

Abstract
Brain-derived neurotrophic factor (BDNF) plays important roles in the development, maintenance, and plasticity of the mammalian forebrain. These functions include regulation of neuronal maturation and survival, axonal and dendritic arborization, synaptic efficacy, and modulation of complex behaviors including depression and spatial learning. Although analysis of mutant mice has helped establish essential developmental functions for BDNF, its requirement in the adult is less well documented. We have studied late-onset forebrain-specific BDNF knockout (CaMK-BDNFKO) mice, in which BDNF is lost primarily from the cortex and hippocampus in early adulthood, well after BDNF expression has begun in these structures. We found that although CaMK-BDNFKO mice grew at a normal rate and can survive more than a year, they had smaller brains than wild type siblings. The CaMK-BDNFKO mice had generally normal behavior in tests for ataxia and anxiety, but displayed reduced spatial learning ability in the Morris water task and increased depression in the Porsolt swim test. These behavioral deficits were very similar to those we previously described in an early-onset forebrain-specific BDNF knockout. To identify an anatomical correlate of the abnormal behavior, we quantified dendritic spines in cortical neurons. The spine density of CaMK-BDNFKO mice was normal at P35, but by P84, there was a 30% reduction in spine density. The strong similarities we find between early- and late-onset BDNF knockouts suggests that BDNF signaling is required continuously in the CNS for the maintenance of some forebrain circuitry also affected by developmental BDNF depletion.
Keywords: BDNF, neurotrophic factor, memory, dendritic spines, visual cortex, behavior
INTRODUCTION
BDNF, a member of the neurotrophin family of secreted proteins, was initially identified based upon its ability to support the survival of peripheral sensory neurons (Barde et al., 1982). Subsequent studies demonstrated the widespread expression of BDNF within the central nervous system (CNS) (Leibrock et al., 1989; Maisonpierre et al., 1990; Hofer et al., 1990), and its ability to influence the survival of many neuron types. BDNF has been shown to affect morphological features of specific neurons, such as soma size and dendritic arborization, as well as biochemical characteristics such as expression of biosynthetic enzymes and transporters for neurotransmitters (McAllister et al., 1999; Bibel, 2000). In addition, BDNF has been demonstrated to play a role in modulating synaptic transmission in many circuits within the brain, often potentiating synaptic strength, but sometimes reducing it (Tao and Poo, 2001; Lu et al., 2008; Waterhouse and Xu, 2009; Cunha et al., 2010; Yoshii and Constantine Paton, 2010). Given that neural activity can regulate BDNF expression and release, the fact that BDNF modulates synaptic transmission provides a molecular mechanism by which neural activity can modify the functional and structural features of neural circuitry (Thoenen, 1995; Cohen and Greenberg, 2008; Kuczewski et al., 2009; Greenberg et al., 2009; Cohen-Cory et al., 2010). Thus, this versatile signaling protein is utilized at many stages of life to accomplish diverse tasks in the development, maintenance, and plasticity of the brain. In addition, reduced BDNF expression has been identified in many neurodegenerative diseases, and augmenting BDNF signaling has received support as a potential therapeutic strategy (see Zuccato and Cattaneo, 2009; Nagahara and Tuszynski, 2011).
BDNF exerts many of its effects by binding to the receptor tyrosine kinase TrkB and triggering an intracellular kinase cascade (see Miller and Kaplan, 2001; Chao, 2003; Reichardt, 2006). BDNF can also exert effects on cells via the pan-neurotrophin receptor p75NTR, a member of the TNF receptor super-family that binds pro-BDNF with much higher affinity than the mature form of BDNF, indicating that the processing of BDNF can be an important regulatory step (see Nykjaer et al., 2005; Underwood and Coulson, 2008; Barker, 2009; Greenberg et al., 2009; Teng et al., 2010). Both TrkB and p75 are expressed throughout the developing CNS (Yan and Johnson, 1988; Buck et al., 1988; Merlio et al., 1992). Expression of p75NTR becomes restricted primarily to basal forebrain cholinergic and striatal neurons in the adult forebrain (see Yan and Johnson, 1988; Kiss et al., 1988; Koh et al., 1989; Sobreviela et al., 1994; Lee et al., 1998), while TrkB remains widely expressed in the adult forebrain (Merlio et al., 1992; Valenzuela et al., 1993). Forebrain-specific mutants of BDNF or TrkB share many similar phenotypes and altering BDNF or TrkB in cultured hippocampal and cortical neurons leads to similar outcomes. These data together suggest that BDNF exerts many of its cortical and hippocampal effects in the intact, healthy brain via TrkB activation (see also Conover and Yancopoulos, 1997; McAllister et al., 1999).
Both BDNF and TrkB null mutant mice lose specific populations of sensory neurons through apoptotic cell death, indicating that BDNF is necessary for the survival of a subset of sensory neurons in the peripheral nervous system (PNS) (Klein et al., 1993; Jones et al., 1994; Ernfors et al., 1994; Huang and Reichardt, 2003). In contrast, only minor cell losses have been reported in the CNS of the null mutants (see Alcantara et al., 1997; Minichiello et al., 1999). However, the essential roles of BDNF and TrkB cannot be fully evaluated in null mutant mice, because they are retarded in their postnatal growth and die within the first few weeks of life, a period during which there is extensive neuronal differentiation, cell death, and synapse formation in the CNS. To circumvent the neonatal lethality of BDNF and TrkB null mutations and allow the study of CNS requirements for BDNF and TrkB signaling, several groups have used the Cre/lox system to engineer conditional BDNF and TrkB mutants that are viable into adulthood, and these mice have been useful in defining the functions of this signaling system in the forebrain (Minichiello et al., 1999; Xu et al., 2000a; 2000b; Rios et al., 2001; Vyssotski et al., 2002; Gorski et al., 2003a; 2003b; Zörner et al., 2003; Baquet et al., 2004; Chen et al., 2005). By selective deletion of BDNF or TrkB in different cell populations using tissue-specific promoters or viruses to direct Cre recombinase expression, it is possible to define the precise pathways in which they act (e.g. Rios et al., 2001; Eisch et al., 2003; Monteggia et al., 2004; Heldt et al., 2007; Unger et al., 2007; Li et al., 2008; Graham et al., 2009; Lobo et al., 2010). Additionally, it is possible to distinguish the developmental versus maintenance roles of BDNF and TrkB in the CNS by using strategies including antisense oligonucleotides, early-onset versus late-onset promoters to direct expression of Cre recombinase, or by using a mutant version of TrkB that can be inhibited with a drug, (e.g. see Ma et al., 1998; Mizuno et al., 2000; Luikart et al., 2005; Chen et al., 2005; Chan et al., 2006; Johnson et al., 2008). The combination of these types of studies continues to shed light on the precise spatial and temporal roles of BDNF-TrkB signaling.
One of the fascinating aspects of the BDNF-TrkB signaling system is its relevance to a huge variety of mammalian behaviors, including learning and memory, depression, obesity, aggression, anxiety, and development of visual acuity (see Duman and Monteggia, 2006; Chen et al., 2006, 2008; Lu et al., 2008; Minichiello, 2009; Heimel et al., 2010; Castren and Rantamäki, 2010; Rios, 2011; Rosas-Vargas et al., 2011). Given the widespread expression of BDNF and TrkB in the central nervous system, their involvement in so many aspects of mammalian life is not surprising. However, the precise neural circuitry in which BDNF and TrkB act to influence specific behaviors is largely unknown. BDNF can potentially signal in anterograde, autocrine, paracrine or the classic retrograde manner (see Altar et al., 1997; Altar and DiStefano, 1998; Nawa and Takei, 2001), thus analysis of both ligand and receptor requirements are essential for understanding signaling direction in any given behavior.
Many of the analyses studying BDNF-TrkB signaling in behavior have relied upon null mutants in which this signaling pathway is affected during development as well as in the adult. There are low levels of BDNF expression in most of the CNS at birth and levels rise during the first few weeks of life, reaching maximum levels in young adulthood (Maisonpierre et al., 1990; Schoups et al., 1995; Katoh-Semba et al., 1998). It is important to determine whether BDNF-TrkB signaling has requirements for maintenance and plasticity of mature brain circuitry that are distinct from those during development. Here, we have performed both behavioral and anatomical analyses of an early adult-onset forebrain-specific BDNF mutant mouse. We find compelling similarities with an embryonic-onset forebrain-specific BDNF mutant mouse we previously described (Gorski et al., 2003a; 2003b), suggesting that sustained adult BDNF expression is essential for maintenance of some of the same brain circuitry that is sensitive to the loss of BDNF during development.
EXPERIMENTAL PROCEDURES
Generation of forebrain-specific BDNF knockout mice
All animal procedures were approved by the University of Colorado Institutional Animal Care and Use Committee and conform to NIH guidelines. To generate mutant mice, CaMKcre mice (Xu et al., 2000a; 2000b) were mated to BDNFneo heterozygous mice (Jones et al., 1994) to create mice that were CaMKcre; BDNFneo. These CaMKcre; BDNFneo mice were heterozygous for the BDNF gene, but, as reported before, (Jones et al., 1994; Ernfors et al., 1994) the mice appeared similar to wild type though they were sometimes obese, consistent with other observations in BDNF null mutant heterozygous mice (Lyons et al., 1999; Kernie et al., 2000). Male CaMKcre; BDNFneo mice were then mated to female BDNFlox mice (Gorski et al., 2003a) to produce mutant mice (CaMKcre; BDNFneo/BDNFlox). These CaMKcre; BDNFneo/BDNFlox mice are late-onset, forebrain specific BDNF knockouts and are referred to as CaMK-BDNFKO mice. As controls we used wild type (this included both true wild-type and CaMKcre/+ mice) and for heterozygous controls we used BDNFneo/BDNFlox and BDNFneo/+ mice. All mice were backcrossed to C57BL/6J (The Jackson Laboratory, Bar Harbor, ME) and mice used in behavioral studies were at least 85% C57BL/6J with remaining contributions of 129/sv, CBA/J and DBA. Mice used for the dendritic spine analyses were greater than 99% C57BL/6J. Mice were genotyped using PCR. Using this PCR assay and standard DNA samples with a known ratio of unrecombined lox: recombined lox, we estimated the amount of recombination in different tissues. Recombination at 8 weeks was primarily in forebrain with highest levels in cortex and hippocampus and there was no apparent recombination elsewhere in the body except for the testes.
Histochemistry
X-gal staining of brain slices was performed as described in previously (Vigers et al., 2000). Adult mice were deeply anesthetized with 20 μl/g 2.5% Avertin in phosphate-buffered saline (PBS; 10 mM sodium phosphate, 150 mM NaCl, pH 7.4: 100% Avertin is 10 g tribromoethanol, 10 ml tert-amyl alcohol). Mice were perfused with ice-cold 4% paraformaldehyde in 0.1 M sodium phosphate, pH 7.4. Brains were removed and postfixed in the same fixative for one hour. Brains were cryoprotected overnight in 22% sucrose in 0.1 M sodium phosphate, pH 7.4 at 4°C, then mounted in OCT compound (VWR International, LLC) and stored at −80 oC until sectioning. Cryostat sections (40μm) were cut and thaw-mounted onto Superfrost plus slides (Fisher Scientific, Fair Lawn, NJ). Slices were dried at room temperature and washed twice (7 minutes each) in X-gal wash (0.1 M sodium phosphate, pH 7.4, 2 mM MgCl2, 5 mM EGTA, 0.01% sodium deoxycholate, 0.02% Nonidet P-40) and stained overnight at 37oC in X-gal staining solution (X-gal wash plus 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal, US Biological, Swampscott, MA)). Slides were then washed twice in X-gal wash for 7 minutes each. After a brief rinse in water, slides were counterstained with neutral red, dehydrated in an ethanol series (70%, 95%, and 100% ethanol), then in xylene, and mounted with Permount (Fisher Scientific).
Protein analyses
We used a quantitative enzyme-linked immunosorbent assay (ELISA, G6981, Promega Inc., Madison, WI) to measure BDNF concentrations in tissue extracts. The method described in the Promega kit (protocol # TB257) was used to prepare extracts, except the lysis buffer was 50 mM Tris-HCl, pH 7.4, 0.6 M NaCl, 0.2% Triton-X100, 1% BSA, 0.1 M benzathonium chloride, 1 mM benzamidine, 0.1 mM PMSF. BDNF was measured in the extracts exactly as described in protocol # TB257.
Behavioral tests
Behavioral tests were performed on adult littermates (aged 8–16 weeks old, unless otherwise stated). Animals were group housed (2–4 per cage) and kept under a 12-hour day-night cycle (7am/7pm), with free access to food and water. For conditioned fear, Porsolt, and Morris water maze tests, the numbers of each genotype (CaMK-BDNFKO = KO, heterozygous controls = HET, wild type controls = WT) and the numbers of each sex (female = F, male = M) are shown in the individual method descriptions. More females were used than males because females can be group housed and most CaMK-BDNFKO males were aggressive, thus preventing group housing. The performance of males and females in behavioral tests was only found to be different in the conditioned fear test as described below. All tests were performed between the hours of 8am and 1pm. Data was plotted and statistical analyses performed using Prism (GraphPad Software, San Diego, CA). Analysis of variance (ANOVA) was performed and results of a Tukey post-hoc analysis are given. Data are presented as the mean ± SEM. Anxiety testing was performed using Black / White box and mirrored chamber as previously described (Gorski et al., 2003b).
Contextual fear conditioning
Contextual fear was measured essentially as described previously (Gorski et al., 2003b), with 14 KO (12 F, 2 M), 15 HET (9 F, 6 M) and 14 WT (9 F, 5 M) mice. On day 1, mice were placed in a conditioning chamber (54cm x 27cm x 30cm high) with a metal grid floor (parallel wire bars 1/16” diameter, separated by 1/4” space) and a plexiglass rectangular cover (26.1cm x 18.5cm) with a wire mesh top. For a total of 5 minutes, freezing behavior (the absence of all but respiratory movements) was monitored every 10 sec. During the first 2 min animals were allowed to explore (baseline behavior), then were exposed to an 80dB, 6 clicks per sec auditory clicker for 30 sec, followed immediately by a 0.35mA foot shock (2 sec duration). There followed another 2 min period of free exploration then another 30 sec of clicker paired with foot shock. Mice spent a final 30 sec in the chamber after the 2nd foot shock, and then were returned to their home cages. On day 2, animals were returned to the conditioning chamber at the same time of day by the same experimenter and allowed to explore for 5 min, during which time freezing behavior was noted (context). Approximately 1 hour later animals were placed in an altered chamber (in a different room, plexiglass floor replaces metal grid floor, addition of a divider to make a triangular shape (234.5 cm2), and addition of orange extract to change the olfactory cue). Mice had 3 min in the altered chamber and then were exposed to the same tone as before (80dB, 6 clicks per sec auditory clicker) for 3 min. Freezing was monitored before the tone (altered context) and during the tone (conditioned stimulus).
Porsolt test
Porsolt described this test as a measure of depression in animals (Porsolt, 2000). Mice (10 KO (6 F, 4 M), 11 HET (7 F, 4 M) and 9 WT (7 F, 2 M)) were placed in a clear glass cylinder (12cm diameter and 40cm high) filled to a height of 25cm with water (22–23° C). Two swim sessions were performed - an initial 10-minute swim on day 1 and a second 5 minute swim on day 2. After each swim session, mice were placed on a dry paper towel to remove excess water before being replaced in their home cages. The swim sessions were videotaped and scored later. Initially animals actively try to escape by swimming, exploring, jumping, rearing, sniffing or diving. This active time is followed by a period of immobility where the mouse stops struggling and merely makes the movements necessary to keep its head above the water. The time spent in these 2 activities (active and immobile) was scored for the 2 swim sessions.
Morris water task
The Morris water task (Morris, 1984) was adapted for mice and performed essentially as described previously (Gorski et al., 2003b). The apparatus was a 121 cm diameter galvanized steel tank which was three-quarters filled with water (22–23°C) made opaque with non-toxic white tempura paint. An 11.4 cm x 11.4 cm square plexiglass platform was placed 1cm under the surface of the water. The tank was located in a room that had visual cues the animals could use in navigating. The visual cues included a cage rack, posters and dark colored poster board cut in asymmetric shapes attached to the walls, a black door, video recording equipment and the experimenter (who always stood in the same location during trials). All cues remained in the same position every day of the experiment. For the non-spatial “visible platform” version of the task a white sail (7.5 x 9 cm) from a child’s toy boat was used as a visible flag to mark the platform. Because the mice performed poorly during this test, the white sail was subsequently covered with black paper for the last of the visible trials to provide better contrast with the white water and white tank walls.
Mice (aged 12–16 weeks; 8 KO (7 F, 1 M), 8 HET (5 F, 3 M), 8 WT (7 F, 1 M)) were trained to swim to the visible platform (4 days with the white sail, then 5 days with the black sail) and then to swim to the hidden platform (3 days). Each training day consisted of three sessions, with four swim trials per session and each session separated by approximately 1 h. The location of the platform was changed to a new position in a random order between sessions for the visible task, but in the hidden task, the platform remained fixed in one quadrant (quadrant 2) of the pool. Each swim trial within a session was initiated at one of four different starting positions at the outer edge of the pool, so the mice had to use visual cues located outside the tank in the room (see above) to reach the hidden platform. Upon reaching the platform, each mouse was allowed to rest for twenty seconds on the platform. If the mouse did not reach the platform within 60 s, the trial was aborted, the mouse was given a score of 60 and it was either guided to the platform or picked up and placed on it. After each session each mouse was returned to its home cage where it rested until the next session. On the last day of spatial training (hidden platform) each mouse was tested in a probe trial which consisted of removing the hidden platform, placing the mouse at the quadrant opposite where the hidden platform had been and following the mouse’s swim path for 1 min. All trials were videotaped and the tape of the probe trial was used to score number of platform crossings, time in each quadrant and distance swum. Thigmotaxis was scored in two ways, either as percent time spent within 20 cm of the wall or percent time spent with nose against wall. Maximum swim speeds were measured on the last trial block of the last day with a black sail and an average swim speed calculated.
Rotarod
Ataxia was measured using a rotarod apparatus (Ugo Basile, Italy). The rotarod was accelerated gradually from 4 to 40 rpm over the course of 5 min. Mice were placed on the apparatus and rotation was initiated. Latency to fall was recorded automatically. Mice that were still on the wheel at 5 min were removed from the wheel and the time was noted as 300s. Trials were given approximately an hour apart, with 3 trials on day 1 and 3 trials on day 2.
Golgi staining
Golgi staining was performed as per manufacturer’s instructions using FD Rapid GolgiStainTM Kit (FD Neurotechnologies Inc., Ellicott City, MD). Animals used were 5 week old CaMK-BDNFKO mice (5KO (2 F, 3M), 5 HET (5 F, 0M), 5 WT (2F, 3M), 12 week old CaMK-BDNFKO mice (5KO (3F, 2M), 5 HET(2F, 3M), WT (3F, 2M) , 5 week old Emx-BDNFKO mice (4 KO (2F, 2M), 4 HET (3F, 1M), 4 WT (2F, 2M). In brief, animals were killed by cervical dislocation, brains removed rapidly, rinsed briefly in water and placed in impregnation solution (mixture of Golgi solutions A and B) for 14 days in the dark at room temperature (the impregnation mixture was replaced once after 12–24 hours of impregnation). The brains were transferred into Golgi solution C and stored at 4oC in the dark for 2–6 days (Golgi solution C was replaced 12–24 hours after immersion). Brains were mounted with minimal amounts of OCT and frozen in dry ice. Sections (100 μm) were cut on a cryostat (at −23oC) and slices mounted on gelatin-coated slides in a drop of Golgi solution C. Excess Golgi solution C was removed from the slide with filter paper and slides were allowed to dry in the dark at room temperature for 24–48 hours. Sections were then stained using Golgi solutions D and E in the kit, dehydrated in an ethanol series followed by xylenes, and mounted in Permount (Fisher Scientific). Slides were stored at room temperature in the dark.
Spine counting
Spines on basal dendrites of layer 2/3 pyramidal neurons in primary monocular visual cortex (V1M) were imaged on a Zeiss Axioplan 2 microscope with a Hamamatsu digital camera. Spines were imaged on segments (20–35μm) of dendrites that contained no branches and were at least 20 μm from the cell body. For each animal, spines were counted using OpenLab software (Improvision, PerkinElmer Inc., Waltham, MA) on 2 segment lengths from each of 7 neurons.
Photography and Anatomy
Photographs were taken using a Nikon Optiphot microscope with a MicroFire camera (Optronics, Goleta, CA). Photos were processed in Adobe Photoshop 5 and anatomical assignments were made from Franklin and Paxinos (1997).
Statistical analyses
Statistical tests were performed on data from spine density studies and behavioral tests using Prism (GraphPad Software Inc., La Jolla, CA). One–way analysis of variance (ANOVA) with a Tukey post-hoc analysis was used to compare three different genotypes and an unpaired t-test was used to compare 2 different genotypes.
RESULTS
CaMKcre can be used to deplete BDNF in the young adult forebrain
Although the CaMKcre transgene used in our studies has been previously characterized (Xu et al., 2000a; 2000b), some reports indicate that Cre transgenes can cause different tissue-specific patterns and timing of recombination with different floxed target genes. Thus, we determined the extent and timing of recombination occurring in CaMKcre; BDNFlox/+ mice at different ages using histochemistry. In BDNFlox mice, Cre-mediated recombination deletes the BDNF coding region and brings E. coli lacZ under control of the BDNF promoters (Gorski et al., 2003a). Thus, the fraction of BDNF-expressing cells that have undergone Cremediated recombination can be readily assessed by comparison of the β-galactosidase distributions in CaMKcre; BDNFlox/+ mice and BDNFlacZ/+ mice (in which lacZ replaces the BDNF coding region in all cells (Bennett et al., 1999)). This comparison showed that recombination occurs extensively in neurons of the cerebral cortex, hippocampus and amygdala, with only sporadic cells recombined in the thalamus, hypothalamus, midbrain and hindbrain (Fig. 1A,B). This regional pattern is similar to the Cre-mediated recombination we observed in early-onset BDNF forebrain-specific mutants generated using Emx1IREScre (Emx-BDNFKO mice, Gorski et al., 2003a). However, the timing is quite different; Emx1IREScre directs recombination during mid-embryogenesis, whereas CaMKcre-directed recombination becomes extensive around the fourth postnatal week, well after the onset of BDNF production in the hippocampus and neocortex (Fig. 1A).
Fig 1.

1A Postnatal development of recombination is driven by CaMKcre in hippocampus and visual cortex (Vis cortex) of BDNFlox/+ mice. Sections from CaMKcre; BDNFlox/+ (CKCBlox) mice (a,c,e,g,i,k) and BDNFlacZ/+ (BLacZ) mice (b,d,f,h,j,l) were stained with X-gal and counter-stained with neutral red. Slices shown are rostral hippocampus (a,b,e,f,i,j), and visual cortex (c,d,g,h,k,l). Panels are from mice aged postnatal day 20 (a,b,c,d), P28 (e,f,g,h), and P56 (i,j,k,l) . Scale bar in panel a is 500μm for all hippocampal images, scale bar in panel c is 100μm for all visual cortex panels.
1B Substantial recombination is apparent in amygdala, but not thalamus, hypothalamus, midbrain or hindbrain of CaMKcre; BDNFlox/+ mice at 8 weeks. Sections are from 8 week old CaMKcre; BDNFlox/+ (CKCBlox) mice (m,o,q,s,u,w) and BDNFlacZ/+ (BLacZ) mice (n,p,r,t,v,x). Slices shown are amygdala (m,n), anterior thalamus (o,p), posterior thalamus (q,r), hypothalamus (s,t), midbrain (u,v) and hindbrain (w,x). Scale bar in panel t is 500μm for amygdala, hypothalamus and thalamus panels, scale bar in panel x is 1mm for midbrain and hindbrain images.
At P20, scattered CA3 cells and a minority of dentate granule cells are recombined in the hippocampus, while in the visual cortex occasional recombined cells were scattered throughout the layers (Fig. 1A panels a & c). The fraction of recombined cells increases substantially in both structures between P20 and P28, and by P56 the density of X-gal-stained cortical and hippocampal neurons in the CaMKcre; BDNFlox/+ and BDNFlacZ/+ mice appears similar, suggesting near-complete recombination has occurred in the BDNF-expressing cells present in these structures (Fig. 1A). Although Xu and co-workers (2000b) found similar timing and widespread neocortical recombination with CaMKcre mice, they found that hippocampal recombination of TrkB was limited to CA1 cells. The pattern of X-gal staining was similar in multiple different individuals at each age, indicating that recombination occurs consistently with respect to timing and tissue specificity. Recombination did not appear to spread further as the mice aged, since staining in sections from 1-year old animals was similar to that from P56 mice (D.S. Amin, unpublished observations).
Although the histochemical analyses provide qualitative evidence that extensive loss of BDNF occurs in neurons of the cortex and hippocampus of CaMK-BDNFKO mice by ~8 weeks of age, we sought to directly measure BDNF amounts. Compared to wild-type mice, the BDNF protein concentration in P84 CaMK-BDNFKO mice is reduced 89% in hippocampus and 97% in visual cortex (Fig. 2A). There is a 50–70% decrease in BDNF protein in most brain areas of P84 BDNFneo/BDNFlox transheterozygotes compared to wild type, which is consistent with previous observations that BDNF expression is reduced about 50% in heterozygous null mutants (Kolbeck et al., 1999) and with reduced BDNF production by the BDNFlox allele in some brain regions. Importantly, and as predicted from the histochemical analyses, BDNF levels in CaMK-BDNFKO mice are significantly lower than those in the BDNFneo/BDNFlox transheterozygotes in regions where CaMKcre is expressed including the cortex (Fig. 2A ViC, p<0.05) and hippocampus (Fig. 2A Hp, p<0.05). In contrast, in brain areas where CaMKcre is not expressed, such as the thalamus/hypothalamus and midbrain/hindbrain, BDNF levels are similar in both BDNFneo/BDNFlox transheterozygotes and CaMK-BDNFKO mice (Fig. 2A and see Fig. 1B also). Thus, CaMK-BDNFKO mice lose BDNF from the vast majority of cortical and hippocampal neurons in early adulthood. Although BDNF levels are reduced elsewhere in these animals, comparison to heterozygotes provides a control for this reduction.
Fig. 2.

A) BDNF protein levels are reduced in the brain of CaMK-BDNFKO mice. Levels of BDNF protein in 12 week old mice were measured in wild-type mice (n=5, black bars), BDNFneo;BDNFlox mice (n=3, gray bars) and CaMK-BDNFKO mice (n=5, white bars). ViC = visual cortex, Hp = hippocampus, T/Hy = thalamus/hypothalamus, M/Hi = mid/hindbrain, DRGs = dorsal root ganglia. B) Body weights of wild type, BDNF heterozygotes (Het), CaMKcre; BDNFlox/+ (CKCBlox) mice and CaMK-BDNFKO mice. Histograms show the body weights of animals aged 12–48 weeks, with the weights binned in 5 gram amounts. (On the abscissa 25 = 20.0–24.9g, 30 = 25.0–29.5g etc.)
Forebrain-specific BDNF mutants appear grossly normal
CaMK-BDNFKO mice are born in Mendelian ratio and are not noticeably smaller than their littermates throughout their postnatal life. Both males and females mate and are fertile, and the females can give birth to viable pups and suckle them. However, most CaMK-BDNFKO mice do not live as long as wild type siblings, and many die by about 10 months of age for unknown reasons. A subset of 3 month-old CaMK-BDNFKO mice and their heterozygous siblings (both BDNFneo/BDNFlox and BDNFneo/+) become obese (Fig. 2B). Haploinsufficiency of BDNF is known to cause obesity in both mice and humans (Lyons et al., 1999; Kernie et al., 2000; Rios et al., 2001; Coppola and Tessarollo, 2004; Gray et al., 2006; Han et al., 2008) and the severity of obesity is positively correlated with the severity of BDNF signaling perturbation (Lyons et al., 1999; Rios et al., 2001; Xu et al., 2003). A hypothalamic source of BDNF has been suggested to be essential in maintenance of normal weight (Xu et al., 2003, Chan et al., 2006, Unger et al., 2007). In contrast, CaMKcre; BDNFlox/+ mice, heterozygous for BDNF only in cells in which CaMKcre causes Cre/lox recombination, have a weight distribution similar to wild type mice (Fig. 2B). This suggests that CaMKcre does not drive recombination in the cells in which BDNF is necessary for maintaining normal body weight, consistent with the paucity of hypothalamic recombination driven by CaMKcre (Fig. 1B, panels s&t). Interestingly others using conditional BDNF mutants having broad forebrain deletion of BDNF did not observe obesity in those animals, suggesting that the neuron-specific enolase promoter used in those mice is also not expressed in the cells that affect obesity (Monteggia et al., 2004).
Evidence for generalized fear in CaMK-BDNFKO mice
As CaMK-BDNFKO mice appeared grossly normal, we tested them in a series of behavioral paradigms to determine how adult loss of forebrain BDNF impacts their behavior. In two different anxiety tests, the light-dark box and mirrored box, CaMK-BDNFKO mice behaved similar to controls suggesting that their baseline anxiety, like that of Emx-BDNFKO mice (Gorski et al., 2003b), is not significantly altered by the loss of forebrain BDNF (Table 1). To gain insight into the learning ability of CaMK-BDNFKO mice, we used a contextual and cued fear-conditioning test. In this assay, an initially neutral environmental context robustly elicits a fear response (measured as immobility) in normal animals after it has been paired with an aversive foot shock stimulus (associated with an auditory cue) during training. Normal animals are able to distinguish between the environmental context in which aversive training has occurred and an altered context (i.e. altered visual and olfactory cues), displaying decreased immobility in the altered context. The fear component of this conditioned behavior is influenced by the amygdala and the contextual component requires the function of the hippocampal formation (Kim and Fanselow, 1992; Phillips and LeDoux, 1995; Logue et al., 1997; Maren et al., 1998). We found that CaMK-BDNFKO mice, as well as heterozygous and wild type controls, displayed high levels of freezing after the auditory cue 24 hours post training, demonstrating robust cued fear conditioning (Fig. 3A).
Table 1.
Anxiety and ataxia testing in CaMK-BDNFKO and control mice Results shown are mean +/− SEM. All measurements are in seconds, except for number of transitions (Black / White box) and number of passage crossings (Mirrored chamber). For Black / White box and Mirrored chamber n = 11 CaMK-BDNFKO mice and wild type mice and n = 12 for heterozygous controls. For rotarod n = 8 for all genotypes.
| Behavioral test Measure |
CamK-BDNFKO | Het | WT |
|---|---|---|---|
| Black / White box | |||
| Latency to enter dark | 29.5 +/− 9.0 | 24.56+/− 4.1 | 12.55 +/− 2.5 |
| Number of transitions | 13.9 +/− 3.3 | 18.2 +/− 2.78 | 22.45 +/− 2.0 |
| Total time in white | 102.2 +/− 25.0 | 89.9 +/− 13.5 | 96.36 +/− 5.6 |
| Mirrored chamber | |||
| Latency to enter mirror | 181.4 +/− 35.8 | 157.4 +/− 31.8 | 165.27 +/− 31.4 |
| Total time in mirror | 20.9 +/− 7.5 | 22.7 +/− 10.0 | 17.18 +/− 4.7 |
| Number of passage crossings | 7.2 +/− 1.5 | 8.0 +/− 1.0 | 6.27 +/− 0.78 |
| Rotarod | |||
| Time on rod - Trial 1 | 119.0 +/− 41.8 | 151.6 +/− 31.9 | 159.88 +/− 36.7 |
| Time on rod - Trial 2 | 170.6 +/− 45.3 | 126.5 +/− 21.7 | 181.75 +/− 31.1 |
| Time on rod - Trial 3 | 204.0 +/− 44.7 | 206.5 +/− 28.5 | 192.00 +/− 27.9 |
| Time on rod - Trial 4 | 197.9 +/− 45.6 | 210.4 +/− 31.2 | 273.75 +/− 14.1 |
| Time on rod - Trial 5 | 210.0 +/− 47.1 | 243.9 +/− 29.2 | 240.75 +/− 19.3 |
| Time on rod - Trial 6 | 214.1 +/− 45.4 | 208.9 +/− 30.5 | 272.00 +/− 15.7 |
Fig. 3.
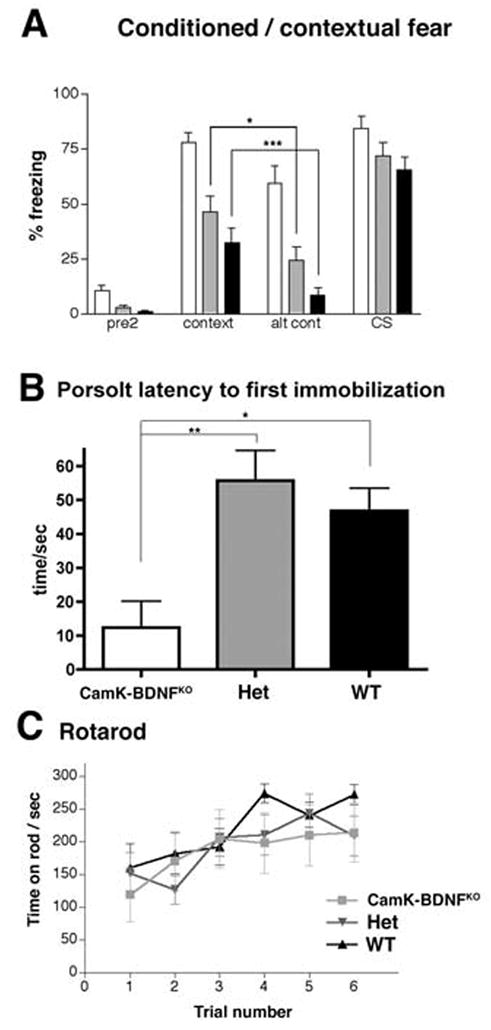
A) CaMK-BDNFKO mice show increased freezing in the conditioned fear test ( n= 14 for wild type and CaMK-BDNFKO mice, n= 15 for heterozygous controls). Percentage freezing is shown for pre2 (before any tone or shock is applied), context, altered context (alt cont) and in the presence of the conditioning stimulus tone (CS).
B) CaMK-BDNFKO mice are impaired in the Porsolt swim test (n = 10 CaMK-BDNFKO mice, n = 11 for heterozygous controls and n = 9 for wild type). Latency to the first immobilization is plotted. In both panels A) and B), wild type (wt) mice are shown with black bars, heterozygous mice (het) with gray bars and CaMK-BDNFKO mice with white bars. * p< 0.05, ** p< 0.01, *** p< 0.001.
C) CaMK-BDNFKO mice are not ataxic (n = 8 for all genotypes). Time spent on the accelerating rotarod is plotted for each of 6 trials. There is no significant difference between any of the genotypes in any of the trials (P> 0.05 in each trial).
Wild type (wt) and heterozygous (het) mice froze significantly more in the context than in the altered context indicating that they can discriminate between these two environments (Fig. 3A). The CaMK-BDNFKO mice displayed elevated levels of freezing in both the context and altered context, so, although they froze less in altered context than in context, the difference was not quite significant (p=0.06). Interestingly, when only female mice were scored in this test, the difference between context and altered context became significant (p = 0.03). These results suggest that the CaMK-BDNFKO mice may be able to discriminate between environments, though their high level of freezing in all environments somewhat obscures this ability. It is also possible there are sex-specific differences in this behavior in CaMK-BDNFKO mice, consistent with observations that lack of forebrain BDNF can lead to increased susceptibility to some depression-like behaviors in female, but not male mice (Autry et al. 2009)
The most striking observation in this test is that CaMK-BDNFKO mice froze significantly more than either wild type or heterozygous controls in both context (P< 0.001 CaMK-BDNFKO mice vs wt, P< 0.01 CaMK-BDNFKO mice vs het) and in altered context (P< 0.001 CaMK-BDNFKO mice vs wt or het). This indicates that CaMK-BDNFKO mice display a generalized freezing response to multiple contexts after aversive training, behavior that was also observed in the Emx-BDNFKO mice (Gorski et al., 2003b). These combined results suggest that the similar region-specific lack of forebrain BDNF occurring in these mice either during development or in adulthood leads to generalized fear responses.
CaMK-BDNFKO mice display signs of increased depression
Upon immersion in water, rodents swim and struggle for some time, before displaying immobility and floating. In the Porsolt test, floating behavior after immersion is measured, and effective anti-depressant drugs typically reduce immobility in this test (Porsolt, 2000). Numerous studies have suggested that BDNF plays a role in depression, and BDNF infused into the midbrain has been shown to alleviate depression by reducing floating behavior in the Porsolt test (Siuciak et al., 1997). We found that CaMK-BDNFKO mice had a significantly shorter latency to the first immobilization compared to heterozygotes and wild types (Fig. 3B), suggesting that CaMK-BDNFKO mice are more depressed than controls.
CaMK-BDNFKO mice have impaired spatial learning
A large body of literature indicates that normal BDNF function is essential for learning and memory in animals and humans (see Minichiello, 2009; Cunha et al., 2010). However, developmental versus ongoing requirements for BDNF in learning and memory processes have not been well defined. The Morris water maze task (MWM) is a test of spatial learning and memory that requires hippocampal function in mice (Morris, 1984; Logue et al., 1997). Mice were tested at 12–16 weeks of age initially using a visible platform version of the Morris water task where a white flag marked the position of the platform. The white flag contrasted weakly with the white colored water and white tank sides, and CaMK-BDNFKO mice performed especially poorly, with significantly longer swim times than wild type or heterozygous mice (Fig. 4A, white). To attempt to improve performance by making the flag more visible, a black flag was substituted, and time to reach the platform decreased for all genotypes (Fig. 4A, black). Although most animals (including CaMK-BDNFKO mice) swam directly and quickly to the black sail, the CaMK-BDNFKO mice as a group did not reach the platform as quickly as control genotypes even after several days of training (Fig. 4A), but there was not a statistically significant difference between the performances of any of the genotypes on the last day of the visible platform training.
Fig. 4.

CaMK-BDNFKO mice are impaired in the Morris water maze. Adult animals (12–16 weeks, n= 8 for all genotypes) were tested in Morris water maze. Trial blocks consisted of 4 swims and three trial blocks were performed each day. Swim times for each day were averaged and plotted in A and times for the final day of the hidden platform are shown in panel B. During the training trials, the hidden platform was in quadrant 2 (quad 2). In the probe trial, the platform was removed and the number of times the animal swam across the area of each possible platform position is shown in C. * p< 0.05, ** p< 0.01, *** p< 0.001. Black asterisks indicate differences between CaMK-BDNFKO mice and wild type mice, gray asterisks indicate differences between CaMK-BDNFKO and heterozygous controls.
The black sail was then removed and, since each swim trial was initiated at one of four different starting positions at the outer edge of pool, the mice had to use visual-spatial cues in the room to navigate to the hidden platform (Fig. 4A, hidden). Wild-type mice reached the hidden platform almost three times as fast as the CaMK-BDNFKO mice (Fig. 4B, P<0.001 wt vs. CaMK-BDNFKO), indicating a profound defect in spatial learning in CaMK-BDNFKO mice. After 3 days of hidden platform trials, the mice were tested in a probe trial (the platform is removed and mice swim, trying to locate the platform). During the probe trial the wild type and heterozygous mice showed a trend towards crossing the former platform site more often than equivalent positions in other quadrants of the tank. The CaMK-BDNFKO mice showed no spatial preference, indicating that they had not learned the position of the hidden platform (Fig. 4C).
Some mice show deficiencies in the Morris water maze due to thigmotaxis, which is a tendency to swim around the outside of the tank, near the wall, but the impaired performance of the CaMK-BDNFKO mice did not appear to be due to increased thigmotaxis (P>0.05 between all genotypes using two measures of thigmotaxis (time spent within 20 cm of tank walls and time spent with nose against tanks walls). The swim speed of CaMK-BDNFKO mice (measured during the last visible trial) was slower than that of wild types or heterozygotes and this may at least partly explain the increased latency to locate the visible platform during the initial training trials (wild type mice 1.24 m/s, CaMK-BDNFKO mice 0.69 m/s, p< 0.001). However, the CaMK-BDNFKO mice did not display abnormalities in the accelerating rotarod assay for ataxia (Fig. 3C and Table 1). Similarly, Emx-BDNFKO mice performed well in a rotarod test (Baquet et al., 2004), indicating that they, like the CaMK-BDNFKO mice do not have severe motor impairments.
CaMK-BDNFKO mice have smaller brains with generally normal cytoarchitecture
Given the behavioral deficits we detected in CaMK-BDNFKO mice as well as the in vivo and in vitro evidence that BDNF supports development of normal neuronal size and dendritic structure in multiple brain regions (McAllister et al., 1999), we sought to determine whether there were neuroanatomical abnormalities that might relate to the behavioral deficits. We found that CaMK-BDNFKO mice had significantly smaller brains than control mice. . Brains from female non-perfused 12–24 week old CaMK-BDNFKO mice weighed 10% less than those of either heterozygous or wild type controls (0.43± 0.03g; n = 19 for CaMK-BDNFKO mice versus 0.48± 0.02g; n = 24 for heterozygous and 0.48± 0.03g; n = 24 for wild-type siblings, p<0.001 between CaMK-BDNFKO and het or wt). A comparison of brain weights from older (24–52 week) CaMK-BDNFKO mice compared to younger CaMK-BDNFKO mice (< 24 wks) showed no significant difference, suggesting no overt loss of brain weight with increasing age (<24 wks n = 9, >24 wks n = 10, p=0.58). The cytoarchitecture of CaMK-BDNFKO in Nissl-stained sections appeared generally normal, although the cortex appeared thinner (compare Fig. 5a & 5c), similar to our findings in Emx-BDNFKO mice (Gorski et al., 2003a) and as described by others in forebrain-specific TrkB mutants (Xu et al., 2000a).
Fig. 5.

Anatomy of the CaMK-BDNFKO brains does not differ grossly from heterozygote or wild type controls. (a–c) Cresyl violet stain of the adult hippocampus from CaMK-BDNFKO mice (a), heterozygous (b) and wild type (c) controls. Immunocytochemistry for calbindin (d–f), met-enkephalin (g-i) and CCK (j–l) was performed. Genotypes are CaMK-BDNFKO (a, d, g and j), heterozygous (b, e, h and k) and wild type (c, f, i and l). Panels m–o show enlargements of CCK-LI in mossy fibers from panels j–l, respectively. Scale bar in panel a is 500μm; panels b–l are the same magnification as a. Scale bar in panel m is 250μm; panel n and o are the same magnification as m.
Although BDNF in the cortex appears to be expressed by excitatory neurons but not inhibitory interneurons (Cellerino et al., 1996), it profoundly affects the maturation and phenotype of interneurons (Marty, 1997; Hong et al., 2008). However, we found no qualitative differences between genotypes in expression of GABA or the calcium-binding proteins calbindin, parvalbumin, calretinin and PEP-19, suggesting a relatively normal abundance of interneurons (Fig. 5d–f and A. J. Vigers, unpublished observations). We also saw no major differences in expression of several neuropeptides expressed by subsets of interneurons including met-enkephalin, vasoactive intestinal protein, somatostatin, neuropeptide Y, and dynorphin (Fig. 5g–I and A.J. Vigers, unpublished observations). Thus, overall interneuron abundance appears relatively normal in both CaMK-BDNFKO and Emx-BDNFKO mice (Gorski et al., 2003b), but it is possible that some alterations in expression of proteins are not readily detectable in our immunocytochemical analyses.
However, we did find an altered pattern of CCK like-immunoreactivity (CCK-LI) in the hippocampus. There are 2 major locations of CCK-LI in the mouse hippocampus; the inner molecular layer of the dentate gyrus is labeled by projections of hilar polymorph neurons that give rise to dentate commissural and ipsilateral associational pathways, and the mossy fiber axons of the dentate granule cells that project to CA3 are also labeled (Gall et al., 1986). We found a consistent decrease in CCK-LI in mossy fibers of the dentate granule cells of CaMK-BDNFKO mice (Fig. 5j-o, arrows). In contrast, CCK-LI in the inner part of the molecular layer of the dentate gyrus from hilar mossy cell axons was unchanged (Fig. 5j-l, arrowheads). Interestingly, CCK-LI is reduced in the hippocampal formation of aged, memory-impaired rats (Croll et al., 1999) and there is a selective reduction of CCK-LI in mouse hippocampal mossy fibers following electrolytic- or kainate-induced seizures (Gall, 1988). Following these seizures, enkephalin-LI was elevated and dynorphin-LI was reduced in mossy fibers (Gall, 1988), but we did not observe alterations in opioid peptides in CaMK-BDNFKO mice, indicating that depletion of forebrain BDNF has a specific effect on the CCK system. Since a similar loss of CCK-LI in mossy fibers was also noted in Emx-BDNFKO mice (J.A. Gorski, unpublished observations), it is possible that the reductions we observe in mossy fiber CCK relate to a pattern of altered hippocampal activity in BDNF forebrain mutants. Notably, the morphology of mossy fiber terminals has been found to display abnormalities in conditional TrkB mutants (Danzer et al., 2008).
BDNF is essential to maintain visual cortical dendritic spine density
The behavioral deficits we detected in CaMK-BDNFKO mice are consistent with sensory and/or hippocampal deficits. Although BDNF is known to modulate hippocampal synaptic function, analyses of several BDNF and TrkB conditional mutants have failed to detect morphological abnormalities in the hippocampus (Baquet et al., 2004, Chakravarthy et al., 2006, Rauskolb et al., 2010). On the other hand, added BDNF can modulate synaptic function and dendritic spine density in cultured cortical neurons (Waterhouse and Xu, 2009) and we found that BDNF is required during postnatal development to stabilize the dendritic arbors of layer 2/3 pyramidal neurons in the visual cortex (Gorski et al., 2003a). In addition, loss of TrkB leads to dendritic tree collapse in layer 2/3 pyramidal neurons in neocortex (Xu et al., 2000a) while overexpression of a truncated TrkB receptor leads to impaired visual acuity and reduced spine density on cortical, but not hippocampal neurons (Chakravarthy et al., 2006, Heimel et al., 2010). Since spine density in layer 2/3 pyramidal neurons of the primary visual cortex is known to be influenced by altered visual experience (Tropea et al., 2011), we chose to analyze whether BDNF affects spine density on basal dendrites of layer 2/3 pyramidal neurons in primary monocular visual cortex (V1M).
We used the Golgi technique to analyze 5 week old CaMK-BDNFKO mice and control mice, an age at which some recombination driven by the CaMKII promoter has occurred but recombination is not yet complete (Fig. 1). We also analyzed mature adult mice at 12 weeks, well after recombination appears complete. Representative Golgi-stained neurons and dendritic segments are shown in Fig. 6A–D. At 5 weeks of age there was no significant difference in spine densities in CaMK-BDNFKO mice compared to wild type mice or heterozygous siblings (Fig. 6E).
Fig. 6.

Spine density in visual cortex is reduced in CaMK-BDNFKO mice at 12 weeks, but not at 5 weeks. A) and B) Typical layer II/III pyramidal cell in visual cortex V1M of wild type (A) and CaMK-BDNFKO mice (B). C) and D) Spines on basal dendrites of layer II/III pyramidal cells in wild type (C) and CaMK-BDNFKO mice (D). E) Spine density is unaltered in 5 week old CaMK-BDNFKO mice compared to wild type controls (n = 70 dendritic segments on 35 neurons from 5 animals of each genotype). F) Spine density is reduced in 12 week old CaMK-BDNFKO mice compared to wild type controls (n = 70 dendritic segments on 35 neurons from 5 animals of each genotype). G) Spine density is reduced in 5 week old Emx-BDNFKO mice compared to wild type controls (n = 56 dendritic segments on 28 neurons from 4 animals of each genotype). *** p< 0.001.
At 12 weeks, the spine densities in V1M in wild-type mice were similar to those in wild-type mice at 5 weeks (Fig. 6E,F). Thus, spine density has reached a mature level by 5 weeks of age, consistent with previous observations (Ballesteros-Yáñez et al., 2006). Strikingly, at 12 weeks of age the spine density in CaMK-BDNFKO mice was significantly lower than in either wild-type or heterozygous siblings (30% and 23% reductions respectively Fig. 6F). In comparison to the wild-type mice, the spine density in the 12-week old CaMK-BDNFKO mice was reduced by 29% compared to 5-week old CaMK-BDNFKO mice. This demonstrates the necessity for continued BDNF signaling in the adult cortex to maintain normal spine density in visual cortical neurons.
In related work, we have analyzed dendritic spine density in layer 2/3 cortical neurons of Emx-BDNFKO mice by labeling mice with diI and have detected developmental defects (S.R. Zeiler et al., unpublished observations). Consistent with the findings obtained by labeling with DiI, we found using Golgi staining that 5 week old Emx-BDNFKO mice have reduced spine density compared to wild type mice (Fig. 6G). Thus, whether BDNF is ablated embryonically, before expression begins, or in P35 mice, when expression has already reached adult levels, a similar reduction in dendritic spine density occurs in the neocortex.
DISCUSSION
Developmental and Adult Loss of BDNF Lead to Behavioral Similarities
Here we have described specific behavioral and anatomical deficits in adult-onset BDNF mutant mice generated using Cre-loxP recombination. The behavioral phenotypes of these mutants are strikingly similar to those of an early-onset forebrain-specific BDNF mutant we previously described (Gorski et al., 2003b). Although the regional specificity of these two conditional mutants within the CNS is similar, it might have been anticipated that loss of BDNF during mid-embryogenesis in the early-onset mutants would lead to more extensive behavioral deficits due to impacts on developmental processes not affected in the adult-onset mutants. The rise of most BDNF expression in the cortex and hippocampus does occur after many cell migration and differentiation events are complete. However, it corresponds with a period of extensive synaptogenesis. It could be speculated that loss of BDNF either before or after it functions in the establishment of a subset of circuitry would lead to similar impairments in that circuitry, resulting in similar behavioral deficits in both the early- and late-onset mutants. Related to this speculation, we found that neocortical dendritic spine density falls when BDNF is removed in adulthood, suggesting that some excitatory circuitry continues to require BDNF in the adult for stabilization. Our findings emphasize the importance of continued BDNF expression in the forebrain, and support the hypothesis that disruptions in BDNF expression can lead to structural changes in the adult brain related to pathological processes.
An Ongoing Requirement for BDNF in Maintaining Dendritic Spines
Most excitatory synapses in the forebrain occur on dendritic spines (see Nimchinsky et al., 2002). Dendritic spine density on cortical and hippocampal neurons rises postnatally as synapse density rises. (see Ballesteros-Yáñez et al., 2006; Alvarez and Sabatini, 2007). Many experiments have shown that BDNF-TrkB signaling can influence dendritic spine density in cortical and hippocampal neurons (see Cohen-Cory et al 2010; Gottman et al, 2009; McAllister et al, 1999). We previously reported decreased spine density on striatal medium spiny neurons in early onset Emx-BDNFKO mice (Baquet et al., 2004). We speculate that losses of dendritic spines and related synapses may lead to larger scale deficits in dendritic branching in these neurons due to an essential function for synapses in stabilizing dendritic arbors, as proposed in the synaptotrophic hypothesis (Cline and Haas, 2008). We previously observed defects in visual cortical pyramidal neuron dendritic branching in Emx-BDNFKO mice (Gorski et al., 2003a), thus, we sought to determine whether a similar reduction of spines on pyramidal neurons in visual cortex occurs in CaMK-BDNFKO mice, consistent with the possibility that visual impairments contribute to the poor performance of both of these mutants in the Morris water task.
Interestingly, we found that although cortical spine density appeared normal in 5 week-old CaMK-BDNFKO mice, it is significantly reduced by 12 weeks, indicating an essential and ongoing role for BDNF in maintaining normal spine density in visual cortical neurons. Notably, although TrkB is required both presynaptically and postsynaptically during development for normal synapse density in the hippocampus, ablation of TrkB after synapse formation did not appear to affect CA1 synapse density (Luikart et al., 2005). There is some controversy regarding the role of BDNF-TrkB signaling in CA1, both no change in spine density after developmental BDNF ablation (Rauskolb et al., 2010), and increased spine density accompanying a lack of BDNF messenger RNAs having a long 3’-untranslated region (An et al., 2008) have been reported. An extensive body of in vitro experiments documents a role for BDNF-TrkB signaling in modulating dendritic spine and synapse density (see Gottmann et al., 2009; Yoshii and Constantine-Paton, 2010). It is possible that the ongoing in vivo requirements for BDNF-TrkB signaling vary in different brain regions and at different developmental stages, perhaps in part due to homeostatic processes (Turrigiano, 2011). It would be of interest to extend the study of spine densities in the CaMK-BDNFKO mice to additional brain regions and cell types within these regions to gain deeper insight into how the ongoing requirements for BDNF may vary, and how structural abnormalities might relate to the behavioral deficits we have observed. Documenting structural defects in these mutants could also lead to further insights into how BDNF expression abnormalities in the adult may lead to pathological conditions. Lower dendritic spine densities have been documented in Alzheimer’s, Huntington’s and Parkinson’s diseases and other pathological states in various brain regions (see (Alvarez and Sabatini, 2007; Harms and Dunaevsky, 2007; Holtmaat and Svoboda, 2009; Bhatt et al., 2009). BDNF levels are also reduced in the brains of patients with these same diseases (Connor et al., 1997; Murer et al., 2001). Our results suggest further effort is warranted in exploring whether BDNF reductions contribute to dendritic spine and synapse losses in these diseases.
We previously found that the early-onset Emx-BDNFKO mice display smaller neuron soma sizes and reduced dendritic arborization in both the cortex and striatum (Gorski et al., 2003a; Baquet et al., 2004). We also found reduced cortical and striatal volumes in the Emx-BDNFKO mice, but hippocampal volumes were not significantly affected at both P35 and P120 (Baquet et al., 2004). Forebrain-specific TrkB mutants directed by CaMKcre transgenes also display cortical thinning and reduced dendritic arborization (Minichiello et al., 1999; Xu et al., 2000a; 2000b). We did not find overt evidence of progressive brain weight loss in CaMK-BDNFKO mice aged up to one year, but we cannot rule out the possibility that there would be additional progressive brain deterioration with age. There is evidence for modest striatal cell losses in Emx-BDNFKO mice over one year old, but this is many months after the initial loss of dendrites and spines were noted (Baquet et al., 2004). Minor cell losses have also been reported in the TrkB null and conditional mutants (Minichiello and Klein, 1996; Alcantara et al., 1997; Xu et al., 2000a). Altogether these results are consistent with the BDNF-TrkB system playing an essential role in maintaining neuronal morphology, particularly in the cortex and striatum. Structural and functional impairments in neurons resulting from BDNF-TrkB signaling dysfunction may create susceptibility that can lead to later neuronal cell losses.
BDNF-TrkB Signaling Requirements in Learning and Memory
BDNF is widely believed to be an important effector in aspects of learning and memory. BDNF levels increase in the rodent brain following activities related to enhanced cognitive performance, such as spatial learning (Kesslak et al., 1998; Hall et al., 2000; Mizuno et al., 2000; Berchtold et al., 2010), exercise (Oliff et al., 1998; Russo-Neustadt et al., 2000; Berchtold et al., 2010) and environmental enrichment (Young et al., 1999; Ickes et al., 2000). MWM is a hippocampal-dependent task (Morris, 1984; Logue et al., 1997), and a variety of evidence supports an important role for hippocampal BDNF. Blockade of endogenous BDNF activity in the hippocampus with antisense (Ma et al., 1998; Mizuno et al., 2000), with anti-BDNF antibodies (Mu et al., 1999), by viral removal (Heldt et al., 2007) or by over-expression of truncated TrkB receptors (Saarelainen et al., 2000) impaired spatial learning in MWM. In contrast, ICV infusion of BDNF had no effect on learning in adult (Cirulli et al., 2000) or aged rats (Fischer et al., 1994).
Supporting a role for BDNF in learning and memory, studies in humans with the BDNF Val66Met polymorphism suggested that the Val66Met variant leads to cognitive impairment including a deficit recalling places and events, and susceptibility to neuropsychiatric diseases such as depression and schizophrenia (see Bath and Lee, 2006). In BDNF mutant mice, conflicting results have been obtained in testing heterozygous BDNF null mutants; one group described a learning deficit (Linarsson et al., 1997), while another found no impairment (Montkowski and Holsboer, 1997). Interestingly, mice over-expressing BDNF also showed learning deficits (Croll et al., 1999; Cunha et al., 2009; Papaleo et al., 2011), suggesting that a setpoint level of BDNF is required for optimal learning, and that too much or too little BDNF may lead to memory impairment. This is intuitively reasonable given that BDNF is important for both LTP and LTD, as well as playing a role in modulating both excitatory and inhibitory circuits (Lu et al., 2008; Cunha et al., 2010). It is possible that BDNF heterozygotes are near the set point, leading to individual variability in their performance.
The CaMK-BDNFKO mice were severely impaired in both the hidden platform and visible platform portions of our MWM test. Similarly, late-onset TrkB mutants are also impaired in both of these tasks (Minichiello et al., 1999), consistent with an ongoing and necessary function for BDNF-TrkB signaling in cortical and/or hippocampal BDNF pathways in learning and memory. Importantly, the performance of the CaMK-BDNFKO mice improved in the visible platform portion of the test when the flag was changed from white to black, presumably because the black flag contrasted better against the white background. Impairments in the visible platform task can result from impaired visual acuity, motor function, and/or motivation. Reduced visual acuity may be a particularly important contributor to this deficit in the CaMK-BDNFKO mice as highlighted by the recent finding that mice over-expressing the truncated form of TrkB (lacking the tyrosine kinase domain) have reduced visual acuity due to a reduction in contrast discrimination ability (Heimel et al., 2010).
Our hidden platform MWM tests, administered after training with the visible flag-marked platform, required the CaMK-BDNFKO mutants to switch to using more distant and abstract visual cues present in the room. It seems likely that visual impairments also hamper performance in this portion of the test. Additionally, the mutants may lack the behavioral flexibility necessary for this switch. The latter possibility is supported by results indicating that BDNF is required for both behavioral flexibility and coping strategies in a novel situation (Alonso et al., 2002; Vyssotski et al., 2002; Cirulli et al., 2004). A lack of flexibility may also play a role in the generalized freezing response that was apparent in CaMK-BDNFKO mice during the conditioned fear test.
Overall, the CaMK-BDNFKO mice display sensory and motor deficits as well as an inability to learn the MWM task. Dissection of the specific role of BDNF-TrkB signaling in learning and memory remains a difficult problem, and further elucidation of its functions will require more specific tools for manipulating this signaling system as well as more sophisticated behavioral tests.
BDNF-TrkB Signaling and Fear
After exposure to training with a footshock, increased freezing of CaMK-BDNFKO mice was observed in all conditions of the contextual fear test, as was also found in Emx-BDNFKO mice (Gorski et al., 2003b). These observations could suggest an overall increased level of fear and/or stress in these mutants, consistent with important functions of amygdala-derived BDNF. Although CaMK-BDNFKO mice display generalized fear expression, they respond to a conditioned stimulus with increased freezing. This suggests that they have retained cue conditioning, an amygdala-dependent behavior (Maren and Fanselow, 1996). BDNF is highly expressed in the amygdala, and both fear-conditioning and conditioned taste aversion paradigms increase levels of BDNF mRNA and protein as well as TrkB phosphorylation in the amygdala (Maren and Fanselow, 1996; Rattiner et al., 2004, Ma et al., 2011). In humans BDNF Val66Met status is associated with an impaired ability to extinguish fearful memories (Frielingsdorf et al., 2010), consistent with the generalized fear we found in CaMK-BDNFKO mice.
Other studies have shown that infusing BDNF antisense cDNA into the hippocampus prevented the consolidation of contextual fear conditioning, an effect that was reversed by co-administration of BDNF (Lee et al., 2004) In contrast, late-onset conditional forebrain TrkB knock-out mice were reported as having retarded acquisition of a freezing response to repeated foot shocks, and impaired freezing to tone in an altered context but normal freezing in the conditioned context at later times of testing (Minichiello et al., 1999). This substantial difference between forebrain-specific BDNF and TrkB mutants could suggest that the increased freezing response we observed is due to removal of forebrain-produced BDNF that normally acts on subcortical TrkB receptors.
BDNF-TrkB Signaling and Depression
BDNF has been shown in a variety of studies to be both necessary and sufficient for anti-depressant drug action (see Duman and Monteggia, 2006; Banasr and Duman, 2008; Schmidt et al., 2008; Castren and Rantamäki, 2010; Yu and Chen, 2011). BDNF levels are decreased during both acute and chronic stress, as well as in monopolar and bipolar depression and many antidepressants as well as electroconvulsive shock (ECS) increase expression of both BDNF and its receptor TrkB in the hippocampus. Early studies in humans with BDNF Val66Met SNPs suggested a susceptibility to neuropsychiatric disorders such as depression, anxiety and bipolar disorder (see Castren and Rantamäki, 2010; Yu and Chen, 2011). There is however, still much controversy in this field, with many conflicting results (see Groves, 2007; Kozisek et al., 2008; Taliaz et al., 2009; Castren and Rantamäki, 2010).
The Porsolt or forced swim test has been used extensively to evaluate potential anti-depressants, and this test was used to show that midbrain injection of BDNF produces an anti-depressant effect and reduces floating behavior (Siuciak et al., 1997). Subsequent studies showed that ventricular administration of BDNF or infusion into the dentate gyrus produced a long-lasting anti-depressant effect in the forced swim test, while transgenic mice over-expressing BDNF in the forebrain also showed decreased depression in the Porsolt test (Shirayama et al., 2002; Hoshaw et al., 2005; Govindarajan et al., 2006). Our finding that that the CaMK-BDNFKO mice appear more depressed than wild type controls in the forced swim test is consistent with these findings and further implicates continued BDNF expression in the mature forebrain as essential for preventing depression.
Interestingly, several other conditional BDNF mutants have performed differently in depression-related behavioral assays. The Rios group found that although their early- and late-onset forebrain-specific BDNF mutants showed increased depressive behavior in the tail suspension test, they had reduced depressive behavior in the Porsolt test (Chan et al., 2006). The latter suggests that the brain regions responsible for depressive behavior in the tail suspension and Porsolt tests may be at least partially distinct. Monteggia et al. (2004) found that their early- and late-onset BDNF mutants performed normally in the Porsolt test, but showed that BDNF is required for the anti-depressant effect of desipramine, and more recently that loss of BDNF increases susceptibility to depression-like behavior in female but not male mice (Autry et al., 2009). Further complicating the effort to identify a specific locus of BDNF modulation of depression, infusion of BDNF to the ventral tegmental area (VTA) or nucleus accumbens (NAc) has a pro-depression effect, whereas blocking BDNF action in the VTA or NAc has an anti-depressant effect (Eisch et al., 2003; Berton et al., 2006). These studies combined suggest that BDNF has an anti-depressant effect in some brain areas but depressive effects in other brain regions, emphasizing how a capability for targeting BDNF-TrkB signaling in a highly region-specific manner is essential in the design of any related therapeutics (see also Nagahara and Tuszynski, 2011).
Conclusion
Our results demonstrate that continued expression of BDNF in the forebrain of the adult mouse is essential for some aspects of behavior, and for the maintenance of dendritic spine density on cortical pyramidal neurons. These observations are consistent with the hypothesis that reduced levels of BDNF in the adult human forebrain may contribute to the sensory and cognitive impairment, synapse loss, and neuronal atrophy that occur in diseases such as Alzheimer’s disease (Arancio and Chao, 2007).
Highlights.
Adult forebrain BDNF knockouts have smaller brains and many normal behaviors
Adult forebrain BDNF knockouts have reduced spatial learning and increased depression
Spine density is reduced by P84 in adult forebrain BDNF knockouts
Neuronal BDNF is necessary in adulthood to maintain normal spine density and behavior
Acknowledgments
This work was supported by the National Institutes of Health (EY014998). AJV was supported by NIH KO1-NS01872 during a portion of this project. BX was supported by the Howard Hughes Medical Institute and NIH NS-P01-16033. We thank Louis Reichardt for supplying the CaMKcre/+ mice and for helpful suggestions. We thank Jeanne Wehner for assistance with behavioral experiments.
Abbreviations
- BDNF
Brain-Derived Neurotrophic Factor
- CNS
Central Nervous System
- PNS
Peripheral Nervous System
- TrkB
Receptor Tyrosine Kinase B
- CaMK-BDNFKO mice
Late-onset forebrain-specific BDNF knockout mice (Cre recombinase driven by CaMKinase II promoter)
- Emx-BDNFKO mice
Early-onset forebrain-specific BDNF knockout mice (using Emx1IREScre mice)
- MWM
Morrris Water Maze
Footnotes
There are no actual or potential conflicts of interest for any of the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
ALISON J. VIGERS, Email: vigers@colorado.edu.
DIPESH S. AMIN, Email: amin.dipesh@gmail.com.
TIFFANY TALLEY-FARNHAM, Email: tctalley@gmail.com.
JESSICA A. GORSKI, Email: Jessica.Gorski@Colorado.EDU.
BAOJI XU, Email: bx3@georgetown.edu.
KEVIN R. JONES, Email: Krjones@Colorado.EDU.
References
- Alcantara S, Frisen J, del Rio J, Soriano E, Barbacid M, Silos-Santiago I. TrkB signaling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy-induced cell death. Journal of Neuroscience. 1997;17:3623. doi: 10.1523/JNEUROSCI.17-10-03623.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso M, Vianna M, Izquierdo I, Medina J. Signaling mechanisms mediating BDNF modulation of memory formation in vivo in the hippocampus. 2002;22:663–674. doi: 10.1023/A:1021848706159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altar C, Cai N, Bliven T, Juhasz M, Conner J, Acheson A, Lindsay R, Wiegand S. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Altar CA, DiStefano PS. Neurotrophin trafficking by anterograde transport. Trends in neurosciences. 1998;21:433–437. doi: 10.1016/s0166-2236(98)01273-9. [DOI] [PubMed] [Google Scholar]
- Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- An JJ, Gharami K, Liao GY, Woo NH, Lau AG, Vanevski F, Torre ER, Jones KR, Feng Y, Lu B, Xu B. Distinct role of long 3' UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell. 2008;134:175–87. doi: 10.1016/j.cell.2008.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancio O, Chao M. Neurotrophins, synaptic plasticity and dementia. Current opinion in neurobiology. 2007;17:325–330. doi: 10.1016/j.conb.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Cheng P, Monteggia LM. Gender Specific Impact of BDNF signaling on Stress-Induced Depression-Like Behavior. Biol Psychiatry. 2009;66:84–90. doi: 10.1016/j.biopsych.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros-Yáñez I, Benavides-Piccione R, Elston G, Yuste R, DeFelipe J. Density and morphology of dendritic spines in mouse neocortex. Neuroscience. 2006;138:403–409. doi: 10.1016/j.neuroscience.2005.11.038. [DOI] [PubMed] [Google Scholar]
- Banasr M, Duman R. Keeping Trk' of Antidepressant Actions. Neuron. 2008;59:349–351. doi: 10.1016/j.neuron.2008.07.028. [DOI] [PubMed] [Google Scholar]
- Baquet Z, Gorski J, Jones K. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. Journal of Neuroscience. 2004;24:4250. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde Y, Edgar D, Thoenen H. Purification of a new neurotrophic factor from mammalian brain. The EMBO Journal. 1982;1:549. doi: 10.1002/j.1460-2075.1982.tb01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker P. Whither proBDNF? Nat Neurosci. 2009;12:105–106. doi: 10.1038/nn0209-105. [DOI] [PubMed] [Google Scholar]
- Bath KG, Lee FS. Variant BDNF (Val66Met) impact on brain structure and function. Cogn Affect Behav Neurosci. 2006;6:79–85. doi: 10.3758/cabn.6.1.79. [DOI] [PubMed] [Google Scholar]
- Bennett J, Zeiler S, Jones K. Patterned expression of BDNF and NT-3 in the retina and anterior segment of the developing mammalian eye. Investigative ophthalmology & visual science. 1999;40:2996. [PubMed] [Google Scholar]
- Berchtold N, Castello N, Cotman C. Exercise and time-dependent benefits to learning and memory. Neuroscience. 2010;167:588–597. doi: 10.1016/j.neuroscience.2010.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton O, McClung C, DiLeone R, Krishnan V, Renthal W, Russo S, Graham D, Tsankova N, Bolanos C, Rios M. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Bhatt D, Zhang S, Gan W. Dendritic spine dynamics. Physiology. 2009;71:261–282. doi: 10.1146/annurev.physiol.010908.163140. [DOI] [PubMed] [Google Scholar]
- Bibel M. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes & Development. 2000;14:2919–2937. doi: 10.1101/gad.841400. [DOI] [PubMed] [Google Scholar]
- Buck CR, Martinez HJ, Chao MV, Black IB. Differential expression of the nerve growth factor receptor gene in multiple brain areas. Brain Res Dev Brain Res. 1988;44:259–268. doi: 10.1016/0165-3806(88)90224-6. [DOI] [PubMed] [Google Scholar]
- Castren E, Rantamäki T. The role of BDNF and its receptors in depression and antidepressant drug action: Reactivation of developmental plasticity. Devel Neurobio. 2010;70:289–297. doi: 10.1002/dneu.20758. [DOI] [PubMed] [Google Scholar]
- Cellerino A, Maffei L, Domenici L. The Distribution of Brain-derived Neurotrophic Factor and its Receptor trkB in Parvlbumin-containing Neurons of the Rat Visual Cortex. European Journal of Neuroscience. 1996;8:1190–1197. doi: 10.1111/j.1460-9568.1996.tb01287.x. [DOI] [PubMed] [Google Scholar]
- Chakravarthy S, Saiepour MH, Bence M, Perry S, Hartman R, Couey JJ, Mansvelder HD, Levelt CN. Postsynaptic TrkB signaling has distinct roles in spine maintenance in adult visual cortex and hippocampus. Proceedings of the National Academy of Sciences. 2006;103:1071–1076. doi: 10.1073/pnas.0506305103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J, Unger T, Byrnes J, Rios M. Examination of behavioral deficits triggered by targeting BDNF in fetal or postnatal brains of mice. Neuroscience. 2006;142:49–58. doi: 10.1016/j.neuroscience.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Chao M. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Chen X, Ye H, Kuruvilla R, Ramanan N, Scangos K, Zhang C, Johnson N, England P, Shokat K, Ginty D. A chemical-genetic approach to studying neurotrophin signaling. Neuron. 2005;46:13–21. doi: 10.1016/j.neuron.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Chen Z-Y, Jing D, Bath KG, Ieraci A, Khan T, Siao C-J, Herrera DG, Toth M, Yang C, McEwen BS, Hempstead BL, Lee FS. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z-Y, Bath K, McEwen B, Hempstead B, Lee F. Impact of genetic variant BDNF (Val66Met) on brain structure and function. Novartis Found Symp. 2008;289:180–8. doi: 10.1002/9780470751251.ch14. discussion 188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli F, Berry A, Alleva E. Intracerebroventricular administration of brain-derived neurotrophic factor in adult rats affects analgesia and spontaneous behaviour but not memory retention in a Morris Water Maze task. Neuroscience Letters. 2000;287:207–210. doi: 10.1016/s0304-3940(00)01173-3. [DOI] [PubMed] [Google Scholar]
- Cirulli F, Berry A, Chiarotti F, Alleva E. Intrahippocampal administration of BDNF in adult rats affects short-term behavioral plasticity in the Morris water maze and performance in the elevated plus-maze. Hippocampus. 2004;14:802–807. doi: 10.1002/hipo.10220. [DOI] [PubMed] [Google Scholar]
- Cline H, Haas K. The regulation of dendritic arbor development and plasticity by glutamatergic synaptic input: a review of the synaptotrophic hypothesis. J Physiol. 2008;586:1509–17. doi: 10.1113/jphysiol.2007.150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Greenberg ME. Communication Between the Synapse and the Nucleus in Neuronal Development, Plasticity, and Disease. Annu Rev Cell Dev Biol. 2008;24:183–209. doi: 10.1146/annurev.cellbio.24.110707.175235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Cory S, Kidane AH, Shirkey NJ, Marshak S. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Devel Neurobio. 2010;70:271–288. doi: 10.1002/dneu.20774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor B, Young D, Yan Q, Faull R, Synek B, Dragunow M. Brain-derived neurotrophic factor is reduced in Alzheimer's disease. Molecular brain research. 1997;49:71–81. doi: 10.1016/s0169-328x(97)00125-3. [DOI] [PubMed] [Google Scholar]
- Conover JC, Yancopoulos GD. Neurotrophin regulation of the developing nervous system: analyses of knockout mice. Reviews in the Neurosciences. 1997;8:13–27. doi: 10.1515/revneuro.1997.8.1.13. [DOI] [PubMed] [Google Scholar]
- Coppola V, Tessarollo L. Control of hyperphagia prevents obesity in BDNF heterozygous mice. Neuroreport. 2004;15:2665–2668. doi: 10.1097/00001756-200412030-00022. [DOI] [PubMed] [Google Scholar]
- Croll S, Suri C, Compton D, Simmons M, Yancopoulos G, Lindsay R, Wiegand S, Rudge J, Scharfman H. Brain-derived neurotrophic factor transgenic mice exhibit passive avoidance deficits, increased seizure severity and in vitro hyperexcitability in the hippocampus and entorhinal cortex. Neuroscience. 1999;93:1491–1506. doi: 10.1016/s0306-4522(99)00296-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha C, Angelucci A, D'Antoni A, Dobrossy M, Dunnett S, Berardi N, Brambilla R. Brain-derived neurotrophic factor (BDNF) overexpression in the forebrain results in learning and memory impairments. Neurobiology of Disease. 2009;33:358–368. doi: 10.1016/j.nbd.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Cunha C, Brambilla R, Thomas KL. A simple role for BDNF in learning and memory? Front Mol Neurosci. 2010;3:1–14. doi: 10.3389/neuro.02.001.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer SC, Kotloski RJ, Walter C, Hughes M, McNamara JO. Altered morphology of hippocampal dentate granule cell presynaptic and postsynaptic terminals following conditional deletion of TrkB. Hippocampus. 2008;18:668–78. doi: 10.1002/hipo.20426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman R, Monteggia L. A neurotrophic model for stress-related mood disorders. Biological Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Eisch AJ, Bolaños CA, de Wit J, Simonak RD, Pudiak CM, Barrot M, Verhaagen J, Nestler EJ. Brain-derived neurotrophic factor in the ventral midbrain-nucleus accumbens pathway: a role in depression. Biological Psychiatry. 2003;54:994–1005. doi: 10.1016/j.biopsych.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- Fischer W, Sirevaag A, Wiegand S, Lindsay R, Björklund A. Reversal of spatial memory impairments in aged rats by nerve growth factor and neurotrophins 3 and 4/5 but not by brain-derived neurotrophic factor. Proceedings of the National Academy of Sciences. 1994;91:8607–8611. doi: 10.1073/pnas.91.18.8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego: Academic Press; 1997. [Google Scholar]
- Frielingsdorf H, Bath KG, Soliman F, Difede J, Casey BJ, Lee FS. Variant brain-derived neurotrophic factor Val66Met endophenotypes: implications for posttraumatic stress disorder. Ann N Y Acad Sci. 2010;1208:150–157. doi: 10.1111/j.1749-6632.2010.05722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall C, Berry LM, Hodgson LA. Cholecystokinin in the mouse hippocampus: localization in the mossy fiber and dentate commissural systems. Exp Brain Res. 1986;62:431–7. doi: 10.1007/BF00238862. [DOI] [PubMed] [Google Scholar]
- Gall C. Seizures Induce Dramatic and Distinctly Different Changes in Enkephalin, Dynorphin, and CCK lmmunoreactivities in Mouse Hippocampal Mossy Fibers. The Journal of Neuroscience. 1988;8(E):1852–l882. doi: 10.1523/JNEUROSCI.08-06-01852.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski J, Zeiler S, Tamowski S, Jones K. Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. Journal of Neuroscience. 2003a;23:6856–6865. doi: 10.1523/JNEUROSCI.23-17-06856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski J, Balogh S, Wehner J, Jones K. Learning deficits in forebrain-restricted brain-derived neurotrophic factor mutant mice. Neuroscience. 2003b;121:341–354. doi: 10.1016/s0306-4522(03)00426-3. [DOI] [PubMed] [Google Scholar]
- Gottmann K, Mittmann T, Lessmann V. BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp Brain Res. 2009;199:203–34. doi: 10.1007/s00221-009-1994-z. [DOI] [PubMed] [Google Scholar]
- Govindarajan A, Rao BSS, Nair D, Trinh M, Mawjee N, Tonegawa S, Chattarji S. Transgenic brain-derived neurotrophic factor expression causes both anxiogenic and antidepressant effects. Proc Natl Acad Sci USA. 2006;103:13208–13213. doi: 10.1073/pnas.0605180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham D, Krishnan V, Larson E, Graham A, Edwards S, Bachtell R, Simmons D, Gent L, Berton O, Bolanos C. Tropomyosin-related kinase B in the mesolimbic dopamine system: region-specific effects on cocaine reward. Biological Psychiatry. 2009;65:696–701. doi: 10.1016/j.biopsych.2008.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J, Yeo GS, Cox JJ, Morton J, Adlam A-LR, Keogh JM, Yanovski JA, Gharbawy El A, Han JC, Tung YL, Hodges JR, Raymond FL, O'rahilly S, Farooqi IS. Hyperphagia, Severe Obesity, Impaired Cognitive Function, and Hyperactivity Associated With Functional Loss of One Copy of the Brain-Derived Neurotrophic Factor (BDNF) Gene. Diabetes. 2006;55:3366–3371. doi: 10.2337/db06-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg M, Xu B, Lu B, Hempstead B. New insights in the biology of BDNF synthesis and release: implications in CNS function. The Journal of Neuroscience. 2009;29:12764–12767. doi: 10.1523/JNEUROSCI.3566-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves J. Is it time to reassess the BDNF hypothesis of depression? Mol Psychiatry. 2007;12:1079–1088. doi: 10.1038/sj.mp.4002075. [DOI] [PubMed] [Google Scholar]
- Hall J, Thomas K, Everitt B. Rapid and selective induction of BDNF expression in the hippocampus during contextual learning. Nat Neurosci. 2000;3:533–535. doi: 10.1038/75698. [DOI] [PubMed] [Google Scholar]
- Han J, Liu Q, Jones M, Levinn R, Menzie C, Jefferson-George K, Adler-Wailes D, Sanford E, Lacbawan F, Uhl G. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. The New England journal of medicine. 2008;359:918–927. doi: 10.1056/NEJMoa0801119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms K, Dunaevsky A. Dendritic spine plasticity: looking beyond development. Brain research. 2007;1184:65–71. doi: 10.1016/j.brainres.2006.02.094. [DOI] [PubMed] [Google Scholar]
- Heimel J, Saiepour M, Chakravarthy S, Hermans JM, Levelt CN. Contrast gain control and cortical TrkB signaling shape visual acuity. Nature. 2010;13:642–648. doi: 10.1038/nn.2534. [DOI] [PubMed] [Google Scholar]
- Heldt S, Stanek L, Chhatwal J, Ressler K. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry. 2007;12:656–670. doi: 10.1038/sj.mp.4001957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer M, Pagliusi SR, Hohn A, Leibrock J, Barde YA. Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. The EMBO Journal. 1990;9:2459–2464. doi: 10.1002/j.1460-2075.1990.tb07423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008;60:610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshaw BA, Malberg JE, Lucki I. Central administration of IGF-I and BDNF leads to long-lasting antidepressant-like effects. Brain research. 2005;1037:204–208. doi: 10.1016/j.brainres.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Ickes BR, Pham TM, Sanders LA, Albeck DS, Mohammed AH, Granholm A-C. Long-Term Environmental Enrichment Leads to Regional Increases in Neurotrophin Levels in Rat Brain. Experimental Neurology. 2000;164:45–52. doi: 10.1006/exnr.2000.7415. [DOI] [PubMed] [Google Scholar]
- Johnson AW, Chen X, Crombag HS, Zhang C, Smith DR, Shokat KM, Gallagher M, Holland PC, Ginty DD. The BDNF receptor TrkB is critical to the acquisition but not expression of conditioned incentive value. Eur J Neurosci. 2008;28:997–1002. doi: 10.1111/j.1460-9568.2008.06383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K, Fariñas I, Backus C, Reichardt L. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh-Semba R, Semba R, Takeuchi I, Kato K. Age-related changes in levels of brain-derived neurotrophic factor in selected brain regions of rats, normal mice and senescence-accelerated mice: a comparison to those of nerve growth factor and neurotrophin-3. Neuroscience research. 1998;31:227–234. doi: 10.1016/s0168-0102(98)00040-6. [DOI] [PubMed] [Google Scholar]
- Kernie S, Liebl D, Parada L. BDNF regulates eating behavior and locomotor activity in mice. The EMBO Journal. 2000;19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesslak JP, So V, Choi J, Cotman CW, Gomez-Pinilla F. Learning upregulates brain-derived neurotrophic factor messenger ribonucleic acid: a mechanism to facilitate encoding and circuit maintenance? Behav Neurosci. 1998;112:1012–1019. doi: 10.1037//0735-7044.112.4.1012. [DOI] [PubMed] [Google Scholar]
- Kim J, Fanselow M. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- Kiss J, McGovern J, Patel AJ. Immunohistochemical localization of cells containing nerve growth factor receptors in the different regions of the adult rat forebrain. Neuroscience. 1988;27:731–748. doi: 10.1016/0306-4522(88)90179-0. [DOI] [PubMed] [Google Scholar]
- Klein R, Smeyne R, Wurst W, Long L, Auerbach B, Joyner A, Barbacid M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75:113–122. [PubMed] [Google Scholar]
- Koh S, Oyler GA, Higgins GA. Localization of nerve growth factor receptor messenger RNA and protein in the adult rat brain. Experimental Neurology. 1989;106:209–221. doi: 10.1016/0014-4886(89)90154-4. [DOI] [PubMed] [Google Scholar]
- Kolbeck R, Bartke I, Eberle W, Barde YA. Brain-derived neurotrophic factor levels in the nervous system of wild-type and neurotrophin gene mutant mice. J Neurochem. 1999;72:1930–1938. doi: 10.1046/j.1471-4159.1999.0721930.x. [DOI] [PubMed] [Google Scholar]
- Kozisek M, Middlemas D, Bylund D. Brain-derived neurotrophic factor and its receptor tropomyosin-related kinase B in the mechanism of action of antidepressant therapies. Pharmacology & therapeutics. 2008;117:30–51. doi: 10.1016/j.pharmthera.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Kuczewski N, Porcher C, Lessmann V, Medina I, Gaiarsa J. Activity-dependent dendritic release of BDNF and biological consequences. Mol Neurobiol. 2009;39:37–49. doi: 10.1007/s12035-009-8050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JLC, Everitt BJ, Thomas KL. Independent cellular processes for hippocampal memory consolidation and reconsolidation. Science. 2004;304:839–843. doi: 10.1126/science.1095760. [DOI] [PubMed] [Google Scholar]
- Lee TH, Kato H, Pan LH, Ryu JH, Kogure K, Itoyama Y. Localization of nerve growth factor, trkA and P75 immunoreactivity in the hippocampal formation and basal forebrain of adult rats. Neuroscience. 1998;83:335–349. doi: 10.1016/s0306-4522(97)00346-1. [DOI] [PubMed] [Google Scholar]
- Leibrock J, Lottspeich F, Hohn A, Hofer M, Hengerer B, Masiakowski P, Thoenen H, Barde YA. Molecular cloning and expression of brain-derived neurotrophic factor. Nature. 1989;341:149–152. doi: 10.1038/341149a0. [DOI] [PubMed] [Google Scholar]
- Li Y, Luikart BW, Birnbaum S, Chen J, Kwon C-H, Kernie SG, Bassel-Duby R, Parada LF. TrkB Regulates Hippocampal Neurogenesis and Governs Sensitivity to Antidepressive Treatment. Neuron. 2008;59:399–412. doi: 10.1016/j.neuron.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linarsson S, Bjorklund A, Ernfors P. Learning Deficit in BDNF Mutant Mice. European Journal of Neuroscience. 1997;9:2581–2587. doi: 10.1111/j.1460-9568.1997.tb01687.x. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Covington HE, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, Mouzon E, Mogri M, Neve RL, Deisseroth K, Han M-H, Nestler EJ. Cell Type-Specific Loss of BDNF Signaling Mimics Optogenetic Control of Cocaine Reward. Science. 2010;330:385–390. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue S, Paylor R, Wehner J. Hippocampal lesions cause learning deficits in inbred mice in the Morris water maze and conditioned-fear task. 1997;111:104–113. doi: 10.1037//0735-7044.111.1.104. [DOI] [PubMed] [Google Scholar]
- Lu Y, Christian K, Lu B. BDNF: A key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiology of learning and memory. 2008;89:312–323. doi: 10.1016/j.nlm.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart B, Nef S, Virmani T, Lush M, Liu Y, Kavalali E, Parada L. TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. Journal of Neuroscience. 2005;25:3774–3786. doi: 10.1523/JNEUROSCI.0041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons W, Mamounas L, Ricaurte G, Coppola V, Reid S, Bora S, Wihler C, Koliatsos V, Tessarollo L. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci USA. 1999;96:15239–15244. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Wang H, Wu H, Wei C, Lee E. Brain-derived neurotrophic factor antisense oligonucleotide impairs memory retention and inhibits long-term potentiation in rats. Neuroscience. 1998;82:957–967. doi: 10.1016/s0306-4522(97)00325-4. [DOI] [PubMed] [Google Scholar]
- Ma L, Wang D, Zhang T, Yu H, Wang Y, Huang S, Lee F, Chen Z. Region-Specific Involvement of BDNF Secretion and Synthesis in Conditioned Taste Aversion Memory Formation. The Journal of Neuroscience. 2011;31:2079–2090. doi: 10.1523/JNEUROSCI.5348-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonpierre P, Belluscio L, Friedman B, Alderson R, Wiegand S, Furth M, Lindsay R, Yancopoulos G. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- Maren S, Anagnostaras S, Fanselow M. The startled seahorse: is the hippocampus necessary for contextual fear conditioning? Trends in Cognitive Sciences. 1998;2:39–41. doi: 10.1016/s1364-6613(98)01123-1. [DOI] [PubMed] [Google Scholar]
- Maren S, Fanselow M. The amygdala and fear conditioning: has the nut been cracked? Neuron. 1996;16:237–240. doi: 10.1016/s0896-6273(00)80041-0. [DOI] [PubMed] [Google Scholar]
- Marty S. Neurotrophins and activity-dependent plasticity of cortical interneurons. Trends in neurosciences. 1997;20:198–202. doi: 10.1016/s0166-2236(96)01026-0. [DOI] [PubMed] [Google Scholar]
- McAllister A, Katz L, Lo D. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- Merlio J-P, Ernfors P, Jaber M, Persson H. Molecular cloning of rat trkC and distribution of cells expressing messenger RNAs for members of the trk family in the rat central nervous system. Neuroscience. 1992;51:513–532. doi: 10.1016/0306-4522(92)90292-a. [DOI] [PubMed] [Google Scholar]
- Miller F, Kaplan D. Neurotrophin signalling pathways regulating neuronal apoptosis. Cellular and Molecular Life Sciences. 2001;58:1045–1053. doi: 10.1007/PL00000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850–860. doi: 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp H, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Klein R. TrkB and TrkC neurotrophin receptors cooperate in promoting survival of hippocampal and cerebellar granule neurons. Genes & Development. 1996;10:2849–2858. doi: 10.1101/gad.10.22.2849. [DOI] [PubMed] [Google Scholar]
- Mizuno M, Yamada K, Olariu A, Nawa H, Nabeshima T. Involvement of brain-derived neurotrophic factor in spatial memory formation and maintenance in a radial arm maze test in rats. Journal of Neuroscience. 2000;20:7116–7121. doi: 10.1523/JNEUROSCI.20-18-07116.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia L, Barrot M, Powell C, Berton O, Galanis V, Gemelli T, Meuth S, Nagy A, Greene R, Nestler E. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci USA. 2004;101:10827–10832. doi: 10.1073/pnas.0402141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montkowski A, Holsboer F. Intact spatial learning and memory in transgenic mice with reduced BDNF. Neuroreport. 1997;8:779–782. doi: 10.1097/00001756-199702100-00040. [DOI] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. Journal of Neuroscience Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Mu J, Li W, Yao Z, Zhou X. Deprivation of endogenous brain-derived neurotrophic factor results in impairment of spatial learning and memory in adult rats. Brain research. 1999;835:259–265. doi: 10.1016/s0006-8993(99)01592-9. [DOI] [PubMed] [Google Scholar]
- Murer M, Yan Q, Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer's disease and Parkinson's disease. Progress in Neurobiology. 2001;63:71–124. doi: 10.1016/s0301-0082(00)00014-9. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011;10:209–219. doi: 10.1038/nrd3366. [DOI] [PubMed] [Google Scholar]
- Nawa H, Takei N. BDNF as an anterophin; a novel neurotrophic relationship between brain neurons. Trends in neurosciences. 2001;24:683–4. doi: 10.1016/s0166-2236(00)01955-x. discussion 684–5. [DOI] [PubMed] [Google Scholar]
- Nimchinsky E, Sabatini B, Svoboda K. Structure and function of dendritic spines. Annu Rev Physiol. 2002;64:313–353. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- Nykjaer A, Willnow TE, Petersen CM. p75NTR–live or let die. Current opinion in neurobiology. 2005;15:49–57. doi: 10.1016/j.conb.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Oliff H, Berchtold N, Isackson P, Cotman C. Exercise-induced regulation of brain-derived neurotrophic factor (BDNF) transcripts in the rat hippocampus. Molecular brain research. 1998;61:147–153. doi: 10.1016/s0169-328x(98)00222-8. [DOI] [PubMed] [Google Scholar]
- Papaleo F, Silverman JL, Aney J, Tian Q, Barkan CL, Chadman KK, Crawley JN. Working memory deficits, increased anxiety-like traits, and seizure susceptibility in BDNF overexpressing mice. Learn Mem. 2011;18:534–544. doi: 10.1101/lm.2213711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R, LeDoux J. Lesions of the fornix but not the entorhinal or perirhinal cortex interfere with contextual fear conditioning. The Journal of Neuroscience. 1995;15:5308–5315. doi: 10.1523/JNEUROSCI.15-07-05308.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsolt RD. Animal models of depression: utility for transgenic research. Reviews in the Neurosciences. 2000;11:53–58. doi: 10.1515/revneuro.2000.11.1.53. [DOI] [PubMed] [Google Scholar]
- Rattiner L, Davis M, French C, Ressler K. Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. The Journal of Neuroscience. 2004;24:4796–4806. doi: 10.1523/JNEUROSCI.5654-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauskolb S, Zagrebelsky M, Dreznjak A, Deogracias R, Matsumoto T, Wiese S, Erne B, Sendtner M, Schaeren-Wiemers N, Korte M, Barde Y-A. Global Deprivation of Brain-Derived Neurotrophic Factor in the CNS Reveals an Area-Specific Requirement for Dendritic Growth. The Journal of Neuroscience. 2010;30:1739–1749. doi: 10.1523/JNEUROSCI.5100-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt L. Neurotrophin-regulated signalling pathways. Philosophical Transactions of the Royal Society B: Biological Sciences. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios M. New Insights into the Mechanisms Underlying the Effects of BDNF on Eating Behavior. 2011;36:368–369. doi: 10.1038/npp.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan R, Jaenisch R. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Molecular Endocrinology. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- Rosas-Vargas H, Martínez-Ezquerro JD, Bienvenu T. Brain-derived Neurotrophic Factor, Food Intake Regulation, and Obesity. Arch Med Res. 2011 doi: 10.1016/j.arcmed.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt A, Beard R, Huang Y, Cotman C. Physical activity and antidepressant treatment potentiate the expression of specific brain-derived neurotrophic factor transcripts in the rat hippocampus. Neuroscience. 2000;101:305–312. doi: 10.1016/s0306-4522(00)00349-3. [DOI] [PubMed] [Google Scholar]
- Saarelainen T, Pussinen R, Koponen E, Alhonen L, Wong G, Sirviö J, Castrén E. Transgenic mice overexpressing truncated trkB neurotrophin receptors in neurons have impaired long-term spatial memory but normal hippocampal LTP. Synapse. 2000;38:102–104. doi: 10.1002/1098-2396(200010)38:1<102::AID-SYN11>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Banasr M, Duman R. Future antidepressant targets: neurotrophic factors and related signaling cascades. Drug Discovery Today: Therapeutic Strategies. 2008;5:151–156. doi: 10.1016/j.ddstr.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoups A, Elliott R, Friedman W. NGF and BDNF are differentially modulated by visual experience in the developing geniculocortical pathway. Dev Brain Res. 1995;86:326–334. doi: 10.1016/0165-3806(95)00043-d. [DOI] [PubMed] [Google Scholar]
- Shirayama Y, Chen A, Nakagawa S, Russell D, Duman R. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. Journal of Neuroscience. 2002;22:3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuciak J, Lewis D, Wiegand S, Lindsay R. Antidepressant-like effect of brain-derived neurotrophic factor (BDNF) Pharmacology Biochemistry and Behavior. 1997;56:131–137. doi: 10.1016/S0091-3057(96)00169-4. [DOI] [PubMed] [Google Scholar]
- Sobreviela T, Clary DO, Reichardt LF, Brandabur MM, Kordower JH, Mufson EJ. TrkA-immunoreactive profiles in the central nervous system: Colocalization with neurons containing p75 nerve growth factor receptor, choline acetyltransferase, and serotonin. J Comp Neurol. 1994;350:587–611. doi: 10.1002/cne.903500407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliaz D, Stall N, Dar D, Zangen A. Knockdown of brain-derived neurotrophic factor in specific brain sites precipitates behaviors associated with depression and reduces neurogenesis. Mol Psychiatry. 2009;15:80–92. doi: 10.1038/mp.2009.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao HW, Poo M. Retrograde signaling at central synapses. Proc Natl Acad Sci USA. 2001;98:11009–11015. doi: 10.1073/pnas.191351698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng KK, Felice S, Kim T, Hempstead BL. Understanding proneurotrophin actions: Recent advances and challenges. In: Chao MV, Ip NY, editors. Devel Neurobio. Vol. 70. 2010. pp. 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoenen H. Neurotrophins and Neuronal Plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- Tropea D, Sur M, Majewska AK. Experience-dependent plasticity in visual cortex: Dendritic spines and visual responsiveness. Commun Integr Biol. 2011;4:216–219. doi: 10.4161/cib.4.2.14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci. 2011;34:89–103. doi: 10.1146/annurev-neuro-060909-153238. [DOI] [PubMed] [Google Scholar]
- Underwood CK, Coulson EJ. The p75 neurotrophin receptor. The International Journal of Biochemistry & Cell Biology. 2008;40:1664–1668. doi: 10.1016/j.biocel.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Unger TJ, Calderon GA, Bradley LC, Sena-Esteves M, Rios M. Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. 2007;27:14265–14274. doi: 10.1523/JNEUROSCI.3308-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela DM, Maisonpierre PC, Glass DJ, Rojas E, Nuñez L, Kong Y, Gies DR, Stitt TN, Ip NY, Yancopoulos GD. Alternative forms of rat TrkC with different functional capabilities. Neuron. 1993;10:963–974. doi: 10.1016/0896-6273(93)90211-9. [DOI] [PubMed] [Google Scholar]
- Vigers AJ, Baquet ZC, Jones KR. Expression of Neurotrophin-3 in the Mouse Forebrain: Insights From a Targeted LacZ Reporter. J Comp Neurol. 2000;416:398–415. doi: 10.1002/(sici)1096-9861(20000117)416:3<398::aid-cne10>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Vyssotski AL, Dell'omo G, Poletaeva II, Vyssotski DL, Minichiello L, Klein RD, Wolfer DP, Lipp H-P. Long-term monitoring of hippocampus-dependent behavior in naturalistic settings: Mutant mice lacking neurotrophin receptor TrkB in the forebrain show spatial learning but impaired behavioral flexibility. Hippocampus. 2002;12:27–38. doi: 10.1002/hipo.10002. [DOI] [PubMed] [Google Scholar]
- Waterhouse EG, Xu B. New insights into the role of brain-derived neurotrophic factor in synaptic plasticity. Mol Cell Neurosci. 2009;42:81–89. doi: 10.1016/j.mcn.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Zang K, Ruff N, Zhang Y, McConnell S, Stryker M, Reichardt L. Cortical Degeneration in the Absence of Neurotrophin Signaling:: Dendritic Retraction and Neuronal Loss after Removal of the Receptor TrkB. Neuron. 2000a;26:233–245. doi: 10.1016/s0896-6273(00)81153-8. [DOI] [PubMed] [Google Scholar]
- Xu B, Gottschalk W, Chow A, Wilson R, Schnell E, Zang K, Wang D, Nicoll R, Lu B, Reichardt L. The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. Journal of Neuroscience. 2000b;20:6888–6897. doi: 10.1523/JNEUROSCI.20-18-06888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Goulding E, Zang K, Cepoi D, Cone R, Jones K, Tecott L, Reichardt L. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Johnson EM. An lmmunohistochemical Study of the Nerve Growth Factor Receptor in DevelopingRats. Journal of Neuroscience. 1988;8:3481–3498. doi: 10.1523/JNEUROSCI.08-09-03481.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii A, Constantine Paton M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Devel Neurobio. 2010;70:304–322. doi: 10.1002/dneu.20765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young D, Lawlor P, Leone P, Dragunow M, During M. Environmental enrichment inhibits spontaneous apoptosis, prevents seizures and is neuroprotective. Nat Med. 1999;5:448–453. doi: 10.1038/7449. [DOI] [PubMed] [Google Scholar]
- Yu H, Chen Z. The role of BDNF in depression on the basis of its location in the neural circuitry. Acta Pharmacol Sin. 2011;32:3–11. doi: 10.1038/aps.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zörner B, Wolfer D, Brandis D, Kretz O, Zacher C, Madani R, Grunwald I, Lipp H, Klein R, Henn F. Forebrain-specific trkB-receptor knockout mice: behaviorally more hyperactive than. Biological Psychiatry. 2003;54:972–982. doi: 10.1016/s0006-3223(03)00418-9. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. 2009;5:311–322. doi: 10.1038/nrneurol.2009.54. [DOI] [PubMed] [Google Scholar]