

Abstract
Non-CG DNA methylation modulates gene expression in the adult brain
Cytosine (C) methylation, or mC, is a modification of DNA that regulates gene expression in various contexts such as development, cancer, and imprinting. In most mammalian somatic tissues, mC arises only when C is in a dinucleotide context followed by the nucleotide guanine (G), and the vast majority of these sites are methylated (mCG). However, in the adult mammalian brain, noncanonical cytosine methylation in a non-CG context occurs at a high level [mCH; H = adenine (A), C, or thymine (T)]. The quantity of non-CG methylation is inversely correlated with gene expression, but the mechanism by which mCH controls transcription has not been clear. Two recent studies (1, 2) suggest a mechanistic link between mCH and gene expression and substantially revise our view of gene regulation by DNA methylation in the brain, which was previously formed only on the basis of studies of mCG.
Non-CG methylation has been found in human embryonic stem cells (3, 4) and more recently in the mammalian brain (5–7), and is present at a much higher level in neurons than in glia, a group of non-neuronal brain cell types. Non-CG methylation predominantly occurs at CA dinucleotides, with mCA accounting for more than 70% of non-CG methylation in the frontal cortex. Intriguingly, mCH is absent or present at very low levels in the brains of newborn mice or humans, respectively, but gradually amasses between birth and adolescence. mCH accumulation coincides temporally with the period of synaptogenesis when connections between neurons proliferate in the developing frontal cortex (7).
Gabel et al. (1) and Chen et al. (2) identify a possible mechanism whereby mCH may regulate neuronal gene expression through its recognition by methyl-CpG binding protein 2 (MeCP2), a factor whose disruption causes the neurological disorder Rett syndrome (8). Gabel et al. observed that human or mouse genes repressed by MeCP2 tend to have greater physical length (number of nucleotides) and are also associated with a greater amount of mCA. The finding was supported by an independent study showing that disruption of MeCP2 is more likely to affect long genes in distinct neuron types (9). With previous reports that MeCP2 can bind mCH (10), Gabel et al. further refined an in vitro binding assay and found that mCAC is a preferred binding target of MeCP2, with comparable or greater affinity than that for mCG. Gabel et al. and Chen et al. performed a series of correlative analyses that led to a model where mCA targets MeCP2 to its genomic binding sites in the adult mammalian brain (see the figure). Chen et al. used transgenic mice expressing a tagged version of MeCP2 and chromatin immunoprecipitation to generate a high-coverage map of MeCP2 binding sites in the mouse genome. Notably, the authors have found that the locations of mCH as well as MeCP2 binding sites are enriched in genes whose expression is both up-regulated and down-regulated when MeCP2 is disrupted, suggesting a more diverse regulatory role of MeCP2. The model in which mCA targets MeCP2 binding is supported by Gabel et al.'s study showing that the elimination of mCH accumulation, through the ablation of de novo DNA methyltransferase Dnmt3a in the nervous system, led to a derepression of long genes similar to that caused by disruption of MeCP2.
Figure. Brain methylation.
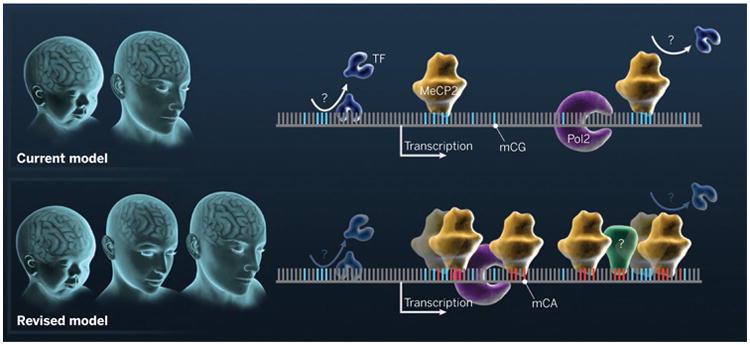
The current model focuses on mCG at discrete regulatory elements in the genome, where it may block the binding of a transcription factor (TF) or bind to MeCP2, which represses transcription. The model does not inform about epigenomic differences between newborn and adult brains. The revised model highlights the accumulation of mCH, which is initiated at birth and continues throughout adolescence, resulting in a distinct state in the adult brain with high-affinity interaction between mCA and MeCP2. The recruitment of MeCP2 (and perhaps other mCH “readers” indicated with “?”) by non-CG methylation represses the expression of genes enriched in mCA, possibly by affecting transcription elongation. Pol2, RNA polymerase 2.
The mechanism uncovered by these two studies provides new insights into the spatial scale and amplitude of the regulation by DNA methylation and the targeting of epigenetic regulatory machinery. Many previous studies focused on correlations between apparent gene expression changes and DNA methylation (primarily mCG) patterns at discrete regulatory elements such as promoters, CpG islands, or distal enhancers. By contrast, MeCP2 preferentially binds mCA deposited along gene bodies (the region of a gene that is transcribed by RNA polymerase) that can span hundreds of kilobases, which may allow fine tuning of gene expression at the level of individual genes. The currently pervasive view, based on correlations observed in large-scale epigenomic profiling analyses between local depletion of mCG and transcription factor binding, is that the initiation or targeting of epigenetic regulation likely involves sequence-specific transcription factors. Although this may well be true, the generality of this view is now challenged by the selective targeting of MeCP2 to mCA on genes with greater length, which is a spatial property of the genome and is unlikely to be associated with specific cis elements. Specifically, the recruitment of MeCP2 may be directly mediated by the epigenomic mark mCA.
Extending the profiling and perturbation of mCA and MeCP2 binding to specific neuron subtypes will be crucial to further unraveling mCA patterns that are currently measured with whole tissues (e.g., cortex) or mixed neuron populations. For example, ablating MeCP2 leads to cell type–specific gene misregulation (9). Such experiments will test whether cell type–specific MeCP2 regulation is associated with differential mCA deposition across distinct brain cell types. The contribution of mCA (versus other possible marks such as mCG) in recruiting MeCP2 remains to be quantified; this could be accomplished by profiling MeCP2 binding in specific neurons that are conditionally depleted of mCH. If mCA is indeed the primary mark for targeting MeCP2 and/or other effectors, what are mechanisms by which mCH is targeted to long genes and excluded from actively expressed neuronal genes? Although a cogent explanation for the former is unknown, for the latter it is possible that DNMT3a is excluded by the elongating RNA polymerase II or certain histone modifications associated with transcription elongation. An opposite mechanism was recently discovered by which DNA methyltransferase DNMT3b interacts with histone mark H3K36me3, which is characteristic of the transcription elongation leading to the preferential deposition of mC at actively transcribed genes in embryonic stem cells and primordial germ cells (11, 12).
It is tempting to speculate on the importance of this epigenetic mechanism in the context of normal brain function or for the understanding of neurological disorders such as Rett syndrome. mCH in the adult brain could provide a docking platform to direct the binding of a variety of factors (e.g., MeCP2) that could mediate distinct functions in the adult versus juvenile brain. Chen et al. have begun to explore this link by finding increased binding of MeCP2 at the brain-derived neurotrophic factor (Bdnf) locus in the adult relative to the juvenile mouse brain. Consistent with the enhanced MeCP2 recruitment, Bdnf expression is misregulated only in adult but not juvenile brains when MeCP2 is disrupted. The authors postulated that the gradual accumulation of mCA between birth and adolescence may provide an explanation of the late onset of Rett syndrome.
The pervasive accumulation of mCH in the adult brain as well as in pluripotent stem cells suggests the possibility that, in addition to MeCP2, other proteins not yet identified may bind to mCAC and mCAG, thereby mediating additional regulatory functions. Finding additional effectors or “readers” of mCH in tissues with abundant non-CG methylation will expand our understanding of the functions of this distinct epigenomic mark. We anticipate that further exploration of epigenomic mechanisms will lead to a more complete picture of the potential exceptionalism of gene regulation in the brain (13).
References
- 1.Gabel HW, et al. Nature. 2015 doi: 10.1038/nature14319. [DOI] [Google Scholar]
- 2.Chen L, et al. Proc Natl Acad Sci U S A. 2015;112:5509. doi: 10.1073/pnas.1505909112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramsahoye BH, et al. Proc Natl Acad Sci U S A. 2000;97:5237. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lister R, et al. Nature. 2009;462:315. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng J, et al. Am J Hum Genet. 2012;91:455. doi: 10.1016/j.ajhg.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie W, et al. Cell. 2012;148:816. doi: 10.1016/j.cell.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lister R, et al. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amir RE, et al. Nat Genet. 1999;23:185. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 9.Sugino K, et al. J Neurosci. 2014;34:12877. doi: 10.1523/JNEUROSCI.2674-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo JU, et al. Nat Neurosci. 2014;17:215. doi: 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baubec T, et al. Nature. 2015;520:243. doi: 10.1038/nature14176. [DOI] [PubMed] [Google Scholar]
- 12.Morselli M, et al. eLife. 2015;4 doi: 10.7554/eLife.06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Insel TR, Landis SC, Collins FS. Science. 2013;340:687. doi: 10.1126/science.1239276. [DOI] [PMC free article] [PubMed] [Google Scholar]