

Abstract
Background
Rice germplasm collections continue to grow in number and size around the world. Since maintaining and screening such massive resources remains challenging, it is important to establish practical methods to manage them. A core collection, by definition, refers to a subset of the entire population that preserves the majority of genetic diversity, enhancing the efficiency of germplasm utilization.
Results
Here, we report whole-genome resequencing of the 137 rice mini core collection or Korean rice core set (KRICE_CORE) that represents 25,604 rice germplasms deposited in the Korean genebank of the Rural Development Administration (RDA). We implemented the Illumina HiSeq 2000 and 2500 platform to produce short reads and then assembled those with 9.8 depths using Nipponbare as a reference. Comparisons of the sequences with the reference genome yielded more than 15 million (M) single nucleotide polymorphisms (SNPs) and 1.3 M INDELs. Phylogenetic and population analyses using 2,046,529 high-quality SNPs successfully assigned rice accessions to the relevant rice subgroups, suggesting that these SNPs capture evolutionary signatures that have accumulated in rice subpopulations. Furthermore, genome-wide association studies (GWAS) for four exemplary agronomic traits in the KRIC_CORE manifest the utility of KRICE_CORE; that is, identifying previously defined genes or novel genetic factors that potentially regulate important phenotypes.
Conclusion
This study provides strong evidence that the size of KRICE_CORE is small but contains high genetic and functional diversity across the genome. Thus, our resequencing results will be useful for future breeding, as well as functional and evolutionary studies, in the post-genomic era.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2734-y) contains supplementary material, which is available to authorized users.
Keywords: Whole-genome resequencing, Rice, Core collection, Germplasm, SNP, INDEL, GWAS
Background
Rice (Oryza sativa L.) is one of the most important staple crops in the world, providing a primary energy source for more than half of the world’s population [38]. It is closely associated with economic and political stability in many developing countries, such as Asia and Africa [38]. Moreover, the amount of land suitable for agriculture is decreasing due to a variety of factors such as rapid climate changes and industrialization, while rice-eating human populations continue to grow [38]. To meet the global nutritional and socio-economic demands, dedicated efforts towards developing superior rice varieties need to be reinforced, such as accumulating and combining beneficial alleles [14, 27, 28, 37].
Rice germplasm collections continue to grow in number and size around the world [22, 35]. The International Rice Research Institute (IRRI) holds more than 11,000 accessions in the collections [22]. Since maintaining and screening such massive resources remains challenging, a significant portion of beneficial alleles in wild relatives and landraces remain under-utilized [7, 12, 22, 28, 35]. Thus, it is important to establish efficient methods to discover and exploit these unused novel alleles to maximize rice-breeding efforts [27, 28, 35].
Developing core collections has been proposed to simplify germplasm conservation and promote their effective utilization [7, 28, 31, 44]. A core collection or set refers to a subset that represents the genetic diversity of an entire genetic resource of a species [7]. A good core set minimizes redundant entries while preserving the majority of available genetic diversity of the entire collection [12], in which 10 % of the entire collection generally constitutes the core collection [7, 12]. However, if the size of the whole collection is too large, a core collection still becomes unmanageable [7, 12]. The mini core collection (about 10 % of the core) can then be subsequently developed from the core using neutral molecular markers to achieve genetic comprehensiveness [7, 12, 39].
The advent of draft genome sequences of two rice subspecies, O. sativa ssp. japonica (Nipponbare) and ssp. indica (93–11), along with subsequent completion of high-quality indica, japonica, and aus reference genomes has accelerated rice functional genomics research [19, 28]. In addition, these reference sequences serve as frameworks for whole-genome resequencing, which is accomplished by alignments of short sequence reads produced by the next-generation sequencing (NGS) technology [18, 20, 28, 51]. Recently, applications of genome resequencing are rapidly expanding toward various rice natural resources, providing the crop research community with unprecedented genomic resolution and scale, as well as relevant functional diversity accumulated in the rice germplasm [18, 20, 28, 51]. Under these circumstances, resequencing the germplasm core collections would be beneficial to the related community.
Here, we report the whole-genome resequencing of the 137 rice mini core collection, potentially representing 25,604 rice germplasms in the Korean genebank of the Rural Development Administration (RDA). Based on the Nipponbare reference genome, our resequencing data yielded more than 15 million (M) SNPs and 1.3 M INDELs. Phylogenetic and population analysis using 2,046,529 high-quality SNPs successfully assigned rice accessions to the relevant rice subgroups, suggesting that the SNPs capture evolutionary signatures present in rice subpopulations. We conducted genome-wide association studies (GWAS) on four agriculturally important traits including ‘grain pericarp color’, ‘amylose content’, ‘protein content’, and ‘panicle number’. Among the detected association peaks, some identified previously discovered genes, indicating that KRICE_CORE can be implemented in GWAS to indentify novel alleles underlying agricultural traits. These results strongly suggest that resequencing results of KRICE_CORE are crucial for future rice breeding, as well as functional and evolutionary studies, in the post-genomic era.
Results
Sequencing of the Korean heuristic rice core set
Of the 166 rice core set selected through a heuristic approach, 137 accessions that can flower in Chungcheong province, South Korea, were selected (Additional file 1: Table S1). The Korean rice core set (KRICE_CORE) included domestically adapted weedy and landrace rice and bred lines, as well as introduced lines from Africa, Europe, and America (Additional file 2: Figure S1) [25, 52].
We sequenced KRICE_CORE using the Illumina Hiseq 2000 or 2500 platform (Table 1). The sequencing quality of the raw reads was generally high (91.6 %) (Table 1 and Additional file 3: Table S2). We next mapped short reads back to the IRGSP 1.0 rice genome (Methods). The mapping rate was generally high (95.48 % in average), varying from 89.5 to 98.86 % (Table 1 and Additional file 3: Table S2). We did not observe any significant correlations between mapping rate and sequencing depth (Additional file 2: Figure S2), indicating that the poor mapping rate was not just derived from the low sequencing depth or errors. Thus, the relative differences in mapping rates may be due to sequence divergence between some accessions of KRICE_CORE and the reference sequence. Overall, effective mapping depth was 9X on average (Table 1).
Table 1.
Summary of sequencing statistics for KRICE_CORE
| Variation range | Sequence read | Clean read rate | Deduplication read | Deduplication rate | Mapping rate | Average depth |
|---|---|---|---|---|---|---|
| Max | 59,334,970 | 95.1 | 54,800,574 | 99.4 | 98.9 | 13.9 |
| Min | 29,090,954 | 86.4 | 25,021,515 | 95.9 | 89.5 | 6.3 |
| Average | 39,753,733.9 | 91.6 | 36,006,200 | 98.8 | 95.5 | 9.0 |
Rice genome variation
We identified ~15 million (M) candidate SNPs from the 137 accessions (Table 2). About 13.5 % (2,046,529) of the SNPs did not contain missing data (hereafter referred to as high-quality SNPs [HQSNPs]; Additional file 4). We found that approximately 40.5 SNPs (5.5 by HQSNPs) were arrayed per kb across the KRICE_CORE genomes (Table 2 and Fig. 1a). We also found that 1.3 M INDELs existed in KRICE_CORE. Among those, 19.3 % were HQINDELs, showing an average density of INDEL per kb of 5.5 (0.7 for HQINDEL, respectively) (Table 2 and Fig. 1a).
Table 2.
Summary of chromosomal SNP and INDEL distribution for KRICE_CORE
| Chromosome | SNP | INDEL | ||||||
|---|---|---|---|---|---|---|---|---|
| Total | High quality | Total | High quality | |||||
| Count | Densitya | Count | Density | Count | Density | Count | Density | |
| 1 | 1,492,529 | 34.5 | 245,749 | 5.7 | 157,701 | 3.6 | 34,662 | 0.8 |
| 2 | 1,194,215 | 33.2 | 219,868 | 6.1 | 123,316 | 3.4 | 28,870 | 0.8 |
| 3 | 1,117,000 | 30.7 | 227,031 | 6.2 | 116,347 | 3.2 | 30,925 | 0.8 |
| 4 | 1,482,884 | 41.8 | 151,464 | 4.3 | 118,490 | 3.3 | 19,101 | 0.5 |
| 5 | 1,136,915 | 37.9 | 190,680 | 6.4 | 98,439 | 3.3 | 23,019 | 0.8 |
| 6 | 1,279,651 | 41.0 | 175,920 | 5.6 | 117,347 | 3.8 | 22,733 | 0.7 |
| 7 | 1,268,980 | 42.7 | 152,153 | 5.1 | 112,520 | 3.8 | 20,766 | 0.7 |
| 8 | 1,323,731 | 46.5 | 148,873 | 5.2 | 113,720 | 4.0 | 18,831 | 0.7 |
| 9 | 969,987 | 42.2 | 128,492 | 5.6 | 85,643 | 3.7 | 16,566 | 0.7 |
| 10 | 1,081,145 | 46.6 | 141,153 | 6.1 | 91,306 | 3.9 | 17,727 | 0.8 |
| 11 | 1,425,491 | 49.1 | 148,897 | 5.1 | 128,449 | 4.4 | 17,902 | 0.6 |
| 12 | 1,352,277 | 49.1 | 116,249 | 4.2 | 115,237 | 4.2 | 15,040 | 0.5 |
| Total | 15,124,805 | 40.5 | 2,046,529 | 5.5 | 1,378,515 | 3.7 | 266,142 | 0.7 |
aDensity = SNP/INDEL count/length of the chromosome
Fig. 1.
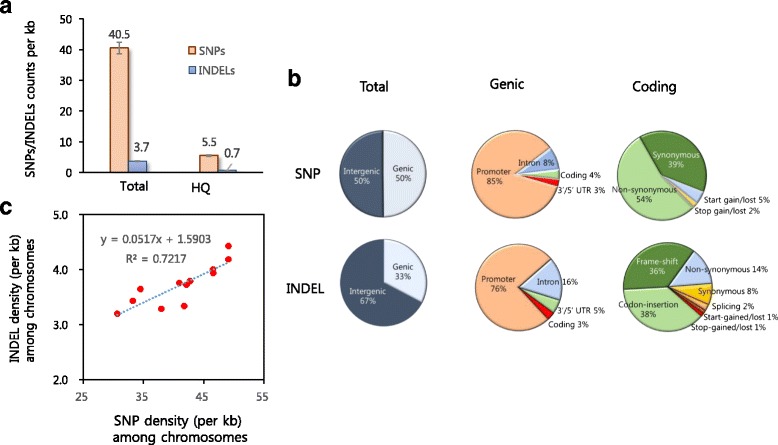
SNP and INDEL frequency in the KRICE_CORE population. a Comparison of the mean density of SNPs and Indels in KRICE_CORE. b SNP and INDEL distribution across various genome regions. The promoter of the genic region refers to the region 2 kb upstream of the transcription start site. c Correlation between SNPs and INDEL occurrences across KRICE chromosomes
About 50 % of SNPs were located in genic and intergenic regions, respectively (Fig. 1b and Additional file 5: Table S3). The majority of genic SNPs were distributed in the promoter region (85 %), followed by intronic (8 %), coding (4 %), 5′/3′UTR (3 %), and splicing (0.02 %) regions (Fig. 1b and Additional file 5: Table S3). In the coding region, we found that SNPs causing non-synonymous amino acid changes (non-synonymous SNPs) were enriched (54 %) (Fig. 1b and Additional file 5: Table S3). The ratio of nonsynonymous to synonymous SNPs was 1.38 (54 % vs. 39 % of coding SNPs), which was similar to previous reports [1, 2].
A lower abundance of INDELs is located in genic regions (33 %) (Fig. 1b and Additional file 5: Table S4). In addition, codon-insertion and frame-shifts are the most abundant classes of INDELs (38 and 36 %, respectively) in the coding region (Fig. 1b and Additional file 5: Table S4).
The density of SNPs and INDELs varied among rice chromosomes, ranging from 30.7 to 49.1 (4.2–6.4 with HQSNPs) and 3.2 to 4.2 (0.5–0.8 with HQINDEL), respectively (Table 2 and Additional file 5: Tables S3 and S4). We also found that the densities of SNPs and INDELs were positively correlated in chromosomes (Fig. 1c). To determine whether this correlation also occurred across local genomic regions, we investigated the correlation between SNP and INDEL densities within 50-kb windows across rice genomes (Fig. 2a). We found that SNPs are clustered in the rice genome (Fig. 2a), suggesting that SNPs were generated non-randomly across the rice genome [2, 15, 33, 47]. Furthermore, we observed a significant positive correlation between SNPs and INDELs across the rice genome, indicaing that INDELs are also clustered (Fig. 2b). Thus, the shared SNP and INDEL clustering patterns may be affected by a genome-wide set of regions that either commonly promote or restrict the genesis of SNPs and INDELs [2, 6].
Fig. 2.
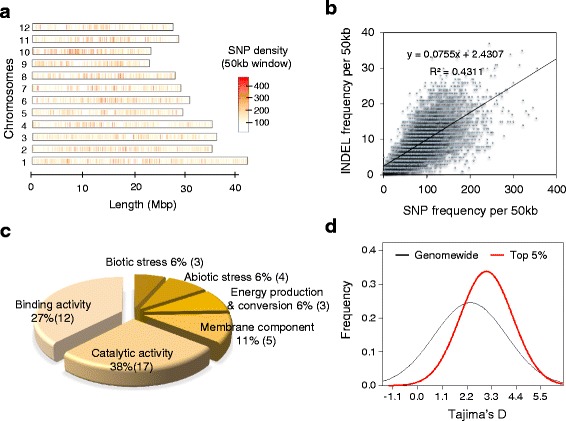
Genome-wide distribution of SNPs and INDELs of KRICE_CORE. a SNP density of the 50-kb window across the KRICE_CORE genome. b Correlation between SNP and INDEL occurrence across KRICE_CORE genome. c Functional category of genes in SNP/INDEL-enriched regions. d Distribution of Tajima’s D values of the genome wide and top 5 % SNP enriched regions
We next investigated functional categories in the regions where SNP and INDEL frequencies were commonly high. Therefore, we performed gene enrichment tests for the top 5 % of regions. Among the genes functionally annotated, catalytic and binding activity were mostly enriched, followed by abiotic/biotic stress responses, energy production & conversion, and protein transport (Fig. 2c and Additional file 5: Table S5), which are important to maintain rice physiology. Considering that the mean Tajima’s D value for the windows of the top 5 % region was significantly higher than that at the genome-wide level, the accumulated SNPs and INDELs in these genes may facilitate creation of functional diversity (Fig. 2d).
Population structure and genetic diversity of KRICE_CORE
Using 2,046,529 HQSNPs, we performed population genetic studies to explore the genetic structures of KRICE_CORE. First, we constructed a neighbor-joining tree (Fig. 3a). To gain legitimate evolutionary insights from the resultant phylogenetic subdivisions, we incorporated the previously defined rice accessions into the tree analysis (Additional file 2: Figure S3) (Methods) [51]. We found that our tree analysis separated into five major subgroups, which partitioned into three major groups corresponding to japonica (81 accessions, 59 %), indica (hereafter IND, 43, 31 %) and aus (AUS, 6 %), with a further subdivision of japonica into temperate (TEJ, 62, 45 %) and tropical (TRJ, 19, 14 %) rice groups (Fig. 3a, b and Additional file 2: Figure S3). However, KRICE_CORE does not reflect Group V since only two accessions belong to the sub group (Fig. 3a, b and Additional file 2: Figure S5).
Fig. 3.
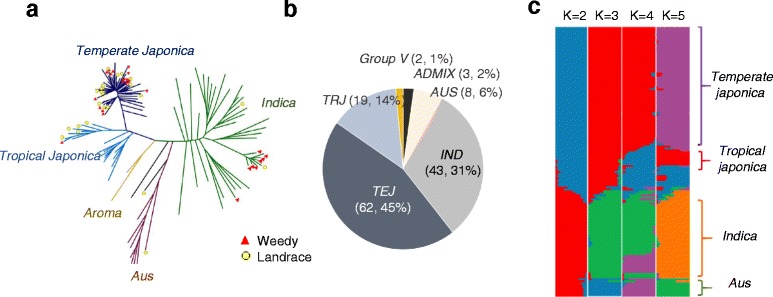
Population structure of KRICE_CORE. a Neighbor-joining analysis among KRICE_CORE using 2,046,529 HQSNPs. Landraces and weedy accessions are denoted as red triangles and yellow circles in the tree, respectively. b Pie graph designating the proportion of each subgroup. c Population structure analysis using FRAFFE. Each color represents one population. Each accession is represented by a horizontal bar, and the size of each colored segment in each horizontal bar represents the proportion contributed by ancestral population. K value represents the number of assumed clusters or populations
We found that Korean landrace and weedy accessions are mostly present in TEJ groups (Fig. 3a). Note that some are assigned to different subgroups, such as IND, AUS and TRJ (Fig. 3a). This led to the hypothesis that independent domestications of TRJ type together with IND (and others) have occurred in the Korean peninsula. However, these results may be derived from the outcrossing between weedy rice and IND and TEJ type rice varieties (Tongil type), which are widely cultivated in Korea. Since we have limited evidence for indica landrace rice, further studies are required to support this hypothesis.
We further investigated plausible population structures using FRAPPE, which estimates individual ancestry and admixture proportions assuming that K populations exist based on a maximum likelihood method (Fig. 3c). We analyzed the data by increasing K (the number of population) from 2 to 5 (Fig. 3c). For K = 2, we observed a division between O. rufipogon/japonica and O. nibara/others, including IND type. When K = 3, AUS subgroups were clearly separated from the IND group. When, K = 4, TRJ and TEJ were separated. When K = 5, a new subgroup including group V emerged from TRJ (Fig. 3c). These results supported the previous hypothesis that the two cultivated rice subspecies have been domesticated independently, forming two ancient populations; one population that gave rise to indica and the other to japonica, respectively. Since we independently confirmed that O. rufipogon and O. nivara were assigned to japonica and indica subgroups, we further supported the hypothesis that one population that gave rise to indica may have evolved from an ancestral population closely related to O. nivara, while the other that gave rise to japonica may have originated from O. rufipogon [51].
We estimated the sequence diversity of KRICE_CORE (Fig. 4 and Additional file 2: Figure S4). Reduction of diversity (ROD) was calculated from the sequence diversity (π) of KRICE_CORE and wild rice, O. rufipogon, across the entire rice genome (Fig. 4 and Additional file 2: Figure S4). We found that the ROD value of KRICE_CORE was highly dynamic across the rice genome, ranging from −30 to 1 (Fig. 4a and Additional file 2: Figure S4). Since HQSNPs were used in the analysis, it is possible that the majority of ROD values were correctly estimated. However, some RODs may be exaggerated, especially when π of wild rice was close to zero but those of others were higher.
Fig. 4.
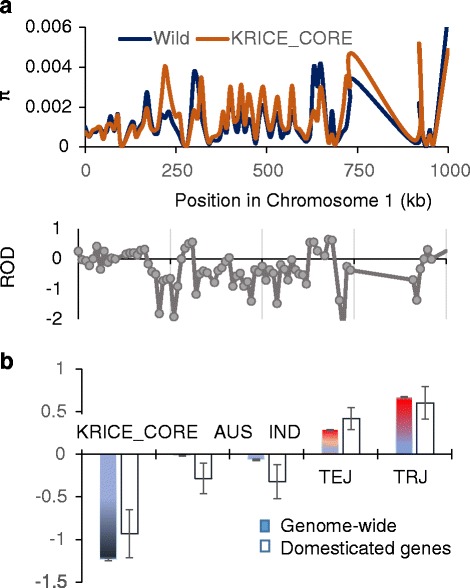
Genetic diversity of KRICE_CORE. a Nucleotide diversity (π) of wild rice (O. rufipogan) vs KRICE_CORE (upper) and the resulting ROD value (Methods). b Mean RODs among the subgroups in genome-wide or domesticated regions. Error bar indicates standard error of the mean (SEM). Sliding window analyses of π or ROD are shown for chromosome 1 with a 10-kb window
We found that KRICE_CORE potentially retains a high diversity level (Fig. 4a, b, Additional file 2: Figure S4). Since TRJ is more closely related to O. rufipogon than TEJ, the average ROD value for TRJ of KRICE_CORE is much higher than that of TEJ (0.67 vs 0.29, P < 0.001 [one-way ANOVA]). However, incorporation of the divergent AUS and IND to KRICE_CORE, which decreased the ROD values greatly, significantly increased the overall diversity, as measured by ROD values (Fig. 4b). We also investigated ROD for regions that contain previously reported domesticated genes (Fig. 4b and Additional file 2: Figure S5). Considering that the amount of genetic diversity for the selected regions were comparable both at the genome-wide level and within each subpopulation (Fig. 4b), KRICE_CORE accommodates abundant functional diversities shaped from diverse external environments. These results will be useful for future rice breeding and fundamental studies in the post-genomic era to address current climate changes. Overall, these genetic and population structure results suggest that the size of KRIC_CORE is small but effectively retains genetic comprehensiveness.
Genome-wide association studies (GWAS)
We performed GWAS to determine whether we could exploit the natural diversity present in the KRICE_CORE to detect agriculturally useful alleles via association genetics (Fig. 5 and Additional file 2: Figure S6). We chose four agriculturally important phenotypes (‘pericarp color’, ‘amylose content’, ‘number of panicle per plant’, and ‘rice seed protein content’), which are representative of the qualitative or quantitative traits in KRICE_CORE (Fig. 5 and Additional file 2: Figure S6). We observed a wide range of phenotypic variation, spanning a 1.7- to 8.6-fold difference (Additional file 2: Figure S7). To determine whether the significant variabilities have a genetic basis, we employed a basic case/contro association test (for qualitative traits) using PLINK or a compressed mixed linear model (for quantitative traits) using GAPIT (Fig. 5 and Additional file 2: Figure S6) (Methods).
Fig. 5.
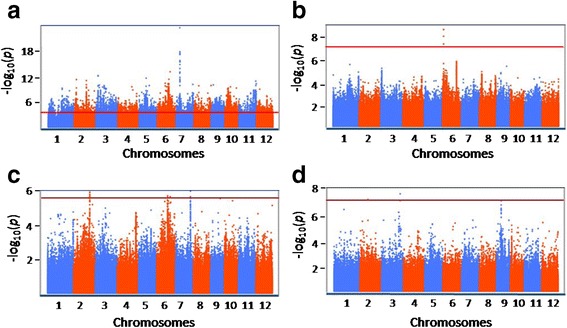
Genome-wide association studies of ‘pericarp color’ (a), ‘amylose content’ (b), ‘rice seed protein content’ (c), and ‘number of panicles per plant’ (d). Manhattan plots of the linear (for a) or mixed linear model (for b–d) are shown from negative log10-transformed P-values, which are plotted against the positions on each of the twelve chromosomes. A red horizontal line indicates the genome-wide significance threshold
We found that GWAS signals associated with pericarp color and grain amylose percentage pinpointed the previously reported Rc locus at chromosome (ch) 7 and Waxy locus (at ch 6), respectively (Table 3, Fig. 5a, b, and Additional file 2: Figure S6). For the red pericarp color, the most significantly associated polymorphism (P = 3.05e-19) that explained ~46 % of the phenotypic variance was the 14 bp INDEL at 6,068,072 bp, which is located in the seventh exon of Rc (Table 3, Fig. 5a, and Additional file 2: Figure S6). For amylose content, one of the most significantly associated polymorphisms was located at the 5′ UTR region of the Wx locus (Os06g013300, at 1,765,761 bp of ch 6), explaining about 52 % of the phenotypic variation (Table 3, Fig. 5b, and Additional file 2: Figure S6).
Table 3.
Genome-wide significant association signals of agronomic traits using the linear and compressed MLM
| Traits | Ch | Position | Major allele (Count) | Minor allele (Count) | MAFa | R2 | P value | Known Loci (Reference) |
|---|---|---|---|---|---|---|---|---|
| Pericarp color | 7 | 6068072 | A(95) | G(41) | 0.30 | 0.465 | 3.05E-19 | Rc(46) |
| 6 | 21304669 | T(93) | C(43) | 0.32 | 0.286 | 4.87E-11 | ||
| 3 | 3640060 | G(116) | A(13) | 0.12 | 0.268 | 2.48E-10 | ||
| 5 | 11479616 | C(108) | T(26) | 0.20 | 0.255 | 7.57E-10 | ||
| Amylose content | 6 | 1765761 | G(73) | T(61) | 0.46 | 0.555 | 5.55E-09 | Wx(21) |
| Protein content | 7 | 24810573 | A(77) | G(59) | 0.43 | 0.348 | 1.14E-06 | |
| 2 | 26791708 | C(110) | T(25) | 0.19 | 0.347 | 1.27E-06 | ||
| 6 | 18133739 | G(93) | A(42) | 0.31 | 0.339 | 2.15E-06 | ||
| Panicle number | 3 | 30877559 | C(128) | T(9) | 0.07 | 0.404 | 3.13E-08 | |
| 9 | 7760141 | C(128) | T(() | 0.07 | 0.386 | 1.09E-07 |
aMinor allele frequency
We observed that the GWAS mapping precision varied between Rc and Wx regions. For pericarp color, the significantly associated SNPs were scattered around the 287-kb region of ch7 (Fig. 6a), but amylose content-related SNPs were dispersed within a 33-kb region (Fig. 6b). To determine whether the mapping accuracy was compounded by different evolutionary processes, such as specific domestication histories, we investigated the genetic diversity level and linkage disequilibrium (LD) structure across the 400-kb region around the association peaks (Fig. 6b–d; Methods). We found that the ROD score of the 400-kb region that included Rc (Rc region) was significantly higher than the Wx including the 400-kb region (Wx region) (mean ROD −0.68 (Rc region) vs-1.8 (Wx region), P < 0.001; one-way ANOVA), indicative of a significant reduction in the genetic diversity in the Rc region (Fig. 6a-c). This substantial genetic reduction of the Rc region in KRICE_CORE may be derived from the strong selection in favor of the 14-bp deletion since the domestication of the white grain rice. We also investigated LD decay across Rc and Wx regions (Fig. 6a–c). We found that the average LD decay of KRICE_CORE was about 85 kb. The LD decay of the Wx region was comparable, but the local LD pattern of the Rc region was atypical, indicating that the LD decay does not neutrally occur against the physical distance (Fig. 6d). To further examine the aberrant LD decay pattern over the Rc region, we constructed LD maps of Rc/Wx genomic regions (Fig. 6e–f). We found that obvious block-like LD structures exist in the Rc region, indicating that more powerful selective pressure might have acted on the Rc region (Fig. 6e, f). In addition, this extended LD structure may cause spurious associations in the Rc region, complicating prediction of the causative gene through GWAS mapping.
Fig. 6.
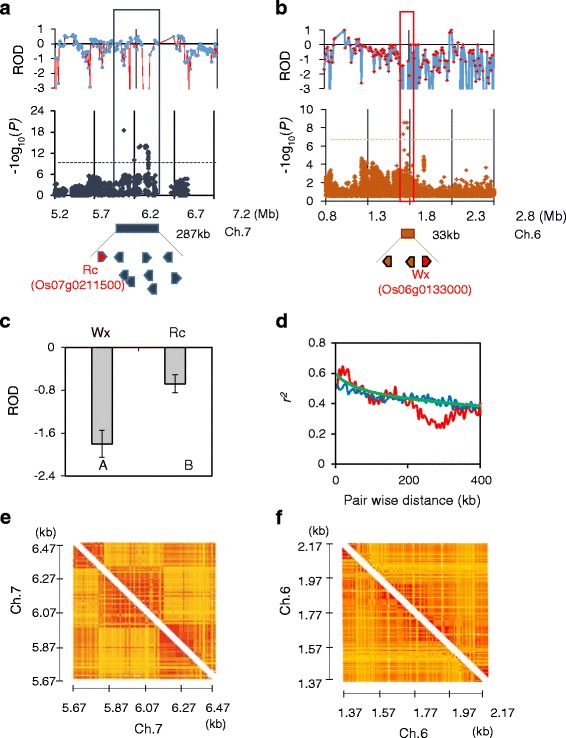
Regions of the associated signals near the Rc (a) and Wx (b) regions. The top of each panel shows the ROD for a 1-Mb window around the peak SNPs. Negative log10P-values for each SNP from the linear or compressed mixed linear model are plotted. Blue or dark orange dashed horizontal lines indicate the genome-wide significance cutoff for the Rc and Wx regions, respectively. The bottom of each panel within the blue or red box denotes the range of SNPs over the cutoff. c Mean ROD values for 400 kb around the Rc and Wx regions. d LD decay plots of the regions compared to genome-wide LD decay. e and f LD blocks of 400 kb on each side of the association peak for the Rc (e) and Wx (f) regions
Several significant association signals were found for seed protein contents and panicle number (Table 3, Fig. 5, and Additional file 2: Figure S6). Although we could not identify any strong candidate genes for those traits, the novel SNPs identified here can be implemented in related breeding studies.
Discussion
To address upcoming challenges for food production, the efficient use of rice germplasms is critical [13, 28]. These rice germplasms, adapted from diverse ecosystems, are valuable for rice breeders/researchers, potentially providing a broad array of useful alleles that enrich gene pools of current cultivated rice varieties [17, 19, 28]. Although ex situ conservation efforts are important for preserving diverse rice genetic resources, identifying novel and favorable genetic variants from the vast genebank collection remains challenging, requiring extensive screening processes [12, 14, 22, 35]. Therefore, the rice core collection is a powerful approach to accelerating utilization of exotic germplasms of the entire population [7, 31, 44]. In addition, the application of whole-genome resequencing technology would establish a potent platform for rapid forward genetic studies and genome-assisted breeding, which would facilitate discovery and exploitation of useful alleles [19, 28, 35].
Genomic variations and population structure of KRICE_CORE
Here, we performed genome-wide resequencing of 137 KRICE_CORE populations originating from the 25,604 rice germplasms in the Korean genebank [52]. We identified ~15 million (M) candidate SNPs from the 137 KRICE_CORE (Table 2); 13.5 % (2,046,529) were HQSNPs at a 9x depth. Our neighbor-joining tree analysis using HQSNPs clearly showed that KRICE_CORE comprises the major rice subgroups, including IND together with TEJ, TRJ, and Aus (Fig. 3 and Additional file 2: Figure S3). Regarding the 3,000 rice genome analyses, approximately 75 % of the SNPs were covered by our study, although only 4 % of rice accessions were sequenced, suggesting that KRICE-CORE represents the diversity of the whole germplasm collection with relatively few accession numbers. These data indicate that the implementation of heuristic approaches using PowerCore software effectively reduced core entry but retained maximum diversity [25].
We found that many Korean landrace and weedy accessions were assigned to distinct ecotype groups (Fig. 3 and Additional file 2: Figure S3). Given that TEJ are dominant in the Korean peninsula, it is possible that TRJ and IND rice may have been introduced or domesticated historically. Recently, South Korea has faced gradual changes in many climate parameters, including annual temperature, rainfall amount, and precipitation [9]. The most significant climate change reported is a massive increase in the range of temperature fluctuations throughout the four seasons. This leads to heavier precipitation during the summer together with elevated temperature [24]. As a result, the Korean Peninsula is predicted to become a subtropical region [23]. To rapidly adapt to on-going climate change, these IND and TRJ weedy and landrace accessions acclimated in Korea would be valuable genetic resources. Still, many interesting evolutionary questions remain for these landrace and weedy lines, including their origins and transferring/fixation processes [42]. Addressing these questions could increase our understanding of the relevant domestication mechanisms, providing information for future improvements of Korean rice cultivars [42].
In KRICE_CORE, we observed a clear separation between japonica and indica type at K = 2. As the K value increased, aus diverged from the indica population. Tropical and temperate japonica types were separated from the japonica population (Fig. 3c). Since O. rufipogon and O. nivara are assigned to the japonica and indica at K = 2 (Fig. 3c and Additional file 2: Figure S3), respectively, we support the hypothesis that two cultivated rice subspecies, indica and japonica, may have originated from two ancient subpopulations [43, 51]; one population that gave rise to indica may have evolved from an ancestral population from O. nivara and the other for japonica may have originated from O. rufipogon [43, 51]. However, further studies are required to determine which model explains the actual domestication processes.
Functional significance of SNPs and INDELs
It is expected that about 40 SNPs and 5.5 INDELs are arrayed per kb across the KRICE_CORE genome (Fig. 1a). However, we found that SNPs and INDELs are not evenly distributed. Rather, significant portions of SNPs and INDELs are clustered in the KRICE_CORE genome (Figs. 1c and 2a). Interestingly, the clustering patterns of those two genetic variants are strongly correlated (Fig. 2b), suggesting that SNPs and INDELs may arise non-randomly and such genesis mechanisms are highly connected [2, 6, 48]. These results are consistent with observations in mammalian and avian genomes [6, 48], indicative of the presence of mutation hot spots or cold spots across the genome [3]. The local recombination variability is reported as one of the key determinants for the mutation hot spot, driving sequence evolutions and genetic diversities [3]. Although high mutation rates may be an important driver underlying the co-clustering pattern, other evolutionary forces could create the non-randomness [2]; among these, natural selection can modulate local variability’s along a chromosome from responding to the ambient environments [11, 49]; balancing selection tends to create regions of increased variability [2, 8, 50], while purifying and directional selection tend to reduce variability [40]. Thus, the effect of mutagenic hot spots in population data may be obscured by a complex interplay of these factors [3]. Varela and Amos (2010) showed that INDEL locations are conserved in the same location between humans and chimpanzees, but SNP density appears highly variable; clusters found in chimpanzees are often not found at the same site in humans, indicating that a species dependent regional context or signature may regulate the surrounding SNP density following the genesis of INDEL [16, 30, 41, 48, 50]. Further studies should explore the major determinants causing SNP/INDEL clustering and its evolutionary consequences in plant/crop species.
We found that many genes in important biological processes, including, catalytic and binding activity, abiotic/biotic stress responses, and energy production are distributed in the high SNP regions (Fig. 2c and Additional file 5: Table S5). Given that the mean Tajima’s D value for the windows of the top 5 % genic SNP region is significantly higher than that at the genome-wide level (3.03 vs 2.36 in mean Tajima, p < 0.01 by bootstrapping method), it is believed that accumulated SNPs and INDELs in these genes are more or less likely to be beneficial in creating multiple alleles, potentially resulting in functional diversity. Thus, resequencing results of KRICE_CORE would increase our understanding of the molecular basis of the functional differences, especially related to important agricultural traits [1].
Genome-wide association studies (GWAS)
GWAS has been implemented to efficiently identify candidate genes related to various useful agricultural traits in many crop species, including rice [19, 29]. Given that significant associations between genetic variations and phenotypic diversity does not require prior knowledge, GWAS with high genome coverage of SNP markers provides a genomics platform to dissect previously unknown adaptive genetic variation accumulated in plant germplasm resources over time [5]. After identifying candidate genes, GWAS allows for informed choice of parents for QTL analysis based on the haplotype information, along with suggesting targets for the following mutagenesis and transgenic studies [26]. Here, we conducted proof-of-concept GWAS analysis using four agriculturally important phenotypes (Fig. 5); the ‘pericarp color’ represents the qualitative trait of KRICE_CORE, while the others are for the quantitative traits.
We found that GWAS signals associated with pericarp color and grain amylose percentage pinpointed the previously reported Red pericarp (Rc) and Waxy (Wx) loci, respectively (Fig. 5). For the red pericarp color, the most significantly associated polymorphism (P = 3.05e-19) is the 14 bp INDEL, which is located in the seventh exon of Rc (Fig. 6a and Table 3). Sweeney et al. (2006) reported that the recessive rc allele by the 14-bp deletion nullifies the regulatory function in the proanthocyanidin synthesis pathway, preventing development of the pigmented pericarp layer [45, 46]. We found that the majority (>94 %, 80 out of 85) of white pericarp KRICE_CORE accessions contain a 14-bp deletion in exon 7 of the gene. The exceptional five white pericarp accessions that do not have the deletion may have been generated through rc-independent mechanisms. For amylose content, one of the most significantly associated polymorphisms is located in the 5′ UTR region of the Wx locus (Fig. 6b). Very close to the association peak, we identified a splice donor site in intron 2 of Wx. Previously, it was reported that Wxb, the GtoT point mutation at the site, is associated with the absence of amylose-characterizing glutinous rice varieties, suggesting that natural variations to the site create functional diversities for the amylose content [21]. Considering that the association peaks are found dominantly at Rc and Wx in KRICE_CORE and others [20], Rc and Wx largely underpin the red pericarp color and amylose content, respectively [20, 45, 46]. To improve the power to recover meaningful but minor associations (especially for amylose content), the sample size should be increased using diverse germplasms [4, 26, 34].
We observed that the GWAS mapping precision varies between Rc and Wx regions. For the pericarp color, the significantly associated SNPs are scattered around the 287-kb region of ch7, but amylose content-related SNPs are dispersed within the 33-kb region (Fig. 6a, b). We concluded that the much stronger LD pattern of Rc may complicate accurate prediction of the causative gene, as observed in other rice GWAS (Fig. 6e, f) [20, 45, 46]. Hence, it is believed that local LD estimations may be required when candidate genes are considered following GWAS. Besides, several significant novel associations were found for seed protein contents and panicle numbers. These results clearly demonstrate that GWAS implemented in KRICE_CORE offer a viable approach for dissecting the causal genetic variants of qualitative and quantitative phenotypes.
Methods
Genomic DNA library construction and sequencing
Quality control (QC) for prepared genomic DNA was performed; A260/280 > 1.6 and A260/230 > 1.6 for UV absorbance using Nanodrop (Tecan) and ≥5 ug fluorescence concentration using the Quant-iT BR assay kit (Invitrogen). DNA was run on 0.7 % agarose gels to assess degradation.
Genomic DNA was fragmented into approximately 100–300-bp insert sizes by sonication. Each DNA fragment was end-repaired using the Paired-End DNA Sample Prep Kit (Illumina; San Diego, CA, USA), followed by the addition of a 3′-A overhang and ligation of the adapters. Size-selection for DNA fragments of 200–300 bp was performed using a gel extraction kit (Qiagen). The constructed DNA samples were quantified using Quant-iT™ dsDNA High Sensitivity Assay Kit (Invitrogen; Carlsbad, CA, USA) on an Agilent 2100 Bioanalyzer (Agilent Technologies; Santa Clara, CA, USA). After qPCR validation, the resulting libraries were subjected to paired-end sequencing with a 100-bp read length using the Illumina HiSeq 2500 platform (Illumina). Raw image files were processed by Illumina Real-Time Analysis (RTA) for image analysis and base calling.
Bioinformatics analysis
Raw sequences were first processed to obtain an average quality score (QS) per read ≤20 by trimming 3′-end of reads using SICKLE (https://github.com/najoshi/sickle). High-quality reads were aligned to the rice reference genome (IRGSP Build 5) using the Burrows-Wheeler Aligner (BWA) (version 0.7.5a) with default parameters [32]. Reads not meeting BWA quality criteria or not matching the reference genome were removed. PCR duplicate reads were removed using PICARD (version 1.88) (http://broadinstitute.github.io/picard/). Using the Genome Analysis Toolkit (GATK) (version 2.3.9 Lite) [36], regional realignment and quality score recalibration were performed, after which SNPs and InDels were identified with ≥3X read depth coverage. The resulting variants were annotated according to genomic position and overlap with current genome annotation using snpEff [10].
Plant materials
Of the 166 accessions in the rice mini core set, selected from a composite population of 4,046 accessions using a heuristic approach (Additional file 2: Figure S1), 137 accessions that can flower in Chungcheong province, South Korea, were included in this study [25, 52].
Population structure inference
To confirm the subgroups of our 137 accessions, we combined our 137 sequence and the published 50 sequence data, which have clear subgroup information, and then performed a phylogenetic study using the PHYLIP 3.68 (http://evolution.genetics.washington.edu/ phylip.html) with 100 replicates for a bootstrap confidence analysis. FigTree v1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/) was used to plot the constructed tree. We used the same method to construct our 137 phylogenetic tree. MEGA6 [5] was used to present the phylogenetic tree.
We further used the maximum-likelihood-based software FRAPPE to investigate the population structure of the 137 accessions. We ran 10,000 iterations and the number of clusters (K) was considered from 2 to 5.
Estimation of population parameters
Linkage disequilibrium parameters (r2) for estimating the degree of LD between pair-wise SNPs (58,008 high quality SNPs with physical distance >5 kb) was calculated using the software TASSEL 5.0 (http://www.maizegenetics.net/#!tassel/c17q9) with 1,000 permutations. The LD decay rate was measured as the chromosomal distance at which the average pairwise correlation coefficient (r2) decreased to half of its maximum value. Nucleotide diversity (π) and Tajima’s D were calculated by Vcftools [6] with a 50-kb slide window. The nucleotide diversity (π) is defined as the average number of nucleotide differences per site between any two DNA sequences chosen randomly from the sample population:
where xi and xj are the respective frequencies of the Ith and jth sequences, πij is the number of nucleotide differences per nucleotide site between the ith and jth sequences, and n is the number of sequences in the sample. The summation is taken over all distinct pairs i,j, without repetition.
Tajima’s D test followed the method developed by Tajima (1989). At equilibrium between genetic drift and selectively neutral mutation, the expected value of D is close to zero.
To measure genetic diversity, we used the reduction of diversity (ROD) as:
where πcul and πwild are the values of π for the cultivated and wild varieties, respectively.
Genome-wide association analysis
For the qualitative trait (pericarp color), we performed GWAS using PLINK (http://pngu.mgh.harvard.edu/~purcell/plink/) software. For the other three quantitative traits (amylose content, protein content and panicle number), we performed GWAS using GAPIT [7] with a mixed linear model, where the PCA matrix and relative kinship matrix were included as fixed and random effects.
Conclusion
This study provides strong evidence that the size of KRICE_CORE is small but contains high genetic and functional diversity across the genome. The genome information through the whole-genome resequencing technology would establish a potent platform for forward/reverse genetic studies as well as genomeassisted breeding, facilitating future discovery and exploitation of useful alleles from rice germplasms.
Acknowledgements
We thank Kyung-Ho Ma and Jong-Wook Chung, RDA, Korea, for preparing materials, technical supports and useful discussions.
Funding
This work is supported by BioGreen 21 Program (No. PJ01116101), Rural Development Administration, Republic of Korea.
We also thank the BioGreen 21 Program (No. PJ01164302), Rural Development Administration, Republic of Korea for project funding.
Availability of data and materials
The data set supporting the results of this article is included within the Supplementary files (Additional files 4 and 6). High-quality SNPs (HQSNPs) in KRICE_CORE from chromosomes 1–12. are included in the zip file chr1_6_KRICE_CORE_HQSNP.zip (TAB delimited txt file format) and zip file chr7_12_KRICE_CORE_HQSNP.zip (TAB delimited txt file format).
The English in this document has been checked by at least two professional editors, both native speakers of English. For a certificate, please see: http://www.textcheck.com/certificate/humtBq.
Authors’ contributions
Conceived and designed the experiments: TK, QH, and YP. Analyzed the data: TK, QH, KK, and YP. Contributed reagents/materials/analysis tools: MY, WR, FPL, WT, JY, EH, WHO, BC, BY, S-JK, S-WK, YC, CL and BP. Wrote the paper: TK, QH, and YP. All authors have read and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable
Ethics approval and consent to participate
Not applicable
Abbreviation
- GWAS
Genome-wide association studies
- HQSNPs
High-quality SNPs
- IND
Indica
- LD
Linkage disequilibrium
- M
Million
- NGS
Next-generation sequencing
- non-synonymous SNPs
Non-synonymous amino acid changes
- Rc
Red pericarp
- RDA
Rural Development Administration
- ROD
Reduction of diversity
- SNPs
Single nucleotide polymorphisms
- TEJ
Temperate japonica
- TRJ
Tropical japonica
- Wx
Waxy
Additional files
List of KRICE_CORE accessions used for whole-genome resequencing. (XLSX 15 kb)
Geographical (a,b) and methodological (c) origins of KRICE_CORE. Figure S2. Scatter plot between mapping rate and sequencing depth of KRICE_CORE. Figure S3. Neighbor-joining tree analysis of previously defined rice accessions. Figure S4. Sliding-window analysis of reduction of diversity (ROD) across KRICE_CORE genome. Figure S5. Genomic positions of previously identified domesticated genes. Figure S6. Quantile-quantile plot of the genome-wide association studies of ‘pericarp color’ (a), ‘amylose content’ (b), ‘rice seed protein content’ (c), and ‘number of panicles per plant’ (d). Figure S7. Phenotypic variation of the target traits for the GWAS study. (PPTX 1389 kb)
Sequencing and mapping statistics for individual KRICE_CORE accessions. (XLSX 36 kb)
High-quality SNPs (HQSNPs) in KRICE_CORE from chromosomes 1–6. Individual HQSNPs (minor allele frequency >0.05) from chromosomes 1–6 of 137 KRICE_CORE accessions are included in the zip file chr1_6_KRICE_CORE_HQSNP.zip (TAB delimited txt file format). (ZIP 24253 kb)
SNP distributions across various KRICE_CORE genomic regions. Table S4. INDEL distributions across various KRICE_CORE genomic regions. Table S5. Functional categories enriched in the high SNP/INDEL region across the KRICE_CORE genomes. (PPTX 54 kb)
HQSNPs of KRICE_CORE from chromosomes 7–12. Individual HQSNPs (minor allele frequency >0.05) from chromosomes 7–12 of 137 KRICE_CORE accessions are included in the zip file chr7_12_KRICE_CORE_HQSNP.zip (TAB delimited txt file format). (ZIP 17471 kb)
Reference
- 1.Alonso-Blanco C, Aarts MG, Bentsink L, Keurentjes JJ, Reymond M, Vreugdenhil D, et al. What has natural variation taught us about plant development, physiology, and adaptation? Plant Cell. 2009;21:1877–1896. doi: 10.1105/tpc.109.068114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amos W. Even small SNP clusters are non-randomly distributed: is this evidence of mutational non-independence? Proc Biol Sci. 2010;277:1443–1449. doi: 10.1098/rspb.2009.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arbeithuber B, Betancourt AJ, Ebner T, Tiemann-Boege I. Crossovers are associated with mutation and biased gene conversion at recombination hotspots. Proc Natl Acad Sci U S A. 2015;112:2109–2114. doi: 10.1073/pnas.1416622112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brachi B, Morris GP, Borevitz JO. Genome-wide association studies in plants: the missing heritability is in the field. Genome Biol. 2011;12:232. doi: 10.1186/gb-2011-12-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brandstrom M, Ellegren H. The genomic landscape of short insertion and deletion polymorphisms in the chicken (Gallus gallus) Genome: a high frequency of deletions in tandem duplicates. Genetics. 2007;176:1691–1701. doi: 10.1534/genetics.107.070805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown AHD. Core Collections: a practical approach to genetic resources management. Genome. 1984;31:818–824. doi: 10.1139/g89-144. [DOI] [Google Scholar]
- 8.Bubb KL, Bovee D, Buckley D, Haugen E, Kibukawa M, Paddock M, et al. Scan of human genome reveals no new Loci under ancient balancing selection. Genetics. 2006;173:2165–2177. doi: 10.1534/genetics.106.055715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung Y, Yoon M, Kim H. On Climate Variations and Changes Observed in South Korea. Clim Change. 2004;66:151–161. doi: 10.1023/B:CLIM.0000043141.54763.f8. [DOI] [Google Scholar]
- 10.Cingolani P, Platts A, Wang IL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corbett-Detig RB, Hartl DL, Sackton TB. Natural selection constrains neutral diversity across a wide range of species. PLoS Biol. 2015;13:e1002112. doi: 10.1371/journal.pbio.1002112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frankel OH, Browon AHD. Current plant genetic resources-a critical appraisal. In Genetics:new frontier. 1984;4:1–11. [Google Scholar]
- 13.Godfray HC, Beddington JR, Crute IR, Haddad L, Lawrence D, Muir JF, et al. Food security: the challenge of feeding 9 billion people. Science. 2010;327:812–818. doi: 10.1126/science.1185383. [DOI] [PubMed] [Google Scholar]
- 14.Govindaraj M, Vetriventhan M, Srinivasan M. Importance of genetic diversity assessment in crop plants and its recent advances: an overview of its analytical perspectives. Genet Res Int. 2015;2015:431487. doi: 10.1155/2015/431487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hellmann I, Prufer K, Ji H, Zody MC, Paabo S, Ptak SE. Why do human diversity levels vary at a megabase scale? Genome Res. 2005;15:1222–1231. doi: 10.1101/gr.3461105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodgkinson A, Ladoukakis E, Eyre-Walker A. Cryptic variation in the human mutation rate. PLoS Biol. 2009;7:e1000027. doi: 10.1371/journal.pbio.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoisington D, Khairallah M, Reeves T, Ribaut JM, Skovmand B, Taba S, et al. Plant genetic resources: what can they contribute toward increased crop productivity? Proc Natl Acad Sci U S A. 1999;96:5937–5943. doi: 10.1073/pnas.96.11.5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang X, Kurata N, Wei X, Wang ZX, Wang A, Zhao Q, et al. A map of rice genome variation reveals the origin of cultivated rice. Nature. 2012;490:497–501. doi: 10.1038/nature11532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang X, Lu T, Han B. Resequencing rice genomes: an emerging new era of rice genomics. Trends Genet. 2013;29:225–232. doi: 10.1016/j.tig.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 20.Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 2010;42:961–967. doi: 10.1038/ng.695. [DOI] [PubMed] [Google Scholar]
- 21.Isshiki M, Morino K, Nakajima M, Okagaki RJ, Wessler SR, Izawa T, et al. A naturally occurring functional allele of the rice waxy locus has a GT to TT mutation at the 5' splice site of the first intron. Plant J. 1998;15:133–138. doi: 10.1046/j.1365-313X.1998.00189.x. [DOI] [PubMed] [Google Scholar]
- 22.Jackson MT. Managing the world’s largest collection of rice genetic resources. Proceedings of the International Symposium on Rice Germplasm Evaluation and Enhancement 1999, 22–28.
- 23.Jeong H. Prospect of Korea Climate change. Rural and Environmental Engineering Journal. 2010;109:22–30. [Google Scholar]
- 24.Kim B, Kripalani R, Oh J, Moon S. Summer monsoon rainfall patterns over South Korea and associated circulation features. Theor Appl Climatol. 2002;72:65–74. doi: 10.1007/s007040200013. [DOI] [Google Scholar]
- 25.Kim K, Chung H, Cho G, Ma K, Chandrabalan D, Gwag J, et al. PowerCore: a program applying the advanced M strategy with a heuristic search for establishing core sets. Bioinformatics. 2007;23. [DOI] [PubMed]
- 26.Korte A, Farlow A. The advantages and limitations of trait analysis with GWAS: a review. Plant Methods. 2013;9:29. doi: 10.1186/1746-4811-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kovach MJ, McCouch SR. Leveraging natural diversity: back through the bottleneck. Curr Opin Plant Biol. 2008;11:193–200. doi: 10.1016/j.pbi.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 28.Kumar GR, Sakthivel K, Sundaram RM, Neeraja CN, Balachandran SM, Rani NS, et al. Allele mining in crops: prospects and potentials. Biotechnol Adv. 2010;28:451–461. doi: 10.1016/j.biotechadv.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 29.Kumar V, Singh A, Mithra SV, Krishnamurthy SL, Parida SK, Jain S, et al. Genome-wide association mapping of salinity tolerance in rice (Oryza sativa) DNA Res. 2015;22:133–145. doi: 10.1093/dnares/dsu046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lercher MJ, Hurst LD. Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet. 2002;18:337–340. doi: 10.1016/S0168-9525(02)02669-0. [DOI] [PubMed] [Google Scholar]
- 31.Leroy T, De BF, Legnate H, Musoli P, Kalonji A, Loor Solorzano RG, et al. Developing core collections to optimize the management and the exploitation of diversity of the coffee Coffea canephora. Genetica. 2014;142:185–199. doi: 10.1007/s10709-014-9766-5. [DOI] [PubMed] [Google Scholar]
- 32.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindblad-Toh K, Winchester E, Daly MJ, Wang DG, Hirschhorn JN, Laviolette JP, et al. Large-scale discovery and genotyping of single-nucleotide polymorphisms in the mouse. Nat Genet. 2000;24:381–386. doi: 10.1038/74215. [DOI] [PubMed] [Google Scholar]
- 34.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCouch SR, McNally KL, Wang W, Sackville HR. Genomics of gene banks: A case study in rice. Am J Bot. 2012;99:407–423. doi: 10.3732/ajb.1100385. [DOI] [PubMed] [Google Scholar]
- 36.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moshelion M, Altman A. Current challenges and future perspectives of plant and agricultural biotechnology. Trends Biotechnol. 2015. [DOI] [PubMed]
- 38.Muthayya S, Sugimoto JD, Montgomery S, Maberly GF. An overview of global rice production, supply, trade, and consumption. Ann N Y Acad Sci. 2014;1324:7–14. doi: 10.1111/nyas.12540. [DOI] [PubMed] [Google Scholar]
- 39.Odong TL, Jansen J, van Eeuwijk FA, van Hintum TJ. Quality of core collections for effective utilisation of genetic resources review, discussion and interpretation. Theor Appl Genet. 2013;126:289–305. doi: 10.1007/s00122-012-1971-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oleksyk TK, Zhao K, De La Vega FM, Gilbert DA, O'Brien SJ, Smith MW. Identifying selected regions from heterozygosity and divergence using a light-coverage genomic dataset from two human populations. PLoS One. 2008;3 doi: 10.1371/journal.pone.0001712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ptak SE, Hinds DA, Koehler K, Nickel B, Patil N, Ballinger DG, et al. Fine-scale recombination patterns differ between chimpanzees and humans. Nat Genet. 2005;37:429–434. doi: 10.1038/ng1529. [DOI] [PubMed] [Google Scholar]
- 42.Sang T, Ge S. Understanding rice domestication and implications for cultivar improvement. Curr Opin Plant Biol. 2013;16:139–146. doi: 10.1016/j.pbi.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 43.Schafleitner R, Nair RM, Rathore A, Wang YW, Lin CY, Chu SH, et al. The AVRDC - The World Vegetable Center mungbean (Vigna radiata) core and mini core collections. BMC Genomics. 2015;16:344. doi: 10.1186/s12864-015-1556-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sweeney MT, McCouch S. The complex history of the domestication of rice. Ann Bot. 2007;100:951–957. doi: 10.1093/aob/mcm128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sweeney MT, Thomson MJ, Pfeil BE, McCouch S. Caught red-handed: Rc encodes a basic helix-loop-helix protein conditioning red pericarp in rice. Plant Cell. 2006;18:283–294. doi: 10.1105/tpc.105.038430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tenaillon MI, Austerlitz F, Tenaillon O. Apparent mutational hotspots and long distance linkage disequilibrium resulting from a bottleneck. J Evol Biol. 2008;21:541–550. doi: 10.1111/j.1420-9101.2007.01490.x. [DOI] [PubMed] [Google Scholar]
- 47.Varela MA, Amos W. Heterogeneous distribution of SNPs in the human genome: microsatellites as predictors of nucleotide diversity and divergence. Genomics. 2010;95:151–159. doi: 10.1016/j.ygeno.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 48.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4 doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vowles EJ, Amos W. Evidence for widespread convergent evolution around human microsatellites. PLoS Biol. 2004;2 doi: 10.1371/journal.pbio.0020199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu X, Liu X, Ge S, Jensen JD, Hu F, Li X, et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat Biotech. 2012;30. [DOI] [PubMed]
- 51.Zhao W, Cho G-T, Ma K-H, Chung J-W, Gwag J-G, Park Y-J. Development of an allele-mining set in rice using a heuristic algorithm and SSR genotype data with least redundancy for the post-genomic era. Mol Breeding 2010, 26.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data set supporting the results of this article is included within the Supplementary files (Additional files 4 and 6). High-quality SNPs (HQSNPs) in KRICE_CORE from chromosomes 1–12. are included in the zip file chr1_6_KRICE_CORE_HQSNP.zip (TAB delimited txt file format) and zip file chr7_12_KRICE_CORE_HQSNP.zip (TAB delimited txt file format).
The English in this document has been checked by at least two professional editors, both native speakers of English. For a certificate, please see: http://www.textcheck.com/certificate/humtBq.