

Abstract
Signal peptides are critical for the initiation of protein transport in bacteria by virtue of their recognition by the SecA ATPase motor protein followed by their transfer to the lateral gate region of the SecYEG protein-conducting channel complex. In this study, we have constructed and validated the use of signal peptide-attached SecA chimeras for conducting structural and functional studies on the initial step of SecA signal peptide interaction. We utilized this system to map the location and orientation of the bound alkaline phosphatase and KRRLamB signal peptides to a peptide-binding groove adjacent to the two-helix finger subdomain of SecA. These results support the existence of a single conserved SecA signal peptide-binding site that positions the signal peptide parallel to the two-helix finger subdomain of SecA, and they are also consistent with the proposed role of this subdomain in the transfer of the bound signal peptide from SecA into the protein-conducting channel of SecYEG protein. In addition, our work highlights the utility of this system to conveniently engineer and study the interaction of SecA with any signal peptide of interest as well as its potential use for X-ray crystallographic studies given issues with exogenous signal peptide solubility.
Graphical abstract
Most proteins are secreted across or integrated into the bacterial plasma membrane utilizing a conserved protein transport (Sec) pathway that employs either cotranslational and post-translational modes of transport that converge on a common channel complex comprising SecYEG protein (for recent reviews, see refs 1 and 2). SecYEG itself forms an hourglass-shaped, gated channel in the plasma membrane that can interact directly with either the translating ribosome or the SecA ATPase, which drives the protein transport process.3 Membrane proteins are generally targeted to SecYEG as ribosome-nascent chain complexes soon after the emergence of their signal-anchor domain, which binds to the signal recognition particle followed by SRP binding to its membrane receptor and transfer of the ribosome-nascent chain complex to SecYEG.4,5 By contrast, most presecretory proteins are targeted to SecYEG via cotranslational or post-translational routes utilizing SecB chaperone and/or SecA protein. SecB recognizes a binding-frame on the mature region of loosely folded preprotein substrates,6 while SecA recognizes the signal peptide and the early mature region of preproteins.7 Signal peptide recognition by SecA allows transfer of preproteins from SecB to SecA utilizing the well-defined interaction between these two proteins.8 Although a kinetic partitioning model has been proposed to explain SecB-preprotein recognition,9 SecA-preprotein recognition remains less well understood. Once preproteins bind SecYEG-bound SecA, they are inserted into and translocated across the SecYEG channel in a processive fashion by the action of SecA ATPase motor protein.10,11 During this process, signal peptide binding to SecA appears to play a number of functions beyond initial preprotein targeting, including allosteric activation of SecA-dependent translocation ATPase activity as well as facilitating the trapping of mature regions of preproteins by the SecYEG-bound SecA complex.12 A number of distinct models have been proposed to explain the SecA-dependent protein translocation mechanism. A recent model of SecA action based on a SecA-SecYEG cocrystal structure posits that the preprotein substrate is captured by a nucleotide-responsive clamp that is formed from the preprotein-binding and nucleotide-binding domains (PPXD and NBD-2, respectively; see Figure 1) of SecA.13 In turn, a two-helix finger subdomain (THF) of SecA that is positioned at the mouth of the channel also interacts with the polypeptide substrate as it moves up and down in a nucleotide-responsive manner in order to push the preprotein into the SecY pore.14 While recent biochemical studies favor this model,15 other studies were unable to substantiate it,16 and direct demonstration of its proposed elements awaits more refined approaches such as those afforded by single molecule studies.
Figure 1.
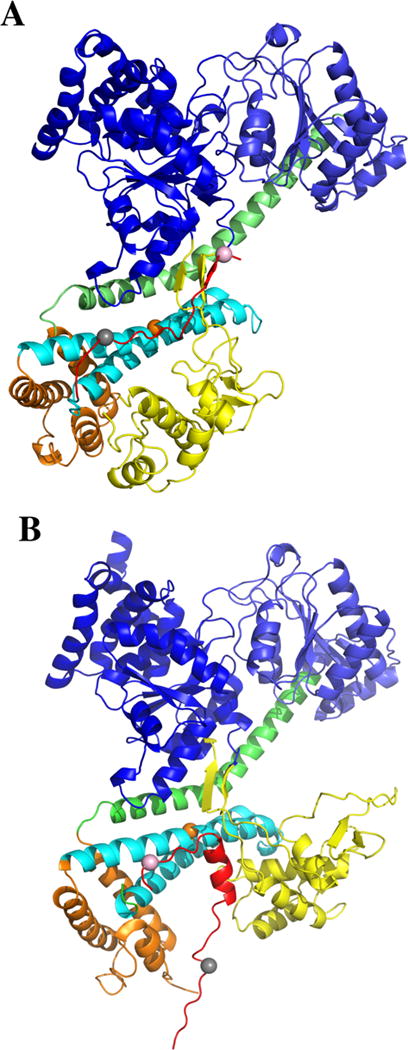
Comparison of SecA signal peptide-binding parallel and perpendicular models. (A) A model where the PhoA signal peptide is bound mainly in a parallel fashion to the THF subdomain of bsSecA is compared to (B) a model where the KRRLamB signal peptide is bound mainly in a perpendicular fashion to the THF subdomain of bsSecA. In B, the ecSecA portion of the peptide bound complex from the NMR structure (PDB ID: 2VDA) was replaced with bsSecA (PDB ID: 1M6N) by superimposing and aligning the major structural domains to create a KRRlamB signal peptide bound bsSecA complex. Color coding of the SecA domains or subdomains is as follows: NBD-I (dark blue), NBD-2 (light blue), PPXD (yellow), HSD excluding THF (green), THF (cyan), and HWD (brown). The bsSecA Trp724 residue is depicted as an orange space-filling sphere. (A) PhoA signal peptide (modeled as the CTL domain of SecA) or (B) KRRLamB signal peptide is shown in red with dye labeled residues at the beginning or end of the corresponding signal peptide depicted by gray or pink space-filling spheres, respectively.
Defining the location(s) of the signal peptide-binding site on SecA is critical to the elucidation of the SecA-dependent translocation mechanism. Historically, genetic and biochemical studies utilizing mutated or truncated SecA proteins with signal peptide binding or cross-linking assays have been utilized to narrow down its location.17–20 More recent studies have employed a structural approach. A nuclear magnetic resonance (NMR) structure of Escherichia coli SecA bound to the KRR-LamB signal peptide was determined where the signal peptide was bound within a groove along the HWD-PPXD interface.21 Another study utilizing Förster resonance energy transfer (FRET) to map 13 distinct distances between SecA and a bound alkaline phosphatase (PhoA) signal peptide placed the binding site within a region bound by the PPXD, HSD, and NBD-1 domains of SecA.22 While both of these studies were in general agreement with the approximate location of the SecA signal peptide-binding site, they differed in its precise location and orientation: the KRRLamB signal peptide was bound mainly perpendicular to the THF subdomain of SecA, while the PhoA signal peptide was bound in a more parallel fashion (Figure 1). We undertook the present studies in order to attempt to resolve this issue as well as to construct and study SecA-signal peptide chimeras as possible biochemical and structural tools to define this system further.
EXPERIMENTAL PROCEDURES
Materials
LB (Miller) broth and agar were obtained from EMD Chemicals and Difco, respectively. Other chemicals were obtained from Sigma or a comparable supplier and were of reagent quality or better. The E. coli alkaline phosphatase signal peptide SP22, MKQSTIALALLPLLFTPVTKAC-NH2, and SP2, MCKQSTIALALLPLLFTPVTKA-NH2 were synthesized (Biomolecules Midwest, Waterloo, IL), HPLC purified, labeled at cysteine with IANBD ester (N-{[2-(iodoacetoxy)ethyl]-N-methyl}- amino-7-nitrobenz-2-oxa-1,3-diazole) or IAEDANS (5-[[[[2-iodoacetyl]amino]ethyl]amino]naphthalene-1-sulfonic acid) (Molecular Probes, Thermo Fisher Scientific), the carboxyl-terminus of the peptides was capped with an amide to prevent an unnatural negative charge, and they were repurified as previously described.18 Peptide identity was verified with electrospray ionization mass spectrometry at the Keck Biotechnology Resource Laboratory at Yale University, and peptide concentration was determined by amino acid analysis at the Keck Biotechnology Resource Laboratory at Yale University or Molecular Biology Core Facilities at Dana Farber Cancer Institute.
Monocysteine and Monotryptophan SecA Chimeric Protein Construction, Expression, and Purification
A series of E. coli SecA-PhoA or B. subtilis SecA-PhoA or SecA-LamB signal peptide chimeras were constructed as described in Supporting Information, Table S1. DH5α [F− ϕ80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 phoA supE44 thi-1 gyrA96 relA1] was used for all plasmid construction, purification, and DNA sequence analysis. The following host strains were utilized for chimera protein expression and in vivo functional analysis: Escherichia coli BLR [F− ompT gal dcm Δ(srl-recA)306::Tn10], BLR containing the pLysS plasmid, BL21.19 [secA13(Am) supF(Ts) trp(Am) zch::Tn10 recA::CAT clpA::KAN],23 or BL21.20 [secA13(Am) supF(Ts) trp(Am) zch::Tn10 recA635::-KAN] containing the pLysS plasmid are all derived from BL21(λDE3).24 Expression of E. coli SecA-signal peptide chimeras utilized BL21.19 at 37 °C, while expression of B. subtilis SecA-signal peptide chimeras employed BLR(pLysS) at 30 °C utilizing freshly transformed colonies. Plasmid-containing strains were subcultured from 50-fold diluted overnight cultures prepared in LB containing appropriate antibiotics (100 μg/mL ampicillin and 25 μg/mL chloramphenicol, where needed) and grown at the indicated temperature until a cell density of A600 of 0.4 was reached, when they were induced with 0.5 mM IPTG for 1 h. Cells were harvested, resuspended in TKM buffer (10 mM Tris-HCl pH 7.5, 50 mM KCL, 10 mM MgOAc, and 1 mM PMSF), and broken in the French press, and chimeric proteins were purified utilizing a His-Bind resin column (Novagen) according to the manufacturer’s protocol as described previously.25 Purified chimeric proteins were dialyzed in TKE buffer (25 mM Tris-HCL pH7.5, 25 mM KCL, 0.5 mM EDTA, and 0.5 mM PMSF) overnight, and protein concentration was measured by the Bradford assay (Bio-Rad) with bovine serum albumin as the standard.
Protein Labeling
Each purified protein was labeled with the selected dye at a protein to dye molar ratio of 1:10 for 2–4 h in TKE buffer on ice with shaking. Free dye was removed using an Amicon Ultra-4 Centrifugal filter (50,000 D cutoff) (Pierce), and protein was stored at −80 °C after the addition of spectroscopic grade glycerol to 5% and rapid freezing in liquid nitrogen. Labeling efficiency was calculated as described in the manufacturer’s protocol (Molecular Probes) and was between 81% and 90% for the different protein preparations. Nonspecific labeling was also evaluated using identical protein constructs lacking cysteine under identical labeling and purification conditions and was found to be less than 2%.
Fluorescence Measurements
Fluorescence anisotropy or spectra were collected on a Fluoromax 4 (Horiba) fluorometer with a programmable water bath (Thermo scientific). Samples of 200 μL were examined in a 3 mm square quartz cuvette at 20 °C. For anisotropy experiments, samples contained 1 μM IANBD-labeled SP22 in TKE buffer, and SecA or SecA chimera protein was titrated over a concentration range from 0 to 40 μM with an incubation time of 45 min prior to data collection. Samples were excited at 465 nm and measured at 550 nm. The spectral bandwidths of the excitation and emission slits were set at 4 and 6 nm, respectively. Data were fit assuming a 1:1 binding interaction using ORIGIN software (version 9) with the following equation:
where [SP] is the total concentration of the signal peptide, [P] is the total concentration of SecA or SecA chimera protein, Kd is the equilibrium dissociation constant, Ao is the anisotropy of the signal peptide in the absence of SecA, and Ai is the anisotropy under saturating binding conditions.
For fluorescence emission spectra, 1 μM IAEDANS labeled or unlabeled chimera or a mixture of SecA protein with 4 μM IAEDANS labeled SP2 or SP22 was incubated in TKE buffer for 15 min at 20 °C. The polarizers were set at 0° and 55° for excitation and emission, respectively, and the samples were scanned at a rate of 1 nm/s at 20 °C. Samples were excited at 295 nm, and emission was measured from 305 to 570 nm. The spectral bandwidths of the excitation and emission slits were set at 4 and 6 nm, respectively.
Steady State FRET Calculation
All spectra were corrected for background. Donor- or acceptor-only spectra were collected in the presence of the unlabeled counterpart to correct for any changes in fluorescence intensity as a consequence of binding. The FRET efficiency, E, was calculated on the basis of the quenching of the donor fluorescence intensity in the FRET complex relative to the donor-only emission in the presence of unlabeled peptide. The efficiency of energy transfer was calculated using the following equation:26
| (1) |
For the dye pair with SecA protein and the exogenous signal peptide, transfer efficiency was calculated from the fluorescence intensity increase of the acceptor upon binding:
| (2) |
where FDA, FD, and FA are the fluorescence intensities of the FRET pair, donor alone, and acceptor alone, and fA and fD are the labeling efficiencies of the acceptor and donor, respectively. εA and εD are the extinction coefficients of the acceptor and donor, respectively. For εA, we used a value of 2100 M−1 cm−1 at 295 nm, and for εD, we used 1449 M−1 cm−1 at 295 nm.12 Acceptor emission intensity was measured at 485 nm. When Trp was used as the donor, fD was taken as 1. The acceptor fluorescence intensities were excited at the donor wavelength of 295 nm and measured over the acceptor emission wavelength range from 440 to 560 nm in the absence and presence of donor and further corrected by the fraction bound, which was 0.86. The efficiency of energy transfer is related to R0, the Förster distance, and R, the distance between donor and acceptor, by the following equation:
| (3) |
R0 is defined as the distance at which the transfer is 50% efficient and was calculated (in angstroms) as follows:26
| (4) |
In eq 4, n is the refractive index (assumed to be 1.4 for biomolecules in aqueous solution),26 κ is the orientation factor (κ2 was assumed to be 2/3 for a randomly oriented, mobile donor and acceptor pair), and QD is the quantum yield of the donor in the absence of acceptor. J(λ), the overlap integral between donor emission and acceptor absorption, was calculated from the spectral data as described.26 The quantum yield of the chimera in the absence of acceptor was measured relative to the standard QST of N-acetyl-L-tryptophamide (Φ = 0.13)27 using the following equation:
where slopeD or slopeST is the plotted graph of fluorescence intensity versus absorbance, n is the refractive index of the solvent (the Tris based donor solvent was corrected to be 1.35, and the standard solvent was taken as 1.33), resulting in a calculated QD of 0.29. The calculated R0 of the Trp-IAEDANS pair was found to be 26.3 Å.
Fluorescence Lifetime Measurements
Tryptophan fluorescence in different chimera constructs was measured using a time-correlated single photon-counting (TCSPC) experimental setup (PTI TimeMaster). Samples were excited with a 280 nm LED laser operating at an average power <1 mW and a repetition rate of 1 MHz. Fluorescence emission was detected at 330 nm. Both excitation and emission slits had a spectral bandpass of 15 nm. Since the laser light is vertically polarized, only one emission polarizer set at 54.7° was used to eliminate any anisotropic bias. Fluorescence decays were collected to a total of 20,000 counts in the peak channel with a time window of 55 ns. Samples were stirred continuously and contained in a 5 mm × 5 mm cuvette. To ensure a sufficient signal and reduce the acquisition time, all decays were collected at a concentration of 20 μM SecA in TKE buffer.
The fluorescence intensity decays were fit to a sum of exponentials using an iterative reconvolution method:
where I(t) represents the fluorescence intensity at time t, and αi represents the fractional subpopulation with a lifetime of τi. All of the data analysis was performed with Globals Unlimited, as previously described.28
RESULTS AND DISCUSSION
Construction and in Vivo Functional Analysis of SecA-Signal Peptide Chimeras
Elucidation of SecA-signal peptide recognition is critical to understand both the initial targeting of preproteins to the Sec-dependent pathway as well as the details of the transfer of signal peptides from SecA to SecYEG protein. Biochemical and particularly structural studies of the SecA-signal peptide interaction are complicated by the relatively poor solubility of hydrophobic signal peptides in aqueous solution, which is roughly only 1 order of magnitude above their measured binding affinity before the onset of aggregation.18,22 Furthermore, given their physical properties, the synthesis and purification of signal peptides can be quite cumbersome, leading investigators to study a more limited subset of signal peptides than might otherwise be prudent. In order to address these concerns, we investigated the feasibility of utilizing SecA-signal peptide chimeras to study this interaction on both a structural and functional level. Toward this end, we constructed a series of SecA-signal peptide chimeras utilizing either the E. coli alkaline phosphatase (PhoA) or lambda receptor protein (LamB) signal peptides or a variant of the latter peptide (KRRLamB) with three additional positively charged residues within the amino-terminal region of the signal peptide.29 Chimeras based on E. coli SecA (ecSecA) were constructed for in vivo functional studies within this host, while parallel chimeras based on B. subtilis SecA (bsSecA) were constructed for biochemical and structural analysis. B. subtilis SecA was deemed superior for these latter studies as it has a limited number of tryptophan and cysteine residues, which are readily engineered for spectroscopic work, and because of its greater propensity to form high quality crystals which better visualize the region around the SecA signal peptide-binding site (e.g., the PPXD domain was poorly structured in ecSecA crystals).30–32
In designing our chimeras, we avoided fusing the signal peptide onto the amino-terminal region of SecA since this region has been shown to be an important SecA dimerization determinant and also promotes SecA interaction with lipids.11,33,34 By contrast, the carboxy-terminal 67 residues of SecA have been shown to be dispensable for SecA function,35 and thus, fusion of peptides onto this end of SecA appeared feasible. Furthermore, the carboxyl-terminal region of SecA has been shown to inhibit SecA-signal peptide interaction,21 and thus, we felt it prudent to investigate the use of both full length (ecSecA901 or bsSecA841) as well as carboxy-terminally truncated SecA derivatives (ecSecA834 or bsSecA783) in our studies. Finally, in order to allow for sufficient flexibility between SecA and a given attached signal peptide, we investigated the effect of adding linkers comprising either 2.5 or 5 repeating glycine-serine residues between these two moieties. On the basis of these considerations, we constructed a series of SecA-signal peptide chimeras (Figure 2; Supporting Information, Table S1).
Figure 2.

SecA-signal peptide chimeras used in the study. The name of each protein chimera is given on the left with a pictorial diagram of the chimera given on the right. bsSecA or ecSecA refers to SecA proteins from B. subtilis or E. coli, respectively, and numbers indicate the residues included in each fusion. PhoAss, LamBss, or KRRLamBss refers to signal peptides derived from E. coli alkaline phosphatase, lambda receptor, or a KRR-variant of the lambda receptor protein, respectively. GS(2.5) or GS(5) refers to glycine-serine linkers with 2.5 or 5 repetitions, respectively (Supporting Information, Table S1). The carboxyl-terminal hexa-histidine tag present on each chimera is not shown. Regions are not drawn to scale.
In order to determine whether the PhoA signal peptide was functional when attached to the carboxyl-terminus of full-length ecSecA or ecSecA834, we made two additional chimeras where the entire alkaline phosphatase gene was fused onto the relevant end of secA, giving rise to ecSecA-PhoA and ecSecA834-PhoA. The alkaline phosphatase mature region has been widely utilized in gene fusion analysis as a topologic determinate to accurately predict the function of signal peptides or signal-anchor domains of secretory or integral membrane proteins, respectively.36 The relevant plasmids were transformed into BL21.20, which contains the secA13(Am) supF(Ts) alleles that allow for the conditional shut off of chromosomal secA gene expression at elevated growth temperatures as well as the pLysS plasmid that limits endogenous expression of T7 RNA polymerase, resulting in little or no overexpression of secA from pT7SecA plasmids in the absence of IPTG induction.24,34 We found that both chimeras were able to complement secA function in this strain when grown at 42 °C. Furthermore, an experiment done in liquid media showed that both chimeras resulted in high alkaline phosphatase activity compared to that of isogenic variants where a strong signal sequence mutation (Leu → Arg14) was introduced into the hydrophobic core region of the relevant alkaline phosphatase signal peptide within the chimera. These latter variants were utilized to show that the high alkaline phosphatase activity observed in the former strains required a functional signal peptide. The defect of this mutant signal peptide has been shown to be at a later stage in protein transport during proofreading by the SecA-SecYEG ternary complex.12 Alkaline phosphatase activity for strains producing ecSecA-PhoA and ecSecA834-PhoA were 38.5 ± 2.7 units and 45.2 ± 0.4 units, respectively, while 0.4 ± 0.3 units and 0.9 ± 1.2 units were obtained for strains producing ecSecA-PhoA Arg14 and ecSecA834-PhoA Arg14, respectively. We did a Western blot analysis of these strains compared to a titration series from a strain constitutive for wild-type alkaline phosphatase (MC1000 phoR−) in order to estimate the specific activity of the chimeras. By comparison of alkaline phosphatase activity to protein amounts, we found that the ecSecA-PhoA chimeras were roughly 4-fold less active than wild-type alkaline phosphatase (Supporting Information, Figure S1). We conclude that the alkaline phosphatase signal peptide is suboptimally active within these two chimeras. Given that signal peptides interact with both SecA and SecY proteins during transport, biochemical analysis was needed to specifically define SecA signal peptide interactions in this system.
Equilibrium Binding Studies
We next turned to a direct measurement of SecA-signal peptide interaction in our bsSecA-PhoAss and bsSecA-LamBss chimeras utilizing an IANBD-labeled PhoA signal peptide, SP22, in a fluorescence anisotropy assay, which has previously been shown to accurately measure SecA-signal peptide binding affinity.18,22 By comparing the relevant bsSecA-signal peptide chimera with its identical bsSecA homologue lacking a signal peptide, we were able to discern the degree of inhibition caused by self-binding of the attached signal peptide. The results of this analysis are given in Table 1, and some representative data are shown in Figure 3. First, we noted that bsSecA783 lacking the CTL domain of SecA had an approximately 4-fold higher affinity for SP22 compared to that of full-length bsSecA841, consistent with the previously reported parallel results for ecSecA and the KRRLamB signal peptide.21 Our measured affinity of bsSecA841 for SP22 of 1.9 ± 0.4 μM was comparable to previously published estimates for ecSecA901 binding to this signal peptide.18,22 Of note, this affinity decreased approximately 2-fold or 7-fold after attachment of the PhoA or KRRLamB signal peptides to SecA, respectively, presumably due to inhibition of SP22 binding from self-binding of the attached signal peptide, although an alternative indirect effect is conceivable as well. Similar results were seen for bsSecA783-derived chimeras, where attachment of the PhoA or KRRLamB signal peptides decreased SP22 binding affinity approximately 9-fold or 26-fold, respectively. Addition of two different lengths of Gly-Ser repeating linkers between the SecA and signal peptide portions of our chimeras did not greatly enhance internal signal peptide binding and in some cases appeared to inhibit it. On the basis of these results, we infer that the attached signal peptides bind to SecA with similar or sometimes significantly increased affinities compared to that of exogenous signal peptides, and therefore, we went forward to further characterize the binding site.
Table 1.
Determination of SecA-Signal Peptide Binding Affinity by Anisotropy
| protein | dissociation constant (μM)a |
|---|---|
| bsSecA841 | 1.9 ± 0.4 |
| bsSecA841-PhoAss | 4.6 ± 0.9 |
| bsSecA841-KRRLamBss | 14 ± 1.8 |
| bsSecA783 | 0.5 ± 0.3 |
| bsSecA783-PhoAss | 4.5 ± 0.6 |
| bsSecA783-GS(2.5)-PhoAss | 2.5 ± 1.4 |
| bsSecA783-GS(5)-PhoAss | 7.2 ± 1.3 |
| bsSecA783-GS(2.5)-LamBss | 6.6 ± 3.2 |
| bsSecA783-GS(5)-LamBss | 16 ± 2.1 |
| bsSecA783-KRRLamBss | 13 ± 3.8 |
| bsSecA783-GS(2.5)-KRRLamBss | 13 ± 6.6 |
| bsSecA783-GS(5)-KRRLamBss | 2.3 ± 1.0 |
The dissociation constant for the indicated protein was measured by fluorescence anisotropy as described in Experimental Procedures.
Figure 3.
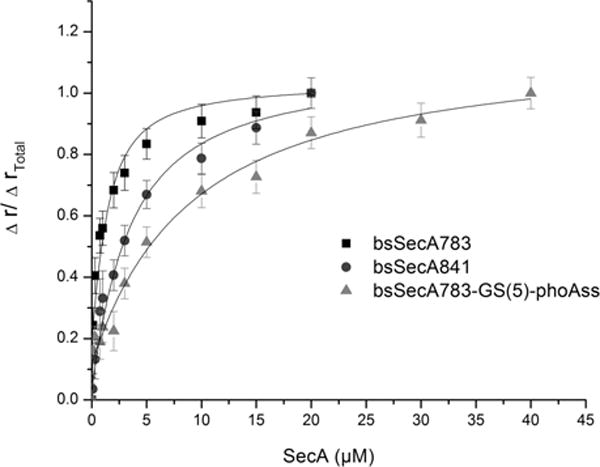
Estimation of SecA-attached signal peptide-binding affinity based on fluorescence anisotropy. One micromolar IANBD-labeled SP22 in TKE buffer was mixed with the indicated final concentration of SecA or chimera protein and incubated at 20 °C for 45 min, when it was subjected to fluorescence anisotropy analysis as described in Experimental Procedures. Data were fit assuming a 1:1 binding interaction.
Both NMR and FRET approaches have been utilized to map either the KRR-LamB or PhoA signal peptides, respectively, to a multidomain interface at the center of ecSecA protein.21,22 While these two studies agreed on the approximate location of the SecA signal peptide-binding site, they differed in its precise location as well as the orientation of the bound signal peptide with respect to the THF subdomain of SecA. In particular, the KRRLamB signal peptide bound in a predominately perpendicular orientation relative to the THF subdomain, while the PhoA signal peptide appeared to bind mainly in a parallel fashion relative to this subdomain (depicted in Figure 1). In order to clarify the bound peptide orientation as well as to validate the use of our chimera approach to study SecA signal peptide interaction, we designed a system to distinguish between these two possibilities.
We took advantage of our earlier studies where single tryptophan and fluorophore-labeled cysteine variants of bsSecA were utilized to characterize the solution-state SecA dimer or to study conformational changes of this protein in the presence of nucleotides and phospholipids.37,38 In particular, we noted that Trp724 was in an ideal position for our current study since it is in the center of the first helix of the THF subdomain (shown as an orange spacefilling sphere in Figure 1A), while the only other tryptophan residue, Trp652, could be functionally substituted with phenylalanine.37,38 In addition, by utilizing the truncated bsSecA783 protein, we were able to avoid the four naturally occurring cysteine residues that lie in the dispensable carboxyl-terminus of the wild-type protein.
Time-resolved fluorescence measurements monitoring the fluorescence from Trp724 support a similar binding geometry for the chimeras and the CTL domain. These studies only monitor fluorescence from Trp724 and report on the factors that quench the fluorescence. As shown in Table 2, the Trp fluorescence in wild-type bsSecA841 exhibited three lifetime components 1.2, 4.5, and 7.1 ns. This observation of three spectroscopically distinct components is consistent with previous measurements of Trp fluorescence in proteins.39 In the case of the truncated bsSecA783, which is missing the CTL domain, only two lifetimes were observed, 2.7 and 6.3 ns. Interestingly, for all four chimera proteins three lifetimes were observed, and all exhibited a 1 ns component similar to that of the wild-type protein. We attribute this 1 ns lifetime component to quenching of the Trp fluorescence either by the unstructured C-terminal tail of the wild-type protein or the signal peptide within the chimera proteins through binding to the groove that runs parallel to the THF subdomain as observed in the X-ray crystal structure. In any case, the similarities in the decay profiles strongly suggest that the signal sequence regions of the chimera proteins bind to the THF subdomain in a manner similar to that of the CTL region of the wild-type protein. These findings support a parallel binding geometry (Figure 1A).
Table 2.
Tryptophan Fluorescence Lifetime Decay Parameters for SecA and SecA-Signal Peptide Chimeras
| protein | α1a | τ1 | α2 | τ2 | α3 | τ3 | τmb |
|---|---|---|---|---|---|---|---|
| bsSecA783 | 0.93 | 2.7 | 0.06 | 6.3 | 3.2 | ||
| bsSecA841 | 0.26 | 1.2 | 0.53 | 4.5 | 0.21 | 7.1 | 5.2 |
| bsSecA783-GS(5)-PhoAss-Cys2-IAEDANS | 0.36 | 1.0 | 0.48 | 3.5 | 0.16 | 6.9 | 4.4 |
| bsSecA783-GS(5)-PhoAss-Cys22-IAEDANS | 0.38 | 1.0 | 0.47 | 3.6 | 0.15 | 7.1 | 4.5 |
| bsSecA783-GS(2.5)-KRRLamBss-Cys2-IAEDANS | 0.24 | 1.3 | 0.44 | 4.1 | 0.32 | 6.5 | 5.1 |
| bsSecA783-GS(2.5)-KRRLamBss-Cys29-IAEDANS | 0.29 | 1.2 | 0.50 | 4.2 | 0.21 | 6.8 | 4.9 |
Fluorescence decays were fit to a sum of exponentials as described, where αi represents the fractional subpopulation associated with lifetime, τi.
The mean lifetime is calculated using the following equation: .
FRET Measurements
To further clarify the orientation of the signal peptides, we measured distances between Trp724 and dyes placed at either the beginning or end of the bound signal peptide in two of our chimeras and compared these results to distances predicted from the parallel or perpendicular-binding models. The perpendicular-binding model was immediately available from the ecSecA-KRRLamB NMR structure.21 In order to construct our parallel-binding model (shown in Figure 1A), we utilized the bsSecA X-ray structure (PDB ID: 1M6N), where the largely unstructured CTL domain is bound for 24 amino acid residues along a peptide-binding groove parallel to the THF subdomain.30 In fact, Hunt et al.30 originally suggested that this region was the mostly likely location for the SecA signal peptide-binding site: a hypothesis supported by our previous FRET mapping and mutagenesis work.22,40 Thus, this region of SecA could be utilized to mimic a signal peptide-bound SecA complex with the parallel orientation. First, the PhoA signal peptide was mapped onto the relevant 21-residue region of the longer KRRLamB signal peptide from the NMR structure, centering it on the critical hydrophobic core region, which was of similar length (13 and 12 residues, respectively). The two peptides were then docked into the CTL-defined groove in the appropriate register as the crystallographically observed CTL to give rise to a parallel-binding model (see Table 3 legend for further details).
Table 3.
Predicted and Measured Distances for SecA-Signal Peptide Bindinga
| experimental FRET
|
parallel-binding modeld
|
perpendicular-binding modele
|
||
|---|---|---|---|---|
| protein with exogenous signal peptide or Sec A chimera | efficiencyb | distance (Å)c | distance (Å) | distance (Å) |
| bsSecA783 F652 with SP2-IAEDANS | 0.60 ± 0.05 | 24.6 ± 3.7 | 27 | 33 |
| bsSecA783 F652 with SP22-IAEDANS | 0.61 ± 0.06 | 24.4 ± 3.6 | 21 | 20 |
| bsSecA783 F652-GS(5)-PhoAss Cys2-IAEDANS | 0.67 ± 0.03 | 23.4 ± 4.3 | 27 | 33 |
| bsSecA783 F652-GS(5)-PhoAss Cys22-IAEDANS | 0.41 ± 0.02 | 27.9 ± 5.8 | 21 | 20 |
| bsSecA783 F652-GS(2.5)-KRRLamBss Cys2-IAEDANS | 0.56 ± 0.05 | 25.3 ± 4.5 | 23 | 45 |
| bsSecA783 F652-GS(2.5)-KRRLamBss Cys29-IAEDANS | 0.45 ± 0.04 | 27.2 ± 5.4 | 27 | 24 |
Distances between Trp724 of bsSecA or Trp775 of ecSecA protein and the indicated residue on the signal peptide were determined utilizing the indicated model and PyMOL or by experimental measurement utilizing FRET.
The calculated energy transfer efficiency from different samples; each value is the average number from at least three individual trials, and standard deviation is reported.
Distances between Trp724 and the indicated IAEDANS-labeled residue on the signal peptide were determined by FRET as described in Experimental Procedures. The calculated error included consideration of the minimal and maximal values of κ2 based on anisotropy measurements as described previously.44
To model the SecA-bound, 21-residue, PhoA signal peptide, we used the NMR structure of the ecSecA-bound, KRRLamB signal peptide21 utilizing the stretch of KRRLamB from Leu11 to Met22 that is the hydrophobic core region. We use the hydrophobic core regions as they are approximately the same length in the two peptides. This 19.4 Å region projected onto the bsSecA X-ray structure corresponds to CTL residues Glu 785 to Gln793. Consequently, in keeping with this alignment, Glu781 and Gln798 correspond to Cys2 and Cys22, respectively, of the bound PhoA signal peptide. Similarly, Met776 and Gln800 correspond to Cys2 and Cys29, respectively, of the bound KRRLamB signal peptide. The predicted distance is measured from the Cγ atom of the identified residue to Cγ of Trp 724.
This model is based on the NMR structure of the ecSecA-bound KRRLamB signal peptide.21 To compensate for the shorter PhoA signal peptide, the 20-residue stretch from Arg6 to Gln25 that centers on the hydrophobic core region was utilized. KRRLamB Lys7 or Ala26 was utilized to mimic Cys2 or Cys22, respectively, of the bound PhoA signal peptide. KRRLamB Met2 or the final residue in that peptide (Ala28) was utilized to mimic Cys2 or Cys29, respectively, of the bound KRRLamB signal peptide. The predicted distance is measured from the Cγ atom of the indicated residue to Cγ of Trp775.
Given the geometry of the system, we noted that probes placed at either the beginning or end of the bound signal peptide were approximately equidistant to Trp724 for the parallel-binding model, while for the perpendicular-binding model, their distances differed by approximately 50% or 100% for the PhoA or KRRLamB signal peptides, respectively (Table 3). Thus, comparison of these two distances for each SecA-bound signal peptide should be informative of their binding orientation, particularly for the KRRLamB chimera. Unfortunately, placement of additional probes centrally within the signal peptide was not feasible, given the convergence of the two models in this region. Furthermore, probes placed within the hydrophobic core region of the signal peptide have previously been shown to disrupt signal peptide binding to SecA protein.22
We collected fluorescence spectra for the appropriate dye-labeled bsSecA783-PhoAss or bsSecA783-KRRLamBss chimeras (Supporting Information, Figure S2), and the relevant distances were determined (Table 3). As a control, we also performed parallel experiments with bsSecA783 and dye-labeled SP2 or SP22, in order to compare the results of SecA binding to an exogenous signal peptide versus one attached within our chimera. The occurrence of energy transfer was readily visualized either as a decrease in donor fluorescence or an increase in acceptor fluorescence. The efficiencies were all in the 0.42–0.68 range and thus reliably measure differences in the distance (Figure 4). The largest source of error in the measurement arose from the relative mobility of the dyes. To account for this mobility in our calculations, the anisotropy of the dyes was measured experimentally and included in the calculation of the error. As reported in Table 3, the distances as measured by FRET yield several insights regarding the binding of the chimeras. Specifically, we compared the results obtained for each chimera protein with those obtained from the exogenous signal peptide from which it is derived. We find that in all cases, the measured distances for the chimeras are in good agreement with those obtained for the exogenous signal peptide. We note that the distances measured for the chimeras based on the SP22 peptide are 3–4 Å longer than those measured for the exogenous peptide itself, but the difference is well within our range of error. Since the dyes are located at approximately the same positions in the chimeras as in the exogenous peptides, these results strongly suggest that the signal peptide regions of the chimeras and the exogenous peptides bind to the THF subdomain in the same manner. From these results, along with our measured binding affinities, we can infer that the chimeras represent a good model system for studying signal peptide-binding interactions with SecA.
Figure 4.
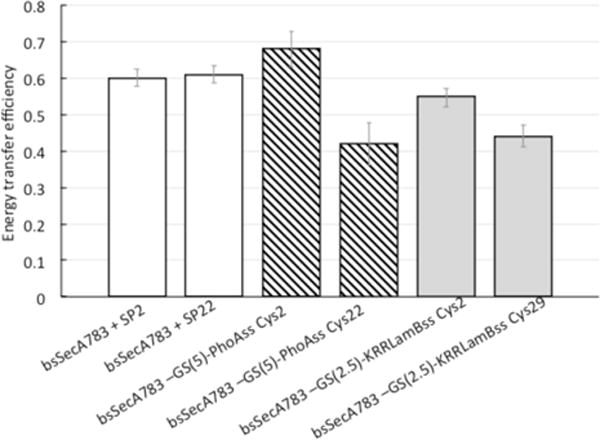
Energy transfer efficiencies of SecA-signal peptide chimeras or SecA-bound signal peptides. The energy transfer efficiency between Trp724 of SecA and the IAEDANS-label on the attached or unattached signal peptide is given. Values were calculated as described in Experimental Procedures.
We further note that the distances measured for chimeras with the dye in the same position but different signal sequences, i.e., PhoAssCys2 and KRRLamBssCys2 or PhoAssCys22 and KRRLamBssCys29, are within one angstrom of each other (24 vs 25 Å and 28 vs 27 Å). Given the equivalence of these measured distances, these findings provide strong evidence that the two signal sequences are binding in the same orientation onto SecA. As shown in Table 3, if the KRRLamBssCys2-IAEDANS chimera were to bind in the same orientation as observed in the NMR study a distance of 44 Å is expected. Given that our measurements yield 25 ± 4.5 Å for this distance, we conclude that the KRRLamB signal sequence binds in the same orientation as the PhoA signal sequence chimera and that both peptides bind to a conserved signal peptide-binding site in a parallel orientation. The distances predicted from our models are measured from Cγ (to approximate the position of the IAEDANS covalently attached to the sulfhydryl group of the Cys residue) to Cγ of the Trp residue. Since we do not include the structure of the dye in our calculations, we do not necessarily expect a one-to-one correspondence between the FRET measured distances and the predicted distances, although we do expect them to be within the error of our measurement. As shown in Table 3, five of our measured FRET distances are within error of the predicted distances from the parallel-binding model.
We found that the measured distance between Trp724 and IAEDANS placed at either the beginning or end of the PhoA signal peptide to be approximately equal whether an exogenous or SecA-attached signal peptide was utilized for this purpose (Table 3, compare lines 1 vs 2 and 3 vs 4). We also found similar distances between Trp724 and IAEDANS placed at either the beginning or end of the longer KRRLamB signal peptide for the appropriate chimeras (Table 3, compare lines 5 vs 6). We further note that for the chimeras with the dyes located at the end of the signal sequence (Cys22/Cys29), the measured distance is slightly longer (2–4 Å) than that measured for the Cys2 dye position. As discussed above, the perpendicular-binding orientation predicts that position 2 of the signal sequence is located further away from Trp724 than the position at the end. Our results in fact indicate that position 2 is located closer to Trp724 and that the end of the signal sequence is approximately the same distance away as position 2 but slightly further, providing greater evidence that the perpendicular-binding orientation does not describe the binding of these peptides. Thus, the parallel-binding model is supported by the relative equivalence of the distances measured between either end of the signal sequence and Trp 724, the agreement between our FRET-measured distances and the distances predicted from the parallel-binding model, and the agreement between the distances measured for the exogenous peptides and the chimeras. Combined with our previous studies where 13 distinct distances between SecA and a bound PhoA signal peptide were determined and pointed to a parallel-binding model,22 these new results indicate that both signal peptides appear to bind SecA at a conserved signal peptide-binding site and in a similar parallel orientation.
To further confirm that the signal peptides and the signal sequence regions of the chimeras were binding to the same region, we performed signal peptide, competition-binding assays with two of the above chimeras, bsSecA783 F652-GS(10)-PhoAss Cys22-IAEDANS or bsSecA783 F652-GS(5)-KRRLamBss Cys29-IAEDANS, and unlabeled SP22. We found that a 5-fold molar excess of unlabeled SP22 reduced our energy transfer efficiency by approximately 50% in both cases (data not shown), indicating that both SecA-attached signal peptides bind in the same manner as the exogenous peptides.
In earlier work defining the SecA signal peptide-binding site, we were not able to eliminate the possibility that the different signal peptides analyzed led to the somewhat different conclusions regarding the precise location of the SecA signal peptide-binding site.21,22 Here, we have addressed this point further by examining both peptides in similar chimeric constructs and find that the results point to a consensus binding site rather than distinct ones for the two signal peptides in question. This result indicates that SecA appears to contain a single universal signal peptide-binding site for interacting with the numerous signal peptides present in the bacterial proteome, although it remains likely that such peptides interact somewhat differently with individual amino acid residues along the surface of the proposed peptide-binding groove (Figure 1A). In conducting these studies, we utilized B. subtilis SecA rather than its E. coli homologue in order to take advantage of the more limited number of tryptophan residues present in the former case. However, we note that SecA from both species is highly homologous (50% identical) and that their protomer structure superimposes well, including the two signal peptide-binding regions in question (Supporting Information, Figure S5). An obvious difference between the two prior studies relates to the protein and peptide concentrations required by the methodology, which for NMR is within a concentration range (≥30 μM) where signal peptides or SecA can aggregate or show nonspecific associations in vitro. By contrast, FRET-based methods can be performed at more standard biochemical concentrations that favor specificity, but the relatively small number of distances measured limits the structural resolution that can be attained by this method. Both methods utilized different probes for ligand mapping (nitroxide spin labels for NMR and IAEDANS for FRET) that could potentially affect the results. Other differences in the two approaches beyond these considerations may also be responsible for the modestly differing results.
While both the NMR and FRET-based models are compatible with the location of particular SecA signal peptide-binding defective mutants within the ascending β-strand “stem” and “bulb” that form the PPXD (e.g., S226A, V310R, and I304A/L306A), other such mutants that are located within the two helix finger region (e.g., I789R, E806R, and F808R) are only consistent with the parallel-binding model.12,21,22,40 However, such genetic results need to be interpreted with caution since mutated residues can have indirect as well as direct effects on the system under study. Both models do support the role of the CTL domain of SecA as an allosteric regulator of signal peptide binding, although the two models differ in the extent of CTL blockage of the relevant signal peptide-binding site.21 Perhaps more importantly, the two models differ significantly regarding recently proposed mechanistic models of SecA-dependent protein transport. In particular, a parallel alignment of the signal peptide relative to the THF-subdomain supports both structural and functional studies suggesting that this latter region of SecA serves as a molecular ratchet to push the signal peptide and mature segments of preproteins into the SecYEG channel.11,13,14 By contrast, the mechanism by which the perpendicularly bound signal peptide could be inserted into the SecYEG channel is unclear since its path into the channel is blocked by the interaction of portions of PPXD with loops of SecY according to the SecA-SecYEG cocrystal.13,41 It could be, however, that in this latter case, the dynamic PPXD domain remains mobile enough to allow for signal peptide insertion in a SecA-SecYEG complex under more physiological conditions. Both models do allow for the binding of the early mature region of the preprotein following the signal peptide to the SecA “clamp” that is formed at the interface of PPXD and NBD-2.15
At present, we cannot rule out that SecA may possess alternative signal peptide-binding modes that function consecutively as part of the translocation cycle. Such considerations indicate that our results need to be interpreted with caution, and they further point to the merit of undertaking additional studies to elucidate this important and complex problem in a more biochemically complete or physiological system. In that regard, our development of a SecA chimera model system that we show to be viable for the study of SecA and any signal peptide of interest extends the available tools for analysis of this system. On a broader level, our study points out the potential feasibility of making similar chimeras between any protein that has a peptide ligand of suitable affinity, particularly for structural studies. We note that X-ray structures were solved for chimeras of both a peptide-bound class II MHC receptor as well as a signal peptide-bound signal-recognition particle utilizing this approach.42,43 Thus, we anticipate that the present chimeras or improved ones with even higher binding affinities should be valuable for future crystallographic studies.
Supplementary Material
Acknowledgments
We thank Yu Chen and Tom Rapoport for preliminary studies on the feasibility of X-ray crystallographic analysis of our chimeras as well as our fellow laboratory members for assistance with the study.
Funding
This work was supported by grants GM110552 from the National Institutes of Health to D.O. and MCB-0843656 from the National Science Foundation awarded to I.M.
ABBREVIATIONS
- CTL
carboxyl-terminal linker domain of SecA
- FRET
Förster resonance energy transfer
- HPLC
high-performance liquid chromatography
- HSD
Helical scaffold domain of SecA
- HWD
helical wing domain of SecA
- IAEDANS
5-((((2-iodoacetyl)-amino)-ethyl)amino)naphthalene-1-sulfonic acid
- IANBD
N-((2-(iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1,3-diazole
- IMV
inverted membrane vesicles
- KRR-LamB
lambda receptor protein signal peptide with an amino-terminal extension
- NBD-1
nucleotide-binding domain-1 domain of SecA
- NBD-2
nucleotide-binding domain-2 domain of SecA
- PPXD
preprotein-cross-linking domain of SecA
- SP2
alkaline phosphatase signal peptide with cysteine at position 2
- SP22
alkaline phosphatase signal peptide with cysteine at position 22
- THF
two helix-finger subdomain of SecA
- TKE
25 mM Tris-HCl, pH 7.5, 25 mM KCl, 1 mM EDTA buffer
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b01115.
List of all the plasmids used in this study; an estimation of translocation efficiency of the SecA-PhoA chimeras based on alkaline phosphatase specific activity; the fluorescence spectra used to determine the efficiency of energy transfer in the protein plus peptide and protein-peptide chimera samples; the effect of GS linker length on the signal peptide binding affinity of the SecA chimeras measured by fluorescence anisotropy; the structure of E. coli SecA with the KRRlamB signal peptide bound in a perpendicular orientation as determined by NMR spectroscopy;21 the superimposed structures of B. subtilis and E. coli SecA structures with the KRRLamB signal peptide bound (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Kudva R, Denks K, Kuhn A, Vogt A, Muller M, Koch HG. Protein translocation across the inner membrane of Gram-negative bacteria: the SecA and Tat dependent protein transport pathways. Res Microbiol. 2013;164:505–534. doi: 10.1016/j.resmic.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 2.du Plessis D, Nouwen N, Driessen A. The Sec translocase. Biochim Biophys Acta, Biomembr. 2011;1808:851–865. doi: 10.1016/j.bbamem.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 3.van den Berg B, Clemons WM, Collinson I, Modls Y, Hartmann E, Harrison SC, Rapoport TA. X-ray structure of a protein-conducting channel. Nature. 2004;427:36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- 4.Lee HC, Bernstein HD. The targeting pathway of Escherichia coli presecretory and integral membrane proteins is specified by the hydrophobicity of the targeting signal. Proc Natl Acad Sci U S A. 2001;98:3471–3476. doi: 10.1073/pnas.051484198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halic M, Beckmann R. The signal recognition particle and its interactions during protein targeting. Curr Opin Struct Biol. 2005;15:116–125. doi: 10.1016/j.sbi.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 6.Crane J, Suo Y, Lilly A, Mao C, Hubbell W, Randall L. Site of interaction of a precursor polypeptide on the export chaperone SecB mapped by site-directed spin labeling. J Mol Biol. 2006;363:63–74. doi: 10.1016/j.jmb.2006.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lill R, Dowhan W, Wickner W. The ATPase activity of SecA is regulated by acidic phospholipids, SecY, and the leader and mature domains of precursor proteins. Cell. 1990;60:271–280. doi: 10.1016/0092-8674(90)90742-w. [DOI] [PubMed] [Google Scholar]
- 8.Suo Y, Hardy JS, Randall L. The basis of asymmetry in the SecA:SecB complex. J Mol Biol. 2015;427:887–900. doi: 10.1016/j.jmb.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Randall LL, Hardy SJS. SecB, one small chaperone in the complex milieu of the cell. Cell Mol Life Sci. 2002;59:1617–1623. doi: 10.1007/PL00012488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van der Wolk JPW, de Wit JG, Driessen AJM. The catalytic cycle of the Escherichia coli SecA ATPase comprises two distinct preprotein translocation events. EMBO J. 1997;16:7297–7304. doi: 10.1093/emboj/16.24.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bauer BW, Shemesh T, Chen Y, Rapoport TA. A “Push and Slide” mechanism allow sequence-insensitive translocation of secretory proteins by the SecA ATPase. Cell. 2014;157:1416–1429. doi: 10.1016/j.cell.2014.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gouridis G, Karamanou S, Gelis I, Kalodimos C, Economou A. Signal peptides are allosteric activators of the protein translocase. Nature. 2009;462:363–367. doi: 10.1038/nature08559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zimmer J, Nam Y, Rapoport TA. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature. 2008;455:936–943. doi: 10.1038/nature07335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erlandson KJ, Miller S, Nam Y, Osborne AR, Zimmer J, Rapoport TA. A role for the two-helix finger of the SecA ATPase in protein translocation. Nature. 2008;455:984–988. doi: 10.1038/nature07439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bauer BW, Rapoport TA. Mapping polypeptide interactions of the SecA ATPase during translocation. Proc Natl Acad Sci U S A. 2009;106:20800–20805. doi: 10.1073/pnas.0910550106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitehouse S, Gold V, Robson A, Allen W, Sessions R, Collinson I. Mobility of the SecA 2-helix-finger is not essential for polypeptide translocation via the SecYEG complex. J Cell Biol. 2012;199:919–926. doi: 10.1083/jcb.201205191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baud C, Karamanou S, Sianidis G, Vrontou E, Politou AS, Economou A. Allosteric communication between signal peptides and the SecA protein DEAD motor ATPase domain. J Biol Chem. 2002;277:13724–13731. doi: 10.1074/jbc.M200047200. [DOI] [PubMed] [Google Scholar]
- 18.Musial-Siwek M, Rusch S, Kendall D. Probing the affinity of SecA for signal peptides in different environments. Biochemistry. 2005;44:13987–13996. doi: 10.1021/bi050882k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Musial-Siwek M, Rusch S, Kendall D. Selective photoaffinity labeling identifies the signal peptide binding domain on SecA. J Mol Biol. 2007;365:637–648. doi: 10.1016/j.jmb.2006.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papanikou E, Karamanou S, Baud C, Frank M, Sianidis G, Keramisanou D, Kalodimos C, Kuhn A, Economou A. Identification of the preprotein binding domain of SecA. J Biol Chem. 2005;280:43209–43217. doi: 10.1074/jbc.M509990200. [DOI] [PubMed] [Google Scholar]
- 21.Gelis I, Bonvin A, Keramisanou D, Koukaki M, Gouridis G, Karamanou S, Economou A, Kalodimos C. Structural basis for signal-sequence recognition by the translocase motor SecA as determined by NMR. Cell. 2007;131:756–769. doi: 10.1016/j.cell.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Auclair S, Moses J, Musial-Siwek M, Kendall D, Oliver D, Mukerji I. Mapping of the signal peptide-binding domain of Escherichia coli SecA using Forster resonance energy transfer. Biochemistry. 2010;49:782–792. doi: 10.1021/bi901446r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitchell C, Oliver DB. Two distinct ATP-binding domains are needed to promote protein export by Escherichia coli SecA ATPase. Mol Microbiol. 1993;10:483–497. doi: 10.1111/j.1365-2958.1993.tb00921.x. [DOI] [PubMed] [Google Scholar]
- 24.Studier WF, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 25.Jilaveanu LB, Zito CR, Oliver D. Dimeric SecA is essential for protein translocation. Proc Natl Acad Sci U S A. 2005;102:7511–7516. doi: 10.1073/pnas.0502774102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2nd. Plenum Publishers; New York: 1999. Energy Transfer; pp. 367–394. [Google Scholar]
- 27.Muino PL, Callis PR. Solvent effects on the fluorescence quenching of tryptophan by amides via electron transfer. Experimental and computational studies. J Phys Chem B. 2009;113:2572–2577. doi: 10.1021/jp711513b. [DOI] [PubMed] [Google Scholar]
- 28.Moreno A, Knee J, Mukerji I. Applying 6-Methylisoxanthopterin-Enhanced Fluorescence to Examine Protein-DNA Interactions in the Picomolar Range. Biochemistry. 2012;51:6847–6859. doi: 10.1021/bi300466d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Triplett TL, Sgrignoli AR, Gao FB, Yang YB, Tai PC, Gierasch LM. Functional signal peptides bind a soluble N-terminal fragment of SecA and inhibit its ATPase activity. J Biol Chem. 2001;276:19648–19655. doi: 10.1074/jbc.M100098200. [DOI] [PubMed] [Google Scholar]
- 30.Hunt JF, Weinkauf S, Henry L, Fak JJ, McNicholas P, Oliver DB, Deisenhofer J. Nucleotide control of interdomain interactions in the conformational reaction cycle of SecA. Science. 2002;297:2018–2026. doi: 10.1126/science.1074424. [DOI] [PubMed] [Google Scholar]
- 31.Papanikolau Y, Papadovasilaki M, Ravelli R, McCarthy A, Cusack S, Economou A, Petratos K. Structure of dimeric SecA, the Escherichia coli preprotein translocase motor. J Mol Biol. 2007;366:1545–1557. doi: 10.1016/j.jmb.2006.12.049. [DOI] [PubMed] [Google Scholar]
- 32.Osborne AR, Clemons WM, Rapoport TA. A large conformational change of the translocation ATPase SecA. Proc Natl Acad Sci U S A. 2004;101:10937–10942. doi: 10.1073/pnas.0401742101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Or E, Boyd D, Gon S, Beckwith J, Rapoport TA. The bacterial ATPase SecA functions as a monomer in protein translocation. J Biol Chem. 2005;280:9097–9105. doi: 10.1074/jbc.M413947200. [DOI] [PubMed] [Google Scholar]
- 34.Das S, Stivison E, Folta-Stogniew E, Oliver D. Reexamination of the role of the amino terminus of SecA in promoting its dimerization and functional state. J Bacteriol. 2008;190:7302–7307. doi: 10.1128/JB.00593-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsuyama S, Kimura E, Mizushima S. Complementation of two overlapping fragments of SecA, a protein translocation ATPase of Escherichia coli, allows ATP binding to its amino-terminal region. J Biol Chem. 1990;265:8760–8765. [PubMed] [Google Scholar]
- 36.Manoil C, Mekalanos JJ, Beckwith J. Alkaline phosphatase fusions: sensors of subcellular location. J Bacteriol. 1990;172:515–518. doi: 10.1128/jb.172.2.515-518.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding H, Hunt JF, Mukerji I, Oliver D. B. subtilis SecA ATPase exists as an antiparallel dimer in solution. Biochemistry. 2003;42:8729–8738. doi: 10.1021/bi0342057. [DOI] [PubMed] [Google Scholar]
- 38.Ding H, Mukerji I, Oliver D. Nucleotide and phospholipid-dependent control of PPXD and C-domain association for SecA ATPase. Biochemistry. 2003;42:13468–13475. doi: 10.1021/bi035099b. [DOI] [PubMed] [Google Scholar]
- 39.Beechem JM, Brand L. Time-Resolved Fluorescence of Proteins. Annu Rev Biochem. 1985;54:43–71. doi: 10.1146/annurev.bi.54.070185.000355. [DOI] [PubMed] [Google Scholar]
- 40.Grady L, Michtavy J, Oliver D. Characterization of the Escherichia coli SecA signal peptide-binding site. J Bacteriol. 2012;194:307–316. doi: 10.1128/JB.06150-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zimmer J, Rapoport TA. Conformational flexibility and peptide interaction of the translocation ATPase SecA. J Mol Biol. 2009;394:606–612. doi: 10.1016/j.jmb.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fremont D, Hendrickson W, Marrack P, Kappler J. Structures of an MHC class II molecule with covalently bound single peptides. Science. 1996;272:1001–1004. doi: 10.1126/science.272.5264.1001. [DOI] [PubMed] [Google Scholar]
- 43.Janda C, Li J, Oubridge C, Hernandez H, Robinson C, Nagai K. Recognition of a signal peptide by the signal recognition particle. Nature. 2010;465:507–510. doi: 10.1038/nature08870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Auclair S, Oliver D, Mukerji I. Defining the solution state dimer structure of Escherichia coli SecA using Forster resonance energy transfer. Biochemistry. 2013;52:2388–2401. doi: 10.1021/bi301217t. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.