

Abstract
A primary cilium is a microtubule‐based sensory organelle that plays an important role in human development and disease. However, regulation of Akt in cilia and its role in ciliary development has not been demonstrated. Using yeast two‐hybrid screening, we demonstrate that Inversin (INVS) interacts with Akt. Mutation in the INVS gene causes nephronophthisis type II (NPHP2), an autosomal recessive chronic tubulointerstitial nephropathy. Co‐immunoprecipitation assays show that Akt interacts with INVS via the C‐terminus. In vitro kinase assays demonstrate that Akt phosphorylates INVS at amino acids 864–866 that are required not only for Akt interaction, but also for INVS dimerization. Co‐localization of INVS and phosphorylated form of Akt at the basal body is augmented by PDGF‐AA. Akt‐null MEF cells as well as siRNA‐mediated inhibition of Akt attenuated ciliary growth, which was reversed by Akt reintroduction. Mutant phosphodead‐ or NPHP2‐related truncated INVS, which lack Akt phosphorylation sites, suppress cell growth and exhibit distorted lumen formation and misalignment of spindle axis during cell division. Further studies will be required for elucidating functional interactions of Akt–INVS at the primary cilia for identifying the molecular mechanisms underlying NPHP2.
Keywords: Akt, Inversin, primary cilia, signal transduction
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Molecular Biology of Disease; Signal Transduction
Introduction
Primary cilia are microtubule (MT)‐based sensory organelles that arise from a basal body surrounded by pericentriolar material and extend from the surface (where the mother centriole is located) during growth arrest in most vertebrate cells (Nigg & Raff, 2009; Hildebrandt et al, 2011) (Suizu et al, 2016). Based on their MT components, cilia are classified as motile (9 + 2) or primary (9 + 0) (Marshall, 2008; Gerdes et al, 2009). The physiological importance of primary cilia is well established given that disruption or malfunction of cilia results in a variety of human diseases called “ciliopathy”, which includes cystic kidney diseases, hydrocephalus, blindness, obesity, polydactyly, diabetes, cognitive impairment, and developmental disorders (Otto et al, 2003; Zariwala et al, 2007; Adams et al, 2008; Marshall, 2008; Gerdes et al, 2009; Nigg & Raff, 2009; Tory et al, 2009; Hildebrandt et al, 2011).
The serine–threonine kinase Akt is a major downstream target of PI3K, which plays important roles in cancer pathogenesis by directly inhibiting apoptosis and maintaining cell metabolism by autophagy. Three isoforms of Akt exist in mammalian cells (Brazil et al, 2004; Manning & Cantley, 2007; Noguchi et al, 2014). Through its biological targets, Akt regulates a wide range of cellular processes, including survival, cell cycle, proliferation, cytoskeletal organization, vesicle trafficking, glucose transport, and platelet function (Noguchi et al, 2007). Therefore, malfunction of Akt contributes to a wide variety of human diseases including cancer, glucose intolerance, schizophrenia, viral infections, and autoimmune diseases (Brazil et al, 2004; Manning & Cantley, 2007; Noguchi et al, 2014).
However, the involvement of Akt in signal transduction mechanisms in primary cilia has been poorly characterized. In this study, Inversin (INVS), a ciliary protein with highly conserved ankyrin repeats and IQ domains, was found to specifically bind both Akt2 and Akt3 by yeast two‐hybrid screening (Mochizuki et al, 1998; Morgan et al, 2002b; Eley et al, 2004). Mutations in the INVS gene result in nephronophthisis type II (NPHP2), an autosomal recessive cystic kidney disease that progresses to end‐stage renal failure during early infancy and is occasionally associated with situs inversus (Otto et al, 2003; Saunier et al, 2005; Badano et al, 2006; Gerdes et al, 2009; Tory et al, 2009). Cystic kidney diseases are caused by an amazingly broad array of genetic mutations and manipulations (Otto et al, 2003; Zariwala et al, 2007; Adams et al, 2008; Marshall, 2008; Gerdes et al, 2009; Nigg & Raff, 2009; Tory et al, 2009; Hildebrandt et al, 2011). However, dysfunction of cilia is suggested to play a role in cystic formation (Oh & Katsanis, 2012). Altered ciliary signaling disoriented cell division in renal tubules, resulting in renal cyst formation (Happe et al, 2011).
INVS localizes to the primary cilia of renal epithelial cells and to the nodal monocilia in mouse embryos within the so‐called Inversin compartment that is located in the proximal part of primary cilia (Morgan et al, 2002b; Watanabe et al, 2003; Shiba et al, 2009). During anaphase, INVS relocalizes to the spindle poles, coinciding with the translocation of HEF1 (human enhancer of filamentation) to the centrosome (Eley et al, 2004). Despite the identification of an increasing number of interacting proteins, the physiological function of INVS has not been fully clarified. INVS interacts with Dishevelled (Dvl), activates PCP (planar cell polarity) signaling, and simultaneously downregulates canonical Wnt signaling by targeting Dvl for proteasome‐mediated degradation (Simons et al, 2005). INVS interacts with anaphase‐promoting complex 2 (APC2) through its D‐box motif, suggesting that it is involved in cell cycle regulation (Morgan et al, 2002b). INVS co‐precipitates with N‐cadherin and catenins, including β‐catenin, as well as with calmodulin in polarized epithelial cells (Yasuhiko et al, 2001; Morgan et al, 2002b; Nurnberger et al, 2002; Saunier et al, 2005). INVS also coordinates with ANKS6 during the pathogenesis of NPHP2 (Hoff et al, 2013). Moreover, INVS interacts with Aurora A, thereby inhibiting phosphorylation and activation, which consequently interferes with HDAC6‐mediated cilia disassembly (Mergen et al, 2013).
Primary cilia are also implicated in the regulation of various signal transduction mechanisms that control a wide variety of cellular events, including sonic hedgehog (Shh) (Corbit et al, 2005), Wnt (Benzing et al, 2007; Lancaster et al, 2011), TGF‐β (Clement et al, 2013a), and platelet‐derived growth factor (PDGF)‐mediated cell signaling (Schneider et al, 2005). PDGF receptor alpha (PDGFRα) signaling in the primary cilium regulates fibroblast migration via the Akt signaling pathway (Clement et al, 2013b). Consistent with this observation, Akt activates a number of growth factors, including PDGF, for mediating downstream signals (Noguchi et al, 2007, 2014). These findings support a possible functional involvement of Akt in the signal transduction of primary cilia. However, the molecular mechanisms underlying the role of INVS in the formation of severe renal cysts in NPHP2 are not fully understood (Otto et al, 2003; Tory et al, 2009).
In the current study, we demonstrate that INVS, located at the basal portion of the primary cilia, interacts with Akt in a PDGF‐AA‐dependent manner. Akt also phosphorylates INVS at amino acid residues 864–866. Introduction of either phosphorylation‐defective or NPHP2‐related mutants of INVS that lack the Akt phosphorylation sites into MDCK (Madin–Darby canine kidney) cells inhibits cell proliferation with aberrant spindle axis orientation during cell division, thereby exhibiting distorted lumen formation. These observations indicate that the interaction between Akt and INVS is of biological significance for primary cilia and dysregulation of this interaction may result in abnormal cyst formation in NPHP2.
Results
INVS associates specifically with Akt in mammalian cells
We conducted yeast two‐hybrid screens using human Akt2 and Akt3 as baits. One molecule that bound specifically to Akt2 was a partial coding sequence (860–1,007) of human INVS. Interestingly, a partially overlapping coding sequence of INVS (755–955) was also found to interact with Akt3. To confirm the interaction of INVS with Akt in mammalian cells, we performed co‐immunoprecipitation assays. Flag‐INVS interacted with HA‐Akt1, but not with HA‐PDK1 or HA‐PrKA, supporting the specificity of the observed interaction (Fig 1A). Since INVS was found to interact with both Akt2 and Akt3 in yeast two‐hybrid screening, we next examined the specificity of the interaction for each of the three Akt isoforms. Flag‐INVS interacted with HA‐Akt1, HA‐Akt2, and HA‐Akt3 in co‐immunoprecipitation assays in mammalian cells (Fig 1B). The interaction of endogenous Akt with INVS was also determined in HEK293 cells that expressed INVS at endogenous levels (Fig 1C).
Figure 1. INVS specifically associates with Akt in mammalian cells.
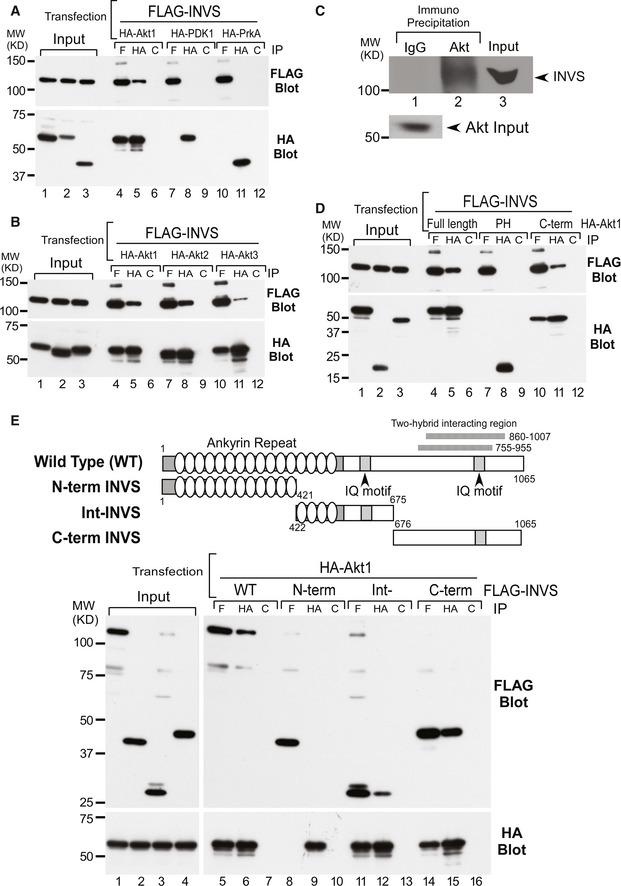
- Flag‐INVS interacted with HA‐Akt1 (lanes 4–6), but not with HA‐PDK1 (lanes 7–9) or HA‐PrKA (lanes 10–12) in HEK293T cells. Expression levels of HA‐Akt1, PDK1, PrKA, and Flag‐INVS were similar, as determined by immunoblotting (HA: anti‐HA antibody; F: anti‐Flag antibody; C: control antibody).
- Flag‐tagged INVS interacted with HA‐tagged Akt1 (lanes 4–6), Akt2 (lanes 7–9), and Akt3 (lanes 10–12) in co‐immunoprecipitation assays. Expressions of each Akt isoform and INVS were similar.
- INVS was co‐immunoprecipitated with endogenous Akt from HEK293 cells (lane 2), in which INVS is endogenously expressed.
- To determine the Akt domain responsible for interacting with INVS, Akt subfragments were generated. The C‐terminus of Akt is the primary domain for INVS interaction (lanes 10–12). Similar levels of each Akt fragment were immunoprecipitated using anti‐HA antibody.
- Schematic showing the structure and the functional domains of Flag‐tagged full‐length and fragmented INVS in mammalian expression vectors (N‐terminal: 1–421, intermediate domain: 422–675, and C‐terminal: 676–1065) used in the current study. Intermediate (lanes 11–13) and C‐terminal (lanes 14–16) fragments of INVS were important for interaction with Akt.
To identify the protein domains required for the Akt–INVS interaction, we generated subfragments of HA‐Akt and Flag‐INVS for additional co‐immunoprecipitation assays. The interaction between HA‐Akt and Flag‐INVS was mediated through the C‐terminal kinase domain of Akt and the middle‐to‐C‐terminal portion of INVS (Int INVS: 421–675, and C‐terminal INVS: 676–1,065) (Fig 1D and E).
Akt phosphorylates INVS at T864, S865, and T866 in vitro
PDGFRα signaling in the primary cilium regulates fibroblast migration via Akt signal transduction mechanisms (Clement et al, 2013b). Akt is phosphorylated during mitosis and is present in the centrosome and basal body (Wakefield et al, 2003; Zhu et al, 2009). Therefore, we next examined whether INVS might be a direct substrate of Akt. Using in vitro Akt kinase assays (IVK) that employed multiple INVS fragments, Akt was able to phosphorylate WT, 1–970, and 1–898 INVS, but failed to phosphorylate the 1–670 fragment of INVS (Fig 2A and Appendix Fig S1A).
Figure 2. INVS is a novel substrate of Akt.
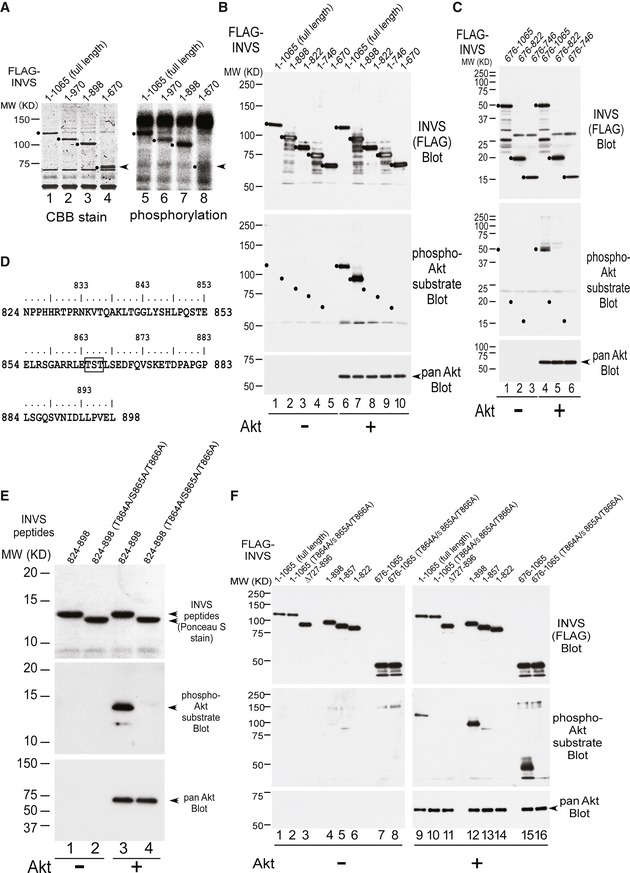
- The indicated recombinant INVS proteins (lanes 1–4) were used for IVK. Akt phosphorylates WT (lane 5), 1–970 (lane 6), and 1–898 (lane 7) INVS, but failed to phosphorylate 1–670 (lane 8) INVS. The arrows (right side) indicated the position of 1‐670 fragment (lane 4), which failed phosphorylation (lane 8).
- The indicated recombinant INVS proteins (lanes 1–10) were used for IVK. Akt phosphorylates WT (lane 6) and 1–898 (lane 7) INVS, but failed to phosphorylate other subfragments of INVS (1–822, 1–746, and 1–670, in lanes 8, 9, and 10, respectively), as detected using a phospho‐Akt substrate antibody.
- Using the indicated subfragments of INVS for IVK, we confirmed that Akt phosphorylates 675–1065 (lane 4), but not 675–822 (lane 5), or 675–746 (lane 6) INVS.
- Amino acid alignment of 824–898. Motif scan analysis using the Scansite3 database identified three amino acids, T864, S865, and T866, as putative Akt phosphorylation targets.
- Purified recombinant WT or 3A INVS were generated (top panel, lanes 1–4). In IVK, Akt phosphorylated the WT fragment (824–898, middle panel, lane 3), but failed to phosphorylate 3A INVS (middle panel, lane 4), as detected by using a phospho‐Akt substrate antibody.
- Akt phosphorylated WT (lane 9), 1–898 (lane 12) and 675–1065 (lane 15) INVS, but failed to phosphorylate full‐length 3A, 675–1065 3A, ∆727–896, 1–857, and 1–822 INVS (lanes 10, 16, 11, 13, and 14, respectively), all of which lacked the T864/S865/T866 target amino acid.
Next, we utilized full‐length or deletion mutants of INVS for further dissecting the phosphorylation site of INVS by Akt via IVK assays. Akt phosphorylated full‐length INVS and 1–898 INVS, but failed to phosphorylate 1–822, 1–746, and 1–670 subfragments of INVS (Fig 2B).
We also used additional INVS protein fragments for further dissecting the Akt phosphorylation site(s) on INVS. In contrast to full‐length and 1–898 INVS, Akt did not phosphorylate the 1–822, 1–746, and 1–670 subfragments of INVS (Fig 2B). Consistent with these results, Akt phosphorylated a 675–1,065 INVS fragment, but not the 675–822 or 675–746 INVS fragments (Fig 2C). Together, these results indicate that Akt phosphorylates INVS in a region between amino acids 823 and 898.
Using the program Scansite (Suizu et al, 2009), we identified three consecutive amino acids (T864, S865, and T866) as putative Akt phosphorylation targets (Fig 2D). These consecutive serine and threonine residues are conserved in mammalian species including Pan troglodytes, Canis lupus familiaris, Bos taurus, or Oryctolagus cuniculus, further supporting the physiological significance of phosphorylation of these residues in vivo (Appendix Fig S1B). Recombinant wild‐type (WT) INVS and a triple‐alanine‐substitution mutant of the three consecutive threonine and serine residues at 864, 865, and 866 (hereafter designated as “3A”) within the 824–898 subfragment of INVS were generated. Akt phosphorylated the WT fragment (824–898), but failed to phosphorylate the 3A mutant (Fig 2E).
Importantly, Akt phosphorylated WT, 1–898, and 675–1,065 INVS, but failed to phosphorylate 3A mutants in both the full‐length and the 675–1,065 subfragment, as well as in the 727–896, 1–857, or 1–822 subfragments, all of which lack the T864/S865/T866 target residues (Fig 2F). Together, these results demonstrate that INVS is a direct substrate of Akt and that Akt phosphorylates INVS at one or several of the serine/threonine residues at T864/S865/T866.
INVS and phosphorylated Akt are located at the basal region of the primary cilia
PDGF controls the migration, differentiation, and activity of a variety of specialized mesenchymal and migratory cell types, both during development and in the adult animal (Hoch & Soriano, 2003; Schneider et al, 2005, 2010; Christensen et al, 2007). Interestingly, PDGFRα signaling in the primary cilium regulates fibroblast migration via Akt signaling (Schneider et al, 2005; Clement et al, 2013b). Primary cilia organize and extend from the basal body (from the mother centriole) where activated Akt is constitutively present (Zhu et al, 2009), suggesting that Akt may be involved in ciliary development.
Using confocal microscopy, we found that active Akt (phospho‐S473 Akt) localized to the basal body of primary cilia or to a centrosome‐like structure in dividing cells (Fig 3A). Consistent with previous reports, INVS also localized to the proximal region in primary cilia, in the so‐called Inversin compartment (Shiba et al, 2009) or ciliary base at the centrosome in dividing cells (Fig 3B). INVS also co‐localized with γ‐tubulin, a centrosome marker, in confluent cells (Fig 3C). Phosphorylated INVS, detected by an anti‐phospho‐Akt substrate antibody in immunoblots (see Fig 2), localized to the basal region of the primary cilia in confluent cells (Fig 3D). Upon PDGF‐AA stimulation, INVS appeared to be the same position with phospho‐Akt substrate at ciliary pocket. PI3K inhibitor (LY294002) inhibited phospho‐S473 Akt localization at ciliary pocket (Appendix Fig S2B‐E).
Figure 3. Both INVS and phosphorylated Akt are located at the basal region of primary cilia.
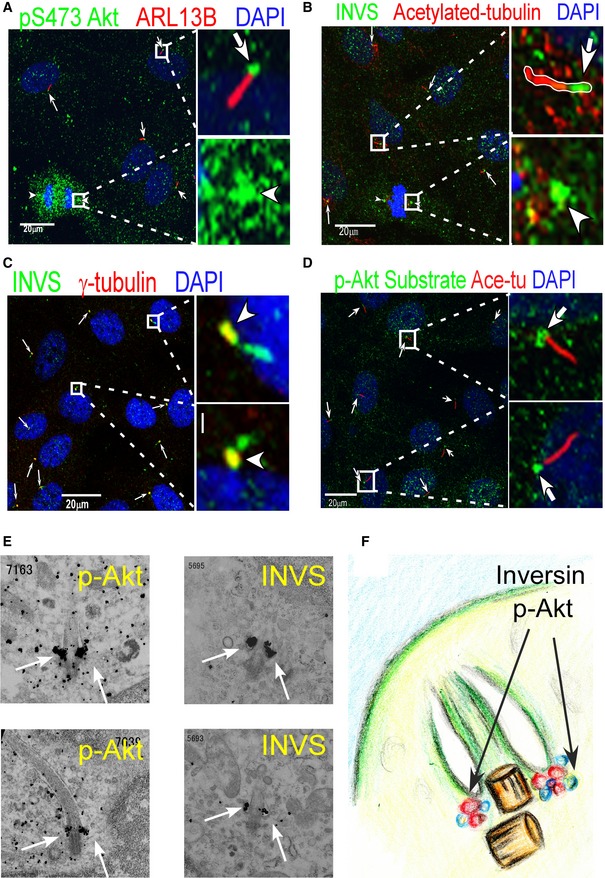
- Confocal microscopy shows that active Akt (phospho‐S473 Akt) is localized in the basal body of primary cilia (arrows) (right‐side panels show higher magnification).
- INVS is localized in the proximal region of the Inversin compartment in primary cilia (arrows) (Inversin compartment, right‐side panels show higher magnification).
- INVS co‐localizes with γ‐tubulin, a centrosome marker (arrows).
- Anti‐phospho‐Akt substrates stain positively at the basal region of the primary cilia (arrows), as determined by anti‐acetylated tubulin staining (right‐side panels show higher magnification).
- Silver‐intensified immunogold electron microscopy shows that both phospho‐Akt (left‐side panels) and INVS (right‐side panels) were localized to the ciliary pocket of primary cilia in the presence of PDGF‐AA (white arrows).
- Schematic representation of the view from an electron microscope.
To further dissect the precise localization of Akt and INVS at the ciliary base, we employed a silver‐intensified immunogold method for electron microscopy. Our results showed that both phospho‐Akt and INVS localized to the basal body, close to the ciliary pocket, only after PDGF‐AA treatment (Fig 3E and F). Phosphorylated Akt, but not INVS, is accumulated at the ciliary pocket prior to PDGF‐AA treatment (Appendix Fig S2A).
PDGF‐AA stimulates co‐localization of INVS and phosphorylated Akt
PDGF‐AA is known to mediate intracellular Akt signaling in cilia (Schneider et al, 2005; Christensen et al, 2007; Clement et al, 2013b). Since Akt interacted with and phosphorylated INVS, we next examined whether PDGF‐AA stimulation affected the co‐localization of INVS and Akt. PDGF‐AA stimulation of cells resulted in a time‐dependent increase in active Akt–INVS interaction at the basal body (Fig 4A and B).
Figure 4. Co‐localization of INVS and phosphorylated Akt increases with PDGF‐AA treatment.
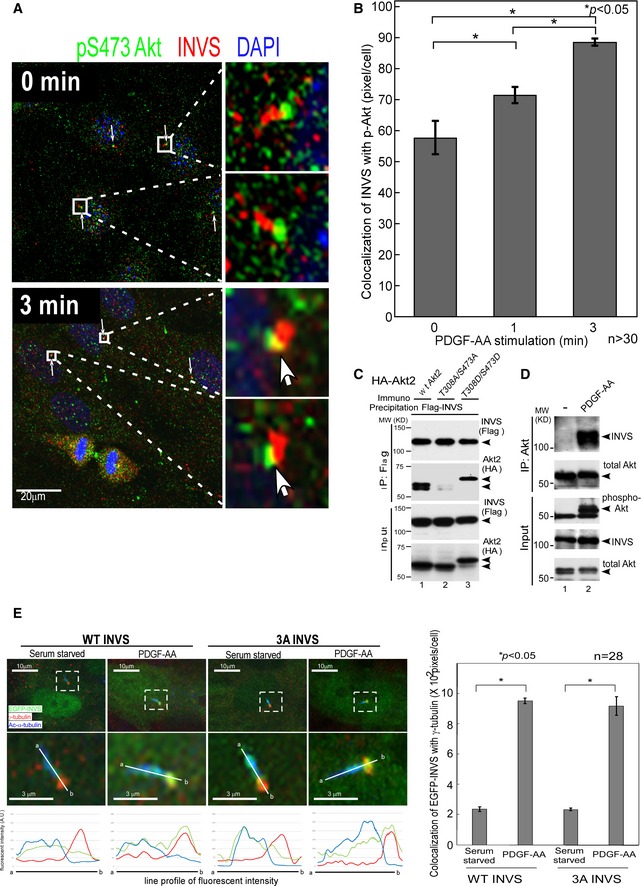
- PDGF‐AA stimulation enhances localization of active Akt (phospho‐S473 Akt) at the basal body of primary cilia (arrows) (right‐side panels show higher magnification).
- Quantification of co‐localization of phosphorylated Akt (green) and INVS (red) at the indicated time points after PDGF‐AA stimulation revealed a time‐dependent increase. Co‐localization was measured by counting yellow pixels (co‐localized area) using Imaris software (Bitplane AG). Results presented are means ± SE (n = 31 at time 0, n = 35 at 1 min, and n = 33 at 3 min). Three independent experiments were analyzed with similar results. Statistical significance was determined by Student's t‐test.
- Co‐immunoprecipitation assays using HEK293T cells show that INVS preferentially binds to WT Akt (second panel, lane 1) or phospho‐mimetic Akt (T308D/S473D, lane 2), but fails to interact with phosphorylation‐defective forms of Akt (T308A/S473A, lane 3).
- PDGF‐AA stimulation of HEK293T cells results in augmented interaction of endogenous INVS with Akt (top panel, comparison between lane 1 and lane 2, untreated and PDGF‐AA‐stimulated conditions, respectively).
- Both WT and 3A‐INVS, which are localized at the Inversin compartment of primary cilia under serum‐starved condition (left‐side panels, lower panels show higher magnification), translocate to the basal body of the cilium after PDGF‐AA stimulation (right‐side panels, lower panels show higher magnification). Results presented are means ± SE of yellow pixels demonstrating co‐localization of EGFP‐INVS (green) with γ‐tubulin (red) in hTERT‐RPE1 cells (n = 28). Three independent experiments showed similar results. Statistical significance was determined by Student's t‐test. Fluorescence intensities of INVS (green), acetylated α‐tubulin (blue), and γ‐tubulin (red) along the line (a‐b) are plotted underneath.
Since Akt phosphorylates INVS, we next examined whether Akt kinase activity affects its interaction with INVS. In co‐immunoprecipitation assays, INVS preferentially bound to WT Akt or a phospho‐mimetic mutant of Akt (T308D/S473D), but failed to interact with a phosphorylation‐deficient Akt mutant (T308A/S473A) (Fig 4C). To further examine the role of Akt activation in Akt–INVS interaction, we stimulated HEK293T cells with PDGF‐AA, which mediates Akt signaling in primary cilia. As expected, the interaction of Akt with INVS was clearly augmented by PDGF‐AA administration (Fig 4D).
Since INVS localizes to the Inversin compartment of primary cilia, we next examined how PDGF‐AA affects the localization of INVS (WT vs. 3A). Consistent with a previous report (Shiba et al, 2009), both WT and 3A INVS localized at the Inversin compartment of primary cilia under serum‐starved conditions (Fig 4E, left‐side panels). After PDGF‐AA stimulation, however, INVS relocalized to the proximal portion of primary cilia around the basal body (Fig 4E, right‐side panels) where phosphorylated Akt was constitutively present (see Fig 3). Both WT and 3A INVS translocated to the basal body after PDGF stimulation.
Akt controls ciliary development through the phosphorylation of INVS at T864/S865/T866
Our observations thus far supported the hypothesis that the interaction between Akt and INVS played an important role in the control of ciliary growth. Therefore, we examined whether Akt phosphorylated INVS at T864/S865/T866 in a physiological context. PDGF‐AA treatment, which activates Akt, induced phosphorylation of WT INVS (Fig 5A, top panel). The phosphorylation of INVS could be suppressed by LY294002, MK2206, or GSK690693 inhibitors for Akt, indicating that INVS phosphorylation at T864/S865/T866 was indeed mediated by Akt in a physiological context. An increase in Akt phosphorylation at both S473 and T308 sites was observed after GSK690693 treatment due to a feedback mechanism (Okuzumi et al, 2009). Similar results were obtained after stimulation of HEK293 cells with serum (Appendix Fig S3A) or stimulation of COS‐7 cells with PDGF‐AA (Appendix Fig S3B).
Figure 5. Decrease in ciliary development by phosphorylation‐defective INVS at T864/S865/T866.
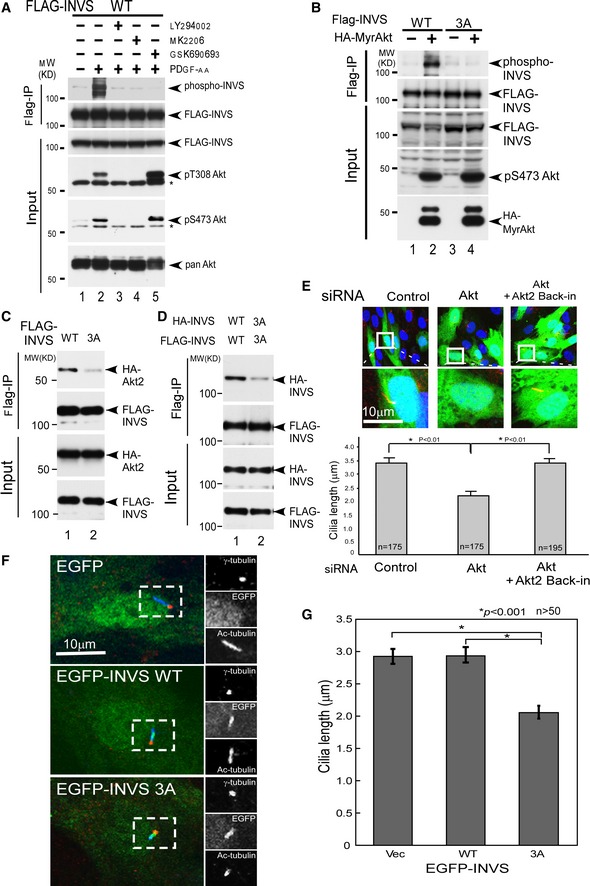
- PDGF‐AA stimulation resulted in the phosphorylation of WT INVS (top panel, lane 2), PDGF‐AA‐stimulated INVS phosphorylation is inhibited by Akt inhibitors (top panel, LY294002, MK2206, and GSK690693, lanes 3, 4, and 5, respectively).
- Myr‐Akt, a constitutively active form of Akt, was expressed together with WT or 3A INVS in HEK293 cells and INVS phosphorylation was quantified. WT, but not 3A INVS, can be phosphorylated in the presence of Myr‐Akt.
- 3A INVS exhibited a weaker interaction with Akt as compared to WT INVS (top panel, compare lanes 1 and 2, WT and phospho‐defective mutant, respectively). Expression of Akt and INVS are shown underneath.
- 3A INVS exhibited weaker dimerization as compared to WT INVS (top panel, compare lanes 1 and 2, WT and 3A, respectively). Expression of Akt and INVS are shown below each.
- Length of primary cilia in confluent hTERT‐RPE1 cells was analyzed after siRNA‐mediated knockdown of Akt1/2. Ciliary length was decreased in cells expressing siRNA. Results presented are means ± SE of the longitudinal length of acetylated tubulin (red), a marker of primary cilia, in siRNA‐transfected (green: EGFP positive) hTERT‐RPE1 cells (n = 175 for control siRNA and Akt siRNA, and n = 195 for Akt siRNA plus siRNA‐resistant Akt). Three independent experiments showed similar results. Statistical significance was analyzed by Student's t‐test. Note that reintroduction of siRNA‐resistant Akt (Matsuda‐Lennikov et al, 2014) in Akt knockdown cells rescued the effect on ciliary length.
- Expression of 3A INVS (bottom panels) but not WT INVS (middle panels) inhibited the development of primary cilia as compared to control cells (top panels).
- Quantification of ciliary length. Results are mean ± SE of longitudinal length of acetylated tubulin (blue), a marker of primary cilia, in EGFP‐INVS (green) transfected in hTERT‐RPE1 cells (n = 58 for EGFP vector and for EGFP‐INVS WT, and n = 52 for EGFP‐INVS 3A mutant). Three independent experiments showed similar results. Statistical significance was determined by Student's t‐test.
Next, we co‐transfected Myr‐Akt, a constitutive active form of Akt (Suizu et al, 2009), to induce Akt kinase activity along with WT or 3A INVS to examine the phosphorylation levels of INVS in a physiological context. WT, but not 3A INVS, was phosphorylated by Myr‐Akt, demonstrating that the T864/S865/T866 residues are phosphorylated by Akt in vivo (Fig 5B). In co‐immunoprecipitation assays, 3A interaction with Akt was weaker as compared to WT INVS indicating that phosphorylation of the T864/S865/T866 residues was important for physical interaction of INVS with Akt (Fig 5C). These observations were consistent not only with the finding that the C‐terminal domain of INVS was responsible for the interaction with Akt (Fig 2), but also with the observation that co‐localization of INVS and Akt was stimulated by PDGF‐AA induced activation of Akt (Fig 4). Moreover, the relatively weaker ability of 3A INVS to dimerize (Fig 5D) highlights the physiological importance of INVS phosphorylation in addition to its C‐terminal domain‐mediated function (Shiba et al, 2009).
These observations prompted us to examine whether Akt‐dependent phosphorylation of INVS at T864/S865/T866 affects ciliary development. For studying the role of Akt in ciliogenesis, we used Akt1/2 siRNA to inhibit Akt expression in a dose‐dependent manner (Appendix Fig S3C). Both ciliary length (Fig 5E and Appendix Fig S3E) and ciliogenesis (Appendix Fig S3D) were suppressed by Akt knockdown. Furthermore, reintroduction of siRNA‐resistant Akt (Matsuda‐Lennikov et al, 2014) rescued these effects on ciliary development (Fig 5E and Appendix Fig S3D). Consistent with these observations, primary cilia development was inhibited in Akt‐null MEFs that lacked Akt1, Akt2, and Akt1/2 (Appendix Fig S3F). Notably, the observed inhibition was stronger in Akt2‐null MEFs than in Akt1‐null MEFs, suggesting that Akt2 plays a more prominent role in ciliary development than Akt1.
The primary cilium typically grows in G1 and is lost before mitosis; however, the physiological regulation of its growth is not fully clarified (Santos & Reiter, 2008). We next examined whether phosphorylation of INVS at T864/S865/T866 had a biological impact on ciliogenesis. We introduced either WT or 3A INVS in cells for examining ciliary development. In contrast to control cells or cells expressing WT INVS, cells expressing 3A exhibited impaired ciliogenesis (Fig 5F and G).
INVS controls proper alignment of the spindle axis during metaphase
Symmetric cell division plays an important role in the maintenance of cellular homeostasis, with regard to cellular shape, structure, and function. Asymmetric cell division can lead to various human diseases (Morrison & Kimble, 2006; Hildebrandt et al, 2009, 2011). Therefore, we next examined whether phosphorylation of INVS affects spindle alignment during cell division. To examine the effect of various mutants of INVS (3A and truncated forms of INVS), each mutant was expressed in MDCK cells and spindle angles (θ) were measured (Fig 6A). Expression of mutant forms of INVS (3A as well as the different truncated forms of INVS found in NPHP2 patients (Tory et al, 2009)) resulted in wider spindle angles, indicative of a disoriented spindle axis, as compared to WT INVS‐expressing cells (Fig 6B). In order to retain correct cell polarity during cell division, the mitotic spindle must align with the axis of polarity. Therefore, these observations suggest that phosphorylation of INVS by Akt regulates the correct formation of renal tubules by spatially controlling the spindle orientation of regenerating tubular cells.
Figure 6. INVS controls spindle axis alignment during cell division.
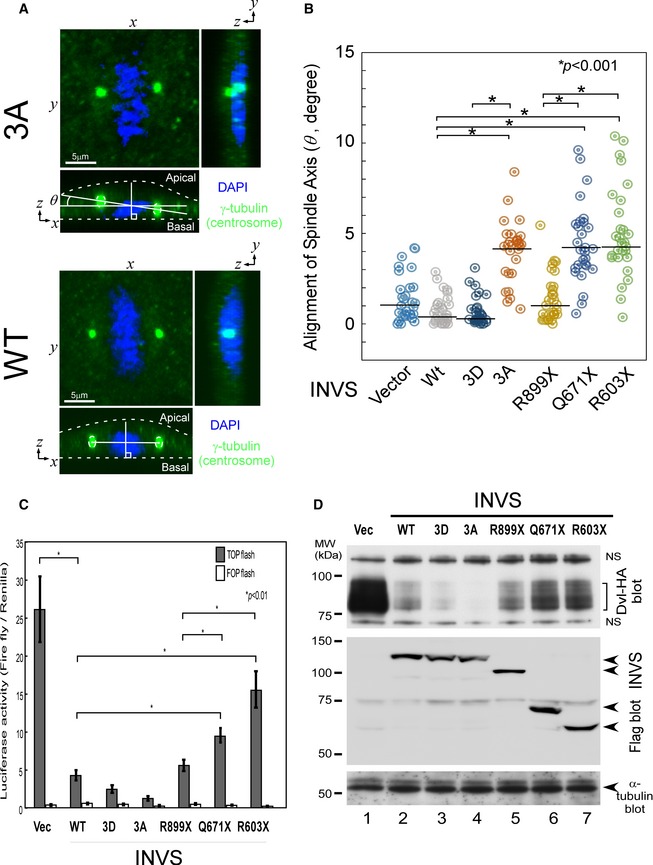
- Schematic describing the measurement of spindle angle (θ) (Kikuchi et al, 2010). Z‐stack images were obtained from 0.45‐μm‐thick sections of metaphase cells and the angle between the axis of metaphase spindle and that of the substrate surface, which is termed the spindle angle, was calculated.
- Spindle angles were measured in MDCK cells transfected with the indicated INVS plasmids (+). The spindle axis was misaligned in cells expressing mutant forms of INVS as compared to cells expressing WT INVS or sham‐transduced cells. Expression of 3A INVS and of clinically relevant truncated forms of INVS, but not WT INVS, resulted in disorientation of the spindle axis during metaphase. Results presented are scatter plots of the angle (θ) of the mitotic spindle (n = 28 for all samples). Three independent experiments showed similar results. Statistical significance was determined by Mann–Whitney nonparametric median test. Similar expression of all INVS constructs was verified by immunoblotting (Appendix Fig S4A).
- WT INVS robustly inhibited the transactivation of Wnt in a luciferase reporter assay, which could be reverted by C‐terminal truncated INVS (R899X, Q671X, or R603X), but not by a phospho‐mimetic mutant or 3A INVS. Results presented are mean ± SE of the ratio of triplicates between sequentially measured firefly TCF/LEF signal and Renilla luciferase signal in HA‐INVS transfected 293T cells. Dual‐Luciferase reporter® Assay System (Promega) was used and experiments were performed in triplicates. Two independent experiments showed similar results. Statistical significance was determined by Student's t‐test.
- The expression of HA‐Dvl (upper panels) and the indicated forms of Flag‐INVS (lower panels) are shown by immunoblots; α‐tubulin was used as an internal control.
Wnt signaling, known to function in primary cilia (Schneider et al, 2005; Christensen et al, 2007), can switch from canonical to noncanonical Wnt signaling pathways based on targeting of cytoplasmic Dvl for ubiquitin‐dependent degradation (Simons et al, 2005; Benzing et al, 2007), which controls the orientation of spindle axis alignment (Kikuchi et al, 2010). Therefore, we examined the effect of INVS on Wnt signaling by using TCF‐reporter assays. Consistent with previous reports (Simons et al, 2005; Benzing et al, 2007), WT INVS inhibited Wnt transactivation, and this effect was reversed by truncation mutants of INVS (Q671X or R603X) found in patients (Tory et al, 2009) (Fig 6C). The Inhibitory effect of Wnt signal by WT INVS was reversed by NPHP2‐related mutant forms of INVS (R899X, Q671X, or R603X), which is correlating with the expression levels of Dvl (Fig 6D). Notably, 3A or phospho‐mimetic INVS showed no effect on Wnt signaling, suggesting that the C‐terminus of INVS, but not Akt‐dependent phosphorylation of INVS at 864–866, plays a major role in Wnt signaling, presumably by degrading Dvl.
Phosphorylation of INVS controls size and proper lumen formation
Mutations in the INVS gene are suggested to cause the malfunctioning of primary cilia, resulting in cystic kidney diseases in NPHP2 (Simons et al, 2005; Tory et al, 2009; Oh & Katsanis, 2012; Ong, 2013). The presence of a cilium is thought to be closely related to cell differentiation and the establishment of proper polarity during development (Simons & Walz, 2006). We examined a model in which disrupted Wnt signaling, which is known to control spindle orientation (Kikuchi et al, 2010), may result in the manifestation of disrupted lumina. We used a 3D culture system of MDCK cells for evaluating cystic formation of the kidney in vivo (Veikkolainen et al, 2012). Therefore, we examined the differences in lumen formation by WT, 3A, phospho‐mimetic, and C‐terminal truncated INVS (R899X, Q671X, and R603X (Tory et al, 2009)) in 3D MDCK cell cultures. Immunoblotting showed that all INVS proteins were equally expressed (Appendix Fig S4A). WT or phospho‐mimetic INVS‐expressing MDCK cells exhibited a precisely formed, round lumen, with a uniform small size, similar to those in control transfected cells (Fig 7A and B and quantifications are shown in Fig 7C and D). However, mutant INVS (3A and NPHP2‐related C‐terminally truncated forms of INVS (R899X, Q671X, and R603X)) exhibited a large, distorted lumen with increased cyst formation. These observations suggested that INVS was important for the correct development of the renal tubular lumen. Interestingly, expression of both Q671X and R603X INVS, which lack the Akt phosphorylation sites, resulted in a more severely distorted lumen than that observed with R899X, which retains the Akt phosphorylation sites, suggesting that phosphorylation of INVS at T864/S865/T866 was important for the proper formation of the renal tubule, thereby explaining the presence of abnormal renal cysts in NPHP2.
Figure 7. The C‐terminus and phosphorylation sites of INVS are required for proper tubular differentiation.
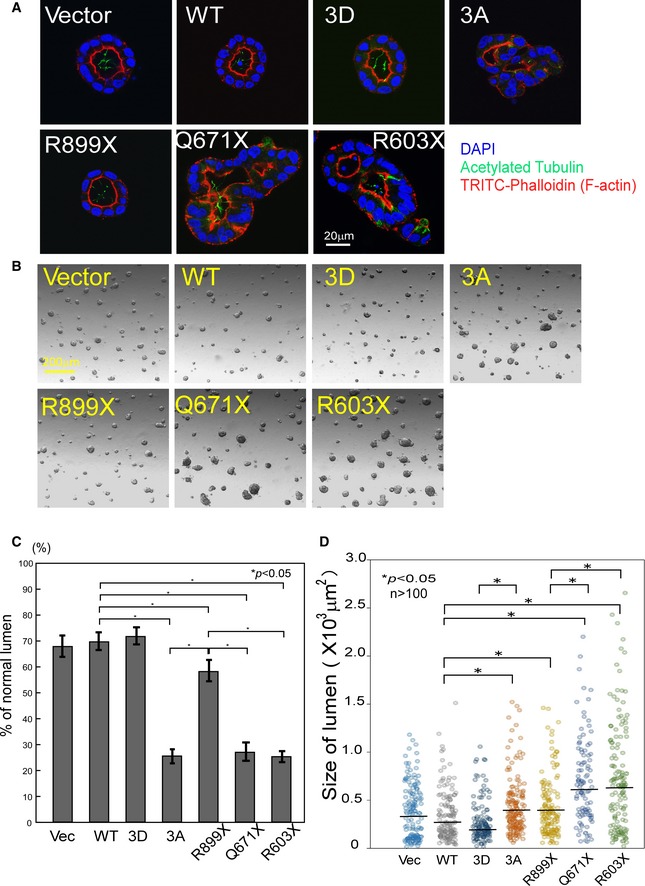
- WT INVS‐transduced MDCK cells in 3D cultures form uniform round tubules. However, expression of 3A or NPHP2‐related truncated INVS mutants, which lack Akt phosphorylation site(s), disrupts this uniformity. Results presented are representative of three independent experiments and show mean ± SE of percent normal lumen in vector‐ or WT INVS‐transduced MDCK cells (n = 422 for vector, n = 398 for HA‐WT, n = 401 for ‐3D, n = 365 for ‐3A, n = 344 for ‐R899X, n = 331 for ‐Q671X, and n = 325 for ‐R603X INVS stably transfected MDCK cells). Three independent experiments showed similar results. Statistical significance was determined by Student's t‐test.
- WT transduced MDCK cells form a relatively smaller lumen; however, expression of 3A INVS or the indicated NPHP2‐related truncation mutants results in relatively larger lumen. Both truncation mutants (Q671X or R603X) lack the Akt phosphorylation site and exhibit a larger lumen size. Three independent experiments showed similar results.
- Quantification of normal lumen in cells transduced with WT or mutant INVS shown in panel (A). Results presented are mean ± SE of percent normal lumen in vector‐ or WT INVS‐transduced MDCK cells (n = 166 for vector, n = 204 for HA‐WT, n = 233 for ‐3D, n = 150 for ‐3A, n = 164 for ‐R899X, n = 128 for ‐Q671X, and n = 137 for ‐R603X INVS stably transfected MDCK cells). Three independent experiments showed similar results. Statistical significance was determined by Student's t‐test.
- Quantification of the size of lumen in cells transduced with WT or mutant INVS is shown from panel (B). Results presented are scatter plots of lumen size (n = 205 for vector, n = 143 for HA‐WT, n = 131 for ‐3D, n = 167 for ‐3A, n = 144 for ‐R899X, n = 128 for ‐Q671X, and n = 144 for ‐R603X INVS stably transfected MDCK cells). Two independent experiments showed similar results. Statistical significance was determined by Mann–Whitney nonparametric median test.
We next examined whether different mutants of INVS (phospho‐mutants or truncated forms of INVS) differentially affected cell proliferation. Cells expressing WT or phospho‐mimetic INVS proliferated more efficiently as compared to cells that expressed 3A or clinically relevant C‐terminal truncated INVS (R899X, Q671X, and R603X) (Appendix Fig S4B). Since tubular epithelial cells are endowed with a predetermined cellular program that controls the orientation of cell division relative to the neighboring cells during lumen formation (Simons & Walz, 2006), our observations support the finding that phosphorylation of INVS by Akt plays an important physiological role in lumen formation.
Discussion
The cilium is a hairlike structure that extends from the cell surface to the extracellular space. Primary cilia are sensory organelles that regulate mechanisms of epithelial cell polarity and cell cycle control, based on mechanosensory, visual, and other stimuli (Marshall, 2008; Gerdes et al, 2009; Nigg & Raff, 2009; Hildebrandt et al, 2011). However, the involvement of Akt in the regulation of ciliary signaling has been poorly characterized.
Using yeast two‐hybrid and co‐immunoprecipitation assays, we have demonstrated that Akt interacts with INVS. INVS mutations result in NPHP2, an autosomal recessive cystic kidney disease, which leads to severe kidney failure in early childhood (Otto et al, 2003; Tory et al, 2009; Oud et al, 2014). Consistent with the phenotype of Inv −/− mice, an animal model for NPHP2 (Mochizuki et al, 1998; Morgan et al, 2002b; Eley et al, 2004), INVS knockdown by antisense morpholino oligonucleotides causes pronephric cysts in zebra fish (Simons et al, 2005).
Genetic analysis of NPHP2 patients revealed a premature stop codon in INVS in most patients, although in some cases single‐amino acid‐substitution mutations were detected (Otto et al, 2003; Tory et al, 2009; Oud et al, 2014). These clinical observations highlight the observation that the C‐terminus of INVS has an important biological function in the mature INVS protein. Since the Akt‐interacting domain of INVS was found by co‐immunoprecipitation experiments to be in the internal/C‐terminal domains of INVS, the truncated forms of INVS present in NPHP2 patients are expected to have a weaker interaction with Akt, hinting at a causative relation between the disrupted Akt–INVS interaction and the manifestation of NPHP2.
The intracellular localization of INVS has been intensively studied in an attempt at unraveling its physiological function. INVS is primarily localized at the so‐called Inversin compartment at the proximal portion of primary cilia (Shiba et al, 2009). However, INVS was found to have a dynamic expression pattern throughout the different phases of the cell cycle in MDCK cells (Morgan et al, 2002b). In polarized interphase cells, INVS localized to primary cilia, basal bodies, nucleus, and cell–cell borders; however, it localized to the spindle poles in metaphase and anaphase (Morgan et al, 2002b; Nurnberger et al, 2002, 2004).
We showed that co‐localization of INVS and phosphorylated Akt at the basal body can be augmented by PDGF‐AA treatment. Furthermore, immunoelectron microscopy revealed that phosphorylated Akt was present at the ciliary pocket in nonstimulated conditions (after 24‐hour serum depletion) to promote the growth of primary cilia.
We showed that PDGF‐AA stimulation induces translocalization of INVS from the Inversin compartment to the basal body, where activated Akt is constitutively present (Zhu et al, 2009). Notably, the levels of phosphorylated Akt at the basal body remain high after serum starvation, which allows primary cilia to grow, even in conditions that are normally sufficient to suppress Akt kinase activity.
In order to efficiently grow cilia, over 48 hours of serum starvation is required in addition to cell confluency on the culture dish. Of note, even after 48 hours of serum starvation for facilitating the growth of primary cilia, phosphorylation levels of Akt and its substrates at the ciliary pocket remained high.
INVS is mainly present at the “Inversin compartment” under normal culture conditions that allow cilia to grow. After PDGF‐AA stimulation, INVS moved from the Inversin compartment of the primary cilia to the ciliary pocket, co‐localized with Akt that was highly phosphorylated, and was phosphorylated by active Akt present at the ciliary pocket. It is noteworthy that while both WT and 3A INVS can relocalize to the basal body of primary cilia upon PDGF‐AA stimulation, phosphorylation of INVS is unlikely to play a major role in this relocalization.
Confocal microscopy revealed that co‐localization of INVS and Akt can be augmented by PDGF‐AA. Further, Akt siRNA or Akt(1/2) ablation in MEFs attenuated ciliary growth, suggesting a role for Akt activity in ciliary development. Since PDGF‐AA signals through the primary cilium and activates Akt for regulating the direction of migration (Hoch & Soriano, 2003; Schneider et al, 2005, 2010), PDGF‐AA can induce INVS to translocalize to the basal body, where it can be phosphorylated by Akt, leading to proper ciliary development. We showed that phosphorylation‐defective INVS inhibited ciliogenesis. The role of INVS in ciliary development appeared to be somewhat controversial in different experimental conditions. For example, an in vitro lentiviral‐mediated inducible knockdown system was used to show that loss of INVS resulted in the reduced presence of cilia in MDCK cells (Mergen et al, 2013). In contrast, NPHP2/Inv mice were reported to have cilia of normal length in postnatal tissues (Phillips et al, 2004). INVS, which interacts with the anaphase‐promoting complex protein‐2, localizes to the primary cilia of kidney, but is not essential for ciliogenesis (Morgan et al, 2002a). The frequency of ciliated Inv −/− MEF or inv/inv mutant mice appears to be the same as in wild‐type mice (Veland et al, 2013).
Malfunction of ciliary function, which results in disoriented cell division in renal tubules, is implicated in the formation of abnormal renal cysts (Happe et al, 2011; Oh & Katsanis, 2012). Cystic kidney diseases are known to be caused by an amazingly broad array of genetic mutations and manipulations. The PI3K–Akt–mTOR pathway has been suggested to play a role in the manifestation of cystic kidney diseases. Cystic epithelia display increased proliferation and apoptotic rates (Simons & Walz, 2006). Akt signaling is a major intracellular mediator of anti‐apoptotic response. Therefore, it is plausible that disruption of Akt signaling correlates with increased levels of apoptosis, which leads to progressive deterioration of renal function manifested in human cystic kidney disease patients (Woo, 1995).
To support this notion, renal cysts form in approximately 30% of patients with tuberous sclerosis, a downstream effector of Akt (Inoki et al, 2005; Huang & Manning, 2009) (Winyard & Jenkins, 2011). Moreover, mice ablated for the anti‐apoptotic protein Bcl‐2, which is activated by Akt phosphorylation of BAD at S136 (Datta et al, 1997), exhibit severe manifestation of cystic kidney disease (Veis et al, 1993). Of note, however, three isoforms of Akt with redundant biological function are present in the human genome, all of which physically interact with INVS. Single or double knockout of Akt 1/2 in mice may not result in the failure of phosphorylation of INVS. Double knockout of Akt1/3 in mice is embryonic lethal by day 12 (Dummler et al, 2006); therefore, triple‐knockout mice were not created. These studies may explain the lack of cyst formation in the kidneys of these animals.
We showed that 3A INVS‐expressing cells proliferated less efficiently than those expressing WT INVS. Furthermore, 3D cultures of MDCK cells, expressing mutant forms of INVS (3A and C‐terminal truncated INVS, R899X, Q671X, and R603X observed in NPHP2) (Otto et al, 2003; Tory et al, 2009; Oud et al, 2014), exhibited distorted and enlarged lumen in contrast to WT (or phosphomimic) INVS‐expressing cells, which have uniformly round lumen of smaller size.
Nephronophthisis type II, caused by mutations of NPHP2, is considered an autosomal recessive disease. Thus, the biological effect presented in MDCK cells was considered as primarily overexpression of the dominant negative effects which are overshadowing on top of endogenous expression derived from the wild‐type allele. Notably, however, the expression levels of endogenous INVS in MDCK cell appeared to be very low (almost undetectable) compared to the other cell lines (293T, HT1080, MCF7, and Cos‐7 cells) we have examined (Appendix Fig S4C). Therefore, the biological effect of the mutant INVS can be relatively clearly observed by the overexpression experiments in MDCK cells although the biological effects of mutant forms of INVS observed in the MDCK overexpression experiments were possibly due to the dominant negative effect of mutant INVS in the presence of minimally expressing endogenous INVS.
Moreover, two cases of human NPHP2 have been reportedly caused by heterozygous mutation in a single chromosome (A10‐1: heterozygous mutation of deletion of 2,908; F75‐1: heterozygous variant mutation of pA650P) (Tory et al, 2009). Although pathogenetic details were not described, three cases of NPHP3, another autosomal recessive type of human nephronophthisis, were associated with only one heterozygous mutation in humans (Tory et al, 2009).
PI3K–Akt signaling controls cell polarity and chemotaxis, in part, through the regulation of PAKα, which is required for myosin II assembly (Chung et al, 2001; Xue & Hemmings, 2013). Recent reports suggest that PI3K p110 delta promotes lumen formation by enhancing apico‐basal polarity and basal membrane organization (Peng et al, 2015). Tubular epithelial cells are endowed with a predetermined cellular program that controls the orientation of cell division relative to the neighboring cells. Therefore, spatially controlled division of regenerating tubular cells is suggested to be required for ensuring nephron integrity (Simons & Walz, 2006). Symmetric cell division plays an important role in the maintenance of cellular homeostasis, and loss of symmetric cell division underlies various human diseases (Morrison & Kimble, 2006). It has been reported that normal tubules in the kidney undergo oriented cell division, as a consequence, resulting in an elongation of the tubule after division (Lubarsky & Krasnow, 2003). When oriented cell division is lost, either through defective PCP or via mutations that give rise to PKD (polycystic kidney diseases), tubules become broader and remain short (McNeill, 2009).
Based on alterations of the angle of the spindle axis during cell division (Toyoshima & Nishida, 2007; Kikuchi et al, 2010), we correlated misalignment of the spindle axis to mutations found in NPHP2 patients. Since spindles can orient along two different axes, apico‐basal and planar, misalignment of the spindle axis in our experimental system may not fully represent or address the pathological nature of the cystogenic formation. Precisely defined structure and architecture at single cell and tissue levels appear to be required for proper nephron function. It is suggested that oriented cell division depends on a correctly positioned spindle axis. Therefore, INVS is suggested to control the orientation of cell division relative to the neighboring cells. When tubules elongate during renal development, tubular cells undergo massive proliferation (Simons & Walz, 2006). Further, Hildebrandt reported that defects in cystoproteins lead to disruption of PCP and formation of renal cysts due to malorientation of the centrosome or the mitotic spindle complex (Hildebrandt et al, 2009, 2011). Consistently, Happe reported that the renal cyst is characterized by a progressive development of fluid‐filled cyst derived from renal tubular epithelial cells. Altered ciliary signaling caused disoriented cell division in renal tubules, which resulted in renal cyst formation (Happe et al, 2011).
Consistent with the observation that INVS acts as a molecular switch between canonical and noncanonical Wnt signaling (Simons et al, 2005; Benzing et al, 2007), WT INVS inhibited Wnt signaling, an effect that was reversed by NPHP2‐related mutant forms of INVS (R899X, Q671X, or R603X), correlating with the expression levels of Dvl. A functional interaction of Wnt signal and Akt via Dvl has previously been suggested (Fukumoto et al, 2001). However, introduction of 3A or phospho‐mimetic INVS did not reverse the effects of WT INVS on the activation of Wnt signals. These observations suggest that the C‐terminus of INVS, rather than the Akt‐dependent phosphorylation of INVS at 864–866, plays an important role in Wnt signaling. Further research may be necessary for precisely defining the regulatory domains of INVS that differentially control Wnt signaling and the orientation of the spindle axis within the context of abnormal cyst formation in NPHP2.
Cell polarity refers to spatial differences in the shape, structure, and function of cells. In order to retain proper cell polarity during cell division, the mitotic spindle must be properly aligned with the axis of polarity (Simons & Walz, 2006; McNeill, 2009). Notably, Akt mediates the activation of glycogen synthase kinase‐3 for orchestrating mitotic spindle dynamics and chromosomal alignment (Wakefield et al, 2003). We showed that cells expressing phosphorylation‐defective INVS (3A) or NPHP2‐related mutant forms of INVS (Q671X, or R603X), which lack Akt phosphorylation sites, have disoriented spindle axes during cell division. This result can be partly explained by altered Wnt signaling, which is known to control spindle orientation (Kikuchi et al, 2010). INVS is localized at the spindle poles during anaphase (Eley et al, 2004) where it controls the orientation of cell division, cell polarity, and cell migration (Simons & Walz, 2006; Veland et al, 2013; Werner et al, 2013). We postulate that loss of INVS phosphorylation possibly underlies the distorted lumen formation observed in NPHP2, perhaps due to disorientation of the spindle axis during metaphase. Abnormal cilia formation and disorganized multi‐lumen structures were linked to the pathogenesis of NPHP2, an autosomal recessive renal disorder (Delous et al, 2009). However, since the spindle axis can be oriented in the apico‐basal or planar directions, the measurement of the baso‐apical angle of the spindle axis in the experimental system may not fully address the pathological nature of the cystogenic formation.
Both our in vitro kinase assays and cellular experiments with PDGF‐AA stimulation, known to transduce signals through primary cilia (Hoch & Soriano, 2003; Schneider et al, 2005, 2010; Christensen et al, 2007), demonstrated that INVS is phosphorylated at its 864–866 serine/threonine amino acid residues. Functionally, we demonstrated that Akt‐dependent phosphorylation of these residues is required for ciliary growth, cell proliferation, and proper lumen formation via proper alignment of the spindle axis. The C‐terminal of INVS determines the localization of INVS (Shiba et al, 2009). Interestingly, truncated forms of INVS, which lack the Akt phosphorylation sites (i.e., Q671X and R603X), appear to exhibit relatively severe clinical symptoms and often lead to end‐stage renal disease (Otto et al, 2003; Tory et al, 2009; Oud et al, 2014).
It is possible that impairment of spatially controlled division of regenerating tubular cells due to deregulation of the Akt–INVS interaction underlies the formation of abnormal lumen, perhaps due to loss of spatial orientation and lack of intercellular communication (Fig 8). Further studies will be required for implicating impaired functional interactions of Akt–INVS at the primary cilia as an underlying mechanism for NPHP2.
Figure 8. Model of Akt–Inversin interaction in nephronophthisis type II .
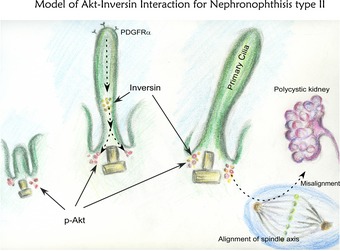
We found that Akt interacts with and phosphorylates the ciliary protein INVS, which is mutated in human NPHP2. Lack of phosphorylation of INVS leads to misalignment of the spindle axis and distorted luminal formation, underscoring the physiological significance of the Akt–INVS interaction in the development of abnormal cysts in NPHP2.
Materials and Methods
Yeast two‐hybrid assays
Yeast two‐hybrid assays were essentially performed as described previously using Akt2 as a bait (Suizu et al, 2009).
Construction of INVS expression vectors
Human INVS cDNA was purchased from RPZ and subcloned into the pFLAG‐CMV2, pME18s‐KOZAK‐HA, pEGFP‐C2, or pCSII‐EF1α. Other forms of INVS construct were generated by restriction digest or PCR.
Co‐immunoprecipitation experiments
Co‐immunoprecipitation experiments were essentially performed as described previously (Suizu et al, 2009). For endogenous Akt–INVS interaction, HEK293 cells were used.
Co‐localization experiment using a confocal microscopy
NIH3T3 cells or hTERT‐RPE1 cells (ATCC) were transfected with indicated plasmid, fixed with indicated antibodies or DAPI, and examined using a confocal microscope (Olympus).
Immunoelectron microscopy (IEM)
NIH3T3 cells were cultured, fixed, dehydrated, embedded, and immunostained for observation under an electron microscope (H‐7100; Hitachi, Tokyo).
Generation of recombinant INVS
Indicated forms of INVS were produced by pGEX6P‐3 according to the manufacturer's protocol with verification of the final products.
Luciferase reporter assays
Luciferase reporter assays were essentially performed using Dual‐Luciferase kit (Promega) using 293T cells transfected with indicated INVS or Dvl1‐HA by PEI (Suizu et al, 2009).
In vitro Akt kinase assays of INVS
IVK were performed essentially using the Akt kinase assay kit (Cell Signaling) with indicated INVS as previously described (Suizu et al, 2009).
Proliferation assays
Proliferation assays were performed essentially using xCelligence, and MDCK cells were transduced by lentivirus containing indicated INVS as previously described (Suizu et al, 2009).
Statistical analysis
Statistical analysis was performed by Student's t‐test or Mann–Whitney test, and P < 0.05 was considered as statistically significant.
Electroporation of siRNA for INVS or Akt
NIH3T3 or hTERT‐RPE1 cells were transfected with indicated siRNA by electroporation, harvested, immunoblotted or stained with anti‐acetylated tubulin and DAPI, and examined by (FLUOVIEW FV‐1000; Olympus). The cilia length was measured by NIH ImageJ.
Measurement of ciliary length
Indicated cells were transduced with indicated lentivirus vectors or plasmids and immunostained, and the ciliary length was measured as described elsewhere.
Measurement of spindle axis
The linear distance and the vertical distance between the two poles of the metaphase spindles of MDCK cells were measured by taking Z‐stack images, and the spindle angle was calculated (Kikuchi et al, 2010).
Three‐dimensional culture
3D culture of MDCK cells was conducted essentially as described elsewhere. The cells were transduced by indicated INVS, mounted into Matrigel, incubated, fixed, permeabilized, stained, and examined using a confocal microscope or differential interference contrast (DIC) microscope.
Further details are provided in Appendix Supplementary Methods.
Author contributions
FS, NH, KK, TE, TT, SI, and TD performed the experiments; TI performed TEM; HN histological analysis; and MN prepared the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Acknowledgements
We thank Drs. Soren Christensen, Hiroyuki Miyoshi, Patricia Salinas, Randall Moon, and Morris Birnbaum for providing valuable reagents used in this study, T. Yoshida for secretary assistance, and Takashi Ohashi and Yuka Morioka for their valuable techinical advice of lentivirus. MN is supported by Takeda Foundation and Naito Foundation. FS is supported by JSPS Kakenhi (25440039, 16K0875, and Japan Rheumatism Foundation).
The EMBO Journal (2016) 35: 1346–1363
References
- Adams M, Smith UM, Logan CV, Johnson CA (2008) Recent advances in the molecular pathology, cell biology and genetics of ciliopathies. J Med Genet 45: 257–267 [DOI] [PubMed] [Google Scholar]
- Badano JL, Mitsuma N, Beales PL, Katsanis N (2006) The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet 7: 125–148 [DOI] [PubMed] [Google Scholar]
- Benzing T, Simons M, Walz G (2007) Wnt signaling in polycystic kidney disease. J Am Soc Nephrol 18: 1389–1398 [DOI] [PubMed] [Google Scholar]
- Brazil DP, Yang ZZ, Hemmings BA (2004) Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci 29: 233–242 [DOI] [PubMed] [Google Scholar]
- Christensen ST, Pedersen LB, Schneider L, Satir P (2007) Sensory cilia and integration of signal transduction in human health and disease. Traffic 8: 97–109 [DOI] [PubMed] [Google Scholar]
- Chung CY, Potikyan G, Firtel RA (2001) Control of cell polarity and chemotaxis by Akt/PKB and PI3 kinase through the regulation of PAKa. Mol Cell 7: 937–947 [DOI] [PubMed] [Google Scholar]
- Clement CA, Ajbro KD, Koefoed K, Vestergaard ML, Veland IR, Henriques de Jesus MP, Pedersen LB, Benmerah A, Andersen CY, Larsen LA, Christensen ST (2013a) TGF‐beta signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep 3: 1806–1814 [DOI] [PubMed] [Google Scholar]
- Clement DL, Mally S, Stock C, Lethan M, Satir P, Schwab A, Pedersen SF, Christensen ST (2013b) PDGFRalpha signaling in the primary cilium regulates NHE1‐dependent fibroblast migration via coordinated differential activity of MEK1/2‐ERK1/2‐p90RSK and AKT signaling pathways. J Cell Sci 126: 953–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF (2005) Vertebrate Smoothened functions at the primary cilium. Nature 437: 1018–1021 [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell‐intrinsic death machinery. Cell 91: 231–241 [DOI] [PubMed] [Google Scholar]
- Delous M, Hellman NE, Gaude HM, Silbermann F, Le Bivic A, Salomon R, Antignac C, Saunier S (2009) Nephrocystin‐1 and nephrocystin‐4 are required for epithelial morphogenesis and associate with PALS1/PATJ and Par6. Hum Mol Genet 18: 4711–4723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dummler B, Tschopp O, Hynx D, Yang ZZ, Dirnhofer S, Hemmings BA (2006) Life with a single isoform of Akt: mice lacking Akt2 and Akt3 are viable but display impaired glucose homeostasis and growth deficiencies. Mol Cell Biol 26: 8042–8051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eley L, Turnpenny L, Yates LM, Craighead AS, Morgan D, Whistler C, Goodship JA, Strachan T (2004) A perspective on inversin. Cell Biol Int 28: 119–124 [DOI] [PubMed] [Google Scholar]
- Fukumoto S, Hsieh CM, Maemura K, Layne MD, Yet SF, Lee KH, Matsui T, Rosenzweig A, Taylor WG, Rubin JS, Perrella MA, Lee ME (2001) Akt participation in the Wnt signaling pathway through Dishevelled. J Biol Chem 276: 17479–17483 [DOI] [PubMed] [Google Scholar]
- Gerdes JM, Davis EE, Katsanis N (2009) The vertebrate primary cilium in development, homeostasis, and disease. Cell 137: 32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happe H, de Heer E, Peters DJ (2011) Polycystic kidney disease: the complexity of planar cell polarity and signaling during tissue regeneration and cyst formation. Biochim Biophys Acta 1812: 1249–1255 [DOI] [PubMed] [Google Scholar]
- Hildebrandt F, Attanasio M, Otto E (2009) Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol 20: 23–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Benzing T, Katsanis N (2011) Ciliopathies. N Engl J Med 364: 1533–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch RV, Soriano P (2003) Roles of PDGF in animal development. Development 130: 4769–4784 [DOI] [PubMed] [Google Scholar]
- Hoff S, Halbritter J, Epting D, Frank V, Nguyen TM, van Reeuwijk J, Boehlke C, Schell C, Yasunaga T, Helmstadter M, Mergen M, Filhol E, Boldt K, Horn N, Ueffing M, Otto EA, Eisenberger T, Elting MW, van Wijk JA, Bockenhauer D et al (2013) ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat Genet 45: 951–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Manning BD (2009) A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans 37: 217–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Corradetti MN, Guan KL (2005) Dysregulation of the TSC‐mTOR pathway in human disease. Nat Genet 37: 19–24 [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Niikura Y, Kitagawa K, Kikuchi A (2010) Dishevelled, a Wnt signalling component, is involved in mitotic progression in cooperation with Plk1. EMBO J 29: 3470–3483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Schroth J, Gleeson JG (2011) Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat Cell Biol 13: 700–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubarsky B, Krasnow MA (2003) Tube morphogenesis: making and shaping biological tubes. Cell 112: 19–28 [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129: 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall WF (2008) The cell biological basis of ciliary disease. J Cell Biol 180: 17–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda‐Lennikov M, Suizu F, Hirata N, Hashimoto M, Kimura K, Nagamine T, Fujioka Y, Ohba Y, Iwanaga T, Noguchi M (2014) Lysosomal interaction of akt with phafin2: a critical step in the induction of autophagy. PLoS One 9: e79795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill H (2009) Planar cell polarity and the kidney. J Am Soc Nephrol 20: 2104–2111 [DOI] [PubMed] [Google Scholar]
- Mergen M, Engel C, Muller B, Follo M, Schafer T, Jung M, Walz G (2013) The nephronophthisis gene product NPHP2/Inversin interacts with Aurora A and interferes with HDAC6‐mediated cilia disassembly. Nephrol Dial Transplant 28: 2744–2753 [DOI] [PubMed] [Google Scholar]
- Mochizuki T, Saijoh Y, Tsuchiya K, Shirayoshi Y, Takai S, Taya C, Yonekawa H, Yamada K, Nihei H, Nakatsuji N, Overbeek PA, Hamada H, Yokoyama T (1998) Cloning of inv, a gene that controls left/right asymmetry and kidney development. Nature 395: 177–181 [DOI] [PubMed] [Google Scholar]
- Morgan D, Eley L, Sayer J, Strachan T, Yates LM, Craighead AS, Goodship JA (2002a) Expression analyses and interaction with the anaphase promoting complex protein Apc2 suggest a role for inversin in primary cilia and involvement in the cell cycle. Hum Mol Genet 11: 3345–3350 [DOI] [PubMed] [Google Scholar]
- Morgan D, Goodship J, Essner JJ, Vogan KJ, Turnpenny L, Yost HJ, Tabin CJ, Strachan T (2002b) The left‐right determinant inversin has highly conserved ankyrin repeat and IQ domains and interacts with calmodulin. Hum Genet 110: 377–384 [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Kimble J (2006) Asymmetric and symmetric stem‐cell divisions in development and cancer. Nature 441: 1068–1074 [DOI] [PubMed] [Google Scholar]
- Nigg EA, Raff JW (2009) Centrioles, centrosomes, and cilia in health and disease. Cell 139: 663–678 [DOI] [PubMed] [Google Scholar]
- Noguchi M, Hirata N, Suizu F (2014) The links between AKT and two intracellular proteolytic cascades: ubiquitination and autophagy. Biochim Biophys Acta 1846: 342–352 [DOI] [PubMed] [Google Scholar]
- Noguchi M, Ropars V, Roumestand C, Suizu F (2007) Proto‐oncogene TCL1: more than just a coactivator for Akt. FASEB J 21: 2273–2284 [DOI] [PubMed] [Google Scholar]
- Nurnberger J, Bacallao RL, Phillips CL (2002) Inversin forms a complex with catenins and N‐cadherin in polarized epithelial cells. Mol Biol Cell 13: 3096–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurnberger J, Kribben A, Opazo Saez A, Heusch G, Philipp T, Phillips CL (2004) The Invs gene encodes a microtubule‐associated protein. J Am Soc Nephrol 15: 1700–1710 [DOI] [PubMed] [Google Scholar]
- Oh EC, Katsanis N (2012) Cilia in vertebrate development and disease. Development 139: 443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuzumi T, Fiedler D, Zhang C, Gray DC, Aizenstein B, Hoffman R, Shokat KM (2009) Inhibitor hijacking of Akt activation. Nat Chem Biol 5: 484–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong AC (2013) Primary cilia and renal cysts: does length matter? Nephrol Dial Transplant 28: 2661–2663 [DOI] [PubMed] [Google Scholar]
- Otto EA, Schermer B, Obara T, O'Toole JF, Hiller KS, Mueller AM, Ruf RG, Hoefele J, Beekmann F, Landau D, Foreman JW, Goodship JA, Strachan T, Kispert A, Wolf MT, Gagnadoux MF, Nivet H, Antignac C, Walz G, Drummond IA et al (2003) Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left‐right axis determination. Nat Genet 34: 413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oud MM, van Bon BW, Bongers EM, Hoischen A, Marcelis CL, de Leeuw N, Mol SJ, Mortier G, Knoers NV, Brunner HG, Roepman R, Arts HH (2014) Early presentation of cystic kidneys in a family with a homozygous INVS mutation. Am J Med Genet A 164A: 1627–1634 [DOI] [PubMed] [Google Scholar]
- Peng J, Awad A, Sar S, Hamze Komaiha O, Moyano R, Rayal A, Samuel D, Shewan A, Vanhaesebroeck B, Mostov K, Gassama‐Diagne A (2015) Phosphoinositide 3‐kinase p110delta promotes lumen formation through the enhancement of apico‐basal polarity and basal membrane organization. Nat Commun 6: 5937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CL, Miller KJ, Filson AJ, Nurnberger J, Clendenon JL, Cook GW, Dunn KW, Overbeek PA, Gattone VH 2nd, Bacallao RL (2004) Renal cysts of inv/inv mice resemble early infantile nephronophthisis. J Am Soc Nephrol 15: 1744–1755 [DOI] [PubMed] [Google Scholar]
- Santos N, Reiter JF (2008) Building it up and taking it down: the regulation of vertebrate ciliogenesis. Dev Dyn 237: 1972–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunier S, Salomon R, Antignac C (2005) Nephronophthisis. Curr Opin Genet Dev 15: 324–331 [DOI] [PubMed] [Google Scholar]
- Schneider L, Cammer M, Lehman J, Nielsen SK, Guerra CF, Veland IR, Stock C, Hoffmann EK, Yoder BK, Schwab A, Satir P, Christensen ST (2010) Directional cell migration and chemotaxis in wound healing response to PDGF‐AA are coordinated by the primary cilium in fibroblasts. Cell Physiol Biochem 25: 279–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST (2005) PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol 15: 1861–1866 [DOI] [PubMed] [Google Scholar]
- Shiba D, Yamaoka Y, Hagiwara H, Takamatsu T, Hamada H, Yokoyama T (2009) Localization of Inv in a distinctive intraciliary compartment requires the C‐terminal ninein‐homolog‐containing region. J Cell Sci 122: 44–54 [DOI] [PubMed] [Google Scholar]
- Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G (2005) Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet 37: 537–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Walz G (2006) Polycystic kidney disease: cell division without a c(l)ue? Kidney Int 70: 854–864 [DOI] [PubMed] [Google Scholar]
- Suizu F, Hiramuki Y, Okumura F, Matsuda M, Okumura AJ, Hirata N, Narita M, Kohno T, Yokota J, Bohgaki M, Obuse C, Hatakeyama S, Obata T, Noguchi M (2009) The E3 ligase TTC3 facilitates ubiquitination and degradation of phosphorylated Akt. Dev Cell 17: 800–810 [DOI] [PubMed] [Google Scholar]
- Suizu F, Hirata N, Ishigaki S, Edamura T, Kimura K, Tanaka T, Noguchi M (2016) Primary cilium‐mediated crosstalk of signaling cascades in ciliogenesis: implications for tumorigenesis and senescence. Cell Commun Insights, in press
- Tory K, Rousset‐Rouviere C, Gubler MC, Moriniere V, Pawtowski A, Becker C, Guyot C, Gie S, Frishberg Y, Nivet H, Deschenes G, Cochat P, Gagnadoux MF, Saunier S, Antignac C, Salomon R (2009) Mutations of NPHP2 and NPHP3 in infantile nephronophthisis. Kidney Int 75: 839–847 [DOI] [PubMed] [Google Scholar]
- Toyoshima F, Nishida E (2007) Integrin‐mediated adhesion orients the spindle parallel to the substratum in an EB1‐ and myosin X‐dependent manner. EMBO J 26: 1487–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veikkolainen V, Naillat F, Railo A, Chi L, Manninen A, Hohenstein P, Hastie N, Vainio S, Elenius K (2012) ErbB4 modulates tubular cell polarity and lumen diameter during kidney development. J Am Soc Nephrol 23: 112–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ (1993) Bcl‐2‐deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 75: 229–240 [DOI] [PubMed] [Google Scholar]
- Veland IR, Montjean R, Eley L, Pedersen LB, Schwab A, Goodship J, Kristiansen K, Pedersen SF, Saunier S, Christensen ST (2013) Inversin/Nephrocystin‐2 is required for fibroblast polarity and directional cell migration. PLoS One 8: e60193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield JG, Stephens DJ, Tavare JM (2003) A role for glycogen synthase kinase‐3 in mitotic spindle dynamics and chromosome alignment. J Cell Sci 116: 637–646 [DOI] [PubMed] [Google Scholar]
- Watanabe D, Saijoh Y, Nonaka S, Sasaki G, Ikawa Y, Yokoyama T, Hamada H (2003) The left‐right determinant Inversin is a component of node monocilia and other 9 + 0 cilia. Development 130: 1725–1734 [DOI] [PubMed] [Google Scholar]
- Werner ME, Ward HH, Phillips CL, Miller C, Gattone VH, Bacallao RL (2013) Inversin modulates the cortical actin network during mitosis. Am J Physiol Cell Physiol 305: C36–C47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winyard P, Jenkins D (2011) Putative roles of cilia in polycystic kidney disease. Biochim Biophys Acta 1812: 1256–1262 [DOI] [PubMed] [Google Scholar]
- Woo D (1995) Apoptosis and loss of renal tissue in polycystic kidney diseases. N Engl J Med 333: 18–25 [DOI] [PubMed] [Google Scholar]
- Xue G, Hemmings BA (2013) PKB/Akt‐dependent regulation of cell motility. J Natl Cancer Inst 105: 393–404 [DOI] [PubMed] [Google Scholar]
- Yasuhiko Y, Imai F, Ookubo K, Takakuwa Y, Shiokawa K, Yokoyama T (2001) Calmodulin binds to inv protein: implication for the regulation of inv function. Dev Growth Differ 43: 671–681 [DOI] [PubMed] [Google Scholar]
- Zariwala MA, Knowles MR, Omran H (2007) Genetic defects in ciliary structure and function. Annu Rev Physiol 69: 423–450 [DOI] [PubMed] [Google Scholar]
- Zhu D, Shi S, Wang H, Liao K (2009) Growth arrest induces primary‐cilium formation and sensitizes IGF‐1‐receptor signaling during differentiation induction of 3T3‐L1 preadipocytes. J Cell Sci 122: 2760–2768 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File