

Abstract
Intramembrane proteases execute fundamental biological processes ranging from crucial signaling events to general membrane proteostasis. Despite the availability of structural information on these proteases, it remains unclear how these enzymes bind and recruit substrates, particularly for the Alzheimer's disease‐associated γ‐secretase. Systematically scanning amyloid precursor protein substrates containing a genetically inserted photocrosslinkable amino acid for binding to γ‐secretase allowed us to identify residues contacting the protease. These were primarily found in the transmembrane cleavage domain of the substrate and were also present in the extramembranous domains. The N‐terminal fragment of the catalytic subunit presenilin was determined as principal substrate‐binding site. Clinical presenilin mutations altered substrate binding in the active site region, implying a pathogenic mechanism for familial Alzheimer's disease. Remarkably, PEN‐2 was identified besides nicastrin as additional substrate‐binding subunit. Probing proteolysis of crosslinked substrates revealed a mechanistic model of how these subunits interact to mediate a stepwise transfer of bound substrate to the catalytic site. We propose that sequential binding steps might be common for intramembrane proteases to sample and select cognate substrates for catalysis.
Keywords: exosite, intramembrane proteolysis, photocrosslinking, substrate recognition, γ‐secretase
Subject Categories: Membrane & Intracellular Transport; Molecular Biology of Disease; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Intramembrane proteases are exceptional proteases that catalyze the cleavage of their substrate proteins by water‐accessible catalytic residues that are deeply embedded in the membrane (Erez et al, 2009). In most but not all cases, they cleave their substrates, which are typically single‐pass membrane proteins, within the transmembrane domain (TMD) (Strisovsky, 2013). Intramembrane proteases are found widespread in all kingdoms of life and play key roles in many important physiological and pathophysiological processes of lower and higher organisms (Erez et al, 2009; Urban, 2009; Lemberg, 2013). For example, by releasing transcription factors retained in the intracellular domain of a substrate controlling lipid biosynthesis (site 2 protease) or cell differentiation in development (γ‐secretase), or by liberating growth factor‐containing substrate ectodomains (rhomboid), intramembrane proteases have been established as key mediators of crucial signaling events. Another important function of intramembrane proteolysis is the maintenance of membrane proteostasis by turnover of membrane protein remnants—probably a major role of γ‐secretase, and of misfolded membrane proteins by certain rhomboids.
Due to its intimate link with Alzheimer's disease (AD), γ‐secretase has been the most intensively studied intramembrane protease (Steiner et al, 2008; De Strooper et al, 2012). The devastating disease is widely believed to occur as a consequence of the proteolytic generation and the accumulation of amyloid β‐peptide (Aβ), a heterogeneous mixture of short peptides, 38–43 amino acids in length (Holtzman et al, 2011). The peptides are released by γ‐secretase from the amyloid precursor protein (APP) following an initial cleavage by β‐secretase that generates a membrane‐retained C‐terminal fragment (CTF) of APP, termed APP CTFβ or C99, as immediate γ‐secretase substrate (Lichtenthaler et al, 2011). Cleavage of C99 proceeds C‐terminally from ε‐ via ζ‐ to γ‐sites, thereby liberating the APP intracellular domain (AICD) and Aβ in a stepwise manner (Qi‐Takahara et al, 2005; Zhao et al, 2005; Takami et al, 2009). The longer Aβ forms released by γ‐secretase such as Aβ42 preferentially oligomerize into toxic species that eventually form insoluble fibrils that are deposited in the brain of patients as senile plaques (Haass & Selkoe, 2007). γ‐Secretase is structurally the most complex intramembrane protease (Bai et al, 2015b) and the only known one, which requires tightly associated partner proteins for activity (Edbauer et al, 2003; Kim et al, 2003; Kimberly et al, 2003; Takasugi et al, 2003). The enzyme complex is composed of four subunits, either presenilin (PS) 1 or PS2, present in the complex as endoproteolytically cleaved fragments, nicastrin (NCT), PEN‐2, and either APH‐1a or APH‐1b (Steiner et al, 2008; De Strooper et al, 2012). Mutations in the catalytic PS subunits of the complex are the major cause of familial AD (FAD) (De Strooper et al, 2012). Apart from the AD‐associated APP (De Strooper et al, 1998; Naruse et al, 1998), γ‐secretase cleaves many other type I membrane protein substrates (Haapasalo & Kovacs, 2011), foremost the Notch1 receptor (De Strooper et al, 1999; Struhl & Greenwald, 1999), a crucial signaling protein, which has profound implications for the enzyme as AD drug target (Imbimbo & Giardina, 2011; De Strooper & Chavez Gutierrez, 2015).
A major unresolved fundamental question that is key to our understanding of intramembrane proteolysis is how intramembrane proteases recognize and recruit their substrates. Due to the membrane immersion of the substrates, this process is fundamentally different from soluble proteases and expected to be governed by different principles (Langosch et al, 2015). Some initial insight was provided by inhibitor and mutational studies showing that γ‐secretase (Esler et al, 2002; Tian et al, 2002; Kornilova et al, 2005) and rhomboids (Strisovsky et al, 2009; Arutyunova et al, 2014) employ exosites for substrate binding, that is, secondary substrate‐binding sites separated from the active site that may aid in substrate recognition and provide substrate specificity (Drag & Salvesen, 2010). For γ‐secretase as a complex, additional subunits may thus be involved in the substrate recognition process besides PS. Whether NCT may possibly fulfill such a role has been highly controversial (Shah et al, 2005; Chavez‐Gutierrez et al, 2008; Dries et al, 2009; Zhao et al, 2010; Zhang et al, 2012; Bolduc et al, 2016) and could also not been clarified from the recently obtained cryo‐EM structure of γ‐secretase at atomic resolution (Bai et al, 2015b). Thus, despite the structural information on the enzyme, the substrate‐binding sites, the mechanism of substrate recognition/recruitment, and the substrate entry path to the active site remained unclear and speculative.
To shed light on the mechanism of substrate recognition by γ‐secretase, we established a chemical biology approach, which, by site‐specific incorporation of a photocrosslinkable phenylalanine derivative into the substrate, allowed us for the first time to precisely map the residues of C99 that are in contact with the protease. Most C99 residues contact the N‐terminal fragment (NTF) of PS1 identifying this subunit as major substrate‐binding site. In addition, substrate binding was also found for the PS1 CTF, PEN‐2, and NCT. Besides mapping the substrate residues that bind in the active site, we identified those, which bind to exosites that were provided by the PS1 NTF, PEN‐2, and NCT. We further found that PS1 FAD mutants displayed an altered binding of the substrate cleavage domain, suggesting substrate mispositioning as a mechanism underlying the generation of the Aβ42(43) species involved in AD pathogenesis. Finally, probing proteolysis of crosslinked substrates suggested a stepwise passage of substrate from the exosites to the active site in PS. Taken together, these data represent a detailed analysis of the enzyme binding sites of a major γ‐secretase substrate and precursor of Aβ. They provide important structural and mechanistic insights into how this intramembrane protease complex binds substrates, suggest altered substrate binding as a mechanistic basis for FAD, and reveal a substrate translocation path from the exosites to the active site.
Results
Identification of APP substrate interaction sites with γ‐secretase
To identify substrate–protease interaction sites of APP with γ‐secretase, we employed a previously described method for site‐specific incorporation of the photocrosslinkable amino acid para‐benzoyl‐L‐phenylalanine (Bpa) into proteins (Chin et al, 2002). Bpa is an unnatural amino acid containing a benzophenone group that crosslinks to neighboring molecules within a ~3 Å distance upon UV irradiation (Dorman & Prestwich, 1994). To this end, amber (TAG) codons were site specifically introduced into the cDNA encoding C100‐His6, a well‐characterized APP C99‐based γ‐secretase substrate containing an N‐terminal methionine and a C‐terminal affinity purification tag (Edbauer et al, 2003). The constructs were expressed in an E. coli strain that allows the incorporation of Bpa at the amber codon sites by a Bpa‐specific aminoacyl‐tRNA synthetase and the co‐expressed amber suppressor tRNA and then affinity‐purified via their C‐terminal His6 tag. By this approach, each single residue from D1 to D68 (Aβ numbering), covering the extracellular domain, the TMD encompassing residues G29 to L52, and 16 additional residues of the intracellular domain, was individually replaced by Bpa (Fig 1A). Residue D68 was chosen as endpoint for the substrate‐binding analysis, because previous studies have shown that C99 constructs with shorter intracellular domains affect the cleavage efficiency and cleavage specificity of γ‐secretase (Iwata et al, 2001; Funamoto et al, 2004). Using CHAPSO‐solubilized γ‐secretase, a widely used experimental system to study all aspects of the biochemistry and enzymology of γ‐secretase (Li et al, 2000a; Tian et al, 2002; Kakuda et al, 2006; Chavez‐Gutierrez et al, 2012), the substrate variants were first assayed for cleavability. As shown in Fig 1B and C, assessment of AICD generation revealed that the Bpa‐containing substrates remained essentially cleavage competent, showing that they are recognized as substrates by γ‐secretase. Only very few of them affected the downstream cleavage steps to cause an increased relative generation of Aβ42 (Appendix Fig S1).
Figure 1. Cleavage of C99‐Bpa constructs by γ‐secretase.
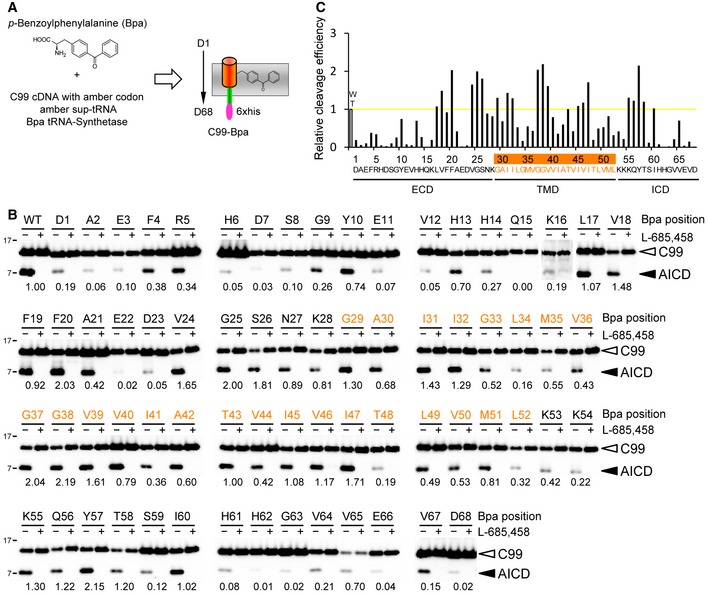
- Schematic representation of the genetic approach to site specifically incorporate the photoactivatable amino acid derivative Bpa into C99.
- C99‐Bpa constructs remain essentially cleavage competent. Numbers denote the relative γ‐secretase cleavage efficiencies of C99‐Bpa substrates as calculated from the AICD/C99‐Bpa ratios relative to that of WT C99, which was set to 1. Inefficiently cleaved substrates (cleavage efficiency below 20%) were mostly found for Bpa substitutions in extramembranous domains. Specificity of substrate cleavage was confirmed by the inhibition of AICD formation in the presence of L‐685,458. Residues of the TMD are highlighted in orange here and elsewhere where appropriate.
- Values obtained in (B) were additionally plotted. Yellow line indicates the cleavage efficiency of WT C99, which was set to 1. ECD, extracellular domain; ICD, intracellular domain.
Next, to identify their molecular targets, the substrates were incubated with CHAPSO‐solubilized γ‐secretase and UV‐irradiated to induce crosslinking. Following the dissociation of the subunits with SDS and urea, crosslinked γ‐secretase subunits were isolated by affinity pulldown via the His6 tag present in the constructs (Fig 2A). As shown in Fig 2B, by scanning the substrate for close contacts with γ‐secretase, the PS1 NTF and CTF, NCT, and PEN‐2 were identified as substrate‐binding subunits. Due to the failure to detect prominent crosslink bands for APH‐1, this subunit was considered to be non‐substrate binding (see also Discussion). The major substrate crosslink sites that were identified were V44 and L49 within the γ‐secretase cleavage domain that crosslinked to the PS1 NTF, M51 and L52 located near the cytoplasmic border of the TMD that crosslinked to the PS1 CTF, H6 in the substrate extracellular domain that crosslinked to NCT, and A30 in the TMD close to its N‐terminal border that crosslinked to PEN‐2. Interestingly, M51 and L52 also weakly crosslinked to the PS1 NTF, showing that two different binding populations existed for these residues. Notably, crosslinking to PS was much more efficient than that to NCT and PEN‐2, consistent with the expectation that the primary substrate‐binding site should locate in the catalytic subunit. Specificity of these interactions was demonstrated by the lack of substrate crosslinking in the absence of UV irradiation and in the presence of Triton X‐100, a detergent that dissociates the γ‐secretase complex (Fig 2B). Moreover, excess amounts of parental C99 WT substrate competed the crosslinking of the respective Bpa derivatives, which provided a further control of binding specificity (Fig 2C). Taken together, these data show that C99 contacts several subunits of γ‐secretase and that V44, L49, M51, and L52 of C99 represent major substrate crosslinking sites with the protease, which apparently cluster in the cleavage domain.
Figure 2. Identification of C99‐Bpa interaction sites in γ‐secretase.
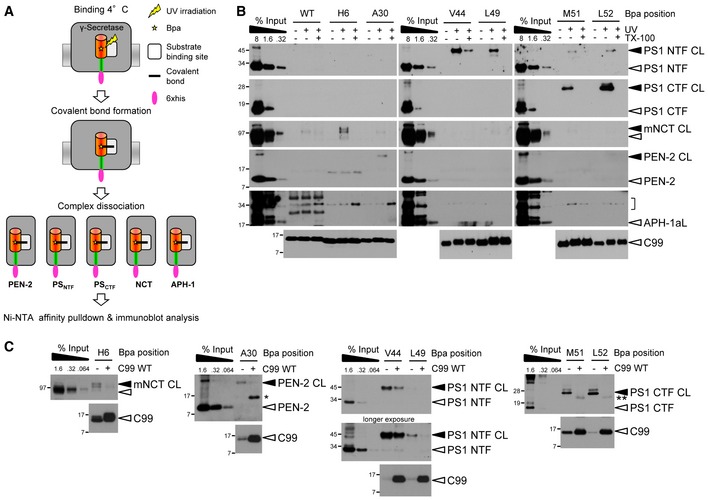
- Schematic representation of the substrate‐photocrosslinking strategy.
- Identification of C99 interaction sites with γ‐secretase. C99‐Bpa substrates were irradiated with UV light in the presence of CHAPSO‐solubilized γ‐secretase. Crosslinked substrates were captured by Ni‐NTA affinity pulldown and bound subunits were identified by a ˜10‐kDa molecular weight increase compared to the input. Results are shown for the major substrate crosslinking residues. Crosslink formation was not observed in the absence of UV irradiation or in the presence of Triton X‐100, which dissociates the γ‐secretase complex, proving crosslink specificity. Bracket indicates the molecular weight range of putative C99‐APH‐1 crosslink bands. APH‐1aL, long splice variant of APH‐1a.
- Excess amounts of parental C99 WT substrate compete crosslinking of C99‐Bpa substrates to γ‐secretase. Single and double asterisks indicate antibody crossreactivities to C99 monomer and dimer, respectively.
Comparing the crosslinking efficiencies over the whole range of the APP sequence showed that the substrates crosslinked primarily to the PS1 NTF (Fig 3). Besides the major interaction sites V44 and L49, many additional residues were identified that crosslinked more weakly to this subunit. Among these, E3 was identified as a prominent crosslink site in the extracellular domain. Likewise, besides H6 and A30, a few additional also weakly crosslinking residues were identified for both NCT and PEN‐2, while crosslinking to the PS1 CTF was limited to M51 and L52 as major sites (Fig 3). Using pharmacological analysis (see next paragraph), only K54 could be further validated of the very few additional residues that weakly crosslinked to this subunit. Thus, we conclude that the primary binding site of C99 is the PS1 NTF.
Figure 3. Quantitative scanning of C99‐Bpa substrate interaction sites with γ‐secretase.
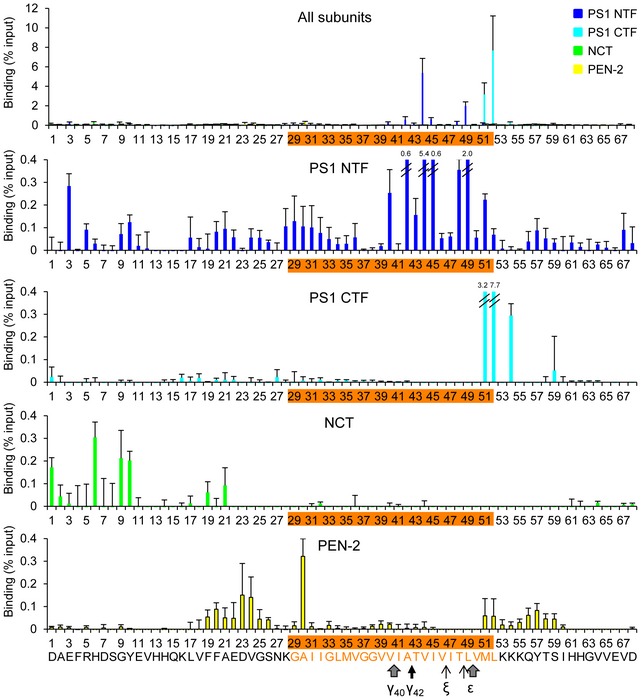
Quantitative crosslinking analysis revealed V44, L49, M51, and L52 as major interactions sites of C99 with γ‐secretase with high crosslinking efficiencies ranging from ~2 to 8% of input. While the PS1 NTF was bound by C99 over nearly the whole range of residues, only very few residues bound to PS1 CTF. Several residues in the substrate N‐terminal extracellular domain bound to NCT. PEN‐2‐binding residues clustered in the membrane‐flanking extracellular and intracellular domains of C99.
Bars denote the mean ± SE (n = 3).
Pharmacological characterization of substrate binding reveals distinct substrate interaction sites
To determine which substrate residues contact the active site of γ‐secretase, we next investigated whether γ‐secretase inhibitors (GSIs) would compete their binding. Two chemically distinct GSIs were used, L‐685,458, an active site‐directed peptidic transition state analog inhibitor, and DAPT, a dipeptide‐type GSI whose binding site partially overlaps with that of L‐685,458 (Wolfe, 2012). As shown in Fig 4A, crosslinking of A42, V44, I45, L49, M51, and L52, that is, residues in the γ‐secretase cleavage domain, was reduced by the GSIs, suggesting that these face binding pockets in the active site region that are not accessible in the presence of the inhibitors. Surprisingly, in contrast to the above residues, crosslinking of E3 and H6 as well as of A30 was increased by both GSIs (Fig 4B). These results indicated that the prevention of access to the active site caused these substrates to be trapped at exosites in the PS1 NTF, NCT, and PEN‐2. By scanning all substrate crosslink positions from residues D1–D68, we noted that crosslinking was increased in the presence of the GSIs for a large number of substrates, primarily with extramembraneously placed Bpa positions, whereas decreased labeling was preferentially observed in the cleavage domain (Figs 4C, EV1 and EV2). Finally, alignment of the interaction sites of active site‐directed inhibitors with the interaction sites of C99 in the PS1 CTF suggested that the active site of γ‐secretase locates at the ε‐cleavage sites of C99 (Fig EV3A and B), supporting the sequential cleavage model of γ‐secretase (Morishima‐Kawashima, 2014). Taken together, these data establish that C99 is in direct contact with distinct substrate‐binding sites of γ‐secretase, that is, with exosites and the active site, and identify the substrate residues directly interacting with these sites. Moreover, they indicate that exosite binding precedes substrate binding at the active site and suggest that substrates accumulate at the exosites when access to the active site is blocked by a GSI (Fig 4D).
Figure 4. Identification of pharmacologically distinct substrate‐binding sites.
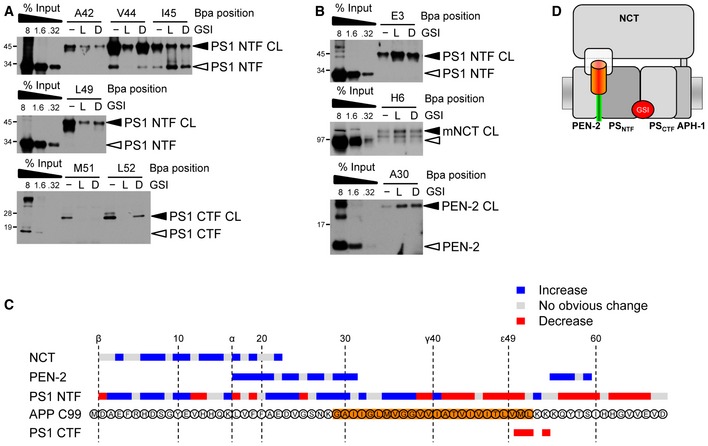
- Inhibition of substrate crosslinking by the GSIs identifies substrate residues binding in the active site. L, L‐685,458; D, DAPT.
- Preventing access to the active site by GSIs identifies substrate residues interacting with the exosites by their increased labeling intensity.
- Summary of effects of L‐685,458 on C99 binding to γ‐secretase. Note that crosslinking of almost all residues of the K54–I60 region to PEN‐2 was increased in the presence of L‐685,458, indicating additional exosites in this subunit binding to the juxtamembrane region of the C99 intracellular domain. See also Figs EV1 and 2 and Discussion.
- Model of substrate binding in the presence of GSIs showing the substrate accumulation at exosites provided by NCT, PEN‐2, and the PS1 NTF.
Figure EV1. Additional pharmacological validation of substrate crosslinking to the PS1 NTF and CTF .
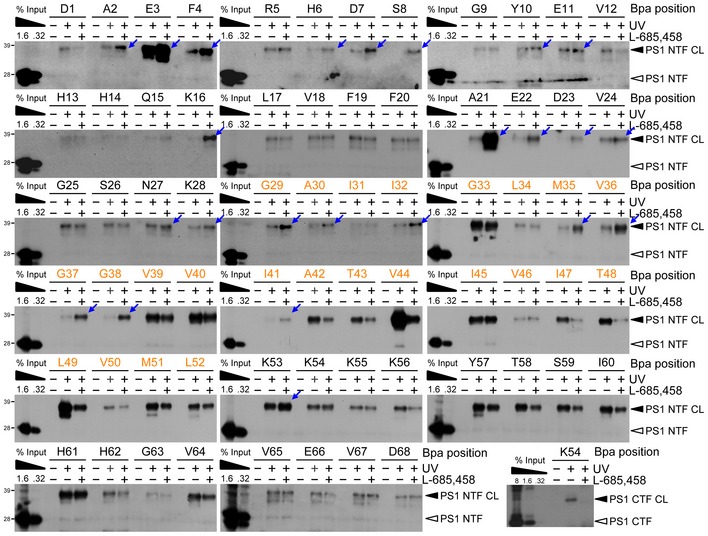
Functional relevance of interactions with the PS1 NTF and CTF was validated by the modulation of substrate crosslinking in the presence of L‐685,458. For the PS1 NTF, with only a few exceptions, L‐685,458 increased the crosslinking efficiency of substrates carrying the Bpa substitutions in the extracellular domain supporting the presence of exosites in the PS1 NTF. Decreased binding to the PS1 NTF of some residues in this region may indicate minor local steric changes caused by L‐685,458. For the PS1 CTF, M51 and L52 as well as residues for which crosslinking could not be detected even in the longest exposures on the films are not shown. TMD residues are highlighted in orange. Blue arrows indicate the residues for which increased labeling was observed.
Figure EV2. Additional pharmacological validation of substrate crosslinking to NCT and PEN‐2.
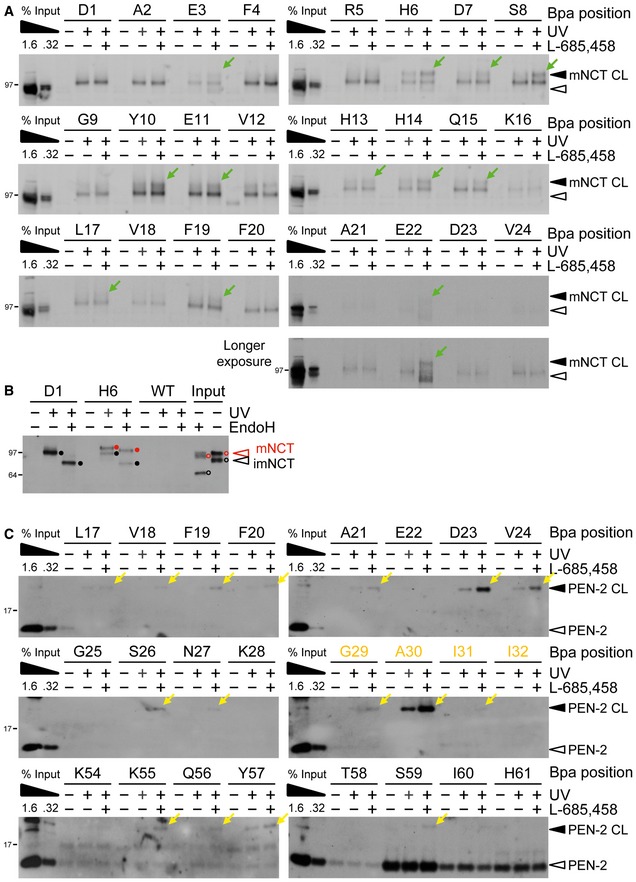
- Validation of functional relevance of C99‐Bpa interactions with NCT by the modulation of substrate crosslinking in the presence of L‐685,458. For a number of substrates binding to mature NCT was increased in the presence of L‐685,458, suggesting that the exclusion of substrate from the active site by the GSI caused substrate trapping at exosites in NCT. Residues for which crosslinking could not be detected even in the longest exposures on the films are not shown. Note that the crosslinking to immature NCT that was observed for all substrate residues did not change in the presence of L‐685,458, suggesting functionally irrelevant binding. See also (B). Green arrows indicate the residues for which increased labeling was observed.
- Deglycosylation by endoH revealed that the crosslink of H6 to NCT originated from binding to complex glycosylated mature NCT, while that of D1 from binding to immature NCT. Closed and open circles indicate partial endoH resistance of mature NCT (red) and endoH sensitivity of immature NCT (black) for the crosslinked samples or the uncrosslinked input control, respectively.
- Validation of functional relevance of C99‐Bpa interactions with PEN‐2 by the modulation of substrate crosslinking in the presence of L‐685,458. Increased substrate crosslinking in the presence of the active site‐targeting GSI is consistent with the presence of exosites in PEN‐2 and supports its role in substrate recruitment. Residues for which crosslinking could not be detected even in the longest exposures on the films are not shown. TMD residues are highlighted in orange. Yellow arrows indicate the residues for which increased labeling was observed.
Figure EV3. Graphic representation of C99 interactions with subsites in the γ‐secretase active site.
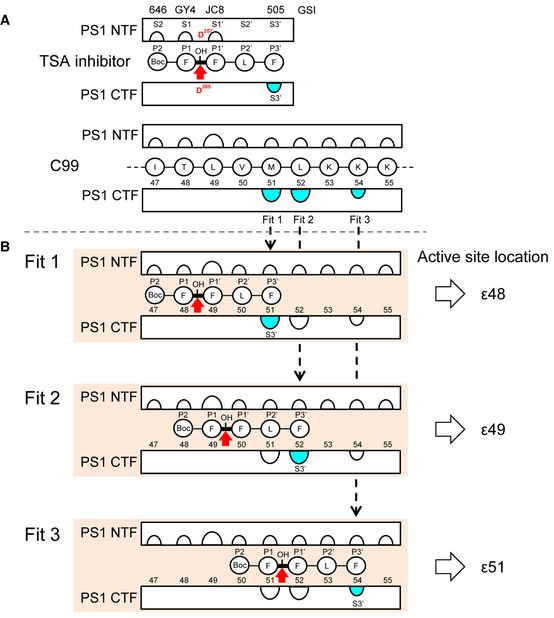
- Comparison of the C99 interaction sites with the known interaction sites of transition state analog GSIs (Li et al, 2000b; Shelton et al, 2009) suggests that the binding site of these GSIs locates at the ε‐cleavage sites of C99. Using differently placed photoreactive benzophenone moieties in the FFLF peptide‐based L‐685,458 GSI derivatives L‐852,646 (646), GY4, JC8, and L‐852,505 (505), the S2‐S1′ pockets were previously located to the PS1 NTF (Li et al, 2000b; Shelton et al, 2009). In addition, the S3′ pocket could almost exclusively be located to the PS1 CTF by GSI 505 (Li et al, 2000b; Shelton et al, 2009). Preferential location of the S3′ pocket to the PS1 CTF was also observed for the related GSI III‐63 (Kornilova et al, 2005). As determined here, the only sites of C99 that crosslinked to the PS1 CTF were residues M51, L52, and K54.
- Combining the information given in (A), we fitted the S3′ pocket in the PS1 CTF determined by GSI 505 with each of these residues (fit 1, 2, and 3). By this analysis, the hydroxyl group of GSI 505 and thus the γ‐secretase active site were found to locate at the ε48, the major ε49, and the very minor ε51 site, respectively.
PS1 FAD mutations cause mispositioning of the substrate cleavage domain
While it is established that FAD mutants in PS cause an imprecision of γ‐secretase cleavage of C99 leading to an increase in the ratios of longer Aβ42(43) species to total Aβ (Scheuner et al, 1996), the molecular cause of these cleavage alterations is still unknown. A plausible reason, which previously has not been possible to directly address experimentally, is that FAD mutants might cause altered substrate binding, which could impact on the downstream processing steps leading to a relative increase in the longer Aβ species. Since our above results identified the substrate residues that are in close contact with γ‐secretase, we could now directly address this question and thus investigated whether substrate binding might be changed for PS1 FAD mutations. Two FAD mutants were chosen for the analysis: the rather mild A246E mutant, which causes only a small increase in the Aβ42/Aβtotal ratio (Scheuner et al, 1996), and the strong PS1 L166P mutant, which causes a dramatic shift in the Aβ42/Aβtotal ratio (Moehlmann et al, 2002). As shown in Fig 5A, exosite binding was if at all not significantly altered for the FAD mutants. In contrast, when scanning the whole γ‐secretase cleavage domain, we found that the substrate crosslink patterns were qualitatively and quantitatively changed for this domain for both mutants in the PS1 NTF (Fig 5B). In particular, crosslinking of L49 and L52 was reduced for both mutants. In addition, the PS1 L166P mutant displayed altered interactions with reduced crosslinking of V40, V44, I45, M51 and increased crosslinking of V46 and T48. Strikingly, the L49 interaction site of PS1 WT was strongly shifted to its neighboring site T48 in the mutant. The alterations in crosslinking thus included apparently major changes at the initial substrate cleavage sites, the ε‐site residues T48 and L49. No shifts were detected for substrate binding to the PS1 CTF, which was strongly reduced for both FAD mutants (Fig 5C). We conclude that FAD mutants in the catalytic subunit of γ‐secretase affect the substrate binding in the active site region causing mispositioning of the C99 cleavage domain (Fig 5D).
Figure 5. Mispositioning of the substrate cleavage domain by PS1 FAD mutations.
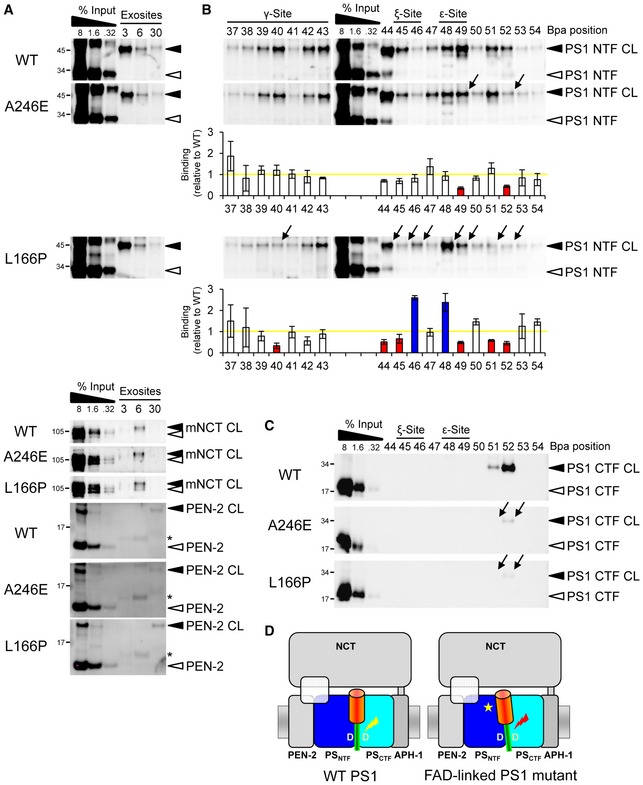
- PS1 FAD mutations display little or no effect on substrate interactions at the exosites. Asterisks indicate antibody crossreactivities to C99 monomer.
- PS1 FAD mutations alter the substrate binding to the PS1 NTF in the γ‐secretase cleavage domain. The weak PS1 A246E FAD mutation alters substrate interactions predominantly at V49 and L52, whereas the highly pathogenic PS1 L166P FAD mutation causes more broad changes over the whole cleavage domain. Arrows denote the changes in binding of individual residues. Corresponding quantitation of substrate binding is shown below the immunoblots. Bars denote the mean ± SE (n = 3). Yellow line indicates the level of substrate binding to PS1 WT, which was set to 1. Blue and red bars indicate the increases or decreases in binding, respectively.
- Both mutants decrease crosslinking to the PS1 CTF.
- Model depicting substrate mispositioning of the PS1 FAD mutants in the active site region. Red lightning mark indicates corresponding substrate miscleavage.
Passage of bound substrate to the active site for substrate cleavage
To get further insight into the mechanism of how γ‐secretase recruits substrates, we next sought to address the question whether or not exosite binding may precede substrate cleavage. To this end, we performed “substrate‐binding chase” experiments and asked whether substrates bound at exosites by crosslinking at 4°C (binding) could be cleaved when the temperature was raised to 37°C (chase) (Fig 6A). As shown in Fig 6B, strongly reduced levels of crosslinked substrates were observed in the chase condition for a number of substrates with extracellular placed Bpa substitutions which crosslinked to exosites in the PS1 NTF. This suggests that these substrates can access the active site to become cleaved. Indeed, the presence of L‐685,458 during the chase reaction prevented the substrate cleavage (Fig 6B). So far, the chase experiments thus indicate that substrates crosslinking to exosites in the PS1 NTF can be, or already are, properly positioned for cleavage at the active site. Surprisingly, however, when we next investigated exosite binding of H6 to NCT and of A30 to PEN‐2 in the same fashion, we noted that both substrates were not cleaved in the chase condition, suggesting that their access to the active site was prevented when they were crosslinked to these subunits (Fig 6C). This unexpected observation indicated that substrates could only be processed when they are bound at the PS1 NTF, suggesting that binding of NCT and/or PEN‐2 may occur before the binding to the catalytic PS subunit. To investigate this possibility further, we took advantage of the observation that some substrates crosslinked to NCT as well as to the PS1 NTF (Figs 3, 4C, EV1 and EV2); that is, two binding populations existed for them. Strikingly, when such substrates (E3, H6, G9, Y10) were further analyzed, we found that they could only be efficiently cleaved in the 37°C chase condition when crosslinked to the PS1 NTF, but not when crosslinked to NCT (Fig 6D). Very similar observations were made for A21 and A30 that crosslinked to the PS1 NTF and PEN‐2, respectively (Figs 3, 4C, EV1 and EV2). These substrates could only be cleaved when they were bound to the PS1 NTF (Fig 6E). Finally, we asked whether another substrate, Notch1, could compete with C99 binding at the exosites. As shown in Fig 6F, excess amounts of the well‐characterized recombinant Notch1‐based γ‐secretase substrate N102‐FmH (Takahashi et al, 2003) competed C99 binding not only as expected at the active site (Fig EV4), but also at the exosites despite having a different primary sequence. This suggests that interaction with the exosites represents a common substrate‐binding step of γ‐secretase and that yet unknown, structural features of substrates are recognized by γ‐secretase at these sites. In summary, these data show that substrate binding to NCT and PEN‐2 occurs before PS1 NTF binding. Together with our data showing that access from the exosites to the active site is prevented in the presence of GSIs, they suggest a mechanism of stepwise substrate binding and transfer from exosites to the active site of γ‐secretase.
Figure 6. Concerted action of exosites and active site in substrate binding and cleavage.
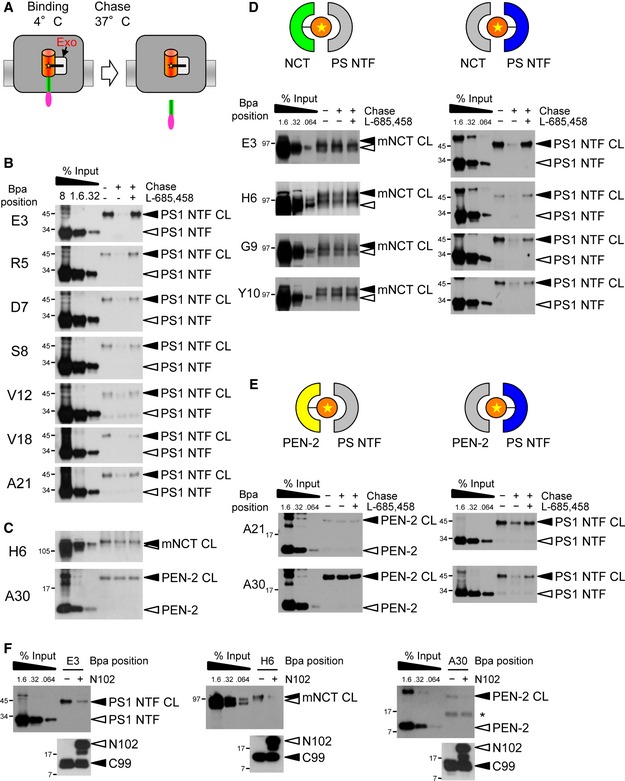
-
ASchematic representation of the substrate‐binding chase experiment assaying whether C99 crosslinked at exosites (Exo) can be cleaved during chase incubation. Note that crosslink product of substrates with Bpa placed in the Aβ region cannot be recovered by Ni‐NTA affinity pulldown after γ‐secretase cleavage in the 37°C chase.
-
BSubstrates bound to exosites in the PS1 NTF at 4°C can be cleaved in the 37°C chase incubation in a L‐685,458‐dependent manner. Note that although many of these substrates crosslinked only weakly at the exosites, their cleavage in the 37°C chase proved that these reflected functional substrate‐binding states.
-
CSubstrates crosslinking in the exosites to NCT and PEN‐2 cannot be cleaved.
-
D, ESubstrates can be cleaved when crosslinked to PS1 NTF, but not when crosslinked to NCT (D) or PEN‐2 (E).
-
FExcess amounts of Notch1 substrate N102‐FmH compete with exosite binding of C99. See Fig EV4 for competition of C99 binding in the active site region.
Figure EV4. Competition of C99 binding to the active site region of γ‐secretase by Notch1.
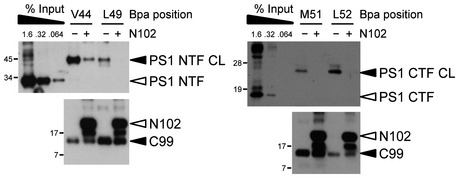
Excess amounts of Notch1‐based N102‐FmH substrate compete with C99‐Bpa substrates crosslinking to γ‐secretase in the active site region.
Discussion
Site‐specific substrate crosslinking provides a powerful approach to identify protease interaction sites
The lack of detailed information on the binding sites of any intramembrane protease for their natural substrates presents a major gap in our mechanistic understanding of intramembrane proteolysis. It is thus unclear whether the substrate recruitment of these proteases follows common principles or whether each class of them employs unique mechanisms. In particular for the AD‐associated γ‐secretase, obtaining information on substrate‐binding sites is also highly relevant for our understanding of the mechanism of FAD mutants and for substrate‐specific drug development. To identify substrate‐binding sites and thereby provide insight into the substrate recognition process of γ‐secretase, we have applied a chemical biology approach to determine the interaction sites of C99, the precursor of Aβ, with γ‐secretase. By site‐specific photocrosslinking of purified C99‐Bpa derivatives with CHAPSO‐solubilized enzyme, we found that most of the substrate interactions occurred within the PS1 NTF, suggesting that this subunit provides the principal substrate‐binding site. The PS1 CTF, NCT, and PEN‐2 were identified as additional substrate‐binding subunits.
Identification of substrate residues binding to exosites in three different subunits, PS1 NTF, NCT, and PEN‐2
Photoaffinity scanning revealed that the extracellular and the N‐terminal TMD region of C99 contacts three different subunits of the γ‐secretase complex: PS1 NTF, NCT, and PEN‐2. The principal interaction sites in these regions of C99 were E3 for the PS1 NTF, H6 for NCT, and A30 for PEN‐2. Since these residues are far away from the cleavage domain spanning residues G37‐L49 of the TMD, the subunits contacted by E3, H6, and A30 should be those that contain exosites. Consistent with this view, binding of these residues was not inhibited when the active site was blocked by L‐685,458, demonstrating that PS1 NTF, NCT, and PEN‐2 provide exosites for substrate binding. Interestingly, rather than inhibiting substrate binding, preventing substrate access to the active site by L‐685,458 increased subunit labeling of E3, H6, and A30. Similar results were obtained with DAPT and all other GSIs of various structural classes that were tested (Appendix Table S1). Thus, substrate residence times at the exosites and/or exposure of the exosites toward the substrate are apparently enhanced when the active site cannot be reached. Our scanning analysis revealed that C99 binding was increased in the presence of GSIs over a broad range, showing that numerous residues of the extracellular domain are in contact with exosites in the PS1 NTF, NCT, and PEN‐2. Interestingly, most Bpa mutations that reduced γ‐secretase cleavage efficiency located in the N‐terminal extracellular domain, indicating that exosite interactions with this domain influence the substrate recognition process. Since the recent high‐resolution structure in the presence of DAPT showed only minor conformational changes compared to the inhibitor‐free form (Bai et al, 2015a), and since GSIs such as DAPT, as small molecules, occupy only little space within the γ‐secretase structure, one might finally also speculate that the cleavage domain of substrates accumulating at the exosites in the presence of a GSI might locate spatially very close to the active site, but can just not access it.
While NCT and PEN‐2 were clearly identified as exosite‐providing non‐catalytic subunits, APH‐1 appeared not to be involved in substrate binding. Although previous data suggested that this subunit could interact with APP CTFs, implying a role in substrate recruitment (Chen et al, 2010), binding of full‐length APP to APH‐1 was reported as well, which conflicted with the known requirement of γ‐secretase substrates to have short ectodomains (Struhl & Adachi, 2000; Bolduc et al, 2016). Unlike observed for all other subunits, we found that potential C99‐APH‐1 crosslink bands were insensitive to L‐685,458 (Appendix Fig S2). This observation, together with the structural arrangement of the γ‐secretase subunits (Sun et al, 2015), in which an APH‐1‐bound substrate would not have an obvious transfer path to the active site, suggests that APH‐1 is not involved in γ‐secretase substrate recruitment.
Previous studies using the short APP TMD‐based helical peptide GSIs D‐10 and D‐13 suggested that a γ‐secretase exosite, termed docking site, locates very close to and overlaps with the active site region in the PS NTF/CTF interface (Kornilova et al, 2005). Accordingly, the exosites in the non‐catalytic subunits PEN‐2 and NCT are distinct from the docking site. Since the C99 residues binding to exosites in the PS1 NTF are distant from those binding in the active site region, these exosites are very likely distinct from the docking site. Consistent with this view, we found that the helical peptide GSIs did not compete exosite substrate binding and rather increased it (Fig EV5A), whereas they strongly inhibited the interaction of substrates targeting the active site (Fig EV5B).
Figure EV5. Helical peptide inhibitors target a substrate‐binding site different from the exosites.
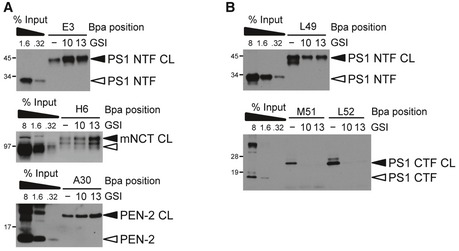
- Helical peptide GSIs D‐10 and D‐13 mimicking TMD segments V36‐I45 (D‐10) and V36‐T48 (D‐13) of APP and targeting a putative exosite termed docking site (D‐10) or both docking site and active site (D‐13) (Kornilova et al, 2005) do not compete for substrate binding at exosites, suggesting that docking site and exosites are distinct. 10, D‐10; 13, D‐13.
- Inhibition of substrate binding in the active site by D‐10 and D‐13 suggests that these GSIs interfere with substrate binding before or at the active site.
The major interaction sites locate in the cleavage domain and face the active site region in the PS1 NTF and CTF
V44, L49, M51, and L52 in the C99 TMD represented the major γ‐secretase interaction sites. These residues displayed extraordinary high crosslink efficiencies compared to the other residues. While V44 and L49 faced the PS1 NTF, M51 and L52 faced the PS1 CTF. Additional more minor interaction sites with the PS1 NTF in this region ranged broadly from V40–L52. Binding of these substrate residues was inhibited by L‐685,458 and DAPT, suggesting that they are in contact with the active site in PS1. M51 and L52 crosslinked also weakly to the PS1 NTF, suggesting that they bind directly at the PS1 NTF/CTF interface. Residues V49–L52 of the cleavage domain are thus very close to the active site interface formed by the PS1 NTF and CTF, which is consistent with the concept that substrate cleavage begins at the ε‐site. Further subsite analysis showed that non‐prime site residues within the cleavage domain upstream of the major ε‐site (ε49) bind the active site in the PS1 NTF and prime‐site residues immediately downstream of ε49 primarily at the PS1 CTF. It should be noted that reduced crosslinking of more distant sites (e.g. A42, V44) in the presence of inhibitor might also be the consequence of inhibitor‐induced conformational changes (Li et al, 2014; Bai et al, 2015a; Elad et al, 2015) that could have reduced the exposure of these crosslink sites.
Residues further downstream in the intracellular domain of the substrate were predominantly bound to the PS1 NTF, but also to PEN‐2. Binding of most residues of the K54–I60 region to PEN‐2 was increased when substrate access to the active site was blocked in the presence of L‐685,458, suggesting that these interacted with additional exosites in this subunit. However, as the crosslinking efficiencies of these residues were extremely low and close to the detection limit, further mechanistic analyses were precluded with our currently available techniques. Finally, it is important to note that from all residues locating between the ε‐ and γ‐sites analyzed, V44 showed the highest crosslinking efficiency, suggesting that it plays a key role in substrate anchoring in the active site region.
Substrate passage from exosites to the active site
A key property of γ‐secretase substrates is that they have very short extracellular domains of about 15–30 amino acids, typically as a result of ectodomain shedding. Our results using C99 suggest that the recognition of these domains involves exosites in the PS1 NTF, PEN‐2, and NCT, supporting a role of the latter subunit in substrate binding (Shah et al, 2005). They further allow important novel mechanistic insight into the functional interplay between the exosites and the active site during substrate binding and catalysis of γ‐secretase substrates. As summarized in Fig 7A, our data are consistent with a model of a stepwise binding path. According to this model, following ectodomain shedding, C99 will initially interact with exosites in NCT and PEN‐2 (stage 1). Release from these sites will then allow the substrate to bind to exosites in the PS1 NTF (stage 2). Following this stage, the substrate can engage the active site for cleavage (stage 3).
Figure 7. Model of a sequential substrate‐binding path of γ‐secretase.
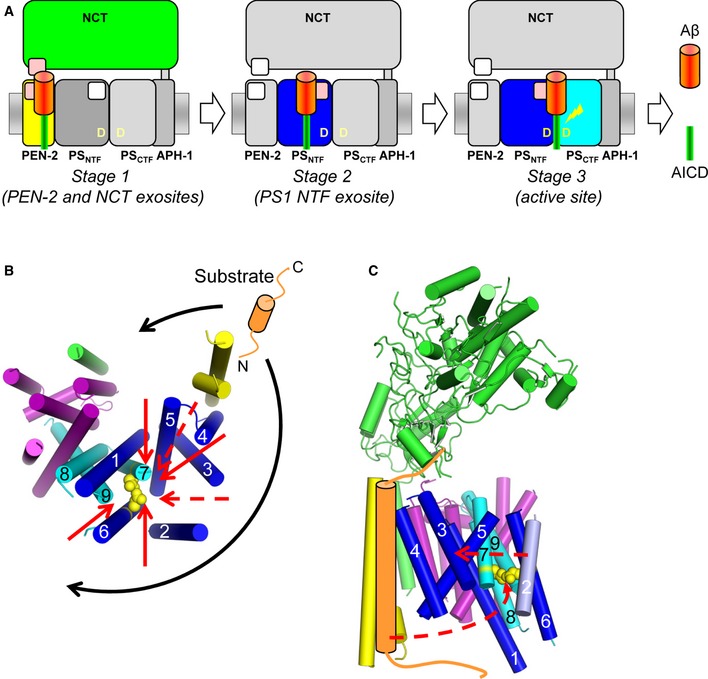
- C99 is first bound at exosites (pink) located in NCT and PEN‐2 (stage 1) and then at exosites in the PS1 NTF (stage 2). When bound at these sites in the PS1 NTF, the cleavage domain of C99 gets access to the γ‐secretase active site formed by the PS1 NTF/CTF heterodimer for peptide bond cleavage (stage 3).
- Potential substrate entry routes to the active site in the context of the γ‐secretase structure. Assuming larger conformational changes upon substrate binding, the access of substrate to the active site appears possible from both convex and concave sides of the horseshoe structure of γ‐secretase (black arrows). Several hypothetical substrate transfer routes (red arrows) from the exosites in PEN‐2 and NCT to the active site are shown in bottom view. Dashed red arrows indicate paths with potential barriers imposed by cytosolic loops between TMDs 2/3 and 4/5 of PS1. Catalytic residues are shown in yellow with side chains as spheres. For clarity, only the TMD domain of NCT is shown.
- Hypothetical conformational movements of PEN‐2 and PS1 TMD2 for the presentation of C99 to the active site from the convex side are shown by dashed red arrows.
Remarkably, although different in primary sequence, Notch1 competed exosite binding of C99, suggesting that it bound to the same exosites as APP. This indicates that structural properties of the substrate rather than specific sequence recognition motifs are recognized by the exosites on γ‐secretase. This flexibility in exosite binding may explain why γ‐secretase can recognize and cleave so many substrates that are diverse in their primary sequence and indicates that the substrate‐binding path deduced here might be common for all γ‐secretase substrates, although it is also possible that the exosite usage is substrate dependent, thus impacting on substrate selectivity.
How do intramembrane protease substrates gain access to the catalytic residues for their cleavage? Based on the currently available structural information, it is possible that intramembrane proteases may switch between open and closed conformations of the active site to allow substrate entry. Access of substrates to the active site may occur by a gating mechanism, which may involve either TMD movement as in the case of site 2 protease or as in the case of rhomboids alternatively by opening of a lid formed by one of the rhomboid loop domains or a combination of both (Strisovsky, 2013). For γ‐secretase, substrate access to the active site may involve movements of TMD2 and TMD6 of PS, which display substantial flexibility in the atomic structure of the enzyme (Bai et al, 2015b) and which, interestingly, had been implicated earlier in substrate binding by mutational analysis (Watanabe et al, 2010). In particular, TMD2 is highly flexible, suggesting that it might be involved in substrate entry to the active site—potentially acting as a lateral gate. This view is supported by latest structural data showing that the flexibility of TMD2 is greatly reduced upon binding of DAPT (Bai et al, 2015a).
Previous biochemical data suggested that PEN‐2 is in close proximity to the PS1 CTF and possibly in the vicinity of the catalytic site (Bammens et al, 2011). However, PEN‐2 locates rather distant from the active site in the atomic γ‐secretase structure (Bai et al, 2015b). Considering our findings that C99 was also bound to PEN‐2 before it could access the active site, it is thus likely that conformational changes in the enzyme will be required for the translocation of exosite‐bound substrates to the active site (Fig 7B and C). Besides a potential lateral movement of PEN‐2 toward the active site, such changes could also involve NCT (Li et al, 2014; Bai et al, 2015b) and would be consistent with the observation that γ‐secretase can exist in different conformations (Bai et al, 2015a; Elad et al, 2015). Conformational changes are also required to bring the catalytic aspartates in TMD6 and TMD7 of PS, which are too distant for an active aspartyl protease in the atomic structure, in closer vicinity to catalyze peptide bond cleavage. Notably, concomitantly occurring movements of PEN‐2 and TMDs 3, 4, 5, and 6 of the PS1 NTF have been reported recently (Bai et al, 2015a). Furthermore, conformational variations of TMDs 2 and 6 were visible in a substrate binding‐mimicking conformation with an additional TMD from a co‐isolated single‐pass membrane protein (Bai et al, 2015a), supporting the concept that conformational changes occur during the substrate recruitment. Interestingly, conformational changes upon the substrate binding to an exosite have also been suggested by kinetic analysis of rhomboid proteases, indicating that these could be a common feature of intramembrane proteases (Arutyunova et al, 2014).
Finally, we suggest that the sequential usage of exosites shown here for γ‐secretase might be a general mechanism of intramembrane proteases for substrate recognition and recruitment. Based on experiments with rhomboid, intramembrane proteases may recognize substrates primarily by TMD instabilities (Moin & Urban, 2012). Rhomboid patrols the membrane for such substrates using an exosite termed “interrogation” site (Dickey et al, 2013). In a similar manner, γ‐secretase may presumably scan substrates for the presence of permissive domains by the exosites in PEN‐2, NCT, and the PS1 NTF. Thus, exosites in intramembrane proteases may help to identify conformationally more flexible substrates and discriminate them from less flexible ones. Substrate sampling by sequential binding to and release from exosites as shown here for γ‐secretase may be a common mechanism of intramembrane proteases to identify their cognate substrates and may also explain why rhomboids and γ‐secretase are very slow enzymes (Dickey et al, 2013; Arutyunova et al, 2014; Kamp et al, 2015).
Altered substrate binding of clinical PS mutants implies a pathogenic mechanism for FAD
PS FAD mutants affect the efficiency of the carboxypeptidase‐like activity with which γ‐secretase processes the TMD of C99 (Quintero‐Monzon et al, 2011; Chavez‐Gutierrez et al, 2012; Okochi et al, 2013; Fernandez et al, 2014; Szaruga et al, 2015). This manifests in an increased ratio of Aβ42 and Aβ43 species over Aβ40, the predominant Aβ species generated by γ‐secretase (Scheuner et al, 1996; Chavez‐Gutierrez et al, 2012). However, the mechanistic basis of altered C99 processing by γ‐secretase caused by the FAD mutants has not yet been established. Several FAD mutations including the aggressive PS1 L166P mutant have been shown to display altered transition state analog GSI binding, suggesting an altered active site topography (Kornilova et al, 2005; Chau et al, 2012). In our study, we made the important observation that the PS1 L166P mutant causes several positional shifts of substrate crosslinking sites in the active site mostly around the ε‐cleavage site. Similar but weaker positional shifts were also found for the milder A246E mutation. The occurrence of prominent binding‐site shifts at the initial ε‐cleavage sites is consistent with altered downstream processing pathways leading to the production of pathogenic Aβ species by the FAD mutants (Morishima‐Kawashima, 2014). Moreover, for both FAD mutants, binding to the PS1 CTF was reduced. On the other hand, substantial changes in exosite binding were not observed for the FAD mutants, suggesting that they primarily affected the positioning of the substrate cleavage domain region. Our data thus suggest that altered interaction of the C99 cleavage domain with γ‐secretase is a decisive mechanism underlying the increased relative levels of Aβ42(43) observed for PS FAD mutants. Future analysis of a larger spectrum of FAD mutations will show whether or not these mutants affect substrate binding in a common way or whether each FAD mutation will show an individual pattern of C99 binding alterations, which, as our data on two different mutations indicate, might be more likely.
Taken together, besides altered C99 positioning in the active site region, changes in the active site conformation, reduced processivity, or combinations of these effects impact mechanistically on the generation of pathogenic Aβ species by PS FAD mutants. Alterations in binding of the C99 cleavage domain region could actually also underlie the reduced processivity that is commonly observed for PS FAD mutants. Independent of whether or not substrate binding of C99 changes at the ε‐site, the ensuing Aβ49/46/43 or Aβ48/45 γ‐secretase substrates may continue to be improperly positioned or not effectively bound for the subsequent processing steps to occur efficiently, ultimately causing increased Aβ42(43)/Aβ40 ratios. This is supported by previous observations that PS FAD mutations weakened the binding of Aβ42 and reduced the efficiency of γ‐secretase for its conversion to Aβ38 (Okochi et al, 2013).
Limitations
The introduction of Bpa into proteins for crosslinking purposes generates mutant proteins. This unavoidable intrinsic limitation of the crosslinking approach could, besides the effects on overall activity, also have caused alterations of C99 processing leading to a relative increase in pathogenic Aβ species as observed for many APP FAD mutants (Weggen & Beher, 2012). However, the large majority of the C99‐Bpa substrates did not show this effect, showing that the introduction of Bpa per se was not causing pathogenic APP processing. Only very few of the C99‐Bpa substrates caused relative increases in Aβ42 generation—mostly at positions known earlier to cause such cleavage specificity changes for synthetic and clinical Bpa‐related phenylalanine mutants of the APP TMD (Lichtenthaler et al, 1999).
It should be further noted that although the use of CHAPSO‐solubilized γ‐secretase allowed the identification of substrate–protease interactions of an active γ‐secretase, we cannot exclude that some crosslink sites and/or intensities might change when alternative complex‐maintaining detergents would be tried. Although the cryo‐EM structure of γ‐secretase is nearly identical in digitonin and amphiphol, ruling out major impacts by the detergent, minor transmembrane domain shifts have been observed (Sun et al, 2015) which could alter the crosslink patterns observed in our study. It is also possible that other substrate residues might be exposed due to conformational adaptations of the complex when the complex is reconstituted into model membranes of various lipid compositions.
Implications for protease research and drug development
To our knowledge, systematic, site‐specific incorporation of the photocrosslinkable amino acid Bpa using a genetic system has been used here for the first time as a novel strategy to identify and map the substrate‐binding sites of a protease. While this powerful approach will be generally applicable for any protease to identify its substrate interaction sites, it could be particularly helpful for the identification of substrate‐binding sites of other intramembrane proteases that have more difficult to analyze membrane‐spanning substrates. As shown here for C99, when authentic full‐length substrates are used, the identification of distantly located exosites will be achievable, which is not possible in classical approaches using short inhibitor‐based substrate mimics that bind in the active site region. Thus, a full spectrum of substrate–protease interaction sites can be obtained with genetically engineered full‐length substrates extending previous analysis possibilities. Furthermore, substrate‐binding chase experiments could also be generally used to elucidate substrate movements and to reveal potential substrate paths from protease exosites to the active site. Finally, besides the prime importance of elucidating the mechanism of substrate recognition by intramembrane proteases for our general understanding of intramembrane proteolysis, especially for γ‐secretase as an AD drug target, information on the substrate–protease interaction sites is also key for the development of substrate‐selective inhibitors, since the lack of sufficient substrate specificity is one of the likely causes of the failure of GSIs in clinical trials (Imbimbo & Giardina, 2011). Here, the powerful substrate crosslinking approach used in this study may be helpful to elucidate and identify differences in the recruitment of diverse γ‐secretase substrates, which could possibly be exploited to develop GSIs with improved selectivity or perhaps even substrate‐targeting molecules. In addition, this approach can be used to shed further light on the mechanism of γ‐secretase modulators as well as to develop drugs that will restore normal substrate positioning of PS FAD mutants.
Materials and Methods
Antibodies
The following polyclonal and monoclonal antibodies were used: N1660 against NCT (Sigma), O2C2 against APH‐1aL (Thermo Scientific), 1638 against PEN‐2 (Steiner et al, 2002), PS1NT against the PS1 NTF (Capell et al, 1997; Covance), 5E12 against the PS1 CTF (Kretner et al, 2016), and penta‐His (Qiagen).
γ‐Secretase inhibitors (GSIs)
L‐685,458 (Shearman et al, 2000) was obtained from Calbiochem. LY‐411575 (May et al, 2001) and III‐31‐C (Esler et al, 2002) were purchased from Sigma. DAPT (Dovey et al, 2001), ELN594 (Liebscher et al, 2014), and begacestat (Mayer et al, 2008) were kind gifts from Boris Schmidt, Technical University Darmstadt, Guriqbal Basi, Elan Pharmaceuticals and Karlheinz Baumann, Roche, respectively. D‐10 and D‐13 (Kornilova et al, 2005) were synthesized by BEX (Tokyo, Japan).
Cell culture and cell lines
Untransfected HEK293 cells and single cell clones of HEK293 cells stably transfected with Swedish APP and WT and mutant PS1 variants were cultured as described (Steiner et al, 2000).
Cloning, expression, purification, and in vitro cleavage of Bpa‐containing γ‐secretase substrates
Amber codons were introduced at the desired sites in the C100‐His6 cDNA using standard cloning techniques. Constructs were co‐expressed in E. coli with suppressor tRNA and tRNA synthetase allowing site‐specific introduction of Bpa at the amber codon sites and affinity‐purified using Ni‐NTA. Analysis of substrate cleavage was performed using CHAPSO lysates of HEK293 cells as enzyme source. Full details are given in the Appendix.
Substrate photocrosslinking
HEK293 cells (three 15‐cm dishes) were lysed in 900 μl of 50 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM CaCl2, 5 mM MgCl2, 1% CHAPSO, 1× PI mix without EDTA (Roche) for 1 h on ice. Following ultracentrifugation at 100,000 × g for 90 min at 4°C, lysates were diluted to 0.35% CHAPSO; 70 μl aliquots of the lysate were mixed with 2 μM purified Bpa substrates and irradiated at 365 nm with a 3UV lamp (8 W, 230 V, 50 Hz; UVP, Upland, CA, USA) in ~1 cm distance for 30 min on ice. Irradiation time was reduced to 15 min for quantitation experiments. To confirm crosslink specificity, 1% Triton X‐100 was added to dissociate γ‐secretase. Water was added to the control samples. To assess competition of substrate binding, 20 μM GSIs was added. DMSO was added to the corresponding vehicle controls. After irradiation, the samples were immediately mixed with 2 volumes of 50 mM Tris–HCl, pH 8.5, 500 mM NaCl, 5 mM CaCl2, 5 mM MgCl2, 1% SDS, and 2 M urea to dissociate the γ‐secretase complex and mixed with Ni‐NTA agarose beads. Following 1‐h incubation at RT with shaking, the beads were washed three times with the same buffer and captured proteins were eluted with 2× SDS–PAGE sample buffer containing 2 M urea and 200 mM imidazole. Samples were separated by SDS–PAGE and immunostained with antibodies against the γ‐secretase complex subunits. For competition experiments using substrates as competitors, 0.4 μM of the respective photocrosslinkable substrates and 2 μM of competitive APP and Notch1 substrates were used to avoid the presence of excess SDS from the purified protein preparations. Negative control samples received the same amount of elution buffer as vehicle. For substrate‐binding chase experiments, following UV irradiation, the samples were supplemented with 20 μM L‐685,458 or DMSO as vehicle and immediately put into a pre‐warmed water bath at 37°C. After 1‐h incubation, the samples were dissociated as above and subjected to Ni‐NTA affinity pulldown as described above.
Quantitative analysis and validation of substrate crosslinking
Intensities of crosslink bands were quantified from immunoblots using luminescent image analyzer (LAS‐4000; Fujifilm, Tokyo, Japan). For all crosslink sites, additional validation experiments were performed by 30 min UV irradiation in the presence or absence of L‐685,458.
Deglycosylation of substrates crosslinked to NCT
Following Ni‐NTA affinity pulldown, the samples were additionally washed twice with 50 mM Tris–HCl, pH 8.5, 500 mM NaCl, 5 mM CaCl2, 5 mM MgCl2, 1% SDS. After the addition of 6 μl of 50 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM CaCl2, 5 mM MgCl2, 1% CHAPSO, 1× PI mix without EDTA (Roche), and 5 mU endoglycosidase H (Roche) and incubation overnight at 37°C, the samples were analyzed by immunoblotting.
Author contributions
AF and HS conceived and designed the experiments. AF performed all experiments. AF and HS analyzed the data and interpreted the results. HS initiated the project and wrote the paper with contributions from AF.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We are grateful to Peter G. Schultz, Guriqbal Basi, Karlheinz Baumann, Boris Schmidt, Taisuke Tomita, and Takeshi Iwatsubo for reagents; Gabriele Basset, Johannes Trambauer, and Edith Winkler for technical help; Masanori Tomioka, Keiro Shirotani, Laura Haas, and Stefan Berchtold for contributing to the project; and Christian Haass for support and advice. We also thank Christian Haass, Dieter Langosch, Marius Lemberg, and Frits Kamp for helpful comments on the manuscript and all members of our group for discussions and suggestions. This work was supported in part by the Hans and Ilse Breuer Foundation and the DFG (SFB596 and FOR2290).
The EMBO Journal (2016) 35: 1628–1643
See also: L Chávez‐Gutiérrez & B De Strooper (August 2016)
References
- Arutyunova E, Panwar P, Skiba PM, Gale N, Mak MW, Lemieux MJ (2014) Allosteric regulation of rhomboid intramembrane proteolysis. EMBO J 33: 1869–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XC, Rajendra E, Yang G, Shi Y, Scheres SH (2015a) Sampling the conformational space of the catalytic subunit of human γ‐secretase. eLife 4: e11182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XC, Yan C, Yang G, Lu P, Ma D, Sun L, Zhou R, Scheres SH, Shi Y (2015b) An atomic structure of human γ‐secretase. Nature 525: 212–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bammens L, Chavez‐Gutierrez L, Tolia A, Zwijsen A, De Strooper B (2011) Functional and topological analysis of Pen‐2, the fourth subunit of the γ‐secretase complex. J Biol Chem 286: 12271–12282 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bolduc DM, Montagna DR, Gu Y, Selkoe DJ, Wolfe MS (2016) Nicastrin functions to sterically hinder γ‐secretase‐substrate interactions driven by substrate transmembrane domain. Proc Natl Acad Sci USA 113: E509–E518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capell A, Saffrich R, Olivo JC, Meyn L, Walter J, Grunberg J, Mathews P, Nixon R, Dotti C, Haass C (1997) Cellular expression and proteolytic processing of presenilin proteins is developmentally regulated during neuronal differentiation. J Neurochem 69: 2432–2440 [DOI] [PubMed] [Google Scholar]
- Chau DM, Crump CJ, Villa JC, Scheinberg DA, Li YM (2012) Familial Alzheimer disease presenilin‐1 mutations alter the active site conformation of γ‐secretase. J Biol Chem 287: 17288–17296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez‐Gutierrez L, Tolia A, Maes E, Li T, Wong PC, de Strooper B (2008) Glu332 in the nicastrin ectodomain is essential for γ‐secretase complex maturation but not for its activity. J Biol Chem 283: 20096–20105 [DOI] [PubMed] [Google Scholar]
- Chavez‐Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B (2012) The mechanism of γ‐secretase dysfunction in familial Alzheimer disease. EMBO J 31: 2261–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AC, Guo LY, Ostaszewski BL, Selkoe DJ, LaVoie MJ (2010) Aph‐1 associates directly with full‐length and C‐terminal fragments of γ‐secretase substrates. J Biol Chem 285: 11378–11391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin JW, Martin AB, King DS, Wang L, Schultz PG (2002) Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli . Proc Natl Acad Sci USA 99: 11020–11024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F (1998) Deficiency of presenilin‐1 inhibits the normal cleavage of amyloid precursor protein. Nature 391: 387–390 [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R (1999) A presenilin‐1‐dependent γ‐secretase‐like protease mediates release of Notch intracellular domain. Nature 398: 518–522 [DOI] [PubMed] [Google Scholar]
- De Strooper B, Iwatsubo T, Wolfe MS (2012) Presenilins and γ‐secretase: structure, function, and role in Alzheimer disease. Cold Spring Harb Perspect Med 2: a006304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Chavez Gutierrez L (2015) Learning by failing: ideas and concepts to tackle γ‐secretases in Alzheimer's disease and beyond. Annu Rev Pharmacol Toxicol 55: 419–437 [DOI] [PubMed] [Google Scholar]
- Dickey SW, Baker RP, Cho S, Urban S (2013) Proteolysis inside the membrane is a rate‐governed reaction not driven by substrate affinity. Cell 155: 1270–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman G, Prestwich GD (1994) Benzophenone photophores in biochemistry. Biochemistry 33: 5661–5673 [DOI] [PubMed] [Google Scholar]
- Dovey HF, John V, Anderson JP, Chen LZ, de Saint Andrieu P, Fang LY, Freedman SB, Folmer B, Goldbach E, Holsztynska EJ, Hu KL, Johnson‐Wood KL, Kennedy SL, Kholodenko D, Knops JE, Latimer LH, Lee M, Liao Z, Lieberburg IM, Motter RN et al (2001) Functional γ‐secretase inhibitors reduce β‐amyloid peptide levels in brain. J Neurochem 76: 173–181 [DOI] [PubMed] [Google Scholar]
- Drag M, Salvesen GS (2010) Emerging principles in protease‐based drug discovery. Nat Rev Drug Discov 9: 690–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dries DR, Shah S, Han YH, Yu C, Yu S, Shearman MS, Yu G (2009) Glu‐333 of nicastrin directly participates in γ‐secretase activity. J Biol Chem 284: 29714–29724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C (2003) Reconstitution of γ‐secretase activity. Nat Cell Biol 5: 486–488 [DOI] [PubMed] [Google Scholar]
- Elad N, De Strooper B, Lismont S, Hagen W, Veugelen S, Arimon M, Horre K, Berezovska O, Sachse C, Chavez‐Gutierrez L (2015) The dynamic conformational landscape of γ‐secretase. J Cell Sci 128: 589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erez E, Fass D, Bibi E (2009) How intramembrane proteases bury hydrolytic reactions in the membrane. Nature 459: 371–378 [DOI] [PubMed] [Google Scholar]
- Esler WP, Kimberly WT, Ostaszewski BL, Ye W, Diehl TS, Selkoe DJ, Wolfe MS (2002) Activity‐dependent isolation of the presenilin–γ‐secretase complex reveals nicastrin and a γ substrate. Proc Natl Acad Sci USA 99: 2720–2725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez MA, Klutkowski JA, Freret T, Wolfe MS (2014) Alzheimer presenilin‐1 mutations dramatically reduce trimming of long amyloid β‐peptides (Aβ) by γ‐secretase to increase 42‐to‐40‐residue Aβ. J Biol Chem 289: 31043–31052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funamoto S, Morishima‐Kawashima M, Tanimura Y, Hirotani N, Saido TC, Ihara Y (2004) Truncated carboxyl‐terminal fragments of β‐amyloid precursor protein are processed to amyloid β‐proteins 40 and 42. Biochemistry 43: 13532–13540 [DOI] [PubMed] [Google Scholar]
- Haapasalo A, Kovacs DM (2011) The many substrates of presenilin/γ‐secretase. J Alzheimer's Dis 25: 3–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β‐peptide. Nat Rev Mol Cell Biol 8: 101–112 [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM (2011) Alzheimer's disease: the challenge of the second century. Sci Transl Med 3: 77sr71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbimbo BP, Giardina GA (2011) γ‐Secretase inhibitors and modulators for the treatment of Alzheimer's disease: disappointments and hopes. Curr Top Med Chem 11: 1555–1570 [DOI] [PubMed] [Google Scholar]
- Iwata H, Tomita T, Maruyama K, Iwatsubo T (2001) Subcellular compartment and molecular subdomain of β‐amyloid precursor protein relevant to the Aβ42‐promoting effects of Alzheimer mutant presenilin 2. J Biol Chem 276: 21678–21685 [DOI] [PubMed] [Google Scholar]
- Kakuda N, Funamoto S, Yagishita S, Takami M, Osawa S, Dohmae N, Ihara Y (2006) Equimolar production of amyloid β‐protein and amyloid precursor protein intracellular domain from β‐carboxyl‐terminal fragment by γ‐secretase. J Biol Chem 281: 14776–14786 [DOI] [PubMed] [Google Scholar]
- Kamp F, Winkler E, Trambauer J, Ebke A, Fluhrer R, Steiner H (2015) Intramembrane proteolysis of β‐amyloid precursor protein by γ‐secretase is an unusually slow process. Biophys J 108: 1229–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Ikeuchi T, Yu C, Sisodia SS (2003) Regulated hyperaccumulation of presenilin‐1 and the “γ‐secretase” complex. Evidence for differential intramembranous processing of transmembrane substrates. J Biol Chem 278: 33992–34002 [DOI] [PubMed] [Google Scholar]
- Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ (2003) γ‐Secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph‐1, and Pen‐2. Proc Natl Acad Sci USA 100: 6382–6387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornilova AY, Bihel F, Das C, Wolfe MS (2005) The initial substrate‐binding site of γ‐secretase is located on presenilin near the active site. Proc Natl Acad Sci USA 102: 3230–3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretner B, Trambauer J, Fukumori A, Mielke J, Kuhn PH, Kremmer E, Giese A, Lichtenthaler SF, Haass C, Arzberger T, Steiner H (2016) Generation and deposition of Aβ43 by the virtually inactive presenilin‐1 L435F mutant contradicts the presenilin loss‐of‐function hypothesis of Alzheimer's disease. EMBO Mol Med 8: 458–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langosch D, Scharnagl C, Steiner H, Lemberg MK (2015) Understanding intramembrane proteolysis: from protein dynamics to reaction kinetics. Trends Biochem Sci 40: 318–327 [DOI] [PubMed] [Google Scholar]
- Lemberg MK (2013) Sampling the membrane: function of rhomboid‐family proteins. Trends Cell Biol 23: 210–217 [DOI] [PubMed] [Google Scholar]
- Li YM, Lai MT, Xu M, Huang Q, DiMuzio‐Mower J, Sardana MK, Shi XP, Yin KC, Shafer JA, Gardell SJ (2000a) Presenilin 1 is linked with γ‐secretase activity in the detergent solubilized state. Proc Natl Acad Sci USA 97: 6138–6143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio‐Mower J, Harrison T, Lellis C, Nadin A, Neduvelli JG, Register RB, Sardana MK, Shearman MS, Smith AL, Shi XP, Yin KC, Shafer JA, Gardell SJ (2000b) Photoactivated γ‐secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405: 689–694 [DOI] [PubMed] [Google Scholar]
- Li Y, Lu SH, Tsai CJ, Bohm C, Qamar S, Dodd RB, Meadows W, Jeon A, McLeod A, Chen F, Arimon M, Berezovska O, Hyman BT, Tomita T, Iwatsubo T, Johnson CM, Farrer LA, Schmitt‐Ulms G, Fraser PE, St George‐Hyslop PH (2014) Structural interactions between inhibitor and substrate docking sites give insight into mechanisms of human PS1 complexes. Structure 22: 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenthaler SF, Wang R, Grimm H, Uljon SN, Masters CL, Beyreuther K (1999) Mechanism of the cleavage specificity of Alzheimer's disease γ‐secretase identified by phenylalanine‐scanning mutagenesis of the transmembrane domain of the amyloid precursor protein. Proc Natl Acad Sci USA 96: 3053–3058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenthaler SF, Haass C, Steiner H (2011) Regulated intramembrane proteolysis–lessons from amyloid precursor protein processing. J Neurochem 117: 779–796 [DOI] [PubMed] [Google Scholar]
- Liebscher S, Page RM, Kafer K, Winkler E, Quinn K, Goldbach E, Brigham EF, Quincy D, Basi GS, Schenk DB, Steiner H, Bonhoeffer T, Haass C, Meyer‐Luehmann M, Hubener M (2014) Chronic γ‐secretase inhibition reduces amyloid plaque‐associated instability of pre‐ and postsynaptic structures. Mol Psychiatry 19: 937–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PC, Altstiel L, Bender MH, Boggs LN, Calligaro DO, Fuson KS, Gitter BD, Hyslop PA, Jordan WH, Li WY, Mabry TE, Mark RJ, Ni B, Nissen JS, Porter WJ, Sorgen SG, Su Y, Audia JE, Dovey HF, Games D et al (2001) Marked reduction of Aβ accumulation and β‐amyloid plaque pathology in mice upon chronic treatment with a functional γ‐secretase inhibitor. Soc Neurosci Abstr 27: 687 [Google Scholar]
- Mayer SC, Kreft AF, Harrison B, Abou‐Gharbia M, Antane M, Aschmies S, Atchison K, Chlenov M, Cole DC, Comery T, Diamantidis G, Ellingboe J, Fan K, Galante R, Gonzales C, Ho DM, Hoke ME, Hu Y, Huryn D, Jain U et al (2008) Discovery of begacestat, a Notch‐1‐sparing γ‐secretase inhibitor for the treatment of Alzheimer's disease. J Med Chem 51: 7348–7351 [DOI] [PubMed] [Google Scholar]
- Moehlmann T, Winkler E, Xia X, Edbauer D, Murrell J, Capell A, Kaether C, Zheng H, Ghetti B, Haass C, Steiner H (2002) Presenilin‐1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Aβ42 production. Proc Natl Acad Sci USA 99: 8025–8030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moin SM, Urban S (2012) Membrane immersion allows rhomboid proteases to achieve specificity by reading transmembrane segment dynamics. eLife 1: e00173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima‐Kawashima M (2014) Molecular mechanism of the intramembrane cleavage of the β‐carboxyl terminal fragment of amyloid precursor protein by γ‐secretase. Front Physiol 5: 463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, Wong PC, Sisodia SS (1998) Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron 21: 1213–1221 [DOI] [PubMed] [Google Scholar]
- Okochi M, Tagami S, Yanagida K, Takami M, Kodama TS, Mori K, Nakayama T, Ihara Y, Takeda M (2013) γ‐Secretase modulators and presenilin 1 mutants act differently on presenilin/γ‐secretase function to cleave Aβ42 and Aβ43. Cell Rep 3: 42–51 [DOI] [PubMed] [Google Scholar]
- Qi‐Takahara Y, Morishima‐Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R, Ihara Y (2005) Longer forms of amyloid β protein: implications for the mechanism of intramembrane cleavage by γ‐secretase. J Neurosci 25: 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero‐Monzon O, Martin MM, Fernandez MA, Cappello CA, Krzysiak AJ, Osenkowski P, Wolfe MS (2011) Dissociation between the processivity and total activity of γ‐secretase: implications for the mechanism of Alzheimer's disease‐causing presenilin mutations. Biochemistry 50: 9023–9035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy‐Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D et al (1996) Secreted amyloid β‐protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med 2: 864–870 [DOI] [PubMed] [Google Scholar]
- Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, Laplant Q, Ball H, Dann CE III, Sudhof T, Yu G (2005) Nicastrin functions as a γ‐secretase‐substrate receptor. Cell 122: 435–447 [DOI] [PubMed] [Google Scholar]
- Shearman MS, Beher D, Clarke EE, Lewis HD, Harrison T, Hunt P, Nadin A, Smith AL, Stevenson G, Castro JL (2000) L‐685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid β‐protein precursor γ‐secretase activity. Biochemistry 39: 8698–8704 [DOI] [PubMed] [Google Scholar]
- Shelton CC, Zhu L, Chau D, Yang L, Wang R, Djaballah H, Zheng H, Li YM (2009) Modulation of γ‐secretase specificity using small molecule allosteric inhibitors. Proc Natl Acad Sci USA 106: 20228–20233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Kostka M, Romig H, Basset G, Pesold B, Hardy J, Capell A, Meyn L, Grim MG, Baumeister R, Fechteler K, Haass C (2000) Glycine 384 is required for presenilin‐1 function and is conserved in polytopic bacterial aspartyl proteases. Nat Cell Biol 2: 848–851 [DOI] [PubMed] [Google Scholar]
- Steiner H, Winkler E, Edbauer D, Prokop S, Basset G, Yamasaki A, Kostka M, Haass C (2002) PEN‐2 is an integral component of the γ‐secretase complex required for coordinated expression of presenilin and nicastrin. J Biol Chem 277: 39062–39065 [DOI] [PubMed] [Google Scholar]
- Steiner H, Fluhrer R, Haass C (2008) Intramembrane proteolysis by γ‐secretase. J Biol Chem 283: 29627–29631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strisovsky K, Sharpe HJ, Freeman M (2009) Sequence‐specific intramembrane proteolysis: identification of a recognition motif in rhomboid substrates. Mol Cell 36: 1048–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strisovsky K (2013) Structural and mechanistic principles of intramembrane proteolysis–lessons from rhomboids. FEBS J 280: 1579–1603 [DOI] [PubMed] [Google Scholar]
- Struhl G, Greenwald I (1999) Presenilin is required for activity and nuclear access of Notch in Drosophila . Nature 398: 522–525 [DOI] [PubMed] [Google Scholar]
- Struhl G, Adachi A (2000) Requirements for presenilin‐dependent cleavage of Notch and other transmembrane proteins. Mol Cell 6: 625–636 [DOI] [PubMed] [Google Scholar]
- Sun L, Zhao L, Yang G, Yan C, Zhou R, Zhou X, Xie T, Zhao Y, Wu S, Li X, Shi Y (2015) Structural basis of human γ‐secretase assembly. Proc Natl Acad Sci USA 112: 6003–6008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaruga M, Veugelen S, Benurwar M, Lismont S, Sepulveda‐Falla D, Lleo A, Ryan NS, Lashley T, Fox NC, Murayama S, Gijsen H, De Strooper B, Chavez‐Gutierrez L (2015) Qualitative changes in human γ‐secretase underlie familial Alzheimer's disease. J Exp Med 212: 2003–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Hayashi I, Tominari Y, Rikimaru K, Morohashi Y, Kan T, Natsugari H, Fukuyama T, Tomita T, Iwatsubo T (2003) Sulindac sulfide is a noncompetitive γ‐secretase inhibitor that preferentially reduces Aβ42 generation. J Biol Chem 278: 18664–18670 [DOI] [PubMed] [Google Scholar]
- Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima‐Kawashima M, Funamoto S, Ihara Y (2009) γ‐Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of β‐carboxyl terminal fragment. J Neurosci 29: 13042–13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T (2003) The role of presenilin cofactors in the γ‐secretase complex. Nature 422: 438–441 [DOI] [PubMed] [Google Scholar]
- Tian G, Sobotka‐Briner CD, Zysk J, Liu X, Birr C, Sylvester MA, Edwards PD, Scott CD, Greenberg BD (2002) Linear non‐competitive inhibition of solubilized human γ‐secretase by pepstatin A methylester, L685458, sulfonamides, and benzodiazepines. J Biol Chem 277: 31499–31505 [DOI] [PubMed] [Google Scholar]
- Urban S (2009) Making the cut: central roles of intramembrane proteolysis in pathogenic microorganisms. Nat Rev Microbiol 7: 411–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Image I II, Takagi S, Tominaga A, Image Image I, Tomita T, Iwatsubo T (2010) Functional analysis of the transmembrane domains of presenilin 1: participation of transmembrane domains 2 and 6 in the formation of initial substrate‐binding site of γ‐secretase. J Biol Chem 285: 19738–19746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weggen S, Beher D (2012) Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal‐dominant Alzheimer's disease. Alzheimers Res Ther 4: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS (2012) γ‐Secretase inhibitors and modulators for Alzheimer's disease. J Neurochem 120(Suppl 1): 89–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Hoey RJ, Lin G, Koide A, Leung B, Ahn K, Dolios G, Paduch M, Ikeuchi T, Wang R, Li YM, Koide S, Sisodia SS (2012) Identification of a tetratricopeptide repeat‐like domain in the nicastrin subunit of γ‐secretase using synthetic antibodies. Proc Natl Acad Sci USA 109: 8534–8539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Cui MZ, Mao G, Dong Y, Tan J, Sun L, Xu X (2005) γ‐Cleavage is dependent on ζ‐cleavage during the proteolytic processing of amyloid precursor protein within its transmembrane domain. J Biol Chem 280: 37689–37697 [DOI] [PubMed] [Google Scholar]
- Zhao G, Liu Z, Ilagan MX, Kopan R (2010) γ‐Secretase composed of PS1/Pen2/Aph1a can cleave notch and amyloid precursor protein in the absence of nicastrin. J Neurosci 30: 1648–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File