability to adapt the model for primates and humans. Therefore, in this work, we developed a mechanistic PK‐PD model that accounts for target‐mediated clearance of anti‐TfR/BACE1 bispecific antibody in plasma and affinity‐dependent uptake into the brain by explicitly accounting for antibody‐TfR interactions in plasma as well as on the brain endothelium. The model was extended to translate PK‐PD predictions from cynomolgus monkey to human and thereby aid in design of a molecule for clinical development. The model was then further applied to explore the impact of a range of target parameters that also play a role in whether an anti‐TfR bispecific platform provides any advantage over a standard anti‐target bivalent antibody approach.
One of the most interesting PK‐PD outcomes of the model is the mechanistic explanation for the existence of an inverse “U” relationship between affinity against TfR and brain uptake of the antibody. Anti‐TfR antibodies with high affinity monovalent binding to TfR clear rapidly from peripheral blood and result in degradation of TfR.14 Both of these phenomena result in low exposure to anti‐TfR antibody in the brain. In contrast, anti‐TfR antibodies with very low affinity to TfR have substantially better peripheral blood exposure because of reduced target‐mediated clearance. However, maximal brain exposure of these antibodies is also low because of the extremely low affinity for TfR results in inefficient transport across the BBB. Correctly identifying the optimal affinity of the anti‐TfR arm is therefore critical to support and optimize sustained exposure in the brain at or above a therapeutic threshold (dependent on the potency of the therapeutic arm; e.g., anti‐BACE1). It is also important to note that, based on our mouse and monkey data, the optimal affinity of the anti‐TfR arm is species‐dependent, as well as target characteristics in the brain.
Our model was extended to simulate expected target engagement by the bispecific and bivalent antibody for a target with given steady‐state levels and turnover rates. Interestingly, the model revealed that there is no universal answer as to whether the bispecific antibody is better than the corresponding bivalent antibody. The answer depends on the dosing regimen as well as the target characteristics. For monthly dosing, TfR bispecifics are better at intermediate levels and turnover rates of the target, whereas for quarterly dosing, bivalent antibodies are better for targets at low level and low turnover rates. Accordingly, the anti‐TfR bispecific platform cannot be treated as a panacea for improving the efficacy of all antibodies against targets in the brain, and should be evaluated for the relative advantage it is likely to confer on a case‐by‐case basis.
Given that bispecific antibodies are typically more expensive to generate and develop than bivalent antibodies, the anti‐TfR bispecific platform would be economically viable only if it offered a significant advantage over its bivalent counterpart. Therefore, the ability of this model to compare different antibody formats can be extremely useful in prioritizing which targets will benefit the most from the bispecific platform and define early experiments (both preclinical and clinical) to verify hypotheses related to drug effects rather than relying entirely on lengthy and expensive clinical trials. It should be noted that the overall paucity of quantitative information on central nervous system targets poses a significant challenge to generating model predictions with high confidence. Accordingly, it would be important to further characterize target biology and design efficient experiments in order to inform appropriate use of the model.
Although the aforementioned analysis can be applied to a generalized target, it cannot be applied if the target is such that antibody activity requires both arms of antibody to be bound to the target simultaneously (e.g., antibodies that potentiate clustering of target receptor). In this case, a bivalent or higher order multivalent antibody approach is clearly required. Alternatively, if the mechanism of action of a candidate antibody involves activation of immune response (e.g., through engagement of Fcγ receptor), then the anti‐TfR bispecific platform would also be excluded based on the requirement for no effector function. Anti‐TfR bispecific antibodies with effector function have been previously shown to have acute and systemic toxicities; therefore this platform is restricted to targets that do not require the activation of Fcγ receptor pathway.12
The modeling and simulation framework described in this article are useful for interpretation of the data from bispecific antibodies within a single model structure and parameter set. This could be a helpful tool for identifying optimal characteristics for antibodies based on the mechanism of action of the therapeutic arm. Finally, the described modeling and simulation framework could predict the profile of expected human response for a specific antibody against a specific target. Thus, this modeling and simulation framework can play a prospectively instrumental role in specifying criteria for designing optimal clinical candidates and efficient clinical studies to enable faster development of the therapeutic bispecific antibodies.
Conflict of Interest. The authors are full time employees of Genentech.
Author Contributions. J.K., K.G., S.R., and S.J. wrote the manuscript. J.K., K.G., R.J.W., M.D., J.A.E., K.S‐L., J.K.A., S.R., and S.J. designed the research. J.K., K.G., D.B., Y.Z., R.K.T., W.L., K.H., Y.L., K.W., and J.C. performed the research. J.K. and K.G. analyzed the data.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
The authors thank Greg Ferl and Paul Fielder for helpful discussions and suggestions for the article and AnshinBiosolutions for editorial support with the preparation of this manuscript.
References
- 1. Pardridge, W.M. Targeted delivery of protein and gene medicines through the blood‐brain barrier. Clin. Pharmacol. Ther. 97, 347–361 (2015). [DOI] [PubMed] [Google Scholar]
- 2. Poduslo, J.F. , Curran, G.L. & Berg, C.T. Macromolecular permeability across the blood‐nerve and blood‐brain barriers. Proc. Natl. Acad. Sci. USA 91, 5705–5709 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mills, E. , Dong, X.P. , Wang, F. & Xu, H. Mechanisms of brain iron transport: insight into neurodegeneration and CNS disorders. Future Med. Chem. 2, 51–64 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zheng, W. & Monnot, A.D. Regulation of brain iron and copper homeostasis by brain barrier systems: implication in neurodegenerative diseases. Pharmacol. Ther. 133, 177–188 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Friden, P.M. , Walus, L.R. , Musso, G.F. , Taylor, M.A. , Malfroy, B. & Starzyk, R.M. Anti‐transferrin receptor antibody and antibody‐drug conjugates cross the blood‐brain barrier. Proc. Natl. Acad. Sci. USA 88, 4771–4775 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jones, A.R. & Shusta, E.V. Blood‐brain barrier transport of therapeutics via receptor‐mediation. Pharm. Res. 24, 1759–1771 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee, H.J. , Engelhardt, B. , Lesley, J. , Bickel, U. & Pardridge, W.M. Targeting rat anti‐mouse transferrin receptor monoclonal antibodies through blood‐brain barrier in mouse. J. Pharmacol. Exp. Ther. 292, 1048–1052 (2000). [PubMed] [Google Scholar]
- 8. Moos, T. & Morgan, E.H. Restricted transport of anti‐transferrin receptor antibody (OX26) through the blood‐brain barrier in the rat. J. Neurochem. 79, 119–129 (2001). [DOI] [PubMed] [Google Scholar]
- 9. Niewoehner, J. et al Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 81, 49–60 (2014). [DOI] [PubMed] [Google Scholar]
- 10. Pardridge, W.M. , Buciak, J.L. & Friden, P.M. Selective transport of an anti‐transferrin receptor antibody through the blood‐brain barrier in vivo. J. Pharmacol. Exp. Ther. 259, 66–70 (1991). [PubMed] [Google Scholar]
- 11. Yu, Y.J. et al Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 3, 84ra44 (2011). [DOI] [PubMed] [Google Scholar]
- 12. Couch, J.A. et al Addressing safety liabilities of TfR bispecific antibodies that cross the blood‐brain barrier. Sci. Transl. Med. 5, 183ra57,1–12 (2013). [DOI] [PubMed] [Google Scholar]
- 13. Bien‐Ly, N. et al Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J. Exp. Med. 211, 233–244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu, Y.J. et al Therapeutic bispecific antibodies cross the blood‐brain barrier in nonhuman primates. Sci. Transl. Med. 6, 261ra154 (2014). [DOI] [PubMed] [Google Scholar]
- 15. Lu, Y. et al Cerebrospinal fluid amyloid‐β (Aβ) as an effect biomarker for brain Aβ lowering verified by quantitative preclinical analyses. J. Pharmacol. Exp. Ther. 342, 366–375 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu, X. et al Mechanistic pharmacokinetic‐pharmacodynamic modeling of BACE1 inhibition in monkeys: development of a predictive model for amyloid precursor protein processing. Drug Metab. Dispos. 41, 1319–1328 (2013). [DOI] [PubMed] [Google Scholar]
- 17. Gadkar, K. et al Mathematical PKPD and safety of bispecific TfR/BACE1 antibodies for the optimization of antibody uptake in brain. Eur. J. Pharm. Biopharm. 101, 53–61 (2016). [DOI] [PubMed] [Google Scholar]
- 18. Iadevaia, S. , Lu, Y. , Morales, F.C. , Mills, G.B. & Ram, P.T. Identification of optimal drug combinations targeting cellular networks: integrating phospho‐proteomics and computational network analysis. Cancer Res. 70, 6704–6714 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kanodia, J. et al Deciphering the mechanism behind fibroblast growth factor (FGF) induced biphasic signal‐response profiles. Cell Commun. Signal. 12, 34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gadkar, K. , Kirouac, D.C. , Mager, D.E. , van der Graaf, P.H. & Ramanujam, S. A six‐stage workflow for robust application of systems pharmacology. CPT Pharmacometrics Syst. Pharmacol.; e-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gutenkunst, R.N. , Waterfall, J.J. , Casey, F.P. , Brown, K.S. , Myers, C.R. & Sethna, J.P. Universally sloppy parameter sensitivities in systems biology models. PLoS Comput. Biol. 3, 1871–1878 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kagan, L. , Abraham, A.K. , Harrold, J.M. & Mager, D.E. Interspecies scaling of receptor‐mediated pharmacokinetics and pharmacodynamics of type I interferons. Pharm. Res. 27, 920–932 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bateman, R.J. , Munsell, L.Y. , Morris, J.C. , Swarm, R. , Yarasheski, K.E. & Holtzman, D.M. Human amyloid‐beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med. 12, 856–861 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cook, J.J. et al Acute gamma‐secretase inhibition of nonhuman primate CNS shifts amyloid precursor protein (APP) metabolism from amyloid‐beta production to alternative APP fragments without amyloid‐beta rebound. J. Neurosci. 30, 6743–6750 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Atwal, J.K. et al A therapeutic antibody targeting BACE1 inhibits amyloid‐β production in vivo. Sci. Transl. Med. 3, 84ra43 (2011). [DOI] [PubMed] [Google Scholar]
- 26. Eberhart, R. & Kennedy, J. A new optimizer using particle swarm theory. MHS'95 Proc. Sixth Int. Symp. Micro. Mach. Hum. Sci. <http://www.ppgia.pucpr.br/∼alceu/mestrado/aula3/PSO_2.pdf> (1995). [Google Scholar]
- 27. Funk, K.E. , Mirbaha, H. , Jiang, H. , Holtzman, D.M. & Diamond, M.I. Distinct therapeutic mechanisms of Tau antibodies: promoting microglial clearance versus blocking neuronal uptake. J. Biol. Chem. 290, 21652–21662 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang, C. & Xiao, S. New developments of clinical trial in immunotherapy for Alzheimer's disease. Curr. Pharm. Biotechnol. 16, 484–491 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information