

Abstract
Activation of Constitutive Androstane Receptor (CAR) protects against bile acid (BA)-induced liver injury. This study was performed to determine the effect of CAR activation on bile flow, BA profile, as well as expression of BA synthesis and transport genes. Synthetic CAR ligand 1,4-bis-[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) was administered to mice for 4 days. BAs were quantified by UPLC-MS/MS (ultraperformance liquid chromatography-tandem mass spectrometry). CAR activation decreases total BAs in livers of male (49%) and female mice (26%), largely attributable to decreases of the 12α-hydroxylated BA taurocholic acid (T-CA) (males (M) 65%, females (F) 45%). Bile flow in both sexes was increased by CAR activation, and the increases were BA-independent. CAR activation did not alter biliary excretion of total BAs, but overall BA composition changed. Excretion of muricholic (6-hydroxylated) BAs was increased in males (101%), and the 12α-OH proportion of biliary BAs was decreased in both males (37%) and females (28%). The decrease of T-CA in livers of males and females correlates with the decreased mRNA of the sterol 12α-hydroxylase Cyp8b1 in males (71%) and females (54%). As a response to restore BAs to physiologic concentrations in liver, mRNA of Cyp7a1 is upregulated following TCPOBOP (males 185%, females 132%). In ilea, mRNA of the negative feedback regulator Fgf15 was unaltered by CAR activation, indicating biliary BA excretion was sufficient to maintain concentrations of total BAs in the small intestine. In summary, the effects of CAR activation on BAs in male and female mice are quite similar, with a marked decrease in the major BA T-CA in the liver.
Keywords: constitutive androstane receptor, bile acids, biliary excretion, TCPOBOP, taurocholic acid, muricholic acid
Introduction
Bile acid (BA) synthesis is a critical function of the liver. BAs synthesized in the liver are termed “primary” BAs (Table 1). Whereas the major primary BAs in humans are cholic acid (CA) and chenodeoxycholic acid (CDCA), mice have 2 additional major primary BAs, α-muricholic acid (αMCA) and βMCA. BAs are viewed as being derived from either the classical or the alternative pathway of synthesis. [AQ7]The classical pathway includes cholesterol 7α-hydroxylase Cytochrome P450 7a1 (Cyp7a1), which is the rate-limiting enzyme of BA synthesis. Cyp8b1 is a 12α-hydroxylase and thus responsible for the formation of CA, the major 12α-hydroxylated (12α-OH) BA. The alternative pathway of BA synthesis is initiated by 27-hydroxylation of cholesterol by mitochondrial Cyp27a1, and includes 7α-hydroxylase Cyp7b1. BAs undergo conjugation with glycine or taurine before excretion into bile for delivery to the small intestine. Bacteria in the intestine metabolize primary BAs to form secondary BAs (Table 1). The steps involved in BA synthesis have been reviewed by Russell (2003). Ninety-five percent of BAs undergo reabsorption from the intestinal lumen via active transport and recirculate to the liver by the portal blood, making them available for efficient reuptake by active transport into the liver (Chiang 2003).
TABLE 1.
Summary of Primary, Secondary, 6-Hydroxy- (6-OH), and 12α-Hydroxylated (12α-OH) Bile Acids (BA)
| Primary | Secondary | 6-OH | 12α-OH |
|---|---|---|---|
| T-αMCA | T-ωMCA | T-αMCA | T-CA |
| T-ßMCA | T-UDCA | T-ßMCA | T-DCA |
| T-CA | T-HDCA | T-ωMCA | CA |
| T-CDCA | T-DCA | αMCA | DCA |
| αMCA | T-LCA | ßMCA | |
| ßMCA | T-HCA | ωMCA | |
| CA | ωMCA | ||
| CDCA | UDCA | ||
| HDCA | |||
| DCA | |||
| LCA | |||
| HCA |
MCA, muricholic acid; CA, cholic acid; UDCA, ursodeoxycholic acid; DCA, deoxycholic acid; HDCA, hyodeoxycholic acid; CDCA, chenodeoxycholic acid; LCA, lithocholic acid; HCA, hyocholic acid.
This process of biliary excretion, intestinal absorption, portal circulation, and reuptake into liver is referred to as enterohepatic circulation (EHC). EHC allows the 2- to 4-g pool of BAs in humans to be conserved for a variety of physiologic functions (Hofmann, 1984). The best known function of BAs is to form micelles and emulsify fatty acids, monoglycerides, and fat-soluble vitamins and aid in their absorption (Monte et al., 2009). These beneficial functions of BAs are largely attributable to the amphipathic nature of BAs. Conversely, accumulation of BAs in hepatocytes and the biliary tract can lead to oxidative stress, inflammation, and necrosis (Perez and Briz, 2009). Considering that the pool of BAs typically circulates 4–10 times daily and BAs are toxic, tight regulation of BA concentrations is thought to be critical (Lindstedt, 1957; Danielsson et al., 1963).
The Farnesoid X Receptor (Fxr) is a BA receptor that regulates BA homeostasis. In liver, Fxr has a negative feedback regulation of BA synthesis. Specifically, activation of hepatic Fxr by the BA ligand induces transcription of Short Heterodimer Partner (Shp), which in turn represses Liver Receptor Homolog-1 (Lrh-1) from promoting Cyp7a1 transcription (Goodwin et al., 2000). In the intestine, more specifically the ileum enterocytes, BAs activate Fxr that induces transcription of Fibroblast Growth Factor (Fgf) 15 (Fgf19 in humans), which also exerts a negative feedback regulation of Cyp7a1 in liver (Lu et al., 2000; Holt et al., 2003; Inagaki et al., 2005). This occurs by the ileal synthesis of Fgf15 that is released into the portal blood and activates the FGF Receptor 4 (Fgfr4) on hepatocytes (Xie et al., 1999; Inagaki et al., 2005; Song et al., 2009). Fxr signaling is not only important for regulating BA synthesis but also in glucose metabolism and energy homeostasis. BA sequestrant therapy has been shown to improve glycemic control in type 2 diabetes (Garg and Grundy, 1994; Yamakawa et al., 2007; Zieve et al., 2007; Bays et al., 2008; Fonseca et al., 2008; Goldberg et al., 2008). BAs have also been shown to function as ligands of the Transmembrane G-protein-coupled Receptor 5 (Tgr5) in mice, leading to increased mitochondrial activity and energy expenditure (Watanabe et al., 2006).
Individual BAs can be distinguished structurally, on the basis of the number, position, and orientation of hydroxyl groups, as well as keto groups and conjugation (glycine, taurine, sulfate, and glucuronide) (Hofmann and Hagey, 2008). Given these differences, there is a gradient of hydrophobicity among the predominant BA species: UDCA < CA < CDCA < DCA < LCA (Heuman, 1989; Thomas et al., 2008). The hydrophobicity of BAs is generally considered to be associated with their degree of hepatotoxicity (Palmer, 1972; Billington et al., 1980), and this has been observed in BA-feeding studies in mice (Song et al., 2011). Individual BAs also differ in terms of capacity to activate receptors such as Fxr (CDCA > DCA > LCA > CA), as well as Tgr5 (LCA > DCA > CDCA > CA) in vitro (Parks et al., 1999; Sato et al., 2008).
Accumulation of BAs in hepatocytes is thought to be a major cause of cholestatic liver injury in certain forms of liver disease, as well as in drug-induced liver injury (Trauner et al., 1998; Bhamidimarri and Schiff, 2013). Accordingly, various cellular targets have been investigated as a means to achieve hepatoprotection from BA-induced injury produced by experimental cholestasis (Halilbasic et al., 2013). One such target in the liver is the nuclear receptor constitutive androstane receptor (CAR, NR1I3). Activation of CAR is well known to induce transcription of genes responsible for hydroxylation, conjugation, and excretion of drugs and other xenobiotics (Assem et al., 2004; Saini et al., 2004; Wagner et al., 2005).
Phenobarbital (PB) is an anticonvulsant drug and a classical CAR activator that has been widely used as a microsomal enzyme inducer (Kakizaki et al., 2003). PB activation of CAR induces the marker gene Cyp2b in rodent and human hepatocytes, and is well known as an inducer of P450 enzyme activities (Honkakoski et al., 1998; Sueyoshi et al., 1999; Madan et al., 2003). PB treatment results in dephosphorylation of threonine 38 of human CAR (serine 202 of mouse CAR), allowing translocation to the nucleus to regulate gene expression (Kawamoto et al., 1999; Hosseinpour et al., 2005; Mutoh et al., 2009). TCPOBOP is a direct ligand and agonist of CAR in mice (Tzameli et al., 2000). In addition to induction of some phase I and II drug metabolizing enzymes, PB and TCPOBOP are known to induce transporters as well (Maher et al., 2005; Buckley and Klaassen, 2009). Studies in mice have shown TCPOBOP to be approximately 650 times more potent than PB, and the extent of maximal induction of P450 activity is slightly greater for TCPOBOP (Poland et al., 1980). Comprehensive comparisons of the PB and TCPOBOP target genes and induction profiles have been reported (Slatter et al., 2006). Both PB and TCPOBOP have been shown to be hepatoprotective against lithocholic acid (LCA)-induced injury in mice by decreasing hepatic concentrations of di- and tri-hydroxyl BAs (Beilke et al., 2009). TCPOBOP has also been shown to increase fecal excretion of muricholic acids (MCAs) (Sberna et al., 2011).
Significant gaps remain in our understanding of the effect of CAR activation on the BA profile in mice. Therefore, the objectives of the present study were to determine the effect of CAR activation on biliary excretion of individual BAs, as well as hepatic and serum content of individual BAs in male and female mice. In both liver and ileum, the effects of CAR activation on major pathways of BA regulation, synthesis, uptake, and excretion were also investigated.
Materials and Methods
Chemicals
Bile acid standards were purchased from Steraloids, Inc. (Newport, Rhode Island) and Sigma-Aldrich (St Louis, Missouri). CAR ligand TCPOBOP or TCPO and corn oil used as vehicle were also purchased from Sigma-Aldrich. All other chemicals were also purchased from Sigma-Aldrich unless otherwise noted.
Animals
Male and female C57BL/6 wild-type (WT) mice were purchased from Charles River Laboratories, Inc. (Wilmington, Massachusetts). Prior to experiments, mice were acclimated for at least 1 week in a standard temperature-, light-, and humidity-controlled facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. Mice had free access to Laboratory Rodent Chow 8604 (Harlan, Madison, Wisconsin) and drinking water. Studies were approved by the University of Kansas Medical Center Institutional Animal Care and Use Committee. Male mice used in experiments were 12–15 weeks of age, whereas females were 16–19 weeks of age, to provide a similar weight of animals for surgery.
Chemical treatment followed by collection of livers, blood, and ilea
Corn oil vehicle or TCPOBOP (3 mg/kg; 5 ml/kg) in corn oil was administered daily for 4 consecutive days by the intraperitoneal (IP) route to male and female mice (n = 5–6 per treatment group). At 24 h after the fourth dose, blood was collected from the retro-orbital sinus of mice anesthetized with 50 mg/kg pentobarbital, and livers and ilea were removed. Serum samples were separated using Microtainer separating tubes (BD Biosciences, San Jose, California). Liver and ileum samples were frozen in liquid nitrogen and stored at −80 °C until further analysis.
Chemical treatment followed by bile collection
Corn oil vehicle or TCPOBOP (3 mg/kg; 5 ml/kg) in corn oil was administered daily IP to male and female mice for 4 consecutive days (n = 5–6 per treatment group). On day 5, mice were anesthetized with a ketamine/midazolam mixture (100 and 5 mg/kg, respectively, IP), and the common bile duct of each mouse was cannulated through a high abdominal incision with the shaft of a 30-gauge needle attached to PE-10 tubing. Bile was collected for 40 min in preweighed 0.6-ml tubes on ice. The volumes of bile were determined gravimetrically, using 1.0 for specific gravity.
RNA extraction
Total RNA of livers and ilea was extracted using RNA-Bee reagent (Tel-Test, Inc., Friendswood, Texas) according to the manufacturer’s protocol. Each RNA pellet was dissolved in 0.2 ml of deionized water containing 0.1% diethyl pyrocarbonate. RNA concentrations were quantified using a NanoDrop1000 Spectrophotometer (NanoDrop Technologies, Wilmington, Delaware) at a wavelength of 260 nm. RNA integrity was confirmed by agarose gel electrophoresis and ethidium bromide staining of 5 µg of total RNA to visualize intact 18S and 28S bands.
Messenger RNA quantification
Messenger RNA of genes in liver and ileum samples was determined using QuantiGene Plex 2.0 Assay (Affymetrix/Panomics, Inc., Fremont, California). Individual bead-based oligonucleotide probe sets, specific for each gene examined, were developed by Affymetrix/Panomics, Inc. Genes and reference sequence numbers are available at https://www.ebioscience.com/application/gene-expression.htm, Accessed March 20, 2016 (sets Nos. 21330 and 21383). Samples were analyzed using a Bio-Plex 200 System Array reader with Luminex 100 xMAP. Data were acquired using Bio-Plex Data Manager version 5.0 (Bio-Rad, Hercules, California).
For some mRNAs, real-time RT-PCR was used. These included Abca1, Abcg5, Abcg8, Atp8b1, breast cancer resistance protein (Bcrp), ß-klotho, Ent1, I-babp, Mate1, Mdr1, Mrp1, Mrp4, Mrp6, Oatp1a4, Oatp2b1, Oat2, Oct1, Ostα, and Ostß (Table 1). Primers were designed with Primer3 software (version 4). Primer sequences for I-babp were published previously (Cui et al., 2012). Total RNA was reverse-transcribed with MultiScribe MuLV Reverse Transcriptase using a High Capacity cDNA Reverse Transcription Kit from Applied Biosystems (Foster City, California). Power SYBR Green Master Mix (Applied Biosystems) was used for real-time RT-PCR analysis. Differences in gene expression between groups were calculated using cycle threshold (Ct) values that were normalized to ribosomal protein L13A (Rpl13a) of the same sample (Rockwell et al., 2012). Relative mRNA levels were calculated with vehicle controls set as 100%.
Ultraperformance liquid chromatography-tandem mass spectrometry analysis of BAs in mouse liver, bile, and serum
Sample extraction and quantification of individual BAs were performed according to methods described previously (Alnouti et al., 2008; Zhang and Klaassen, 2010).
Calculations of primary, secondary, 6-hydroxylated (6-OH), and 12α-OH BAs
Concentrations of various BA groups (primary, secondary, 6-hydroxylated (6-OH), and 12α-OH) were determined by calculating the sum concentration of the group members, as shown in Table 1.
Statistical analysis
Individual values were log-transformed to obtain a normal distribution before performing Student’s t tests. Statistical significance was determined using a P-value of 0.05. Asterisks (*) denote differences between vehicle- and TCPOBOP-treated mice.
Results
Effects of CAR Activation on BA Concentrations and Composition in Mouse Serum
In both male and female mice, CAR activation did not produce alterations in serum concentrations of the major BA categories, including total (Σ) BAs, conjugated BAs, unconjugated BAs (U-BAs), primary BAs, secondary BAs, 6-OH BAs, and 12α-OH BAs (Figure 1, top graphs). In male mice, increases in T-α+ßMCA (307%), T-UDCA (tauro-ursodeoxycholic acid) (70%), T-HDCA (tauro-hyodeoxycholic acid) (148%), and αMCA (130%) were observed, whereas T-DCA and deoxycholic acid (DCA) were decreased 95% and 83%, respectively (Figure1, middle left and bottom left). In female mice, the only statistically significant alteration was a 17-fold increase in T-CDCA, a minor BA in mice (Figure 1, middle right).
FIG. 1.

Effect of TCPOBOP on bile acid concentrations in sera of male and female mice. Corn oil vehicle or TCPOBOP (3 mg/kg) was administered daily (IP) for 4 consecutive days to male and female mice (n = 5–6 per treatment group). At 96 h after the first dose, blood was collected and serum separated. Individual BAs were quantified by UPLC-MS/MS. Data represent means ± SEM. Asterisks (*) denote statistically different from vehicle control (P < 0.05). Primary bile acids (1° BAs), secondary bile acids (2° BAs), 6-hydroxylated (6-OH), 12α-hydroxylated (12α-OH), bile acid (BA), cholic acid (CA), chenodeoxycholic acid (CDCA), deoxycholic acid (DCA), females (F), hyodeoxycholic acid (HDCA), lithocholic acid (LCA), males (M), muricholic acid (MCA), total bile acids (Σ-BAs), taurine-conjugated bile acids (T-BAs), 1,4-bis(2-[3,5-dichloropyridyloxy])benzene (TCPOBOP), unconjugated bile acids (U-BAs), ursodeoxycholic acid (UDCA), vehicle (veh). Color image is available in the online version of the article.
The relative proportion of each BA in the serum is shown in Figure 2. The most abundant BA is indicated at the top of the graph, with BAs being in clockwise order of most abundant to least abundant in vehicle-treated males and females. Percentages are shown for BAs with serum composition of ≥ 5%, as well as for those that were significantly altered by CAR activation. Taurocholic acid (T-CA) was the most abundant BA in serum of control male mice (29%). CAR activation resulted in T-α+ßMCA becoming the most abundant (35%) BA in male mouse serum, whereas it was fourth most abundant in controls (10%). Additionally, CAR activation decreased T-DCA from 3.9% to 0.15% and DCA from 5.2% to 0.82%.
FIG. 2.

Effect of TCPOBOP on serum composition of individual bile acids in male and female mice. Corn oil vehicle or TCPOBOP (3 mg/kg) was administered daily (IP) for 4 consecutive days to male and female mice (n = 5–6 per treatment group). At 96 h after the first dose, blood was collected and serum separated. Individual BAs were quantified by UPLC-MS/MS. Each section in pie charts was calculated to represent the mean proportion of an individual BA relative to the total BA concentration. Asterisks (*) denote statistically different from vehicle control (P < 0.05). Primary bile acids (1° BAs), secondary bile acids (2° BAs), 6-hydroxylated (6-OH), 12α-hydroxylated (12α-OH), bile acid (BA), cholic acid (CA), chenodeoxycholic acid (CDCA), deoxycholic acid (DCA), females (F), hyodeoxycholic acid (HDCA), lithocholic acid (LCA), males (M), muricholic acid (MCA), total bile acids (Σ-BAs), taurine-conjugated bile acids (T-BAs), 1,4-bis(2-[3,5-dichloropyridyloxy])benzene (TCPOBOP), unconjugated bile acids (U-BAs), ursodeoxycholic acid (UDCA), vehicle (veh). Color image is available in the online version of the article.
In serum of female mice, the effect of CAR activation was more modest than in male mice (Figure 2). T-CA was the most abundant BA in serum of both control (34%) and TCPOBOP-treated (19%) mice. CAR activation increased the proportion of T-ωMCA from 7.4% to 16%, as well as T-CDCA from 0.03% to 0.48%. TCPOBOP decreased T-DCA from 2.3% to 0.77%. The effect of CAR activation on relative composition for each BA in serum of male and female mice is listed in Supplementary Table 2. In serum of vehicle-treated mice, the U-BAs are 39% (males) and 38% (females) of total bile acids (Σ-BAs). CAR activation in male and female mice did not alter the percent of Σ-BAs that are unconjugated. To summarize, the overall effect of TCPOBOP on serum BA concentrations was relatively small, but a 48% increase in 6-OH BAs (MCAs) was observed in male mice, with a similar tendency (29% increase) in female mice.
Effects of CAR Activation on BA Concentrations and Composition in Mouse Liver
The effects of TCPOBOP-induced CAR activation on the concentrations of BAs in liver were generally also more pronounced in male than female mice (Figure 3). In males, CAR activation decreased Σ-BAs (49%), T-BAs (48%), U-BAs (68%), primary BAs (47%), and secondary BAs (61%) (Figure 3, top left). In female mice, CAR activation tended to decrease total BAs (26%) and primary BAs (26%), although not statistically significant (Figure 3, top right). CAR activation decreased 12α-OH BAs in livers of both male (68%) and female mice (46%), whereas 6-OH BAs (MCAs) were not altered in livers of either sex. In male mice, CAR activation decreased hepatic concentrations of the 12α-OH BAs T-CA (65%), CA (96%), T-DCA (87%), and DCA (92%), as well as the non-12α-hydroxylated (non12α-OH) BAs T-LCA (75%), αMCA (72%), ßMCA (47%), CDCA (74%), and UDCA (94%) (Figure 3, middle left and bottom left). In female mice, CAR activation decreased hepatic concentrations of the 12α-OH BAs T-CA (45%), T-DCA (53%), CA (34%), and DCA (58%). The only BA increased in liver by TCPOBOP was T-CDCA (38%) (Figure 3, middle right).
FIG. 3.

Effect of TCPOBOP on concentrations of bile acids in livers of male and female mice. Corn oil vehicle or TCPOBOP (3 mg/kg) was administered daily (IP) for 4 consecutive days to male and female mice (n = 5–6 per treatment group). At 96 h after the first dose, livers were collected and individual BAs were quantified by UPLC-MS/MS. Data represent means ± SEM. Asterisks (*) denote statistically different from vehicle control (P < 0.05). Primary bile acids (1° BAs), secondary bile acids (2° BAs), 6-hydroxylated (6-OH), 12α-hydroxylated (12α-OH), bile acid (BA), cholic acid (CA), chenodeoxycholic acid (CDCA), deoxycholic acid (DCA), females (F), lithocholic acid (LCA), males (M), muricholic acid (MCA), total bile acids (Σ-BAs), taurine-conjugated bile acids (T-BAs), 1,4-bis(2-[3,5-dichloropyridyloxy])benzene (TCPOBOP), unconjugated bile acids (U-BAs), ursodeoxycholic acid (UDCA), vehicle (veh). Color image is available in the online version of the article.
The relative proportion of each BA in liver is shown in Figure 4. T-CA was the most abundant BA in liver of control male mice (52%). However, CAR activation decreased the proportion of the 12α-OH BAs, including T-CA (52% to 36%), T-DCA (5.4% to 1.4%), and DCA (0.04% to 0.01%). Thus, CAR activation resulted in T-α+ßMCA being the most abundant BA (42%) in livers of male mice, such that the 6-OH (MCA) proportion of BAs in liver increased by 51%. TCPOBOP also increased T-CDCA (2.6% to 6.4%), whereas it decreased the proportion of UDCA (0.07% to 0.01%).
FIG. 4.

Effect of TCPOBOP on liver composition of individual bile acids in male and female mice. Corn oil vehicle or TCPOBOP (3 mg/kg) was administered daily (IP) for 4 consecutive days to male and female mice (n = 5–6 per treatment group). At 96 h after the first dose, livers were collected and individual BAs were quantified by UPLC-MS/MS. Each section in pie charts was calculated to represent the mean proportion of an individual BA relative to the total BA concentration. Asterisks (*) denote statistically different from vehicle control (P < 0.05). Primary bile acids (1° BAs), secondary bile acids (2° BAs), 6-hydroxylated (6-OH), 12α-hydroxylated (12α-OH), bile acid (BA), cholic acid (CA), chenodeoxycholic acid (CDCA), deoxycholic acid (DCA), females (F), lithocholic acid (LCA), males (M), muricholic acid (MCA), total bile acids (Σ-BAs), taurine-conjugated bile acids (T-BAs), 1,4-bis(2-[3,5-dichloropyridyloxy])benzene (TCPOBOP), unconjugated bile acids (U-BAs), ursodeoxycholic acid (UDCA), vehicle (veh). Color image is available in the online version of the article.
As in males, T-CA was the most abundant BA in female mouse livers (53%) (Figure 4). Although CAR activation decreased the proportion of T-CA to 39% in livers of female mice, T-CA remained the most abundant. The proportion of the other major 12α-OH BA, T-DCA, was also decreased (7.5% to 4.7%). Accordingly, CAR activation increased the proportion of 6-OH BAs (by 34%), TωMCA (7.9% to 11%), and T-CDCA (2.6% to 8.4%) in female mouse livers. The effect of CAR activation on relative composition of each BA in livers of male and female mice is listed in Supplementary Table 3. In livers of vehicle-treated mice, the proportion of total BAs that are U-BAs is 6% in males and 4% in females, which means a decrease of 85% (males) and 90% (females) relative to the proportions in serum. CAR activation did not alter the proportion of U-BAs in males or females. In summary, activating the CAR receptor in mice by TCPOBOP decreases the total concentration of BAs in liver primarily by decreasing the 12α-OH BAs.
Effects of CAR Activation on Biliary Excretion of BAs
CAR activation did not alter the biliary excretion of Σ-BAs or T-BAs, but increased the biliary excretion of U-BAs in both male (174%) and female (169%) mice (Figure 5, top graphs). However, the biliary excretion of 6-OH BAs was increased, but only in male mice (101%), and this increase was largely attributable to 260% and 82% increases in the quantitatively major 6-OH BAs, T-αMCA and T-ßMCA, respectively (Figure 5, middle left graph). Additionally, biliary excretion was increased for the non12α-OH BAs T-CDCA (224%), T-LCA (150%), T-HCA (tauro-hyocholic acid) (68%), αMCA (482%), ßMCA (134%), and ωMCA (189%).
FIG. 5.

Effect of TCPOBOP on biliary excretion of bile acids in male and female mice. Corn oil vehicle or TCPOBOP (3 mg/kg) was administered daily (IP) for 4 consecutive days to male and female mice (n = 5–6 per treatment group). On day 5, bile was collected for 40 min. Bile acids (BAs) were quantified by UPLC-MS/MS. Data represent means ± SEM. Asterisks (*) denote statistically different from vehicle control (P < 0.05). Primary bile acids (1° BAs), secondary bile acids (2° BAs), 6-hydroxylated (6-OH), 12α-hydroxylated (12α-OH), bile acid (BA), cholic acid (CA), chenodeoxycholic acid (CDCA), deoxycholic acid (DCA), females (F), hyocholic acid (HCA), hyodeoxycholic acid (HDCA), lithocholic acid (LCA), males (M), muricholic acid (MCA), total bile acids (Σ-BAs), taurine-conjugated bile acids (T-BAs), 1,4-bis(2-[3,5-dichloropyridyloxy])benzene (TCPOBOP), unconjugated bile acids (U-BAs), ursodeoxycholic acid (UDCA), vehicle (veh). Color image is available in the online version of the article.
In female mice, CAR activation increased the biliary excretion of T-αMCA (154%), αMCA (394%), and ßMCA (307%), as well as the 12α-OH BA CA (193%) (Figure 5, middle and bottom graphs). Just as CAR activation decreased T-DCA concentration in female mouse liver (53%), biliary excretion of T-DCA (71%) was also decreased.
The relative proportion of each BA in bile is shown in Figure 6. The most abundant BA in bile of male mice was T-CA (56%), and CAR activation decreased it to 37%. T-ßMCA became the most abundant BA (41%) following CAR activation, and T-αMCA was increased from 5.9% to 15%. Thus, the 6-OH proportion of BAs in bile increased by 51%, as it did in liver of male mice. CAR activation also produced a number of increases in BAs that represent smaller proportions of bile composition, including increases in T-CDCA (0.88% to 2.0%), ßMCA (0.07% to 0.12%), ωMCA (0.07% to 0.14%), T-LCA (0.03% to 0.05%), and αMCA (0.01% to 0.04%). These changes are consistent with the overall increase in biliary excretion rate of non12α-OH BAs in male mice.
FIG. 6.

Effect of TCPOBOP on bile composition of individual bile acids in male and female mice. Corn oil vehicle or TCPOBOP (3 mg/kg) was administered daily (IP) for 4 consecutive days to male and female mice (n = 5–6 per treatment group). On day 5, bile was collected for 40 min. Each section in pie charts was calculated to represent the mean proportion of an individual BA relative to the total BA concentration. Asterisks (*) denote statistically different from vehicle control (P < 0.05). Primary bile acids (1° BAs), secondary bile acids (2° BAs), 6-hydroxylated (6-OH), 12α-hydroxylated (12α-OH), bile acid (BA), cholic acid (CA), chenodeoxycholic acid (CDCA), deoxycholic acid (DCA), females (F), hyocholic acid (HCA), hyodeoxycholic acid (HDCA), lithocholic acid (LCA), males (M), muricholic acid (MCA), total bile acids (Σ-BAs), taurine-conjugated bile acids (T-BAs), 1,4-bis(2-[3,5-dichloropyridyloxy])benzene (TCPOBOP), unconjugated bile acids (U-BAs), ursodeoxycholic acid (UDCA), vehicle (veh). Color image is available in the online version of the article.
In bile of female mice, T-CA (63%) was also the most abundant BA (Figure 6). CAR activation decreased the fraction of T-CA in bile to 39%. T-ßMCA became the most abundant (41%) following CAR activation, and T-αMCA was increased from 5.4% to 15%. The 6-OH proportion of BAs in bile increased by 75%. As in males, CAR activation produced a number of increases in BAs in female mice that are quantitatively low in bile, including increases in T-CDCA (1.3% to 2.5%), T-HCA (0.11% to 0.26%), ßMCA (0.02% to 0.08%), and αMCA (0.01% to 0.05%). The effect of CAR activation on relative composition for each BA in bile is listed in Supplementary Table 4. In bile of vehicle-treated mice, the proportion of U-BAs is < 0.2% in males (0.16%) and females (0.08%). CAR activation increased the proportion of U-BAs in bile by 94% (males) and 188% (females). Nonetheless, the relative proportion of U-BAs remained quite small (0.31% in males, 0.23% in females). Comparing bile with serum, the proportion of U-BAs in bile is > 99.5% lower than in serum. This difference was observed in males and females. In summary, the CAR activator had no effect on the biliary excretion of total BAs, but there was an increase in unconjugated BAs and MCAs, and a decrease in CAs.
Effects of CAR Activation on Liver Weight and Bile Flow
The CAR activator TCPOBOP increased liver weights of male and female mice by 61% and 105%, respectively (Figure 7). CAR activation increased bile flow relative to bodyweight in both male (64%) and female mice (50%), whereas bile flow was not increased when calculated per unit liver weight. Therefore, CAR increased liver weight and bile flow in both male and female mice.
FIG. 7.
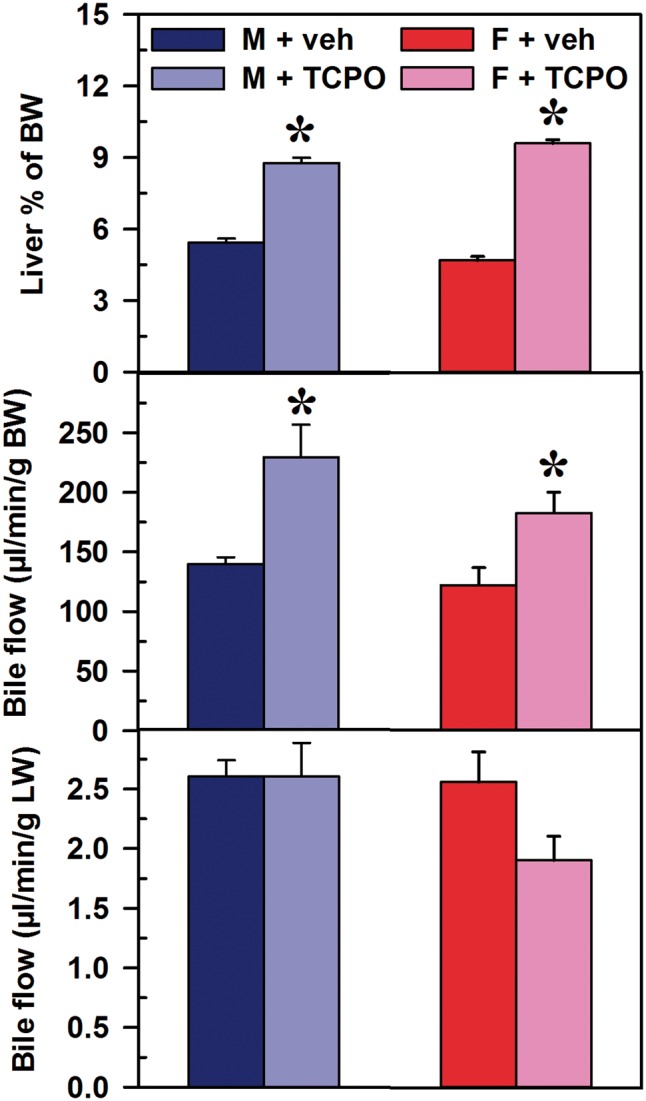
Effect of TCPOBOP on liver weight and bile flow in male and female mice. Corn oil vehicle or TCPOBOP (3 mg/kg) was administered daily (IP) for 4 consecutive days to male and female mice (n = 5–6 per treatment group). On day 5, mice were anesthetized and bile was collected for 40 min. Liver weight is expressed as a percent of bodyweight (BW) (upper panel). Bile flow rates were normalized to BW (middle panel) and liver weight (lower panel). Data represent means ± SEM. Asterisks (*) denote statistically different from vehicle control (P < 0.05). Females (F), males (M), 1,4-bis(2-[3,5-dichloropyridyloxy])benzene (TCPO). Color image is available in the online version of the article.
Effects of CAR Activation on mRNAs of BA and Xenobiotic Transporters in Mouse Liver
In male mice, TCPOBOP-induced CAR activation increased mRNA of the conjugated BA uptake transporter Ntcp (222%), as well as Oatp1b2 (132%), which is the major uptake transporter of unconjugated BAs (Csanaky et al., 2011) (Figure 8, top left). CAR activation also increased mRNA of bile salt export pump (Bsep) (136%) and Mrp2 (523%), which are major drivers of BA-dependent and BA-independent bile flow, respectively (Figure 8, middle left). Further, CAR activation increased Mdr2 (138%), an ATP-dependent “floppase” of phosphatidylcholine from the inner- to outer-leaflet of canalicular membranes. Sinusoidal efflux transporters of BAs into the blood, Mrp3 (795%) and Mrp4 (1388%), were increased, whereas there were decreases in Mrp6 (89%), Ostα (61%), and Abca1 (44%) (Figure 8, bottom left).
FIG. 8.

Effect of TCPOBOP on mRNA of transporters in livers of male and female mice. Corn oil vehicle or TCPOBOP (3 mg/kg) was administered daily (IP) for 4 consecutive days to male and female mice (n = 5–6 per treatment group). At 96 h after the first dose, livers were collected. Total RNA was analyzed by QuantiGene Plex 2.0 Assay, as well as by RT-qPCR. Relative mRNA levels were calculated with vehicle controls set as 100%. Data represent means ± SEM. Asterisks (*) denote statistically different from vehicle control (P < 0.05). Breast cancer resistance protein (Bcrp), Bile salt export pump (Bsep), Equilibrative nucleoside transporter (Ent), females (F), males (M), Multidrug and toxin extrusion transporter (Mate), Multidrug resistance protein (Mdr), Multidrug resistance-associated protein (Mrp), Na(+)-taurocholate cotransporting polypeptide (Ntcp), Organic anion transporting polypeptide (Oatp), Organic cation transporter (Oct), Organic solute transporter (Ost), 1,4-bis(2-[3,5-dichloropyridyloxy])benzene (TCPOBOP). Color image is available in the online version of the article.
In female mice, CAR activation decreased mRNA of the sinusoidal uptake transporters Oatp1a1 (98%), Oatp2b1 (42%), and unconjugated BA uptake transporter Oatp1b2 (42%) (Figure 8, top right). CAR activation also increased the canalicular efflux transporter Mrp2 (123%), whereas it decreased Bsep (38%), Mdr2 (58%), Atp8b1 (56%), as well as sterol half-transporters Abcg5 (70%) and Abcg8 (74%) (Figure 8, middle right). As in males, CAR activation in female mice also increased the sinusoidal BA efflux transporters Mrp3 (150%) and Mrp4 (1043%), whereas it decreased Abca1 (48%), which mediates canalicular efflux of cholesterol (Figure 8, bottom right). Further, CAR activation in female mice also decreased Mrp1 (59%) and Ostß (45%). Overall, the effects of the CAR activator TCPOBOP in both male and female mice were an increase in the biliary drug efflux transporter Mrp2, and the sinusoidal efflux transporters Mrp3 and Mrp4.
Effects of CAR Activation on mRNAs of BA Synthesis and Regulation
In male mice, CAR activation induced the mRNA of Cyp7a1 (185%), whereas the sterol 12α-hydroxylase Cyp8b1 was decreased 71% (Figure 9, top left). Cyp7a1, also known as cholesterol 7α-hydroxylase, is the rate-limiting enzyme of the classic (neutral) BA synthesis pathway. Cyp27a1, which functions in both the alternative (acidic) and classic pathways, was increased 107%, whereas Cyp7b1 of the alternative pathway was decreased (61%). The enzymes Baat and Bal, which mediate taurine-conjugation of BAs, were increased 119% and 57%, respectively. The mRNAs of the transcription factors known to regulate BA synthesis were also quantified. The mRNA of the major BA “sensor” Fxr in liver was increased 273%, as well as the Fxr transcriptional target gene Shp (174%) (Figure 9, bottom left). The mRNA of Lrh-1 and Hepatocyte nuclear factor 4α (Hnf4α), both of which have been shown to be positive regulators of Cyp7a1, were increased 196% and 76%, respectively.
FIG. 9.

Effect of TCPOBOP on mRNA of BA synthesis and regulation in livers of male and female mice. Corn oil vehicle or TCPOBOP (3 mg/kg) was administered daily (IP) for 4 consecutive days to male and female mice (n = 5–6 per treatment group). At 96 h after the first dose, livers were collected. Total RNA was analyzed by QuantiGene Plex 2.0 Assay, as well as by RT-qPCR. Relative mRNA levels were calculated with vehicle controls set as 100%. Data represent means ± S.E.M. Asterisks (*) denote statistically different from vehicle control (P < 0.05). Bile acid CoA:amino acid N-acyltransferase (Baat), Bile acid CoA ligase (Bal), Cytochrome p450 (Cyp), Farnesoid x receptor (Fxr), females (F), Fibroblast growth factor receptor (Fgfr4), Hepatocyte nuclear factor 4α (Hnf4α), Liver receptor homolog-1 (Lrh-1), males (M), Small heterodimer partner (Shp), 1,4-bis(2-[3,5-dichloropyridyloxy])benzene (TCPOBOP). Color image is available in the online version of the article.
In female mice, as in males, TCPOBOP increased Cyp7a1 mRNA (132%), whereas Cyp8b1 was decreased 54% (Figure 9, top right). These changes occurred despite downregulation of Lrh-1 (by 35%) and Hnf4α (by 34%) mRNAs, which are generally considered positive regulators of Cyp7a1 and Cyp8b1 (Figure 9, bottom right). However, Shp mRNA was decreased 77%, and Shp protein is known to inhibit Lrh-1-mediated upregulation of Cyp7a1 (Figure 9, bottom right). Thus, the consistent effects that CAR activation had on BA synthesis and regulation genes in the liver of both male and female mice were a marked increase in Cyp7a1, the rate-limiting enzyme of BA synthesis, and a decrease of sterol 12α-hydroxylase Cyp8b1.
Effects of CAR Activation on mRNAs of BA Transport and Regulation in Mouse Ileum
In ilea of male mice, the only alteration following CAR activation was a 133% increase in mRNA of Mrp2 (Figure 10, bottom left). In female mouse ileum, CAR activation also increased Mrp2 (146%), whereas it decreased Mrp3 by 29% (Figure 10, bottom right). Further, CAR activation in female mouse ileum increased mRNAs of Nieman-Pick c1-like 1 (Npc1l1) (37%), which participates in intestinal cholesterol absorption. CAR activation also increased the mRNA of the oxysterol sensor Liver x receptor α (Lxrα) (30%), the BA sensor Tgr5 (34%), and Shp (362%). Thus, the only consistent effect of the CAR activator TCPOBOP on the mRNA of transporters and regulators in ilea of male and female mice was an increase in Mrp2, which transports drugs rather than BAs.
FIG. 10.

Effect of TCPOBOP on mRNA of BA regulators and transporters in ilea of male and female mice. Corn oil vehicle or TCPOBOP (3 mg/kg) was administered daily (IP) for 4 consecutive days to male and female mice (n = 5–6 per treatment group). At 96 h after the first dose, ilea were collected. Total RNA was analyzed by QuantiGene Plex 2.0 Assay, as well as by RT-qPCR. Relative mRNA levels were calculated with vehicle controls set as 100%. Data represent means ± SEM. Asterisks (*) denote statistically different from vehicle control (P < 0.05). ATP-binding cassette (Abc), Apical sodium-dependent bile acid transporter (Asbt), Farnesoid X Receptor (Fxr), females (F), Fibroblast growth factor (Fgf), I-babp (ileal bile acid binding protein), Liver x receptor α (Lxrα), males (M), Multidrug resistance-associated protein (Mrp), Nieman-Pick c1-like 1 (Npc1l1), Organic solute transporter (Ost), total bile acids (Σ-BAs), Small heterodimer partner (Shp), 1,4-bis(2-[3,5-dichloropyridyloxy])benzene (TCPOBOP), Transmembrane G protein-coupled receptor 5 (Tgr5). Color image is available in the online version of the article.
Discussion
It has been shown in BA-feeding and bile duct-ligation models of cholestasis that CAR activators, such as TCPOBOP and PB, activate pathways of BA detoxification and protect against BA-induced liver injury (Guo et al., 2003; Wagner et al., 2005; Beilke et al., 2009; Sberna et al., 2011). Although these studies provide considerable insight into how CAR activation prevents cholestatic injury, significant gaps regarding the effect of CAR activation on BA homeostasis need to be addressed, including a complete characterization of the various BAs in serum, liver, and bile after CAR activation in both male and female mice, as well as to determine whether these changes are due to alterations of the BA transporters and BA synthesis enzymes mediated by the Fxr receptor.
The importance of quantifying and understanding the role of individual BAs is underscored by several aspects. Each BA has a different critical micellar concentration, which is attributable to the hydroxyl groups on the steroid ring that are unique to each BA (Hofmann and Roda, 1984). This means that shifts in the concentration and composition of the BAs can directly influence solubilization and absorption of lipids and fat-soluble vitamins into intestinal enterocytes (Carey and Small, 1972; Westergaard and Dietschy, 1976; Shiau, 1990; Wang et al., 2003). Hydrophobicity varies substantially among individual BAs, resulting in differences in hepatotoxicity (Billington et al., 1980; Heuman, 1989; Song et al., 2011). For this reason, the BA UDCA has long been used as a drug to treat primary biliary cirrhosis, as well as for the dissolution and prevention of gallstones (May et al., 1993; Guarino et al., 2013; Tabibian and Lindor, 2015). Each BA has a different affinity for the BA receptors Fxr and Tgr5, and thus the various BAs have different effects on BA homeostasis, as well as glucose and energy metabolism (Parks et al., 1999; Sato et al., 2008). Accurate identification and quantification of individual BA concentrations is also a critical step in the diagnosis and therapeutic monitoring of BA malabsorption and inborn errors of BA metabolism (Daugherty et al., 1993; Heubi et al., 2015; Orekoya et al., 2015; Zhang et al., 2015). Quantification of individual BAs has also revealed an association between insulin resistance and decreased plasma ratio of 12α-OH/non12α-OH BAs (Haeusler et al., 2013). Therefore, consideration of concentrations of individual BAs is critical in order to have a complete understanding of the BA phenotype.
Numerous reports have described the effects of the widely studied CAR activator PB on the biliary excretion of BAs in various rodents. However, these reports focused on either the total concentrations of BAs (Berthelot et al., 1970; Okuda et al., 1988); tri-, tetra-, and penta-hydroxylated BAs (Wagner et al., 2005); did not differentiate conjugated from U-BAs (Sberna et al., 2011); or reported on select major BAs known at the time, eg, T-CA and T-CDCA (Klaassen, 1970, 1971a, b; Paumgartner et al., 1971). Following treatment of cholestatic mice with CAR activators, PB and TCPOBOP, Beilke et al. (2009) used an analytical method that accounted for approximately 78% of the BA composition in serum and liver that we report here. Additionally, the previous method did not chromatographically resolve the secondary BA DCA from the primary BA CDCA. Therefore, in quantifying 8 different BAs and the taurine-conjugate of each in serum, liver, and bile, the ultraperformance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) method used here (Alnouti et al., 2008; Zhang and Klaassen, 2010) provides the most comprehensive analysis of the BA profile in response to CAR activation.
The present study indicates that the effect of the CAR activator TCPOBOP on the profile of BAs in male and female mice is generally quite similar, with the effects in males more prominent than in female mice. This is evident in the similar effects of TCPOBOP in male and female mice in decreasing the concentration of total BAs and 12α-OH BAs (primarily T-CA) in liver, as well as increasing the proportion of MCAs excreted into bile. In attempting to determine the mechanism of these changes in BA homeostasis after CAR activation, the mRNA of transporters, including increases in Mrp2, Mrp3, Mrp4, and Ntcp was also similar in male and female mice. TCPOBOP treatment increases Cyp7a1 and decreases Cyp8b1, in both sexes of mice. However, the mRNA expression of some genes is different in male and female following TCPOBOP. These genes likely have a very minor contribution to the similar changes in BA disposition in the response to CAR activation observed in male and female mice. It should be noted that the analyses of BAs and gene expression were performed on samples collected on day 5 following 4 daily TCPOBOP treatments. Thus, this study does not characterize the series of events relating to gene regulation and metabolism that occur prior to day 5. Additionally, the age of the adult mice used was slightly different between males (12–15 weeks) and females (16–19 weeks). These 2 factors may help explain some of the unanticipated responses of gene expression, as well as some differences of the BA profile between the sexes.
Bile flow in male and female mice increases in response to the synthetic CAR ligand TCPOBOP (Figure 7). This is consistent with the well-documented observation that the classical CAR activator PB increases bile flow in various rat strains (Roberts and Plaa, 1967; Klaassen and Plaa, 1968; Klaassen, 1969, 1971b; Berthelot et al., 1970; Slater and Delaney, 1970; Paumgartner et al., 1971). Also consistent with the studies using PB, the CAR activator TCPOBOP does not increase the biliary excretion of total BAs in mice (Figure 5) (Klaassen 1971a). These results indicate that the CAR-stimulated increase in bile flow is BA-independent.
The total BAs in mice after administration of the CAR activator TCPOBOP decrease (25–50%) in liver (Figure 3), whereas total serum BA concentrations and total biliary excretion of BAs do not change appreciably (Figs. 1 and 5). Given the substantial decrease in liver concentrations of BAs, one would anticipate an increase in mRNA of the rate-limiting enzyme for BA synthesis, Cyp7a1, which was demonstrated (Figure 9). In both liver and ileum, the nuclear receptor Fxr plays a central role in negative feedback regulation of BA synthesis (Inagaki et al., 2005; Kong et al., 2012). In liver, BAs bind to the Fxr receptor, which responds by increasing the expression of Shp. Shp protein binds to Lrh-1 protein to inhibit Lrh-1 activation of Cyp7a1 transcription. Thus, Fxr activation decreases BA synthesis. Thus, one would anticipate in the present study a decrease in Shp; however, this was not observed. Fxr is also expressed in ileum, where BAs can bind to Fxr to upregulate the expression of Fgf15, a protein that enters the blood and is taken to the liver to decrease BA synthesis. However, there was not a decrease in Fgf15, probably because TCPOBOP does not alter the net biliary excretion of BAs, and thus there is an adequate amount of BAs entering the ileum. Thus, although TCPOBOP decreases the total BAs in liver, there is a subsequent increase in the rate-limiting enzyme of BA synthesis, in an attempt to restore BA concentrations in the liver. This increase in Cyp7a1 appears to be due to a non-Fxr pathway.
A decrease in 12α-OH BAs occurs in the liver after CAR activation by TCPOBOP (Figure 3). This is primarily due to a decrease in T-CA, but also to decreases in T-DCA. Because the enzyme that hydroxylates the 12α position of BAs is Cyp8b1, one would anticipate a decrease in Cyp8b1 (Li-Hawkins et al., 2002). Therefore, it was not surprising that a decrease in Cyp8b1 was observed in the TCPOBOP-treated mice (Figure 9). Similarly, TCPOBOP has been shown to decrease Cyp8b1 approximately 50%, 24 h following a single dose (BALB/c mice, 0.3 mg/kg IP) (Miao et al., 2006).
The biliary excretion of total BAs into bile is not altered appreciably by TCPOBOP, even though the liver concentration of total BAs is decreased appreciably. This suggests that the liver might not only be the site of BA synthesis but also a site to store BAs to maintain a consistent source of BAs in the intestine for the absorption of lipids and lipid-soluble vitamins. The fraction of BAs that is T-CA in the bile decreases after TCPOBOP, whereas the fraction of MCAs increases, similar to the ratio of these 2 BAs in liver. The canalicular transporter Mrp2, although induced by TCPOBOP, seems unlikely to contribute to excretion of BAs in the present study, as rat Mrp2 does not transport T-CA (Madon et al., 1997; Gerloff et al., 1998; Stieger et al., 2000). In contrast, Bsep is essential for the biliary excretion of BAs, as Bsep-null mice have extremely low biliary excretion of CA (5% of WT) (Wang et al., 2001).
Concentrations of total BAs in the serum do not decrease as they do in the liver after TCPOBOP administration, and thus total serum BA concentrations cannot be used to approximate total BA concentrations in liver. In contrast, there is an increase in the fraction of MCAs in serum, as occurs in liver and that is excreted into bile. Thus, the fraction of T-CA and MCA is relatively similar in the 3 compartments. However, the fraction of BAs in the serum that are unconjugated is much higher (18% in males; 42% in females) than in the liver (approximately 3% in both sexes) and bile (approximately 0.25% in both sexes). TCPOBOP-treated mice have an increase in the mRNA of the conjugated BA uptake transporter Ntcp and also marked increases in the mRNA of the sinusoidal BA efflux transporters, Mrp3 and Mrp4 (Figure 8). The role of the increase in these transporters in the BA homeostasis after TCPOBOP is not clear.
As described in the introduction, CAR activation enhances fecal excretion of MCAs, as well as protects the liver during LCA-induced cholestasis (Beilke et al., 2009; Sberna et al., 2011). Consistent with these studies, the present study indicates that CAR activation by TCPOBOP decreases total BA concentration in liver. In addition, the present study indicates that the effect of TCPOBOP in a noncholestatic model is specific to the Cyp8b1 pathway, which is required for formation of CA. This results in an increase in the proportion of MCAs. Thus, the effect of CAR activation on BAs is primarily due to a decrease in T-CA in the liver.
The overall effects of CAR activation on BA homeostasis appear to be beneficial. The activity of TCPOBOP to decrease total BA concentrations in liver could be beneficial because high concentrations of BAs are known to be toxic to the liver. Although CAR activation in mice increases bile flow, it does not increase biliary excretion of BAs. Thus, CAR activation would not be expected to interfere with biliary excretion of BAs by Bsep. As CAR activation induces the canalicular transporter Mrp2, which is responsible for the biliary excretion of many drugs, this pathway would be expected to be enhanced. Beyond rodent models, it is extremely difficult at this stage to project the human relevance of the findings of the current study. For example, in contrast to mice, MCAs are an extremely minor component of BA composition of humans. Additionally glycine-conjugates of BAs are the predominant amino acid conjugate in humans, rather than taurine-conjugates as in mice.
In summary, the CAR activator TCPOBOP decreases total BAs in mouse liver, mainly by a decrease in the 12α-OH BA T-CA. The decrease in T-CA appears to be due to a decrease of the sterol 12α-hydroxylase Cyp8b1. Cyp7a1 is upregulated in liver following TCPOBOP, as a response by the liver in an attempt to return the BAs to physiological concentrations. CAR activation also increases bile flow by increasing BA-independent flow, but the biliary excretion of BAs is not altered. In liver, bile, and serum, there is a decrease in the fraction that is T-CA and an increase in MCAs.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
ACKNOWLEDGMENT
The authors would like to thank all members of the Klaassen laboratory for technical assistance with blood and tissue collection.
FUNDING
This work was supported by National Institute of Environmental Health Sciences (Grant/Award Number: R01 ES009649, R01 ES025708); National Institute of Diabetes and Digestive and Kidney Diseases (Grant/Award Number: F32 DK092069).
REFERENCES
- Alnouti Y., Csanaky I. L., Klaassen C. D. (2008). Quantitative-profiling of bile acids and their conjugates in mouse liver, bile, plasma, and urine using LC-MS/MS. J. Chromatogr. B, Analyt. Technol. Biomed. Life Sci. 873, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assem M., Schuetz E. G., Leggas M., Sun D., Yasuda K., Reid G., Zelcer N., Adachi M., Strom S., Evans R. M., et al. (2004). Interactions between hepatic Mrp4 and Sult2a as revealed by the constitutive androstane receptor and Mrp4 knockout mice. J. Biol. Chem. 279, 22250–22257. [DOI] [PubMed] [Google Scholar]
- Bays H. E., Goldberg R. B., Truitt K. E., Jones M. R. (2008). Colesevelam hydrochloride therapy in patients with type 2 diabetes mellitus treated with metformin: Glucose and lipid effects. Arch. Intern. Med. 168, 1975–1983. [DOI] [PubMed] [Google Scholar]
- Beilke L. D., Aleksunes L. M., Holland R. D., Besselsen D. G., Beger R. D., Klaassen C. D., Cherrington N. J. (2009). Constitutive androstane receptor-mediated changes in bile acid composition contributes to hepatoprotection from lithocholic acid-induced liver injury in mice. Drug Metab. Dispos. 37, 1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthelot P., Erlinger S., Dhumeaux D., Preaux A. M. (1970). Mechanism of phenobarbital-induced hypercholeresis in the rat. Am. J. Physiol. 219, 809–813. [DOI] [PubMed] [Google Scholar]
- Beysen C., Murphy E. J., Deines K., Chan M., Tsang E., Glass A., Turner S. M., Protasio J., Riiff T., Hellerstein M. K. (2012). Effect of bile acid sequestrants on glucose metabolism, hepatic de novo lipogenesis, and cholesterol and bile acid kinetics in type 2 diabetes: A randomised controlled study. Diabetologia 55, 432–442. [DOI] [PubMed] [Google Scholar]
- Bhamidimarri K. R., Schiff E. (2013). Drug-induced cholestasis. Clin. Liver Dis. 17, 519–531. vii. [DOI] [PubMed] [Google Scholar]
- Billington D., Evans C. E., Godfrey P. P., Coleman R. (1980). Effects of bile salts on the plasma membranes of isolated rat hepatocytes. Biochem. J. 188, 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. B., Klaassen C. D. (2009). Induction of mouse UDP-glucuronosyltransferase mRNA expression in liver and intestine by activators of aryl-hydrocarbon receptor, constitutive androstane receptor, pregnane X receptor, peroxisome proliferator-activated receptor alpha, and nuclear factor erythroid 2-related factor 2. Drug Metab. Dispos. 37, 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey M. C., Small D. M. (1972). Micelle formation by bile salts. Physical-chemical and thermodynamic considerations. Arch. Intern. Med. 130, 506–527. [PubMed] [Google Scholar]
- Chiang J. Y. (2003). Bile acid regulation of hepatic physiology: III. Bile acids and nuclear receptors. Am. J. Physiol. Gastrointest. Liver Physiol. 284, G349–G356. [DOI] [PubMed] [Google Scholar]
- Csanaky I. L., Lu H., Zhang Y., Ogura K., Choudhuri S., Klaassen C. D. (2011). Organic anion-transporting polypeptide 1b2 (Oatp1b2) is important for the hepatic uptake of unconjugated bile acids: Studies in Oatp1b2-null mice. Hepatology 53, 272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J. Y., Aleksunes L. M., Tanaka Y., Fu Z. D., Guo Y., Guo G. L., Lu H., Zhong X. B., Klaassen C. D. (2012). Bile acids via Fxr initiate the expression of major transporters involved in the enterohepatic circulation of bile acids in newborn mice. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G979–G996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielsson H., Eneroth P., Hellstrom K., Lindstedt S., Sjovall J. (1963). On the turnover and excretory products of cholic and chenodeoxycholic acid in man. J. Biol. Chem. 238, 2299–2304. [PubMed] [Google Scholar]
- Daugherty C. C., Setchell K. D., Heubi J. E., Balistreri W. F. (1993). Resolution of liver biopsy alterations in three siblings with bile acid treatment of an inborn error of bile acid metabolism (delta 4-3-oxosteroid 5 beta-reductase deficiency). Hepatology 18, 1096–1101. [PubMed] [Google Scholar]
- Fonseca V. A., Rosenstock J., Wang A. C., Truitt K. E., Jones M. R. (2008). Colesevelam HCl improves glycemic control and reduces LDL cholesterol in patients with inadequately controlled type 2 diabetes on sulfonylurea-based therapy. Diabetes Care 31, 1479–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A., Grundy S. M. (1994). Cholestyramine therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. A short-term, double-blind, crossover trial. Ann. Intern. Med. 121, 416–422. [DOI] [PubMed] [Google Scholar]
- Gerloff T., Stieger B., Hagenbuch B., Madon J., Landmann L., Roth J., Hofmann A. F., Meier P. J. (1998). The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J. Biol. Chem. 273, 10046–10050. [DOI] [PubMed] [Google Scholar]
- Goldberg R. B., Fonseca V. A., Truitt K. E., Jones M. R. (2008). Efficacy and safety of colesevelam in patients with type 2 diabetes mellitus and inadequate glycemic control receiving insulin-based therapy. Arch. Intern. Med. 168, 1531–1540. [DOI] [PubMed] [Google Scholar]
- Goodwin B., Jones S. A., Price R. R., Watson M. A., McKee D. D., Moore L. B., Galardi C., Wilson J. G., Lewis M. C., Roth M. E., et al. (2000). A regulatory cascade of the nuclear receptors Fxr, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 6, 517–526. [DOI] [PubMed] [Google Scholar]
- Guarino M. P., Cocca S., Altomare A., Emerenziani S., Cicala M. (2013). Ursodeoxycholic acid therapy in gallbladder disease, a story not yet completed. World J. Gastroenterol.19, 5029–5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G. L., Lambert G., Negishi M., Ward J. M., Brewer H. B., Jr., Kliewer S. A., Gonzalez F. J., Sinal C. J. (2003). Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J. Biol. Chem. 278, 45062–45071. [DOI] [PubMed] [Google Scholar]
- Haeusler R. A., Astiarraga B., Camastra S., Accili D., Ferrannini E. (2013). Human insulin resistance is associated with increased plasma levels of 12alpha-hydroxylated bile acids. Diabetes 62, 4184–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halilbasic E., Baghdasaryan A., Trauner M. (2013). Nuclear receptors as drug targets in cholestatic liver diseases. Clin. Liver Dis. 17, 161–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heubi J. E., Setchell K. D., Jha P., Buckley D., Zhang W., Rosenthal P., Potter C., Horslen S., Suskind D. (2015). Treatment of bile acid amidation defects with glycocholic acid. Hepatology 61, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuman D. M. (1989). Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions. J. Lipid Res. 30, 719–730. [PubMed] [Google Scholar]
- Hofmann A. F. (1984). Chemistry and enterohepatic circulation of bile acids. Hepatology 4, 4S–14S. [DOI] [PubMed] [Google Scholar]
- Hofmann A. F., Hagey L. R. (2008). Bile acids: Chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell. Mol. Life Sci. 65, 2461–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann A. F., Roda A. (1984). Physicochemical properties of bile acids and their relationship to biological properties: An overview of the problem. J. Lipid Res. 25, 1477–1489. [PubMed] [Google Scholar]
- Holt J. A., Luo G., Billin A. N., Bisi J., McNeill Y. Y., Kozarsky K. F., Donahee M., Wang D. Y., Mansfield T. A., Kliewer S. A., et al. (2003). Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 17, 1581–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkakoski P., Zelko I., Sueyoshi T., Negishi M. (1998). The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol. Cell Biol. 18, 5652–5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseinpour F., Moore R., Negishi M., Sueyoshi T. (2005). Serine 202 regulates the nuclear translocation of constitutive active/androstane receptor. Mol. Pharmacol. 69, 1095–1102. [DOI] [PubMed] [Google Scholar]
- Inagaki T., Choi M., Moschetta A., Peng L., Cummins C. L., McDonald J. G., Luo G., Jones S. A., Goodwin B., Richardson J. A., et al. (2005). Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2, 217–225. [DOI] [PubMed] [Google Scholar]
- Kakizaki S., Yamamoto Y., Ueda A., Moore R., Sueyoshi T., Negishi M. (2003). Phenobarbital induction of drug/steroid-metabolizing enzymes and nuclear receptor CAR. Biochim. Biophys. Acta 1619, 239–242. [DOI] [PubMed] [Google Scholar]
- Kawamoto T., Sueyoshi T., Zelko I., Moore R., Washburn K., Negishi M. (1999). Phenobarbital-responsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol. Cell Biol. 19, 6318–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen C. D. (1969). Biliary flow after microsomal enzyme induction. J. Pharmacol. Exp. Ther. 168, 218–223. [PubMed] [Google Scholar]
- Klaassen C. D. (1970). Effects of phenobarbital on the plasma disappearance and biliary excretion of drugs in rats. J. Pharmacol. Exp. Ther. 175, 289–300. [PubMed] [Google Scholar]
- Klaassen C. D. (1971a). Does bile acid secretion determine canalicular bile production in rats? Am. J. Physiol. 220, 667–673. [DOI] [PubMed] [Google Scholar]
- Klaassen C. D. (1971b). Studies on the increased biliary flow produced by phenobarbital in rats. J. Pharmacol. Exp. Ther. 176, 743–751. [PubMed] [Google Scholar]
- Klaassen C. D., Plaa G. L. (1968). Studies on the mechanism of phenobarbital-enhanced sulfobromophthalein disappearance. J. Pharmacol. Exp. Ther. 161, 361–366. [PubMed] [Google Scholar]
- Kong B., Wang L., Chiang J. Y., Zhang Y., Klaassen C. D., Guo G. L. (2012). Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 56, 1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Hawkins J., Gafvels M., Olin M., Lund E. G., Andersson U., Schuster G., Bjorkhem I., Russell D. W., Eggertsen G. (2002). Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J. Clin. Invest. 110, 1191–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstedt S. (1957). The turnover of cholic acid in man: Bile acids and steroids. Acta Physiol. Scand. 40, 1–9. [DOI] [PubMed] [Google Scholar]
- Lu T. T., Makishima M., Repa J. J., Schoonjans K., Kerr T. A., Auwerx J., Mangelsdorf D. J. (2000). Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 6, 507–515. [DOI] [PubMed] [Google Scholar]
- Madan A., Graham R. A., Carroll K. M., Mudra D. R., Burton L. A., Krueger L. A., Downey A. D., Czerwinski M., Forster J., Ribadeneira M. D., et al. (2003). Effects of prototypical microsomal enzyme inducers on cytochrome P450 expression in cultured human hepatocytes. Drug Metab. Dispos. 31, 421–431. [DOI] [PubMed] [Google Scholar]
- Madon J., Eckhardt U., Gerloff T., Stieger B., Meier P. J. (1997). Functional expression of the rat liver canalicular isoform of the multidrug resistance-associated protein. FEBS Lett. 406, 75–78. [DOI] [PubMed] [Google Scholar]
- Maher J. M., Cheng X., Slitt A. L., Dieter M. Z., Klaassen C. D. (2005). Induction of the multidrug resistance-associated protein family of transporters by chemical activators of receptor-mediated pathways in mouse liver. Drug Metab. Dispos. 33, 956–962. [DOI] [PubMed] [Google Scholar]
- May G. R., Sutherland L. R., Shaffer E. A. (1993). Efficacy of bile acid therapy for gallstone dissolution: A meta-analysis of randomized trials. Aliment. Pharmacol. Ther. 7, 139–148. [DOI] [PubMed] [Google Scholar]
- Miao J., Fang S., Bae Y., Kemper J. K. (2006). Functional inhibitory cross-talk between constitutive androstane receptor and hepatic nuclear factor-4 in hepatic lipid/glucose metabolism is mediated by competition for binding to the DR1 motif and to the common coactivators, GRIP-1 and PGC-1alpha. J. Biol. Chem. 281, 14537–14546. [DOI] [PubMed] [Google Scholar]
- Monte M. J., Marin J. J., Antelo A., Vazquez-Tato J. (2009). Bile acids: Chemistry, physiology, and pathophysiology. World J. Gastroenterol. 15, 804–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutoh S., Osabe M., Inoue K., Moore R., Pedersen L., Perera L., Rebolloso Y., Sueyoshi T., Negishi M. (2009). Dephosphorylation of threonine 38 is required for nuclear translocation and activation of human xenobiotic receptor CAR (NR1I3). J. Biol. Chem. 284, 34785–34792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda H., Sorrentino D., Alpini G., Tavoloni N., Jones M. J., Kiang C. L., Berk P. D. (1988). Bile acid secretion and pool size during phenobarbital induced hypercholeresis. Proc. Soc. Exp. Biol. Med. Exp. Biol. Med. 187, 202–208. [DOI] [PubMed] [Google Scholar]
- Orekoya O., McLaughlin J., Leitao E., Johns W., Lal S., Paine P. (2015). Quantifying bile acid malabsorption helps predict response and tailor sequestrant therapy. Clin. Med. (London, England) 15, 252–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer R. H. (1972). Bile acids, liver injury, and liver disease. Arch. Intern. Med. 130, 606–617. [PubMed] [Google Scholar]
- Parks D. J., Blanchard S. G., Bledsoe R. K., Chandra G., Consler T. G., Kliewer S. A., Stimmel J. B., Willson T. M., Zavacki A. M., Moore D. D., and., et al. (1999). Bile acids: Natural ligands for an orphan nuclear receptor. Science 284, 1365–1368. [DOI] [PubMed] [Google Scholar]
- Paumgartner G., Horak W., Probst P., Grabner G. (1971). Effect of phenobarbital on bile flow and bile salt excretion in the rat. Naunyn-Schmied. Archiv. Pharmacol. 270, 98–101. [DOI] [PubMed] [Google Scholar]
- Perez M. J., Briz O. (2009). Bile-acid-induced cell injury and protection. World J. Gastroenterol. 15, 1677–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland A., Mak I., Glover E., Boatman R. J., Ebetino F. H., Kende A. S. (1980). 1,4-Bis[2-(3,5-dichloropyridyloxy)]benzene, a potent phenobarbital-like inducer of microsomal monooxygenase activity. Mol. Pharmacol. 18, 571–580. [PubMed] [Google Scholar]
- Poland A., Mak I., Glover E. (1981). Species differences in responsiveness to 1,4-bis[2-(3,5-dichloropyridyloxy)]-benzene, a potent phenobarbital-like inducer of microsomal monooxygenase activity. Mol. Pharmacol. 20, 442–450. [PubMed] [Google Scholar]
- Roberts R. J., Plaa G. L. (1967). Effect of phenobarbital on the excretion of an exogenous bilirubin load. Biochem. Pharmacol. 16, 827–835. [DOI] [PubMed] [Google Scholar]
- Rockwell C. E., Zhang M., Fields P. E., Klaassen C. D. (2012). Th2 skewing by activation of Nrf2 in CD4(+) T cells. J. Immunol. 188, 1630–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell D. W. (2003). The enzymes, regulation, and genetics of bile acid synthesis. Ann. Rev. Biochem. 72, 137–174. [DOI] [PubMed] [Google Scholar]
- Saini S. P., Sonoda J., Xu L., Toma D., Uppal H., Mu Y., Ren S., Moore D. D., Evans R. M., Xie W. (2004). A novel constitutive androstane receptor-mediated and CYP3A-independent pathway of bile acid detoxification. Mol. Pharmacol. 65, 292–300. [DOI] [PubMed] [Google Scholar]
- Sato H., Macchiarulo A., Thomas C., Gioiello A., Une M., Hofmann A. F., Saladin R., Schoonjans K., Pellicciari R., Auwerx J. (2008). Novel potent and selective bile acid derivatives as TGR5 agonists: Biological screening, structure-activity relationships, and molecular modeling studies. J. Med. Chem. 51, 1831–1841. [DOI] [PubMed] [Google Scholar]
- Sberna A. L., Assem M., Gautier T., Grober J., Guiu B., Jeannin A., Pais de Barros J. P., Athias A., Lagrost L., Masson D. (2011). Constitutive androstane receptor activation stimulates faecal bile acid excretion and reverse cholesterol transport in mice. J. Hepatol. 55, 154–161. [DOI] [PubMed] [Google Scholar]
- Shiau Y. F. (1990). Mechanism of intestinal fatty acid uptake in the rat: The role of an acidic microclimate. J. Physiol. 421, 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater T. F., Delaney V. B. (1970). Liver adenosine triphosphate content and bile flow rate in the rat. Biochem. J. 116, 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatter J. G., Cheng O., Cornwell P. D., de Souza A., Rockett J., Rushmore T., Hartley D., Evers R., He Y., Dai X., et al. (2006). Microarray-based compendium of hepatic gene expression profiles for prototypical ADME gene-inducing compounds in rats and mice in vivo. Xenobiotica 36, 902–937. [DOI] [PubMed] [Google Scholar]
- Song K. H., Li T., Owsley E., Strom S., Chiang J. Y. (2009). Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology 49, 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song P., Zhang Y., Klaassen C. D. (2011). Dose-response of five bile acids on serum and liver bile acid concentrations and hepatotoxicity in mice. Tox. Sci. 123, 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieger B., Fattinger K., Madon J., Kullak-Ublick G. A., Meier P. J. (2000). Drug- and estrogen-induced cholestasis through inhibition of the hepatocellular bile salt export pump (Bsep) of rat liver. Gastroenterology 118, 422–430. [DOI] [PubMed] [Google Scholar]
- Sueyoshi T., Kawamoto T., Zelko I., Honkakoski P., Negishi M. (1999). The repressed nuclear receptor CAR responds to phenobarbital in activating the human CYP2B6 gene. J. Biol. Chem. 274, 6043–6046. [DOI] [PubMed] [Google Scholar]
- Tabibian J. H., Lindor K. D. (2015). Primary biliary cirrhosis: Safety and benefits of established and emerging therapies. Expert. Opin. Drug Saf. 14, 1435–1444. [DOI] [PubMed] [Google Scholar]
- Thomas C., Pellicciari R., Pruzanski M., Auwerx J., Schoonjans K. (2008). Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 7, 678–693. [DOI] [PubMed] [Google Scholar]
- Trauner M., Meier P. J., Boyer J. L. (1998). Molecular pathogenesis of cholestasis. N. Engl. J. Med. 339, 1217–1227. [DOI] [PubMed] [Google Scholar]
- Tzameli I., Pissios P., Schuetz E. G., Moore D. D. (2000). The xenobiotic compound 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene is an agonist ligand for the nuclear receptor CAR. Mol. Cell. Biol. 20, 2951–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M., Halilbasic E., Marschall H. U., Zollner G., Fickert P., Langner C., Zatloukal K., Denk H., Trauner M. (2005). CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology 42, 420–430. [DOI] [PubMed] [Google Scholar]
- Wang D. Q., Tazuma S., Cohen D. E., Carey M. C. (2003). Feeding natural hydrophilic bile acids inhibits intestinal cholesterol absorption: Studies in the gallstone-susceptible mouse. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G494–G502. [DOI] [PubMed] [Google Scholar]
- Wang R., Salem M., Yousef I. M., Tuchweber B., Lam P., Childs S. J., Helgason C. D., Ackerley C., Phillips M. J., Ling V. (2001). Targeted inactivation of sister of P-glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc. Nat. Acad. Sci. USA. 98, 2011–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M., Houten S. M., Mataki C., Christoffolete M. A., Kim B. W., Sato H., Messaddeq N., Harney J. W., Ezaki O., Kodama T., et al. (2006). Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439, 484–489. [DOI] [PubMed] [Google Scholar]
- Westergaard H., Dietschy J. M. (1976). The mechanism whereby bile acid micelles increase the rate of fatty acid and cholesterol uptake into the intestinal mucosal cell. J. Clin. Invest. 58, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M. H., Holcomb I., Deuel B., Dowd P., Huang A., Vagts A., Foster J., Liang J., Brush J., Gu Q., et al. (1999). FGF-19, a novel fibroblast growth factor with unique specificity for FGFR4. Cytokine 11, 729–735. [DOI] [PubMed] [Google Scholar]
- Yamakawa T., Takano T., Utsunomiya H., Kadonosono K., Okamura A. (2007). Effect of colestimide therapy for glycemic control in type 2 diabetes mellitus with hypercholesterolemia. Endocr. J. 54, 53–58. [DOI] [PubMed] [Google Scholar]
- Zhang W., Jha P., Wolfe B., Gioiello A., Pellicciari R., Wang J., Heubi J., Setchell K. D. (2015). Tandem mass spectrometric determination of atypical 3beta-hydroxy-Delta5-bile acids in patients with 3beta-hydroxy-Delta5-C27-steroid oxidoreductase deficiency: Application to diagnosis and monitoring of bile acid therapeutic response. Clin. Chem. 61, 955–963. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Klaassen C. D. (2010). Effects of feeding bile acids and a bile acid sequestrant on hepatic bile acid composition in mice. J. Lipid Res. 51, 3230–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieve F. J., Kalin M. F., Schwartz S. L., Jones M. R., Bailey W. L. (2007). Results of the glucose-lowering effect of WelChol study (GLOWS): A randomized, double-blind, placebo-controlled pilot study evaluating the effect of colesevelam hydrochloride on glycemic control in subjects with type 2 diabetes. Clin. Ther. 29, 74–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.