

Abstract
Purpose
Distinguishing independent primary tumors from intrapulmonary metastases in non–small-cell carcinoma remains a clinical dilemma with significant clinical implications. Using next-generation DNA sequencing, we developed a chromosomal rearrangement–based approach to differentiate multiple primary tumors from metastasis.
Methods
Tumor specimens from patients with known independent primary tumors and metastatic lesions were used for lineage test development, which was then applied to multifocal tumors. Laser capture microdissection was performed separately for each tumor. Genomic DNA was isolated using direct in situ whole-genome amplification methodology, and next-generation sequencing was performed using an Illumina mate-pair library protocol. Sequence reads were mapped to the human genome, and primers spanning the fusion junctions were used for validation polymerase chain reaction.
Results
A total of 41 tumor samples were sequenced (33 adenocarcinomas [ADs] and eight squamous cell carcinomas [SQCCs]), with a range of three to 276 breakpoints per tumor identified. Lung tumors predicted to be independent primary tumors based on different histologic subtype did not share any genomic rearrangements. In patients with lung primary tumors and paired distant metastases, shared rearrangements were identified in all tumor pairs, emphasizing the patient specificity of identified breakpoints. Multifocal AD and SQCC samples were reviewed independently by two pulmonary pathologists. Concordance between histology and genomic data occurred in the majority of samples. Discrepant tumor samples were resolved by genome sequencing.
Conclusion
A diagnostic lineage test based on genomic rearrangements from mate-pair sequencing demonstrates promise for distinguishing independent primary from metastatic disease in lung cancer.
INTRODUCTION
Given the reduction in lung cancer mortality demonstrated in the National Lung Screening Trial, a number of organizations have endorsed chest computed tomography screening for high-risk individuals. Compared with previous reports estimating that 0.8% to 4% of patients with lung cancer present with or develop multiple lesions, more recent data indicate that up to 20% of patients in screening populations will be diagnosed with multiple synchronous or metachronous tumors.1–9
The question of whether multiple lesions represent true metastases versus independent primary lesions remains a difficult clinical problem. This distinction typically represents the difference between aggressive local therapy for patients deemed to have independent primary tumors versus less aggressive or even palliative systemic therapy for patients thought to have metastatic disease.10–18 Currently, the determination of independent primary tumors versus metastasis is largely based on provider-specific clinical and/or pathologic assumptions. There are presently no ancillary tests that allow this distinction to be made with any reasonable clinical accuracy.
Next-generation sequencing of several cancers has revealed that solid tumors harbor tens to hundreds of somatic chromosomal rearrangements and thousands of single-nucleotide variations (SNVs). Both of these types of alterations have been used to investigate lineage relationships in tumors from the same individual.19–22 Given that the average genome contains 3 to 6 × 106 single-nucleotide polymorphisms and given the extensive numbers of background SNVs within the local environment of a tissue, the derivation of tumor-specific somatic SNVs remains challenging. Although germline rearrangements exist, the numbers are low and easier to determine through polymerase chain reaction (PCR) validation or bioinformatics.22 The additional commonality of many recurrent SNV mutations, such as EGFR and KRAS, means they are often shared even between independent tumors.23 In contrast, the probability of detecting an identical somatic chromosomal breakpoint in two unrelated tumors is extremely unlikely, if not zero. The common TMPRSS2-ERG fusion is observed in approximately 50% of patients with prostate cancer. Although the resultant fusion products are often identical at the RNA and protein level, identical fusion junctions at the DNA level have never been reported between tumors of different patients.21,24–26 Similarly, for the common EML4-ALK translocation observed in approximately 5% of lung adenocarcinomas (ADs), no duplicate breakpoints have been reported in the literature to date, despite overlap in protein products and functional consequence.27 Furthermore, recent publications used rearrangements to successfully demonstrate lineage of adjacent lepidic and invasive components of lepidic predominant ADs22 and adjacent Gleason patterns in prostate cancer21 using next-generation DNA sequencing with a mate-pair (MP) library approach. Given these observations, we used similar methodology to identify lineage-defining genomic alterations in the setting of multiple pulmonary lesions, hypothesizing that intrapulmonary metastases would share such unique genomic alterations, whereas independent primary tumors would not.
METHODS
Sample Selection
The following three groups of samples were identified as controls: multiple tumor samples from different tissue blocks of a solitary tumor (n = 4), 2- synchronous primary lung cancers of different histologic subtype (n = 3), and primary lung cancers with corresponding distant metastasis to brain or kidney (n = 4). The study group (Table 1) comprised 11 synchronous lung cancers of similar histologic subtype. ADs and squamous cell carcinomas (SQCCs) were diagnosed according to WHO classifications,28 and ADs were classified according to new proposed classifications.29 Two pulmonary pathologists performed independent reviews, blinded to clinical and genomic data. Based on morphology, using criteria as suggested by Girard et al,30 a sample was predicted to be an independent primary or metastasis, or to favor one of these predictions, or indeterminate if the pathologists did not agree. This study was approved by the Mayo Clinic Institutional Review Board.
Table 1.
Correlation Between Histologic Prediction and Genomic Data in Multiple Lung Carcinomas of Similar Histologic Subtype
| Sample No. | Tumor 1 |
Tumor 2 |
Histologic Prediction | Genomic Prediction | ||
|---|---|---|---|---|---|---|
| Type | Location | Type | Location | |||
| LU26 | RLL | LUL | Unrelated | |||
| R1 | AD pred cribriform (C, 80%; A, 10%; S, 10%) | AD pred acinar (A, 50%; L, 40%; P, 10%) | Independent primary tumors | |||
| R2 | AD pred solid (S, 50%; C, 50%) | AD pred papillary (P, 40%; A, 30%; L, 10%; C, 10%; S, 10%) | Independent primary tumors | |||
| LU28 | RML | LUL | Unrelated | |||
| R1 | AD pred solid (S, 90%; A, 10%) | AD pred solid (S, 90%; A, 10% + giant cells) | Independent primary tumors | |||
| R2 | AD pred solid (S, 80%; A, 10%; MP, 10%) | AD pred solid (S, 100% with signet ring or large-cell features) | Favor independent primary tumors | |||
| LU32 | RLL | LUL | Unrelated | |||
| R1 | AD pred acinar (A, 70%; L, 30%) | MIA | Independent primary tumors | |||
| R2 | AD pred acinar (A, 50%; P, 50%) | AD pred lepidic (L, 70%; A, 30%) | Independent primary tumors | |||
| LU43 | RLL | RLL | Unrelated | |||
| R1 | AD mucinous | AD pred acinar (A, 70%; C, 20%; P, 10%) | Independent primary tumors | |||
| R2 | AD mucinous | AD pred acinar (A, 70%; P, 30%) | Favor independent primary tumors | |||
| LU45 | LUL | RUL | Unrelated | |||
| R1 | AD pred solid (S, 45% A 35% L 20%) | AD pred acinar (A, 70%; L, 20%; S, 10%) | Indeterminate: independent primary tumors | |||
| R2 | AD pred acinar (A, 50%; S, 20%; L, 20%; C, 10%) | AD pred acinar (A, 40%; L, 30%; S, 20%; P, 10%) | Favor metastasis | |||
| LU27 | RLL | RML | Related | |||
| R1 | AD mucinous | AD mucinous | Metastasis | |||
| R2 | AD mucinous | AD mucinous | Metastasis | |||
| LU29 | RUL | Lingula | Related | |||
| R1 | SQCC G3 | SQCC G3 | Metastasis | |||
| R2 | SQCC G3 | SQCC G3 | Favor metastasis | |||
| LU30 | RML | LLL | Related | |||
| R1 | AD mucinous | AD mucinous | Metastasis | |||
| R2 | AD mucinous | AD mucinous | Favor metastasis | |||
| LU40 | RML/RLL | LLL | Related | |||
| R1 | SQCC G3 | SQCC G3 | Favor metastasis | |||
| R2 | SQCC G2-3 | SQCC G2-3 | Metastasis | |||
| LU41 | LUL | RUL | Unrelated | |||
| R1 | SQCC G2 | SQCC G2 with CIS | Favor metastasis | |||
| R2 | SQCC G2 | SQCC G1-2 with CIS | Metastasis | |||
| LU44 | RLL | LUL | Unrelated | |||
| R1 | AD pred solid (S, 75%; C, 20%; A, 5%) | AD pred solid (S, 50%; C, 25%; A, 25%) | Indeterminate metastasis | |||
| R2 | AD pred solid (S, 90%; A, 10%) | AD pred solid (S, 80%; A, 20%) | Independent primary tumors | |||
Abbreviations: A, acinar; AD, adenocarcinoma; C, cribriform; G, grade; L, lepidic; LLL, left lower lung; LUL, left upper lung; MIA, minimally invasive adenocarcinoma; P, papillary; pred, predominant; RLL, right lower lung; RML, right middle lung; RUL, right upper lung; S, solid; SQCC, squamous cell carcinoma.
Laser Capture Microdissection Frozen Tissue Specimens
Hematoxylin and eosin–stained fresh-frozen sections were reviewed for quality control. Laser capture microdissection (LCM) was performed on 10-μm frozen sections, and pure cell populations of tumor and non-neoplastic lung (NL) were isolated using the Arcturus PixCell II microscope and CapSure Macro LCM caps (Arcturus, Carlsbad, CA; LCM 0211). DNA was extracted directly from LCM captured cells using a previously described single-step whole-genome amplification (WGA) procedure.20–22 Four individual 50-μL WGA reactions were pooled for each sample. DNA was quantified by Quant-iT-PicoGreen analysis (Invitrogen, Eugene, OR; P7581). Where available, germline blood extracted DNA was obtained from the Mayo Lung Specimen Registry.
Next-Generation Sequencing
MP sequencing tiles the genome with larger spanning (approximately 3 kb) fragments than conventional paired-end next-generation sequencing to increase the probability of spanning a genomic breakpoint. MP libraries were assembled from WGA DNA, according to a previously published protocol,20–22 using the Illumina MP kit (Illumina, San Diego, CA). Two multiplexed libraries were loaded per lane of an Illumina flow cell and sequenced to 101 × 2 paired-end reads on an Illumina HiSeq. Base calling was performed using Illumina Pipeline v1.5.
Data Analysis
Bioinformatics protocols to rapidly and efficiently process next-generation sequencing MP data using a 32-bit binary indexing of the Hg19 reference genome have been previously published from our laboratory.31,32 The algorithm maps both MP reads successively to the whole genome, selecting reads less than 15 kb apart allowing up to 10 mismatches, with the lowest cumulative mismatch count sent to the output. Discordant MPs mapping more than 30 kb apart or in different chromosomes were selected for further analysis. Algorithmic filters to determine lineage relationships were set to minimize the effects of both false-positive and false negative results. Namely, the lowest limit of MP associates to call an event was set at seven, where the false-positive rate was practically zero, and a mask of breakpoints was used to eliminate common variants and discordant MPs from experimental or algorithmic errors.20,21,31,32 The minimum gap between the last read from each event to the first read of the following event was set to 3,000, thereby eliminating closely related but not identical breakpoints from being called as shared. Breakpoints near gaps of reference genome sequence were also eliminated. The false-negative rate was estimated to be less than 15%, dictated by the incompleteness of the reference genome and by regions that are difficult to map. Using a probability statistic, we estimated that the probability of relatedness between two samples is P < .15n, where the expected number of shared breakpoints is n and no shared events are found. Sequencing data for each sample are listed in Appendix Table A1 (online only).
Validation of Genomic Rearrangements
Primers spanning the detected fusion junctions were used in PCR validations (25-μL reaction volumes, 50-ng template, 35 cycles) using the EasyA high-fidelity polymerase (Stratagene, La Jolla, CA; No. 600404). PCR validations were performed on tumor DNA, NL, and a human genomic DNA control (Promega, Madison, WI; G304A), as well as germline blood DNA where available. Glyceraldehyde 3-phosphate dehydrogenase control PCRs were performed using standard primers.
RESULTS
MP sequencing on the 41 tumors (33 ADs and eight SQs) discovered 2,201 unique rearrangements that passed lineage filters, ranging from three (LU31A) to 276 (LU45A) breakpoints per tumor. The average number of breakpoints was 54, with eight tumor samples presenting with less than 10 breakpoints and eight tumors with more than 100.
The control group of lung ADs, LU23, LU33, LU45, and LU47, with two different frozen tumor blocks sequenced from the same tumor (Fig 1A), had a range of four to 276 genomic breakpoints detected. The number of shared events between the different regions of the tumor ranged from 50% to 100% (Fig 1B). Rearrangements unique to different tumor regions were also identified. From the 529 total breakpoints detected in these four samples, 45% were shared with adjacent sampling of the same tumor. Significantly, no common breakpoints were identified as shared between different individual tumors. A selection of PCR validations yielded identical breakpoints in related tumors but not in NL tissue collected from the same patient (Fig 1C).
Fig 1.

Control group 1: multiple tumor samples from different tissue blocks of a solitary tumor. (A) Schematic illustrating a single primary tumor (AD1) from which the two tumor regions (A/B) were collected. (B) Numbers of genomic breakpoints detected in each tumor pair (both y-axes). Total numbers of breakpoint events are listed as unique to tumor AD1A (gold) or AD1B (gray) or shared between both tumors (blue). (C) Polymerase chain reaction validation bands of selected breakpoints from the tumor pairs (AD1A and AD1B), non-neoplastic lung tissue from the same patient (NL), and a mixed-population independent genomic DNA control (C) run on agarose gels (1%). The two genomic loci and the impacted genes at the breakpoint are in addition listed at the left and right of each gel panel, respectively. AD, adenocarcinoma; ng, no gene involved.
The paired lung synchronous primary tumors of different histologic subtype (LU29, LU31, and LU46) had 162, 39, and 82 total breakpoints, respectively (Figs 2A to 2C). One shared large genomic rearrangement was observed between tumors in LU31. This was subsequently demonstrated to be a germline variation, validating evenly in the NL and blood DNA of that patient (Fig 2D). This event was also present in the multiple human genome control sample. In summary, no shared somatic genomic rearrangements were identified among samples with definitive histopathologic evidence of identity as independent primary tumors.
Fig 2.
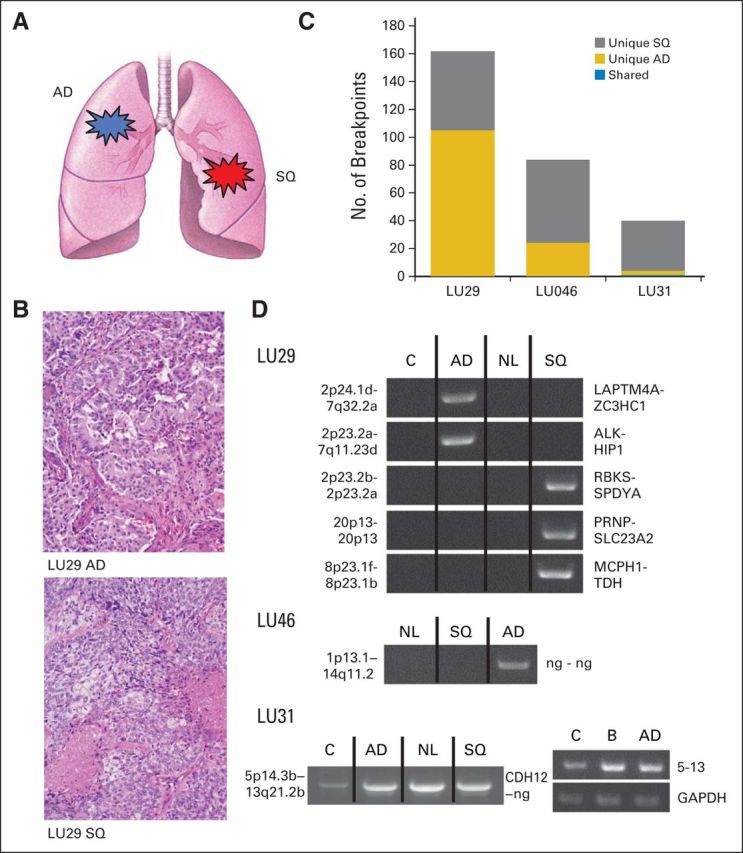
Control group 2: synchronous primary lung cancers of different histologic subtype. (A) Schematic of tumor pairs of different histologic subtype. (B) Hematoxylin and eosin images of adenocarcinoma (AD; upper panel) and squamous cell carcinoma (SQ; lower panel) from LU29. (C) Numbers of genomic breakpoints detected in each tumor pair (y-axis). Total numbers of breakpoint events are listed as unique to tumor AD (gold) or SQ (gray) or shared between both tumors (blue). (D) Polymerase chain reaction (PCR) validation bands of selected breakpoints from the tumor pairs (AD and SQ), non-neoplastic lung tissue from the same patient (NL), and a mixed-population independent genomic DNA control (C) run on agarose gels (1%). The two genomic loci and the impacted genes at the breakpoint are listed at the left and right of each gel panel, respectively. For the LU31 event, an additional germline blood (B) PCR validation is presented for the chromosome 5 to 13 translocation and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) control. ng, no gene involved.
In contrast, shared rearrangements were identified within individual primary tumor/metastasis pairs in all tumor pairs (LU33, LU34, LU38, and LU39), with the frequency of shared breakpoints detected varying from 8% to 75%. The number of breakpoints ranged between four and 62 breakpoints per tumor (Figs 3A to 3C). PCR successfully validated selected breakpoints in all four tumor pairs (Fig 3D). Two selected events also validated in surrounding NL at similar or reduced densities.
Fig 3.

Control group 3: primary lung cancer with corresponding distant metastasis to brain or kidney. (A) Schematic of tumor pairs, indicating origin from lung and extrapulmonary metastases (met). (B) Hematoxylin and eosin images of primary lung adenocarcinoma (AD) and brain metastasis of LU33 (upper panels) and LU34 (lower panel). (C) Numbers of genomic breakpoints detected in each tumor pair (y-axis). Total numbers of breakpoint events are listed as unique to primary lung tumor AD (gold) or met (gray) or shared between both tumors (blue). (D) Polymerase chain reaction (PCR) validation bands of selected breakpoints from the primary tumor (AD) or brain metastasis (BrM), non-neoplastic lung tissue from the same patient (NL), and a mixed-population independent genomic DNA control (C) run on agarose gels (1%). For LU33, two samples of the solitary AD tumor were extracted from adjacent blocks (AD1A and AD1B) and used for PCR validation. The two genomic loci and the affected genes at the breakpoint are listed at the left and right of each gel panel, respectively. ng, no gene involved.
In conclusion, samples clearly classified as either independent primary tumors or metastases, based on clinical and histologic parameters, can be accurately separated into independent primary tumors and metastases based on genomic data. Most significantly, none of the detected breakpoints were shared between tumors from different patients, emphasizing the patient and tumor specificity of identified breakpoints.
For the study group (Table 1), pathologists agreed on the histologic subtype in all tumors, and for AD, they agreed on the predominant pattern in 12 (75%) of 16 ADs. Figures 4A to 4C presents hematoxylin and eosin images for six of the 11 tumor pairs selected, with two each predicted as independent primary tumors and metastases and the remaining two predicted as indeterminate. The total number of breakpoints detected for each tumor sample is presented in Figure 5A.
Fig 4.
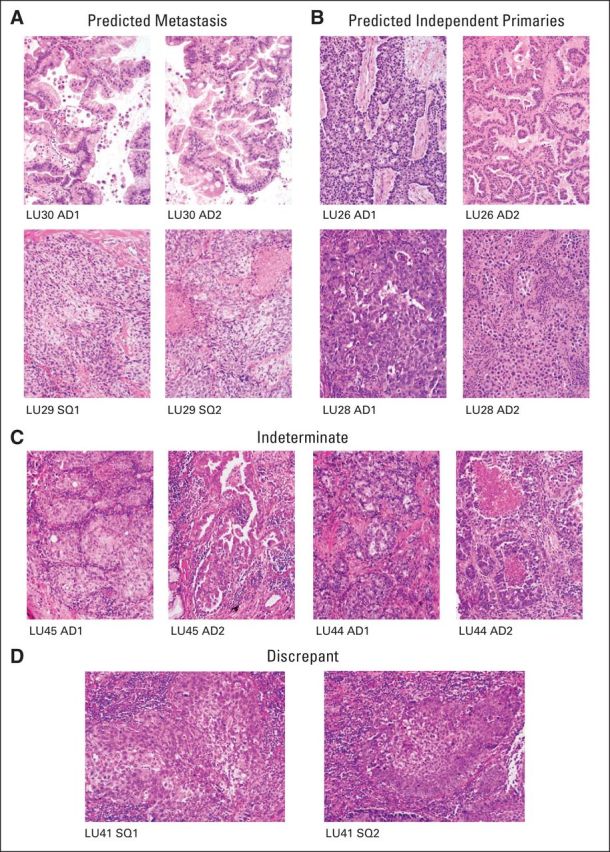
Hematoxylin and eosin (H&E) images of tumor pairs from the study group. (A) H&E images of two tumor pairs of predicted metastases: LU30 (upper panels) and LU29 (lower panels). (B) H&E images of two tumor pairs of predicted independent primary tumors: LU26 (upper panels) and LU28 (lower panels). (C) H&E images of two tumor pairs of indeterminate lineage: LU45 (left panels) and LU44 (right panels). (D) H&E images of tumor pairs of LU41 with discrepancy between histologic and genomic prediction. AD, adenocarcinoma; SQ, squamous cell carcinoma.
Fig 5.
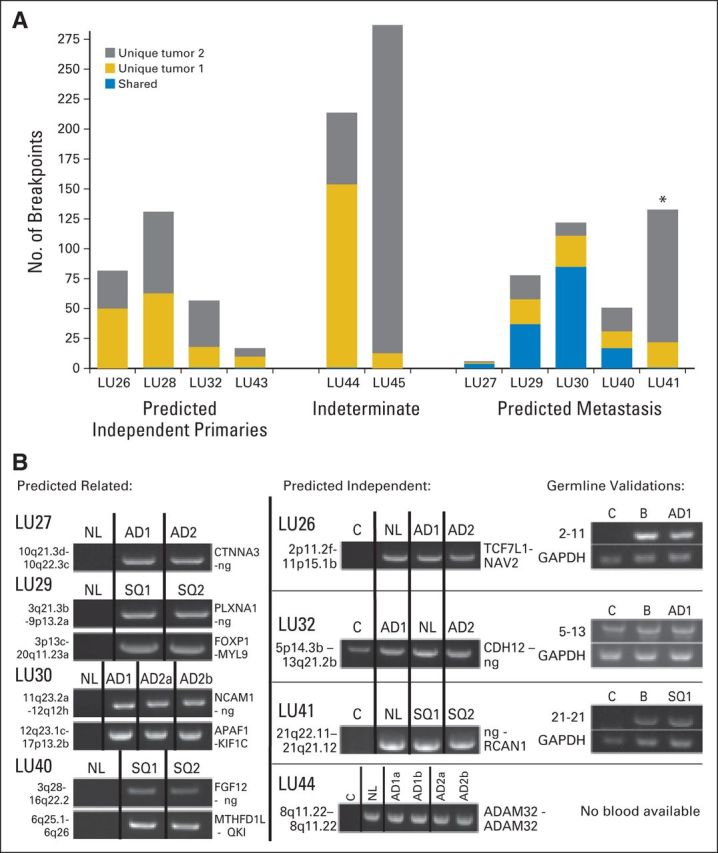
Breakpoint numbers and polymerase chain reaction (PCR) validations of tumor pairs from the study group. (A) Numbers of genomic breakpoints detected in each tumor pair (y-axis). Total numbers of breakpoint events are listed as unique to either tumor 1 (AD1 or SQ1; gold) or tumor 2 (AD2 or SQ2; gray) of the tumor pairs or shared between both tumors (blue). (B) PCR validation bands of selected breakpoint events on samples predicted as related (left panels) or independent (central panels). PCR on non-neoplastic lung tissue from the same patient (NL) and a mixed-population independent genomic DNA control (C) were also run on the same agarose gels (1%). Germline validations on patient-derived blood DNA (B) are presented in the right panel, together with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) controls. AD, adenocarcinoma; ng, no gene involved; SQ, squamous cell carcinoma. (*) Indicates the discrepant case, LU41, where the genomic and clinical predictions are discordant.
For the four cases predicted as independent primary tumors (LU26, LU28, LU32, and LU43), the number of detected genomic rearrangements ranged from eight to 69, with no shared somatic rearrangements identified (Fig 5A). Single shared germline events were observed in tumor samples LU26 and LU32, which validated evenly in the associated NL and germline DNA from each patient (Fig 5B). Four of five tumor pairs predicted to be metastases (LU27, LU29, LU30, and LU40) shared between four and 85 genomic breakpoints (46% to 89%). PCR validation confirmed a shared somatic mutation, absent from NL, in each sample (Fig 5B). Conversely, the fifth tumor pair, predicted to be a metastasis (LU41; Fig 4D), had only a single shared germline rearrangement between the two tumors and, therefore, was predicted independent by the genomic data (Fig 5B). Concordance between the agreed histologic prediction of both pathologists and the genomic data occurred in eight of nine tumor pairs. Two tumor pairs were indeterminate (LU44 and LU45) and contained 217 and 287 total breakpoints, respectively, with no shared somatic DNA rearrangements, predicting for independent primaries (Fig 5A).
There was disagreement between the clinical assessment and histologic prediction in three tumor pairs (LU27, LU29, and LU41), with the genomic data supporting the histologic prediction in two of these tumor pairs (Appendix Table A2, online only). In one tumor pair (LU45), there was no definitive clinical assessment and a disagreement between pathologists, with genomic data suggesting independent primary tumors.
DISCUSSION
Next-generation sequencing using an MP protocol is a robust, efficient, and cost-effective method to identify genomic rearrangements. Our results from the current and prior studies20–22,31–33 suggest that chromosomal breakpoints are unique enough for individual patients and tumors to be able to determine tumor lineage. Significantly, of the 2,201 breakpoints identified in our current 41-tumor study, all somatic mutations were unique to an individual patient, with the only shared rearrangements observed in the setting of metastatic disease. This observation is further supported in other publications from our group20–22,31–33 and the more than 500 tumors we have MP sequenced, where an exact breakpoint has never been observed between samples from two different patients (unpublished data). In addition, to our knowledge, identical fusions across tumors, even in the highly recurring lung cancer EML4-ALK or prostate TMPRSS2-EFG fusions, have not been reported.24–27
The presence of large numbers of shared breakpoints in multiple tumor blocks within single lung cancers further confirms that MP sequencing can detect lineage associations despite tumor heterogeneity. This is an important feature of an approach based on sequencing genomic rearrangements that would not be feasible using the targeted mutation panels that are emerging as an important diagnostic test for lung cancer care.34 Using rearrangements as identifiers of lineage, unlike single-nucleotide mutations, also benefits from the fact that rearrangements are much less frequently found in the germline. In our study, few samples predicted to be independent primary tumors demonstrated shared events, which we subsequently demonstrated as germline through PCR validation in non-neoplastic lung and blood DNA. Large genomic rearrangements are known to exist in the germline of the human population, which differ from the reference genome applied to bioinformatically detect rearrangements. In contrast to single-nucleotide polymorphisms, germline rearrangements have not been well defined in the reference genome. The extent of genomic variation from large genomic rearrangements has been more extensively mapped through copy number variation (CNV) studies. A study on the genomes of 385 healthy African American and 435 healthy white patients predicted averages of 3.5 and 4.8 CNV per genome, respectively.35 In these and other germline CNV studies,36–37 the majority of events arise from smaller intrachromosomal events spanning less than 30 kb. Through our extensive MP sequencing data of more than 500 tumors, we have been able to assemble a robust database of common germline variations to efficiently filter out the majority of polymorphic germline events. By applying filters masking discordant mapping events spanning less than 30 kb, we are also able to mask the majority of additional unique germline events. Although unique germline events passing our current masking filters will be expected to continually arise, the data from this study show that the frequency of such events remains low and easily evaluated by PCR. In addition, as more genomes are sequenced and our database of germline events increases, filtering will become increasingly efficient at masking out false-positive germline events. Thus, through the cumulative positive properties, including uniqueness of fusion junctions, low levels of germline events, and the enhanced ability to filter out many false-positive results (including germline events), we believe breakpoints present a valuable mechanism to determine tumor lineage.
The group of patients with independent primary tumors had no shared identical breakpoints between tumors within the same patient. Consistent with our hypothesis, we believe the identification of shared large chromosomal rearrangements between two tumors supports the concept of tumor lineage or relatedness. In our study, when many events were shared between two tumors, PCR validation was performed systematically on events until a shared event, absent from the non-neoplastic lung, was observed. Thus, the identification of metastasis worked on the preface that just a single somatic shared event was diagnostic of lineage. The number of observed events present within each tumor would theoretically strengthen such inference. However, to further define thresholds for clinical applications of an MP lineage test will require analysis of a larger sample set.
For the purpose of determining tumor lineage, the function of a mutation is less important than the fact that the mutation occurs early and is retained in the multiple tumor tissues. In this study, all genes had an equal role in the calling of tumor lineage. Although a number of genes were hit by rearrangements in multiple samples in this study, including the well-known lung tumor gene ALK, the sample size of this study was too small to predict driver versus passenger gene functions.
It is important to clarify that sensitivity of lineage detection is somewhat different from the sensitivity of complete breakpoint detection, although it is influenced by it. Sensitivity of lineage detection between two samples is dominated by recurrent high-coverage breakpoints, whereas the sensitivity of complete breakpoint detection is influenced by tumor heterogeneity, sequencing coverage for the region, and contamination by normal cells. In tumors where foci are determined to be in-lineage via the presence of many shared breakpoints validated by Sanger sequencing, the false-negative result will not influence the determination. Therefore, the positive predictive value of lineage detection for 12 tumor pairs (Figs 1D, 3D, and 5B) is essentially 100%. However, in the 10 paired tumors where the two tumors were rendered independent because no shared breakpoints were found (Figs 2D and 5B), the potential false-negative rate is an issue and dependent on algorithmic filters. To achieve a balanced compromise between false-negative and false-positive rates, we set the lower limit of breakpoint-supporting MPs to seven. Concordant mapping MP sequence coverage for all samples was more than 25 reads in regions where breakpoints were detected. With the conservative estimate of a 15% false-negative rate (dictated by the incompleteness of the reference genome and by regions that are difficult to map), we estimated that the probability of relatedness between any two samples is P < .15n, where n is the lowest number of total breakpoints between the two samples and no commonality is found. For the proposed test to be translated into a clinical setting, a precise criterion for calling lineage will be required. Although just a single shared somatic breakpoint could be considered strong evidence for lineage, this parameter still needs to be addressed with a larger sample study. In addition, the lower limit of total breakpoints present within a tumor, which can still allow an accurate lineage call, will also need to be addressed. Adaptation of testing to small biopsies or cytology specimens will also improve clinical translation.
An important consideration for a diagnostic test of lineage determination in cancer is whether it would be limited to lung cancer alone. Many vexing clinical dilemmas, including synchronous/metachronous SQCC of the head and neck and lung or multiple synchronous tumors of the female genital tract, would all be scenarios where a lineage test based on the detection of unique genomic rearrangements may add value for patient care. Our data suggest this may be an alternative and potentially more accurate and specific route to lineage determination than through the analysis of driver mutations alone. Although more work is required, the data also suggest the potential to improve on clinical and pathologic analysis alone.
Acknowledgment
We thank the Mayo Clinic Biobank for providing normal genomic samples used in establishing our germline rearrangement mask; the efforts of Kathy Laivell and Eric Edell, MD, in selection and acquisition of clinical samples from the Mayo Lung Specimen Registry; and Robert Sikkink and Bruce Eckloff of the Mayo Medical Genome Facility for next-generation sequencing.
Appendix
Table A1.
Sequencing Data
| Sample | Machine | Total No. of Fragments | Fragments Not Mapped (%) | Replication (%) | Bridged Coverage (×) | Base Coverage (×) | Average Mate-Pair Fragment (bp) |
|---|---|---|---|---|---|---|---|
| LU23_AD1a | HiSeq | 78,261,948 | 12.9 | 19.7 | 25.5 | 3.6 | 2,048 |
| LU23_AD1b | HiSeq | 69,571,432 | 27.8 | 26.9 | 18.4 | 2.4 | 2,185 |
| LU26_AD1 | HiSeq | 117,629,621 | 6.3 | 20.5 | 35.6 | 5.6 | 1,982 |
| LU26_AD2 | HiSeq | 106,004,409 | 14.4 | 20.8 | 40.7 | 4.7 | 2,642 |
| LU27_AD1 | HiSeq | 102,885,161 | 26.2 | 11.3 | 32.0 | 4.4 | 2,105 |
| LU27_AD2 | HiSeq | 74,356,244 | 15.3 | 9.9 | 30.4 | 3.7 | 2,246 |
| LU28_AD1 | HiSeq | 151,862,895 | 19.3 | 16.8 | 54.6 | 6.7 | 2,332 |
| LU28_AD2 | HiSeq | 159,939,374 | 17.3 | 35.9 | 36.2 | 5.1 | 2,136 |
| LU29_AD | HiSeq | 107,234,794 | 15.6 | 17.9 | 29.1 | 4.9 | 1,810 |
| LU29_SQ1 | HiSeq | 124,425,025 | 13.5 | 12.0 | 48.9 | 6.2 | 2,374 |
| LU29_SQ2 | HiSeq | 122,561,843 | 15.2 | 12.3 | 38.3 | 6.0 | 1,873 |
| LU30_AD1 | HiSeq | 135,020,898 | 10.3 | 12.9 | 51.0 | 6.8 | 2,223 |
| LU30_AD2 | HiSeq | 124,259,334 | 14.6 | 17.1 | 37.3 | 5.7 | 1,859 |
| LU31_AD | HiSeq | 103,281,231 | 16.3 | 8.7 | 34.8 | 5.2 | 1,927 |
| LU31b_ADb | HiSeq | 107,285,631 | 25.1 | 16.7 | 34.9 | 4.5 | 2,261 |
| LU32_AD1 | HiSeq | 123,168,647 | 22.5 | 11.4 | 46.1 | 5.5 | 2,408 |
| LU32_AD2 | HiSeq | 170,782,535 | 13.5 | 17.9 | 50.9 | 7.9 | 1,861 |
| LU33_AD1a | HiSeq2000 | 78,482,640 | 3.5 | 17.7 | 25.5 | 4.1 | 1,695 |
| LU33_AD1b | HiSeq2000 | 90,518,578 | 3.4 | 31.1 | 27.4 | 4.0 | 1,968 |
| LU33_BrM | HiSeq2000 | 87,887,485 | 3.6 | 36.4 | 10.5 | 2.3 | 1,355 |
| LU34_AD | HiSeq2000 | 78,018,388 | 3.2 | 20.8 | 25.8 | 3.9 | 1,798 |
| LU34_BrM | HiSeq2000 | 90,569,506 | 3.6 | 8.4 | 26.6 | 5.1 | 1,502 |
| LU38_AD | HiSeq2500 | 87,588,479 | 2.4 | 4.2 | 66.4 | 5.0 | 3,680 |
| LU38_BrM | HiSeq2000 | 81,965,528 | 4.0 | 22.8 | 25.7 | 3.9 | 1,838 |
| LU39_AD | HiSeq2000 | 120,412,216 | 2.2 | 15.7 | 81.0 | 6.0 | 3,765 |
| LU39_BrM | HiSeq2000 | 149,153,283 | 2.2 | 17.6 | 94.3 | 7.5 | 3,487 |
| LU40_SQ1 | HiSeq2000 | 139,718,870 | 2.5 | 9.4 | 97.6 | 7.9 | 3,402 |
| LU40_SQ2 | HiSeq2000 | 170,908,065 | 2.9 | 12.3 | 108.2 | 9.2 | 3,324 |
| LU41_SQ1 | HiSeq2000 | 140,145,548 | 2.5 | 9.4 | 104.5 | 7.8 | 3,679 |
| LU41_SQ2 | HiSeq2000 | 140,555,903 | 5.6 | 13.6 | 92.1 | 7.3 | 3,472 |
| LU43_AD1 | HiSeq2000 | 114,529,674 | 2.6 | 33.2 | 64.4 | 4.7 | 3,813 |
| LU43_AD2 | HiSeq2000 | 143,789,100 | 2.8 | 14.4 | 97.0 | 7.8 | 3,501 |
| LU44_AD1 | HiSeq2000 | 146,515,354 | 2.5 | 13.8 | 94.3 | 7.9 | 3,273 |
| LU44_AD2 | HiSeq2000 | 151,949,088 | 2.5 | 14.1 | 96.8 | 8.0 | 3,283 |
| LU45_AD1a | HiSeq2000 | 131,646,178 | 2.5 | 13.5 | 88.9 | 6.8 | 3,660 |
| LU45_AD1b | HiSeq2000 | 122,183,666 | 2.7 | 12.2 | 84.1 | 6.4 | 3,587 |
| LU45_AD2 | HiSeq2000 | 154,298,674 | 2.9 | 14.0 | 94.6 | 7.9 | 3,264 |
| LU46_AD | HiSeq2500 | 55,384,261 | 2.3 | 3.1 | 44.9 | 3.3 | 3,657 |
| LU46_SQ | HiSeq2500 | 80,884,315 | 2.3 | 3.7 | 62.9 | 4.7 | 3,624 |
| LU47_AD1a | HiSeq2500 | 65,046,795 | 2.3 | 3.7 | 47.4 | 3.6 | 3,621 |
| LU47_AD1b | HiSeq2500 | 103,749,753 | 2.4 | 4.0 | 71.8 | 5.6 | 3,552 |
| Average | 116,482,463 | 7 | 14 | 61 | 6 | 2,800 |
Table A2.
Correlation Between Histologic Prediction, Genomic Prediction, and Clinical Assessment
| Sample No. | Clinical Assessment | Histologic Prediction | Genomic Prediction |
|---|---|---|---|
| LU26 | Independent primary tumors | Independent primary tumors | Unrelated |
| LU27 | Independent primary tumors | Metastasis | Related |
| LU28 | Independent primary tumors | Independent primary tumors | Unrelated |
| LU29 | Independent primary tumors | Metastasis | Related |
| LU30 | Metastasis | Metastasis | Related |
| LU32 | Independent primary tumors | Independent primary tumors | Unrelated |
| LU40 | Metastasis | Metastasis | Related |
| LU41 | Independent primary tumors | Metastasis | Unrelated |
| LU43 | Independent primary tumors | Independent primary tumors | Unrelated |
| LU44 | Independent primary tumors | Indeterminate | Unrelated |
| LU45 | Indeterminate | Indeterminate | Unrelated |
Footnotes
See accompanying editorial on page 4029
Supported by the Mayo Clinic Center for Individualized Medicine Biomarker Discovery Program and grants from Uniting Against Lung Cancer (D.A.W.).
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Disclosures provided by the authors are available with this article at www.jco.org.
AUTHOR CONTRIBUTIONS
Conception and design: Stephen J. Murphy, Marie-Christine Aubry, Tobias Peikert, Ping Yang, George Vasmatzis, Dennis A. Wigle
Administrative support: Marie-Christine Aubry, George Vasmatzis, Dennis A. Wigle
Provision of study materials or patients: Ping Yang
Collection and assembly of data: Stephen J. Murphy, Marie-Christine Aubry, Faye R. Harris, Geoffrey C. Halling, Simone Terra, Travis M. Drucker, Michael K. Asiedu, Ping Yang, George Vasmatzis, Dennis A. Wigle
Data analysis and interpretation: Stephen J. Murphy, Marie-Christine Aubry, Faye R. Harris, Geoffrey C. Halling, Sarah H. Johnson, Simone Terra, Travis M. Drucker, Tobias Peikert, Benjamin R. Kipp, Ping Yang, Eunhee S. Yi, George Vasmatzis, Dennis A. Wigle
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Identification of Independent Primary Tumors and Intrapulmonary Metastases Using DNA Rearrangements in Non-Small-Cell Lung Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Stephen J. Murphy
Research Funding: Oncospire (Inst)
Marie-Christine Aubry
Research Funding: Oncospire (Inst)
Faye R. Harris
Research Funding: Oncospire (Inst)
Geoffrey C. Halling
Research Funding: Abbott Molecular (I), Oncospire (Inst)
Patents, Royalties, Other Intellectual Property: Abbott Molecular (I)
Sarah H. Johnson
Research Funding: Oncospire (Inst)
Simone Terra
Research Funding: Oncospire (Inst)
Travis M. Drucker
Research Funding: Oncospire (Inst)
Michael K. Asiedu
No relationship to disclose
Benjamin R. Kipp
Honoraria: Qiagen
Research Funding: Abbott Molecular
Patents, Royalties, Other Intellectual Property: Receive royalties for FISH probe set not used in this study
Eunhee S. Yi
Research Funding: Pfizer (Inst), Oncospire (Inst)
Tobias Peikert
Research Funding: Oncospire (Inst)
Ping Yang
No relationship to disclose
George Vasmatzis
Research Funding: Oncospire (Inst)
Dennis A. Wigle
Research Funding: Oncospire (Inst)
REFERENCES
- 1.Deschamps C, Pairolero PC, Trastek VF, et al. Multiple primary lung cancers: Results of surgical treatment. J Thorac Cardiovasc Surg. 1990;99:769–777. [PubMed] [Google Scholar]
- 2.Verhagen AF, Tavilla G, van de Wal HJ, et al. Multiple primary lung cancers. Thorac Cardiovasc Surg. 1994;42:40–44. doi: 10.1055/s-2007-1016453. [DOI] [PubMed] [Google Scholar]
- 3.Carey FA, Donnelly SC, Walker WS, et al. Synchronous primary lung cancers: Prevalence in surgical material and clinical implications. Thorax. 1993;48:344–346. doi: 10.1136/thx.48.4.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pommier RF, Vetto JT, Lee JT, et al. Synchronous non-small cell lung cancers. Am J Surg. 1996;171:521–524. doi: 10.1016/S0002-9610(96)00019-0. [DOI] [PubMed] [Google Scholar]
- 5.Nakata M, Sawada S, Yamashita M, et al. Surgical treatments for multiple primary adenocarcinoma of the lung. Ann Thorac Surg. 2004;78:1194–1199. doi: 10.1016/j.athoracsur.2004.03.102. [DOI] [PubMed] [Google Scholar]
- 6.Chang YL, Wu CT, Lee YC. Surgical treatment of synchronous multiple primary lung cancers: Experience of 92 patients. J Thorac Cardiovasc Surg. 2007;134:630–637. doi: 10.1016/j.jtcvs.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Aguiló R, Macià F, Porta M, et al. Multiple independent primary cancers do not adversely affect survival of the lung cancer patient. Eur J Cardiothorac Surg. 2008;34:1075–1080. doi: 10.1016/j.ejcts.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Kim TJ, Goo JM, Lee KW, et al. Clinical, pathological and thin-section CT features of persistent multiple ground-glass opacity nodules: Comparison with solitary ground-glass opacity nodule. Lung Cancer. 2009;64:171–178. doi: 10.1016/j.lungcan.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Arai J, Tsuchiya T, Oikawa M, et al. Clinical and molecular analysis of synchronous double lung cancers. Lung Cancer. 2012;77:281–287. doi: 10.1016/j.lungcan.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Shen KR, Meyers BF, Larner JM, et al. Special treatment issues in lung cancer: ACCP evidence-based clinical practice guidelines. Chest. 2007;132:290S–305S. doi: 10.1378/chest.07-1382. [DOI] [PubMed] [Google Scholar]
- 11.Fleisher AG, McElvaney G, Robinson CL. Multiple primary bronchogenic carcinomas: Treatment and follow-up. Ann Thorac Surg. 1991;51:48–51. doi: 10.1016/0003-4975(91)90446-w. [DOI] [PubMed] [Google Scholar]
- 12.Rosengart TK, Martini N, Ghosn P, et al. Multiple primary lung carcinomas: Prognosis and treatment. Ann Thorac Sug. 1991;52:773–778. doi: 10.1016/0003-4975(91)91209-e. [DOI] [PubMed] [Google Scholar]
- 13.Shimizu N, Ando A, Date H, et al. Prognosis of undetected intrapulmonary metastases in resected lung cancer. Cancer. 1993;71:3868–3872. doi: 10.1002/1097-0142(19930615)71:12<3868::aid-cncr2820711213>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 14.Adebonojo SA, Moritz DM, Danby CA. The results of modern surgical therapy for multiple primary lung cancers. Chest. 1997;112:693–701. doi: 10.1378/chest.112.3.693. [DOI] [PubMed] [Google Scholar]
- 15.Yano M, Arai T, Inagaki K, et al. Intrapulmonary satellite nodule of lung cancer as a T factor. Chest. 1998;114:1305–1308. doi: 10.1378/chest.114.5.1305. [DOI] [PubMed] [Google Scholar]
- 16.Trousse D, Barlesi F, Loundou A, et al. Synchronous multiple primary lung cancer: An increasing clinical occurrence requiring multidisciplinary management. J Thorac Cardiovasc Surg. 2007;133:1193–1200. doi: 10.1016/j.jtcvs.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 17.Okada M, Tsubota N, Yoshimura M, et al. Operative approach for multiple primary lung carcinomas. J Thorac Cardiovasc Surg. 1998;115:836–840. doi: 10.1016/S0022-5223(98)70364-2. [DOI] [PubMed] [Google Scholar]
- 18.Rea F, Zuin A, Callegaro D, et al. Surgical results for multiple primary lung cancers. Eur J Cardiothorac Surg. 2001;20:489–495. doi: 10.1016/s1010-7940(01)00858-2. [DOI] [PubMed] [Google Scholar]
- 19.Navin NE, Hicks J. Tracing the tumor lineage. Mol Oncol. 2010;4:267–283. doi: 10.1016/j.molonc.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy SJ, Cheville JC, Zarei S, et al. Mate pair sequencing of whole-genome-amplified DNA following laser capture microdissection of prostate cancer. DNA Res. 2012;19:395–406. doi: 10.1093/dnares/dss021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kotvun IV, Cheville JC, Murphy SJ, et al. Lineage relationship of Gleason patterns in Gleason score 7 prostate cancer. Cancer Res. 2013;73:3275–3284. doi: 10.1158/0008-5472.CAN-12-2803. [DOI] [PubMed] [Google Scholar]
- 22.Murphy SJ, Wigle DA, Lima JF, et al. Lineage relationships from genomic rearrangements in lung adenocarcinoma in situ and invasive adenocarcinoma. Cancer Res. 2014;74:3157–3167. doi: 10.1158/0008-5472.CAN-13-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cai X, Sheng J, Tang C, et al. Frequent mutations in EGFR, KRAS and TP53 genes in human lung cancer tumors detected by ion torrent DNA sequencing. PLoS One. 2014;9:e95228. doi: 10.1371/journal.pone.0095228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.St John J, Powell K, Conley-Lacomb MK, et al. TMPRSS2-ERG fusion gene expression in prostate tumor cells and its clinical and biological significance in prostate cancer progression. J Cancer Sci Ther. 2012;4:94–101. doi: 10.4172/1948-5956.1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark J, Merson S, Jhavar S, et al. Diversity of TMPRSS2-ERG fusion transcripts in the human prostate. Oncogene. 2007;26:2667–2673. doi: 10.1038/sj.onc.1210070. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Cai Y, Ren C, et al. Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer Res. 2006;66:8347–8351. doi: 10.1158/0008-5472.CAN-06-1966. [DOI] [PubMed] [Google Scholar]
- 27.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 28.Travis WD World Health Organization, International Agency for Research on Cancer, et al. Pathology and Genetics of Tumours of the Lung, Pleura, Thymus and Heart. Lyon, France: International Agency for Research on Cancer Press; 2004. [Google Scholar]
- 29.Travis WD, Brambilla E, Noguchi M, et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society International Multidisciplinary Classification of Lung Adenocarcinoma. J Thorac Oncol. 2011;6:244–285. doi: 10.1097/JTO.0b013e318206a221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Girard N, Deshpande C, Lau C, et al. Comprehensive histologic assessment helps to differentiate multiple lung primary nonsmall cell carcinomas from metastases. Am J Surg Pathol. 2009;33:1752–1764. doi: 10.1097/PAS.0b013e3181b8cf03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feldman AL, Dogan A, Smith DI, et al. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood. 2011;117:915–919. doi: 10.1182/blood-2010-08-303305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vasmatzis G, Johnson SH, Knudson RA, et al. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas. Blood. 2012;120:2280–2289. doi: 10.1182/blood-2012-03-419937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murphy SJ, Hart SN, Lima JF, et al. Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology. 2013;145:1098–1109. doi: 10.1053/j.gastro.2013.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McElroy JP, Nelson MR, Caillier SJ, et al. Copy number variation in African Americans. BMC Genet. 2009;10:15. doi: 10.1186/1471-2156-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yim SH, Kim TM, Hu HJ, et al. Copy number variations in East-Asian population and their evolutionary and functional implications. Hum Mol Genet. 2010;19:1001–1008. doi: 10.1093/hmg/ddp564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Li X, Zhang F, et al. A preliminary study of copy number variation in Tibetans. PLoS One. 2012;7:e41768. doi: 10.1371/journal.pone.0041768. [DOI] [PMC free article] [PubMed] [Google Scholar]