

Yeast cells know whether or not they have a bud. The kinase Elm1 and the septin cytoskeleton are key transducers of cell shape information.
Abstract
Bud formation by Saccharomyces cerevisiae must be coordinated with the nuclear cycle to enable successful proliferation. Many environmental stresses temporarily disrupt bud formation, and in such circumstances, the morphogenesis checkpoint halts nuclear division until bud formation can resume. Bud emergence is essential for degradation of the mitotic inhibitor, Swe1. Swe1 is localized to the septin cytoskeleton at the bud neck by the Swe1-binding protein Hsl7. Neck localization of Swe1 is required for Swe1 degradation. Although septins form a ring at the presumptive bud site before bud emergence, Hsl7 is not recruited to the septins until after bud emergence, suggesting that septins and/or Hsl7 respond to a “bud sensor.” Here we show that recruitment of Hsl7 to the septin ring depends on a combination of two septin-binding kinases: Hsl1 and Elm1. We elucidate which domains of these kinases are needed and show that artificial targeting of those domains suffices to recruit Hsl7 to septin rings even in unbudded cells. Moreover, recruitment of Elm1 is responsive to bud emergence. Our findings suggest that Elm1 plays a key role in sensing bud emergence.
INTRODUCTION
Cell cycle progression is orchestrated by a regulatory network centered on cyclin-dependent kinases (CDKs), whose activity oscillates during the cell cycle, sequentially triggering DNA replication, chromosome segregation, and cytokinesis (Morgan, 1997). The proper order of cell cycle events is further enforced by checkpoint controls, which are surveillance pathways that can detect errors or delays in key cell cycle events (Hartwell and Weinert, 1989). Most cells have checkpoints that delay entry into mitosis if DNA replication is incomplete (or if there is DNA damage) and delay the metaphase–anaphase transition if sister chromatids have not attained a bipolar attachment to the mitotic spindle. By delaying the later event, checkpoints prevent the potentially catastrophic effects of proceeding with the cell cycle when an early event fails to occur in a timely manner.
The budding yeast Saccharomyces cerevisiae has served as a tractable model for studies of cell cycle control. Yeast cells are surrounded by a rigid cell wall, and daughter cells are produced as buds adjacent to the mother cell. After bud formation, the mitotic spindle aligns along the mother–bud axis so that mother and daughter both inherit a full complement of chromosomes during nuclear division. Bud formation and progression of the nuclear cycle are coupled by two cell cycle checkpoints in addition to those discussed so far. The morphogenesis checkpoint (Lew, 2003) delays nuclear division in cells that have not yet formed a bud, and the spindle orientation checkpoint (Lew and Burke, 2003) delays exit from mitosis until one pole of the anaphase spindle has penetrated into the bud. Together, these checkpoints prevent the formation of binucleated cells.
The morphogenesis checkpoint delays nuclear division via inhibitory phosphorylation of the mitotic CDK at Tyr-19 (Lew and Reed, 1995; Sia et al., 1996), which is regulated by the Wee1-family kinase Swe1 and the Cdc25-family phosphatase Mih1 (Russell et al., 1989; Booher et al., 1993). Both Swe1 and Mih1 are targets of regulatory pathways, and regulation of one enzyme is only effective in combination with regulation of the other (McMillan et al., 1999a; Harrison et al., 2001; Anastasia et al., 2012). One of the regulatory pathways impinges on the abundance of Swe1. SWE1 is transcribed only in late G1/early S phase, and Swe1 is then degraded before nuclear division (Lim et al., 1996; Sia et al., 1996, 1998). However, stresses that perturb bud formation promote stabilization of Swe1, leading to phosphorylation of the CDK (Sia et al., 1998).
Degradation of Swe1 is believed to be a multistep process involving 1) priming of phosphorylation of Swe1 by mitotic CDK, 2) further phosphorylation of Swe1 by the Polo-family kinase Cdc5, 3) ubiquitination of hyperphosphorylated Swe1 by the ubiquitin ligases Dma1 and Dma2, and 4) degradation by the proteasome (Sakchaisri et al., 2004; Asano et al., 2005; Raspelli et al., 2011). This delicately balanced pathway, involving mutual antagonism between mitotic CDK and Swe1, is tilted in favor of Swe1 when cells are exposed to a variety of stresses that compromise the actin cytoskeleton and hence bud formation (King et al., 2013). A central question is how (perturbation of) bud formation influences Swe1/Mih1 and hence mitotic progression.
Strikingly, Swe1 degradation is coupled to its localization (Longtine et al., 2000; King et al., 2012). Swe1 transits in and out of the nucleus, and in budded cells, a fraction of the Swe1 is localized to the mother–bud neck, where it is phosphorylated to promote its degradation (Longtine et al., 2000; Keaton et al., 2008). Neck localization of Swe1 is mediated by a cascade of interactions: Swe1 is localized via binding to Hsl7, which is localized via binding to Hsl1, which is localized via binding to septins (Barral et al., 1999; Shulewitz et al., 1999; Longtine et al., 2000; Cid et al., 2001). Septins are a family of cytoskeletal filament-forming proteins important for cytokinesis: they assemble into a ring at the presumptive bud site before bud emergence and then reorganize to form an hourglass-shaped collar at the mother–bud neck (Ong et al., 2014). Curiously, Hsl7 (and hence Swe1) localizes to the septins only after a bud has formed (Longtine et al., 2000; Cid et al., 2001; Theesfeld et al., 2003), suggesting that Hsl7 localization might serve as a “bud sensor,” enabling Swe1 degradation only once a bud had been formed.
Given that septins assemble into a ring before bud emergence, why does Hsl7 not accumulate there until after a bud has formed? The Nim1-family kinase Hsl1 links Hsl7 to the septins (Shulewitz et al., 1999; Longtine et al., 2000). HSL1 is transcribed in late G1, and Hsl1 is targeted for degradation by the anaphase-promoting complex during mitotic exit, so one obvious reason why Hsl7 does not localize could be the absence of Hsl1 until S phase, when a bud has formed (McMillan et al., 1999a; Burton and Solomon, 2000). In cells treated with the actin-depolymerizing drug latrunculin to block bud emergence, Hsl1 accumulates and becomes localized to the septin ring, but Hsl7 localization in such cells is significantly delayed (Theesfeld et al., 2003; King et al., 2013). Thus Hsl7 localization probably requires additional factors beyond the presence of Hsl1. In this study, we investigated Hsl7 localization using live-cell imaging. We show that Hsl7 localization requires another kinase, Elm1, in addition to Hsl1. Like Hsl7, Elm1 localization is only detectable after bud emergence, and our findings suggest that Elm1 localization is affected by the local membrane geometry in the vicinity of the septins. We speculate that local membrane curvature affects septin filament organization, so that bud formation creates a structure with higher affinity for Elm1. In this way, the septins may act as sensors for bud emergence, transmitting that information through Elm1 and Hsl7 to degrade Swe1 and hence allow nuclear division once a bud has formed.
RESULTS
Live-cell imaging of Hsl7 localization
The foundational studies of Hsl7 localization were performed using immunofluorescence on fixed cells (Shulewitz et al., 1999; Longtine et al., 2000; Theesfeld et al., 2003). Using a functional green fluorescent protein (GFP)–Hsl7 probe (King et al., 2013), we confirmed that Hsl1-GFP and GFP-Hsl7 localized to septin rings (marked using a functional Cdc3-mCherry probe; Tong et al., 2007) only in budded cells (Figure 1A). However, the GFP-Hsl7 signal was too weak to be reliably detected in time-lapse imaging. Using the strong constitutive TEF1 promoter, we overexpressed GFP-Hsl7, allowing us to quantify Hsl7 localization through the cell cycle (Figure 1, B–D). As previously reported (McMillan et al., 1999a), overexpression of Hsl7 did not impair cell growth or morphology. Time-lapse imaging showed that overexpressed GFP-Hsl7 localized to septin rings after a delay of ∼10 min, once buds were present (Figure 1, B–D, and Supplemental Video S1). Similar results were obtained in strains expressing a nondegradable version of Hsl1 (Burton and Solomon, 2001; Supplemental Figure S1 and Supplemental Video S2). Thus the delay in Hsl7 recruitment to septins is not due to absence of Hsl1 or low expression of Hsl7.
FIGURE 1:
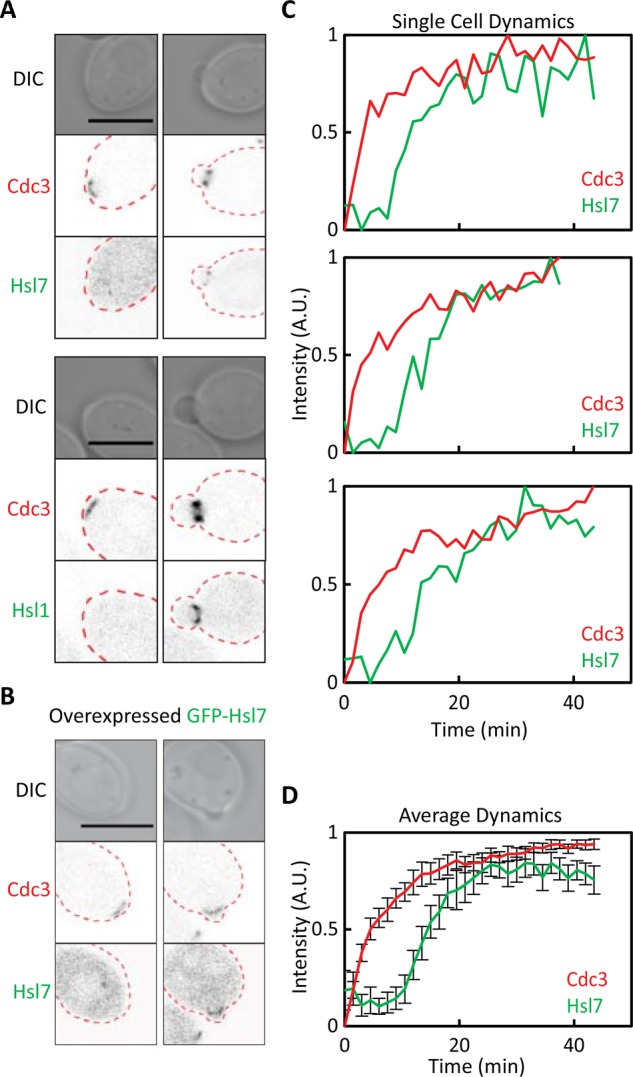
Imaging Hsl7 recruitment to the septin ring. (A) Hsl1 and Hsl7 recruitment occurs after bud emergence. Images of cells expressing the septin marker Cdc3-mCherry and GFP-Hsl7 (DLY14838) or Hsl1-GFP (DLY18904) from their endogenous promoters. (B) Overexpression of Hsl7 does not advance the timing of Hsl7 recruitment. Images of Cdc3-mCherry and overexpressed GFP-Hsl7 (DLY17799). (C) Quantification of septin and Hsl7 recruitment with time in individual cells overexpressing Hsl7. The fluorescence intensity of the two probes was quantified from time-lapse videos of three individual cells. (D) Average fluorescence intensities from 23 cells aligned to the first time point at which septins became detectable. Error bars, SD. Scale bar, 5 μm.
Role of Hsl1 in the timing of Hsl7 localization
Hsl1 has an N-terminal kinase domain and a large C-terminal regulatory domain with conserved motifs that mediate septin, Hsl7, and membrane interactions (Crutchley et al., 2009; Moravcevic et al., 2010). Although a C-terminal region (residues 1138–1307) is sufficient to bind Hsl7 in vitro, efficient Hsl7 localization in vivo also appears to require Hsl1 kinase activity (Theesfeld et al., 2003; Crutchley et al., 2009). Confirming the importance of Hsl1 kinase activity, we found that strains expressing a kinase-dead version of Hsl1 failed to recruit detectable amounts of overexpressed GFP-Hsl7 to the bud (Figure 2, A–C). This observation suggests that Hsl1 phosphorylates a substrate that enhances Hsl1–Hsl7 interaction.
FIGURE 2:

Hsl1 kinase activity is required for Hsl7 recruitment but can be bypassed by tethering the Hsl7-binding domain of Hsl1 to the septin ring. (A) Impaired Hsl7 recruitment in cells with kinase-dead Hsl1K110R. Images of Cdc3-mCherry and overexpressed GFP-Hsl7 (DLY21067). (B) Quantification of septin and Hsl7 recruitment with time in individual cells. (C) Average fluorescence intensities from 11 cells aligned to the first time point at which septins became detectable. GFP-Hsl7 intensity was normalized to that in wild-type (WT) cells (DLY21066) using the peak Hsl7:Cdc3 intensity ratio. Peak Hsl7:Cdc3 intensity ratio was 10.0 ± 3.3 in WT cells and 1.5 ± 0.36 in Hsl1K110R cells. (D) Schematic of Hsl1 indicating domains that were fused to the septin. (E) Images of Cdc3-mCherry and the indicated Cdc3-GFP-Hsl1 fusions (DLY14978, DLY14921), showing that fusions coassemble into septin rings of unbudded cells. (F) Expression of Cdc3-Hsl1879-1307 in cells with kinase-dead Hsl1K110R restored Hsl7 recruitment in budded cells. Images of Cdc3-mCherry and overexpressed GFP-Hsl7 (DLY21065). (G) Quantification of septin and Hsl7 recruitment with time in individual cells. (H) Average fluorescence intensities from 18 cells aligned to the first time point at which septins became detectable. (I) Expression of Cdc3-Hsl1879-1307 does not alter Hsl7 recruitment in wild-type cells. Images of Cdc3-mCherry and overexpressed GFP-Hsl7 (DLY21066). (J) Quantification of septin and Hsl7 recruitment with time in individual cells. (K) Average fluorescence intensities from 21 cells aligned to the first time point at which septins became detectable. Error bars, SD. Scale bar, 5 μm.
The most obvious candidate substrates are Hsl1 itself and Hsl7: both are documented substrates for Hsl1 kinase (Barral et al., 1999; McMillan et al., 1999a). One possibility is that autophosphorylation by Hsl1 induces a conformational change that unmasks the Hsl7-binding region. If that were the case, then targeting an “unmasked” Hsl7-binding region of Hsl1 to septins might bypass the need for Hsl1 kinase activity. To test this, we fused either the minimal Hsl7-binding domain (residues 1138–1307) or a larger region of Hsl1 (residues 879–1307) to the septin Cdc3 (Figure 2D). Both fusions lack the kinase domain and localize to septin rings (Figure 2E). The larger construct led to stronger Hsl7 recruitment (see later discussion), so in most subsequent experiments, we used Cdc3-Hsl1879-1307.
Expression of Cdc3-Hsl1879-1307 restored Hsl7 recruitment in strains expressing a kinase-dead version of Hsl1 (Figure 2, F–H) to levels comparable to those in wild-type strains expressing the construct (Figure 2, I–K). Because the kinase-dead Hsl1 itself contains residues 879–1307 and localizes well to the mother–bud neck (Theesfeld et al., 2003), these findings suggest that the major role of Hsl1 kinase activity in Hsl7 recruitment is to unmask the Hsl7-binding domain of Hsl1. Thus, when that domain is targeted to septins by direct fusion, Hsl1 kinase activity becomes dispensable for Hsl7 recruitment.
Both Cdc3-Hsl11138-1307 and Cdc3-Hsl1879-1307 were able to recruit Hsl7 to the mother–bud neck even in cells deleted for endogenous HSL1 (Figure 3, A and B). However, there was still a delay in Hsl7 recruitment to the septin rings, similar to the delay observed in wild-type cells (Figure 3, C and D, and Supplemental Video S3). In contrast, Cdc3-Hsl11138-1307 and Cdc3-Hsl1879-1307 were coassembled into initial septin rings with no delay (Figure 2E and Supplemental Video S4). These observations indicate that additional factors beyond the unmasking of Hsl1 must regulate the ability of Hsl11138-1307 to recruit Hsl7.
FIGURE 3:
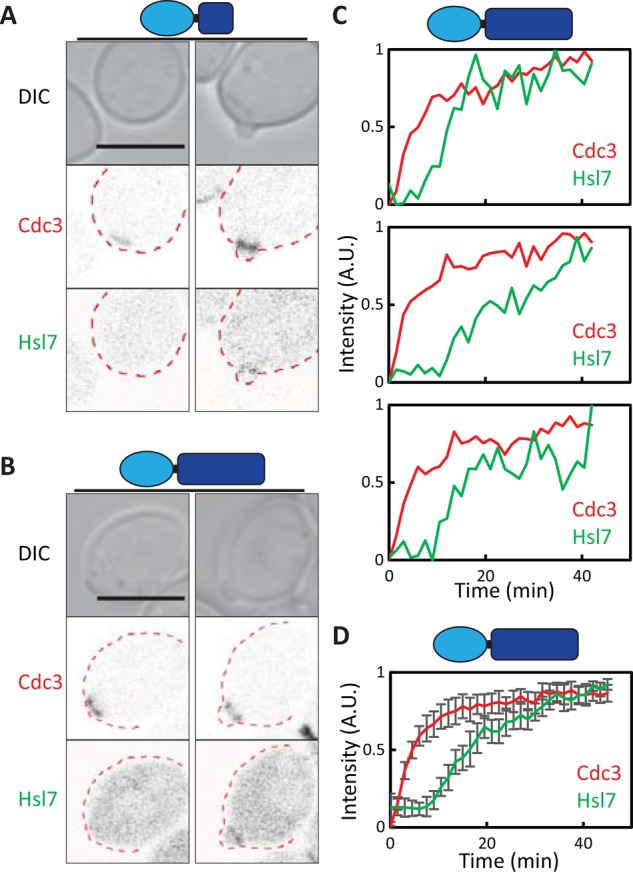
Tethering of the Hsl7-binding domain of Hsl1 to the septins promotes Hsl7 recruitment only in budded cells. (A, B) Cdc3-Hsl1 fusions recruit Hsl7 to septin rings in budded but not unbudded cells. Images of Cdc3-mCherry and overexpressed GFP-Hsl7 (DLY14895, DLY17674). (C) Quantification of septin and Hsl7 recruitment with time in individual cells (DLY17674). (D) Average fluorescence intensities from 19 cells aligned to the first time point at which septins became detectable. Error bars, SD. Scale bar, 5 μm.
Role of Elm1 in the timing of Hsl7 localization
The kinase Elm1 is an attractive candidate for an Hsl7-recruitment factor. Elm1 is localized to the septin collar and has been implicated in targeting Swe1 for degradation by phosphorylating the activation-loop threonine in the kinase domain of Hsl1 (Bouquin et al., 2000; Szkotnicki et al., 2008). Moreover, early reports suggested that Elm1 localization, like that of Hsl7, occurred only after bud emergence (Bouquin et al., 2000). We confirmed that a functional Elm1-GFP localized to septin rings (marked using Cdc3-mCherry) only in budded cells (Figure 4A). Similar to GFP-Hsl7, the Elm1-GFP signal was too weak to be reliably detected in time-lapse imaging, so we overexpressed Elm1-GFP from the ADH1 promoter. However, unlike overexpressed Hsl7, overexpressed Elm1-GFP was localized to the septin rings of unbudded cells (Figure 4B). Time-lapse imaging also indicated rapid recruitment of overexpressed Elm1 to forming septin rings (Figure 4, C and D, and Supplemental Video S5). These findings suggest that the delayed recruitment of Elm1 to septin rings stems from its low level of expression.
FIGURE 4:
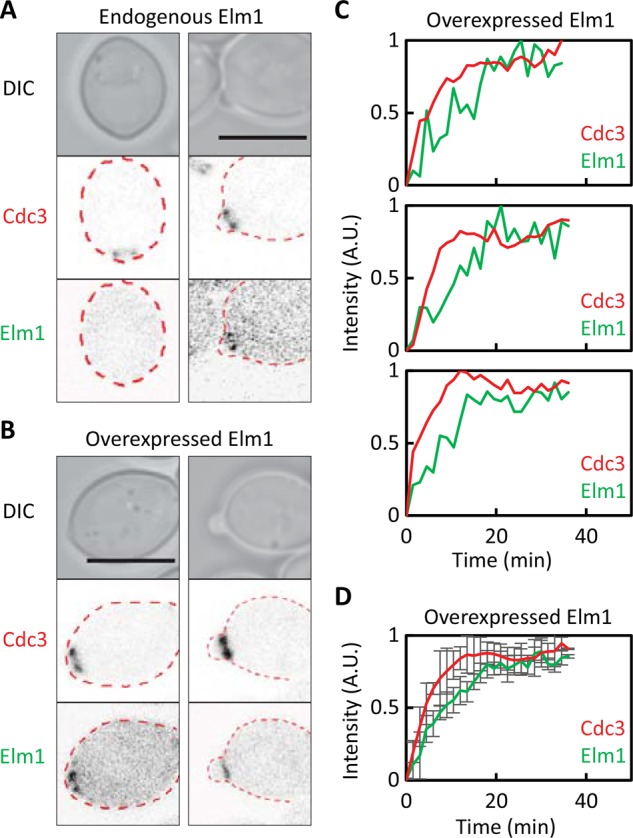
Localization of Elm1 to septin rings is advanced upon overexpression. (A) Elm1 recruitment occurs after bud emergence. Images of cells expressing Cdc3-mCherry and Elm1-GFP (DLY16705) from their endogenous promoters. (B) Overexpression of Elm1 advances the timing of Elm1 recruitment. Images of Cdc3-mCherry and overexpressed Elm1-GFP (DLY18285). (C) Quantification of septin and overexpressed Elm1 recruitment with time in individual cells. (D) Average fluorescence intensities from 21 cells aligned to the first time point at which septins became detectable. Error bars, SD. Scale bar, 5 μm.
To ask whether earlier recruitment of Elm1 would suffice to promote early recruitment of Hsl7 to septin rings, we monitored GFP-Hsl7 localization in cells overexpressing Elm1. Hsl7 recruitment was still delayed in cells overexpressing Elm1 (Figure 5, A–C, and Supplemental Video S6). To ask whether that was due to the absence of unmasked Hsl1 (see earlier discussion), we combined Cdc3-Hsl1879-1307 with overexpression of Elm1. That combination promoted early recruitment of Hsl7 to septin rings (Figure 5, D–F, and Supplemental Video S7), suggesting that Hsl7 recruitment requires both Hsl1 and Elm1 at the septin ring.
FIGURE 5:
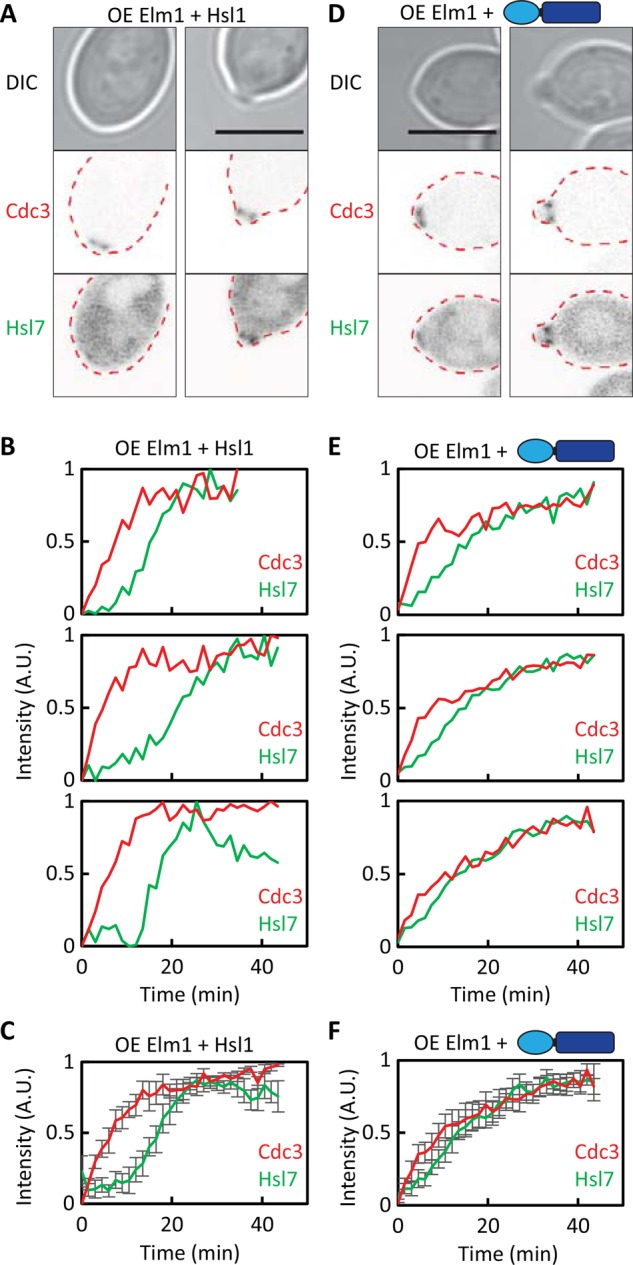
Elm1 overexpression and Hsl7 recruitment. (A) Elm1 overexpression is not sufficient to advance Hsl7 recruitment. Images of Cdc3-mCherry and overexpressed GFP-Hsl7 in cells overexpressing Elm1 (DLY18541). (B) Quantification of septin and Hsl7 recruitment with time in individual cells. (C) Average fluorescence intensities from 18 cells aligned to the first time point at which septins became detectable. (D) A combination of Elm1 overexpression and Cdc3-Hsl1879-1307 advances Hsl7 recruitment. Images of Cdc3-mCherry and overexpressed GFP-Hsl7 (DLY18387). (E) Quantification of septin and Hsl7 recruitment with time in individual cells. (F) Average fluorescence intensities from 22 cells aligned to the first time point at which septins became detectable. Error bars, SD. Scale bar, 5 μm.
Because overexpression of Elm1 might have indirect effects beyond advancing the timing of Elm1 localization, we sought to confirm this conclusion by using an alternative strategy. Inspired by a similar approach used by others (Moore et al., 2010), we fused the Elm1 kinase domain to the septin-binding protein Bni4, which is targeted to newly formed septin rings in unbudded cells (DeMarini et al., 1997; Figure 6A). Bni4-Elm1 was targeted to septin rings in unbudded cells (Figure 6B), but on its own, this construct did not affect the timing of Hsl7 recruitment (Figure 6, C–E). However, in combination, Bni4-Elm1 and Cdc3-Hsl1879-1307 advanced the timing of Hsl7 recruitment to initial septin rings (Figure 6, F–H, and Supplemental Video S8). This was dependent on Elm1 kinase activity, because a similar strain expressing a kinase-dead Bni4-Elm1 fusion did not display early Hsl7 recruitment (Figure 6, I–K).
FIGURE 6:

Targeting the Elm1 kinase domain to the septin ring. (A) Schematic of Bni4-Elm1 constructs. (B) Fusion of the Elm1 kinase domain to Bni4 causes recruitment of the fusion to septin rings in unbudded cells. Images of Cdc3-mCherry and Bni4-GFP-Elm1 (DLY16156). (C) Bni4-Elm1 is not sufficient to advance Hsl7 recruitment. Images of Cdc3-mCherry and overexpressed GFP-Hsl7 in cells expressing Bni4-Elm1 (DLY16162). (D) Quantification of septin and Hsl7 recruitment with time in individual cells. (E) Average fluorescence intensities from 21 cells aligned to the first time point at which septins became detectable. (F) A combination of Bni4-Elm1 and Cdc3-Hsl1987-1307 advances Hsl7 recruitment. Images of Cdc3-mCherry and overexpressed GFP-Hsl7 (DLY17723). (G) Quantification of septin and Hsl7 recruitment with time in individual cells. (H) Average fluorescence intensities from 19 cells aligned to the first time point at which septins became detectable. (I) Expression of a kinase-dead Bni4-Elm1K117R does not advance Hsl7 recruitment by Cdc3-Hsl1879-1307. Images of Cdc3-mCherry and overexpressed GFP-Hsl7 (DLY18597). (J) Quantification of septin and Hsl7 recruitment with time in individual cells. (K) Average fluorescence intensities from 17 cells aligned to the first time point at which septins became detectable. Error bars, SD. Scale bar, 5 μm.
In another approach, we directly fused the Elm1 kinase domain to Cdc3-Hsl1879-1307 (Figure 7A) and found that this single fusion construct was sufficient to advance the timing of Hsl7 recruitment (Figure 7, B–D, and Supplemental Video S9). However, a similar construct with a kinase-dead mutation in Elm1 did not advance Hsl7 recruitment (Figure 7, E–G). These findings indicate that Hsl7 recruitment to septins responds to the combined presence of a C-terminal Hsl7-binding domain from Hsl1 and an active Elm1 kinase domain.
FIGURE 7:
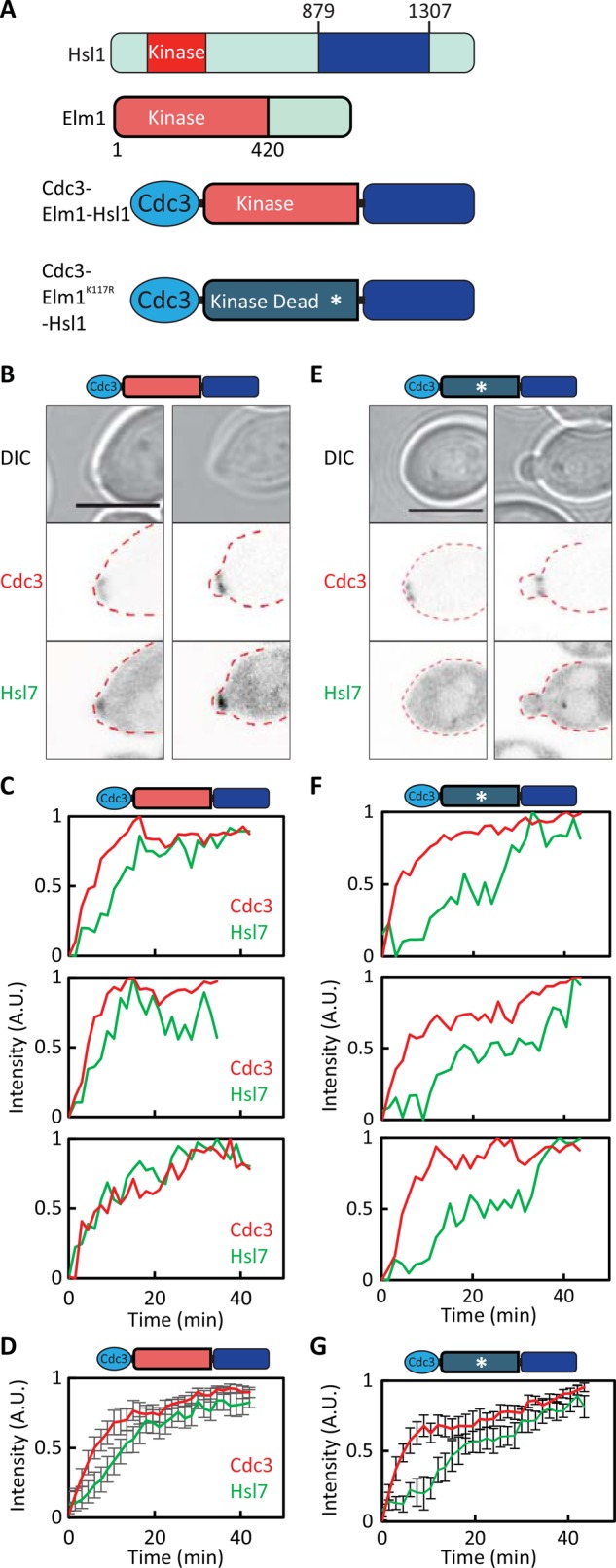
Colocalization of the Hsl7-binding domain from Hsl1 and the Elm1 kinase domain suffices to promote localization of Hsl7 to the septin ring. (A) Schematic of the Cdc3-Elm1-Hsl1879-1307 construct. (B) Cdc3-Elm1-Hsl1879-1307 advances Hsl7 recruitment. Images of Cdc3-mCherry and overexpressed GFP-Hsl7 (DLY18288). (C) Quantification of septin and Hsl7 recruitment with time in individual cells. (D) Average fluorescence intensities from cells aligned to the first time point at which septins became detectable. (E) Kinase-dead Cdc3-Elm1 K117R-Hsl1879-1307 does not advance Hsl7 recruitment. Images of Cdc3-mCherry and overexpressed GFP-Hsl7 (DLY21088). (F) Quantification of septin and Hsl7 recruitment with time in individual cells. (G) Average fluorescence intensities from 17 cells aligned to the first time point at which septins became detectable. Error bars, SD. Scale bar, 5 μm.
Effect of local cortical geometry on Elm1 localization
Because Elm1 and Hsl7 are recruited to the septins only after a bud has emerged, it is possible that their recruitment reflects the local geometry of the cell cortex in the vicinity of the septin ring (Lew, 2003; Theesfeld et al., 2003). To test this hypothesis, we used a pheromone arrest–release protocol to generate “shmoos” (cells with mating projections). After washout of the pheromone, the cells enter the cell cycle and generate septin rings either within the projection or away from the projection. Previous studies showed that Hsl7 was recruited earlier to rings that formed within the projections (local geometry similar to the bud neck) than rings that formed away from the projection (local geometry as in unbudded cells; Theesfeld et al., 2003; King et al., 2013). We next investigated whether geometry might affect Hsl1 or Elm1 recruitment to the septin ring.
Haploid MATa cells were arrested by a 3-h treatment with a high dose of α-factor. We then washed out the pheromone, incubated the cells for 15 min to allow recovery from the arrest, and plated the cells onto a slab containing latrunculin B to depolymerize actin and prevent bud formation. As cells proceeded into the cell cycle, they formed septin rings (detected using Cdc3-mCherry), to which they recruited Elm1-GFP (Figure 8A) and Hsl1-GFP (Figure 8B). Hsl1 was recruited with similar timing regardless of whether septin rings formed near the tip of the mating projection or away from the projection (Figure 8, B and C). However, there was a significant difference in the timing of Elm1 recruitment: septin rings inside the projection recruited Elm1 much faster than those outside the projection (Figure 8, A and C). Thus Elm1 recruitment to the septin ring is sensitive to local geometry, potentially accounting for the previously documented responsiveness of Hsl7 recruitment to local geometry.
FIGURE 8:

Cortical geometry and recruitment of Hsl1 and Elm1. (A) Recruitment of Elm1 to septin rings in shmoos. Cells expressing Elm1-GFP from the endogenous promoter (DLY16728) were arrested in G1 with pheromone, released, and treated with latrunculin B to block bud formation. Images of Cdc3-mCherry and Elm1-GFP for representative cells that formed septin rings within the mating projection (left) or elsewhere (right). Time (minutes) is from the first time point at which septins became detectable. Quantification of septin and Elm1 localization with time in individual cells. (B) Recruitment of Hsl1 to septin rings in shmoos. Cells expressing Hsl1-GFP from the endogenous promoter (DLY18964) were treated as in A. Quantification of septin and Hsl1 localization with time in individual cells. (C) Timing of Hsl1 and Elm1 recruitment to septin rings, classified according to whether the rings formed within or outside the mating projection. Elm1 recruitment is responsive to local geometry, whereas Hsl1 recruitment is not. Intervals between first detection of the septin ring and first detection of Elm1 or Hsl1 are plotted (box report 25–75% quartiles, line is median, and whiskers report full range). Statistical significance was tested by a two-tailed Student’s t test (ns, not significantly different). Note that because Hsl1-GFP is newly synthesized, fluorophore maturation may contribute to the timing with which septin-localized signal became detectable in this case. (D) Model of how local membrane curvature might affect septin organization. Scale bar, 5 μm.
DISCUSSION
Activation of the mitotic CDK in budding yeast can be delayed by a variety of stresses that affect bud formation (Lew and Reed, 1995; McMillan et al., 1998; Barral et al., 1999; Alexander et al., 2001; Theesfeld et al., 2003; Uesono et al., 2004; McNulty and Lew, 2005; Clotet et al., 2006; King et al., 2013). The delay results from Swe1-mediated inhibitory phosphorylation of the CDK, and the duration of the delay reflects the balance between the activities of Swe1 and the counteracting phosphatase Mih1 (Sia et al., 1996), which is regulated by a cell wall integrity stress-response pathway involving Pkc1 (Harrison et al., 2001; Anastasia et al., 2012). The Swe1-regulatory pathway requires tethering of Swe1 at the septin ring (King et al., 2012), which in turn requires localization of Hsl7 to the septin ring. We now show that Hsl7 localization reflects the combined presence of two septin-localized kinases: Hsl1 and Elm1.
Roles of Hsl1 and Elm1 in recruiting Hsl7 to the septin collar
Both Elm1 and Hsl1 contain N-terminal kinase domains and C-terminal regulatory domains that mediate their localization to the septin ring. Although the kinase activity of endogenous Hsl1 was important for Hsl7 recruitment, tethering of Hsl1 residues 1138–1307 to the septins was sufficient to bypass the need for Hsl1 kinase activity (and all other Hsl1 domains). We infer that the only critical role of Hsl1 kinase activity in Hsl7 recruitment is to enable Hsl7 interaction with residues 1138–1307, and we speculate that the extensive Hsl1 autophosphorylation documented previously (Barral et al., 1999; McMillan et al., 1999a) alters Hsl1 conformation so as to unmask the Hsl7-binding domain.
Although sufficient to recruit Hsl7 to the septin collar in budded cells, tethering of the Hsl7-binding domain of Hsl1 to a septin was not sufficient to recruit Hsl7 to the septin ring in unbudded cells. This finding indicates that additional factors are needed to recruit Hsl7, and we show that recruitment of the Elm1 kinase domain to septins is sufficient to provide this function. Elm1 kinase activity was required to promote Hsl7 recruitment in this context.
Elm1 is believed to activate Hsl1 kinase activity via phosphorylation of the T-loop threonine in the Hsl1 kinase domain (Szkotnicki et al., 2008). However, this cannot fully explain Elm1’s role in Hsl7 recruitment because we found that septin-tethered Elm1 could promote Hsl7 recruitment even in strains in which the Hsl1 kinase domain was absent. The simplest way to account for our findings is that Elm1 can phosphorylate either Hsl7 or the Hsl7-binding domain of Hsl1 to promote their interaction.
Our findings suggest that the C-terminal Hsl7-binding domain of Hsl1 and the kinase domain of Elm1, when present in combination, suffice for Hsl7 recruitment. However, previous studies indicated that the role of Elm1 in cell cycle control could be bypassed by a phosphomimic mutation in the Hsl1 kinase domain (Szkotnicki et al., 2008), suggesting that the only essential role of Elm1 was to activate Hsl1 kinase. How can this be reconciled with our observations that Elm1 can promote Hsl7 recruitment even in strains with no Hsl1 kinase domain? One possibility is that the phosphorylation(s) promoted by Elm1 in this context can also be carried out by an active Hsl1 kinase domain. If both Elm1 and Hsl1 can phosphorylate the same targets, then the only essential role for Elm1 would be to activate Hsl1 kinase: then either Elm1 or Hsl1 kinase could promote Hsl1–Hsl7 interaction and hence recruitment of Hsl7 to the septin ring.
Regulation of Hsl7 recruitment in response to bud emergence: a model
Hsl7 recruitment to the septins (and therefore Swe1 degradation) does not begin until the cell has formed a bud. However, Hsl7 recruitment can occur in unbudded cells, particularly if the cell shape is altered so that the septin ring forms in a locally tubular geometry similar to that at the mother–bud neck. Based on these findings, it was proposed that local membrane geometry influenced the organization of septin filaments and that septin organization was then somehow transduced into the recruitment of Hsl7 (Theesfeld et al., 2003; King et al., 2013). We found that, like Hsl7, Elm1 was recruited to the septin ring in budded but not unbudded cells. Also like Hsl7, Elm1 could be recruited to septin rings in unbudded cells if they formed in a locally tubular geometry. Thus our findings implicate Elm1 as the transducer in this model.
Although Elm1 did not localize detectably to the septin ring before budding, overexpression of Elm1 led to immediate recruitment of some Elm1 to the initially formed septin ring. This may indicate that excess Elm1 can induce the organization of septins that Elm1 normally detects. Alternatively, it may be that Elm1 has a basal affinity for the septin organization found in the initial ring and a higher affinity for the septin organization in the hourglass at the neck (or mating projection).
What is the “organization” of septins in the ring, and how might that be influenced by local cortical geometry? A recent landmark study elucidated the septin architecture in the “early hourglass” (shortly after budding)–stage using correlative light and platinum replica electron microscopy (Ong et al., 2014). At this stage, the hourglass consists of short septin filaments linked together in a parallel array. The organization of the septins before bud emergence has not been elucidated. However, recent findings suggest that septin assembly is influenced by the local micrometer-scale membrane curvature: they have a preference for positive curvature over negative curvature (Bridges et al., 2016). Before bud emergence, the cortex of a spherical cell would have isotropic (mild) negative curvature in all directions (Figure 8D). However, formation of a bud or other protrusion such as a mating projection would lead to regions of zero or positive curvature along the axis of the protrusion and enhanced negative curvature along the circumference of the protrusion (Figure 8D). This curvature asymmetry may induce septin filaments to align in parallel along the mother–bud (or projection) axis (Figure 8D). We speculate that this switch to a parallel filament alignment may strengthen Elm1 interaction with septins. For example, if Elm1 were to bind between two neighboring septin filaments, then there would be many more Elm1-binding sites in a parallel array of septin filaments than in a less-ordered array.
Materials and Methods
Yeast strains and plasmid constructs
Yeast strains used in this study are in the BF264-15DU (ade1, his2, leu2-3112, trp1-1a, ura3Δns; Richardson et al., 1989) background. Relevant genotypes are listed in Table 1. The following alleles have been described in previous studies: cdc28E12K (McMillan et al., 1999b), hsl1, HSL1K110R, HSL1-13myc (Crutchley et al., 2009), CDC3-mCherry and CDC3-GFP (Tong et al., 2007), Gal4BD-hER-VP16 (Takahashi and Pryciak, 2008), Hsl1mdb/mkb (Burton and Solomon, 2001), GFP-Hsl7 (King et al., 2013), and ELM1K117R (Caydasi et al., 2010).
TABLE 1:
Strains used in this study.
| Strain | Relevant genotype | Source |
|---|---|---|
| DLY7318 | a bar1 Hsl1pr-HSL1-13myc::TRP1 hsl1::URA3 | Crutchley et al. (2009) |
| DLY14838 | a/α CDC28E12K/CDC28E12K bar1/bar1 GFP-HSL7/HSL7 hsl1::kanR/HSL1 | This study |
| CDC3-mCherry::URA3/CDC3 | ||
| DLY14895 | a/α CDC28E12K/CDC28E12K bar1/bar1 hsl1::kanR/hsl1::kanR CDC3-mCherry::URA3/CDC3 GAL1pr-CDC3-HSL11138-1307-12xMyc::URA3/ura3 ADH1pr-Gal4BD-hER-VP16::TRP1/trp1 TEF1pr-GFP-Hsl7::LEU2/leu2 | This study |
| DLY14921 | a/α hsl1::kanR/hsl1::kanR CDC28E12K/CDC28E12K bar1/bar1 SWE1pr-CDC3-GFP-HSL1879-1307-12xMyc::URA3/ura3 CDC3-mCherry::URA3/CDC3 | This study |
| DLY14978 | a/α hsl1::kanR/hsl1::kanR CDC28E12K/CDC28E12K bar1/bar1 ADH1pr-Gal4BD-hER-VP16::TRP1/trp1 GAL1pr-CDC3-GFP- HSL11138-1307-12xMyc::URA3/ura3 CDC3-mCherry::URA3/CDC3 | This study |
| DLY16156 | a/α hsl1::kanR/hsl1::kanR CDC28E12K/CDC28E12K bar1/bar1 CDC3-mCherry::URA3/CDC3 GAL1pr-CDC3-HSL1879-1307-12xMyc::URA3/ura3 ADH1pr-Gal4BD-hER-VP16::TRP1/trp1 GAL1pr-BNI4-GFP-ELM11-420::LEU2/leu2 | This study |
| DLY16162 | a/α CDC28E12K/CDC28E12K bar1/bar1 CDC3-mCherry::URA3/CDC3 | This study |
| hsl1::kanR/HSL1 GAL1pr-BNI4-ELM11-420::LEU2/TEF1pr-GFP-HSL7::LEU2 ADH1pr-Gal4BD-hER-VP16::TRP/trp1 | ||
| DLY16705 | a/α CDC3-mCherry::URA3/CDC3 bar1/bar1 ELM1-GFP::TRP1/ELM1 | This study |
| DLY16728 | a CDC3-mCherry::URA3 bar1 ELM1-GFP::TRP1 | This study |
| DLY17668 | a CDC28E12K bar1 hsl1::kanR HSL1pr-HSL1mdb/mkb-13myc::TRP1 | This study |
| DLY17674 | a/α CDC28E12K/CDC28E12K bar1/bar1 hsl1::kanR/hsl1::kanR CDC3-mCherry::URA3/CDC3 GAL1pr-CDC3-HSL1879-1307-12xMyc::URA3/ura3 ADH1pr-Gal4BD-hER-VP16::TRP1/trp1 TEF1pr-GFP-HSL7::LEU2/leu2 | This study |
| DLY17723 | a/α hsl1::kanR/hsl1::kanR CDC28E12K/CDC28E12K bar1/bar1 GAL1pr-CDC3- HSL1879-1307-12xMyc::URA3/ura3 ADH1pr-Gal4BD-hER-VP16::TRP1/trp1 TEF1pr-GFP-HSL7::LEU2/GAL1pr-BNI4-ELM11-420::LEU2 CDC3-mCherry::URA3/CDC3 | This study |
| DLY17799 | a/α CDC28E12K/CDC28E12K bar1/bar1 CDC3-mCherry::URA3/CDC3 | This study |
| hsl1::kanR/HSL1 TEF1pr-GFP-Hsl7:LEU2/leu2 | ||
| DLY17800 | a/α CDC28E12K/CDC28E12K bar1/bar1 hsl1::kanR/hsl1::kanR CDC3-mCherry::URA3/CDC3 HSL1pr-HSL1mdb/mkb-13myc::TRP1/trp1 | This study |
| TEF1pr-GFP-HSL7::LEU2/leu2 | ||
| DLY18285 | a/α ADH1pr-GFP-ELM1::LEU2/leu2 ADH1pr-Gal4BD-hER-VP16::TRP1/trp1 CDC3-mCherry::URA3/CDC3 bar1/bar1 | This study |
| DLY18288 | a/α hsl1::kanR/hsl1::kanR CDC28E12K/CDC28E12K bar1/bar1 ADH1pr-Gal4BD-hER-VP16::TRP1/trp1 GAL1pr-CDC3-Elm11-420-HSL1879-1307-12xMyc::URA3/ura3 CDC3-mCherry::LEU2/TEF1pr-GFP-HSL7::LEU2 | This study |
| DLY18387 | a/α hsl1::kanR/hsl1::kanR CDC28E12K/CDC28E12K bar1/bar1 CDC3-mCherry::URA3/CDC3 ADH1pr-HA-ELM1::LEU2/TEF1pr-GFP-HSL7::LEU2 GAL1p-CDC3- HSL1879-1307-12xMyc::URA3/ura3 ADH1pr-Gal4BD-hER-VP16::TRP1/trp1 | This study |
| DLY18541 | a/α hsl1::kanR/hsl1::kanR CDC28E12K/CDC28E12K bar1/bar1 CDC3-mCherry::URA3/CDC3 ADH1pr-HA-ELM1::LEU2/TEF1pr-GFP-HSL7::LEU2 HSL1pr-HSL1-11Myc::TRP1/trp1 | This study |
| DLY18597 | a/α hsl1::kanR/hsl1::kanR CDC28E12K/CDC28E12K bar1/bar1 GAL1pr-CDC3- HSL1879-1307-12xmyc::URA3/ura3 ADH1pr-Gal4BD-hER-VP16::TRP1/trp1 TEF1pr-GFP-HSL7::LEU2/GAL1pr-BNI4-elm11-420K117R::LEU2 CDC3-mCherry::URA3/CDC3 | This study |
| DLY18904 | a/α CDC3-mCherry::URA3/CDC3 bar1/bar1 HSL1-GFP::kanR/HSL1 | This study |
| DLY18964 | a/α CDC3-mCherry::URA3 HSL1-GFP::kanR bar1 | This study |
| DLY21065 | a/α CDC28E12K/CDC28E12K bar1/bar1 HSL1K110R/hsl1::kanR | This study |
| CDC3-mCherry::URA3/CDC3 ADH1pr-Gal4BD-hER-VP16::TRP1/trp1 | ||
| GAL1pr-CDC3-HSL1879-1307-12xmyc::URA3/ura3 | ||
| TEF1pr-GFP-HSL7::LEU2/leu2 SWE1-myc::HIS2/SWE1 | ||
| DLY21066 | a/α CDC28E12K/CDC28E12K bar1/bar1 HSL1/hsl1::kanR GAL1pr-CDC3- HSL1879-1307-12xMyc::URA3/ura CDC3-mCherry::URA3/CDC3 ADH1pr-Gal4BD-hER-VP16::TRP1/trp1 TEF1pr-GFP-HSL7::LEU2/leu2 | This study |
| DLY21067 | a/α CDC28E12K/CDC28E12K bar1/bar1 HSL1K110R/hsl1::kanR | This study |
| CDC3-mCherry::URA3/CDC3 ADH1pr-Gal4BD-hER-VP16::TRP1/trp1 | ||
| TEF1pr-GFP-HSL7::LEU2/leu2 SWE1-myc::HIS2/SWE1 | ||
| DLY21088 | a/α hsl1::kanR/hsl1::kanR CDC28E12K/CDC28E12K bar1/bar1 ADH1pr-Gal4BD-hER-VP16::TRP1/trp1 GAL1pr-CDC3-elm11-420K117R-HSL1879-1307-12xMyc:URA3/ura3 CDC3-mCherry::LEU2/TEF1pr-GFP-HSL7::LEU2 | This study |
To fluorescently label Hsl1 and Elm1 at their genomic loci, we used the PCR-based gene modification method (Longtine et al., 1998). Briefly, primers with 50 base pairs of Hsl1 or Elm1 C-terminal and 3′ untranslated region homology were used to amplify the pFA6 GFP transformation module from pDLB53 and pDLB51. The PCR product was then purified and transformed into strains with wild-type Hsl1 and Elm1 via standard methods. Proper integration was confirmed by sequencing and fluorescence microscopy.
To generate fusion constructs with Cdc3 and Hsl1 fragments, we first generated a suitable recipient plasmid by replacing the polylinker in PRS306 with the KpnI-SacI polylinker GGTACCgctGCATGCgctCTCGAGgctgctGCGGCCGCagctACTAGTgctGTCGACgctGGATCCgctGAGCTC. Then we cloned the GAL1 promoter into the KpnI and SphI sites, the Cdc3 open reading frame into the SphI and XhoI sites, Hsl1 fragments into the SpeI and SalI sites, and a 12xmyc tag into the SalI and BamHI sites to generate plasmids pDLB3690 (PRS306-GAL1pr-CDC3-HSL11138-1307-12xMyc) and DLB3703 (PRS306-GAL1pr-CDC3-HSL1879-1307-12xMyc). Similar constructs with GFP inserted in Cdc3 (Tong et al., 2007) were generated by subcloning a Cdc3-GFP fragment from pDLB3137 into the SphI and HindIII sites. These plasmids were linearized with PstI to target integration at ura3.
To generate the fusion construct GAL1pr-CDC3-ELM11-420-HSL1879-1307-12xMyc, a PCR-generated ELM11-420 fragment was amplified from genomic DNA and ligated into pDLB3703 (above) using EagI, yielding pDLB4165 (PRS306-GAL1pr-CDC3-Elm11-420-HSL1879-1307-12xMyc). The same strategy was used to generate GAL1pr-CDC3-elm11-420K117R-HSL1879-1307-12xMyc, except that the elm11-420K117R fragment was amplified from pDLB3988 (pMG1-ELM11-420K117R, a gift from Gislene Pereira (German Cancer Research Center, Heidelberg, Germany). Integration at ura3 was performed as described.
With regard to the expression of fusion constructs containing Hsl1 fragments, note that to avoid confounding effects of endogenous Hsl1, all experiments expressing fusion constructs were conducted in strains deleted for the endogenous HSL1. Because removing Hsl1 stabilizes Swe1, we also modified these strains to express Cdc28E12K, a Swe1-resistant allele of the CDK Cdc28 (McMillan et al., 1999b; King et al., 2013). This avoids any potential feedback from Cdc28, as well as any cell cycle delays due to misregulation of Swe1. Fusion constructs containing Hsl1 or Elm1 fragments fused to the septin Cdc3 were expressed using an artificial transcription factor, Gal4BD-hER-VP16, which activates the GAL1 promoter in a titratable manner upon addition of β-estradiol (Takahashi and Pryciak, 2008).
To express Bni4-GFP-Elm11-420, we cloned the GAL1 promoter into the ApaI and HindIII sites, GFP into the BamHI site, BNI4 into the HindIII and XmaI sites, and ELM11-420 into the SpeI and SacII sites of PRS305, yielding pDLB3793 (pRS305-GAL1pr-BNI4-GFP-ELM11-420). To express Bni4-Elm11-420 without the GFP, we digested pDLB3793 with XmaI and SpeI and replaced the intervening GFP sequence with ccgggGGTGGGGGTGGTTCTGGTGGTGGTGGTa by annealing in oligonucleotides to yield pDLB3841 (pRS305-GAL1pr-BNI4-ELM11-420). We used a similar strategy to generate GAL1pr-BNI4-elm11-420K117R. We linearized plasmids with AflII to target integration at leu2.
To overexpress GFP-Hsl7, we subcloned GFP-Hsl7 from pDLB3591 into pDLB4087 (pRS405-TEF1pr-YFP) using PacI and AscI, yielding pDLB4102 (pRS405-TEF1pr-GFP-HSL7). We linearized the plasmid with EcoRI to target integration at leu2.
To overexpress Elm1, we cloned the ADH1 promoter into the SacI and XmaI sites, GFP into the PacI and BamHI sites, and ELM1 into the XbaI and AscI sites of pRS405, yielding pDLB4167 (pRS405-ADH1pr-GFP-ELM1). We generated a similar ADH1pr-HA-ELM1 construct by subcloning an ADH1pr-HA fragment from pDLB764 (ADH1pr-HA-CDC24, 2 μ; a gift from M. Peter, Swiss Institute for Experimental Cancer Research, Vaud, Switzerland) into the SacI and XbaI sites of pDLB4167, yielding pDLB4166 (pRS405-ADH1pr-HA-Elm1). We linearized plasmids with AgeI to target integration at leu2.
Microscopy and image analysis
Cells were grown in Complete Synthetic Media (CSM; MP Biomedicals, Santa Ana, CA) with 0.67% yeast nitrogen base, 2% dextrose, and 0.01% adenine to mid log phase at 30°C before imaging. Expression of fusion constructs (see prior description of plasmid constructs) was induced by treatment with β-estradiol for 3 h: Cdc3-Hsl1 fusions were induced with 30 nM β-estradiol and Elm1-containing fusions with 10 nM β-estradiol. Then cells were mounted on 2% agarose (Denville Scientific, Holliston, MA) slabs with growth medium and β-estradiol. Slab edges were sealed with petroleum jelly. Cells were imaged at 30°C. Time-lapse movies were acquired using an Andor Revolution XD spinning-disk confocal microscope (Olympus, Center Valley, PA) with Andor Ixon3 897 512 electron-multiplying charge-coupled device (EMCCD) camera (Andor, Belfast, United Kingdom). A 100×/1.4 UplanSApo oil-immersion objective was used. Images (30 z-stacks with 0.25-μm spacing) were captured at 90-s intervals, using 10% maximal output of the diode laser, 100-ms exposure, and 200 gain on the EMCCD camera. Single-plane images were captured using a Zeiss 780 confocal microscope with an argon/2 and 561-nm diode laser, a 63×/1.4 Oil plan-Apochromat 44 07 62 (02) WD 0.19-mm objective, and a GaAsP high-QE 32-channel spectral array detector.
Image analysis was performed using a custom MATLAB graphical user interface (GUI; NucTrackV3.3). The GUI uses either a fixed user-defined threshold value or an automated variable threshold given by a standard Otsu algorithm to identify the polarized Cdc3-mCherry signal. The identified regions are tracked over the entire time-lapse movie. Then the fluorescence intensity of both Cdc3-mCherry and GFP-Hsl7 (or Elm1-GFP) within the thresholded region is calculated and normalized to the peak intensity in that region for each track. Misidentified regions (usually due to residual Cdc3-mCherry signal after cytokinesis) are manually discarded. For Figure 8B, the timing of Cdc3 and Elm1 localization was scored visually. The GUI is available upon request from Denis Tsygankov (denis.Tsygankov@bme.gatech.edu).
Images of representative cells and supplemental movies were generated using ImageJ (FIJI). Image stacks were compiled to a single plane by maximum projection, scaled, and inverted. Images within the same figure were scaled the same unless otherwise noted.
Pheromone arrest–release and latrunculin treatment
Cells were grown to mid log phase in CSM + dextrose medium at 30°C, arrested with 2 μM α-factor (Genway Biotech, San Diego, CA) for 3 h, washed, and released into fresh CSM + dextrose. For the experiment of Figure 2, α-factor was added back 1 h after release so that cells would rearrest after a single cycle. Samples were taken, and budding percentage was scored at 15-min intervals.
For the experiment of Figure 8, cells were mounted onto a slab with 100 μM latrunculin B (Enzo Life Sciences, Farmingdale, NY) after a 10-min recovery period in fresh medium after release from arrest. Images were acquired as stated but at 2-min instead of 1.5-min intervals.
Western blotting
Samples were prepared using trichloroacetic acid from 107 cells. SDS–PAGE and Western blotting were performed by standard procedures (Keaton et al., 2008). Blots were probed using mouse monoclonal c-myc (9E10) antibody at 1:5000 dilution and rabbit polyclonal anti-Cdc11 antibody at 1:5000 dilution (Santa Cruz Biotechnology, Dallas, TX). Fluorophore-conjugated secondary antibodies for mouse (IRDye800-conjugated anti-mouse immunoglobulin G [IgG]; Rockland Immunochemicals, Pottstown, PA) or rabbit (Alexa Fluor 680 goat anti-rabbit IgG; Invitrogen, Carlsbad, CA) were used at 1:5000 dilution. Western blots were visualized using the ODYSSEY imaging system (LI-COR Biosciences, Lincoln, NE).
Supplementary Material
Acknowledgments
We thank Gislene Pereira for the kinase-dead Elm1 and Drew Bridges and Amy Gladfelter for communicating results before publication. We thank Lew lab members for stimulating discussions and anonymous reviewers for insightful comments. This work was funded by National Institutes of Health/National Institute of General Medical Sciences Grants GM62300 and GM103870 to D.J.L.
Abbreviations used:
- CDK
cyclin-dependent kinase
- CSM
complete synthetic media
- GFP
green fluorescence protein.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-01-0014) on April 6, 2016.
REFERENCES
- Alexander MR, Tyers M, Perret M, Craig BM, Fang KS, Gustin MC. Regulation of cell cycle progression by swe1p and hog1p following hypertonic stress. Mol Biol Cell. 2001;12:53–62. doi: 10.1091/mbc.12.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasia SD, Nguyen DL, Thai V, Meloy M, MacDonough T, Kellogg DR. A link between mitotic entry and membrane growth suggests a novel model for cell size control. J Cell Biol. 2012;197:89–104. doi: 10.1083/jcb.201108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano S, Park JE, Sakchaisri K, Yu LR, Song S, Supavilai P, Veenstra TD, Lee KS. Concerted mechanism of Swe1/Wee1 regulation by multiple kinases in budding yeast. EMBO J. 2005;24:2194–2204. doi: 10.1038/sj.emboj.7600683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral Y, Parra M, Bidlingmaier S, Snyder M. Nim1-related kinases coordinate cell cycle progression with the organization of the peripheral cytoskeleton in yeast. Genes Dev. 1999;13:176–187. doi: 10.1101/gad.13.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booher RN, Deshaies RJ, Kirschner MW. Properties of Saccharomyces cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J. 1993;12:3417–3426. doi: 10.1002/j.1460-2075.1993.tb06016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouquin N, Barral Y, Courbeyrette R, Blondel M, Snyder M, Mann C. Regulation of cytokinesis by the Elm1 protein kinase in Saccharomyces cerevisiae. J Cell Sci. 2000;113:1435–1445. doi: 10.1242/jcs.113.8.1435. [DOI] [PubMed] [Google Scholar]
- Bridges AA, Jentzsch MS, Oakes PW, Occhipinti P, Gladfelter AS. Micron-scale plasma membrane curvature is recognized by the septin cytoskeleton. J Cell Biol. 2016;213:23–32. doi: 10.1083/jcb.201512029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JL, Solomon MJ. Hsl1p, a Swe1p inhibitor, is degraded via the anaphase-promoting complex. Mol Cell Biol. 2000;20:4614–4625. doi: 10.1128/mcb.20.13.4614-4625.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JL, Solomon MJ. D box and KEN box motifs in budding yeast Hsl1p are required for APC-mediated degradation and direct binding to Cdc20p and Cdh1p. Genes Dev. 2001;15:2381–2395. doi: 10.1101/gad.917901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caydasi AK, Kurtulmus B, Orrico MI, Hofmann A, Ibrahim B, Pereira G. Elm1 kinase activates the spindle position checkpoint kinase Kin4. J Cell Biol. 2010;190:975–989. doi: 10.1083/jcb.201006151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid VJ, Shulewitz MJ, McDonald KL, Thorner J. Dynamic localization of the Swe1 regulator Hsl7 during the Saccharomyces cerevisiae cell cycle. Mol Biol Cell. 2001;12:1645–1669. doi: 10.1091/mbc.12.6.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clotet J, Escote X, Adrover MA, Yaakov G, Gari E, Aldea M, de Nadal E, Posas F. Phosphorylation of Hsl1 by Hog1 leads to a G(2) arrest essential for cell survival at high osmolarity. EMBO J. 2006;25:2338–2346. doi: 10.1038/sj.emboj.7601095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crutchley J, King KM, Keaton MA, Szkotnicki L, Orlando DA, Zyla TR, Bardes ES, Lew DJ. Molecular dissection of the checkpoint kinase Hsl1p. Mol Biol Cell. 2009;20:1926–1936. doi: 10.1091/mbc.E08-08-0848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMarini DJ, Adams AEM, Fares H, De Virgilio C, Valle G, Chuang JS, Pringle JR. A septin-based hierarchy of proteins required for localized deposition of chitin in the Saccharomyces cerevisiae cell wall. J Cell Biol. 1997;139:75–93. doi: 10.1083/jcb.139.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JC, Bardes ES, Ohya Y, Lew DJ. A role for the Pkc1p/Mpk1p kinase cascade in the morphogenesis checkpoint. Nat Cell Biol. 2001;3:417–420. doi: 10.1038/35070104. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- Keaton MA, Szkotnicki L, Marquitz AR, Harrison J, Zyla TR, Lew DJ. Nucleocytoplasmic trafficking of G2/M regulators in yeast. Mol Biol Cell. 2008;19:4006–4018. doi: 10.1091/mbc.E08-03-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King K, Jin M, Lew D. Roles of Hsl1p and Hsl7p in Swe1p degradation: beyond septin tethering. Eukaryot Cell. 2012;11:1496–1502. doi: 10.1128/EC.00196-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King K, Kang H, Jin M, Lew DJ. Feedback control of Swe1p degradation in the yeast morphogenesis checkpoint. Mol Biol Cell. 2013;24:914–922. doi: 10.1091/mbc.E12-11-0812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew DJ. The morphogenesis checkpoint: how yeast cells watch their figures. Curr Opin Cell Biol. 2003;15:648–653. doi: 10.1016/j.ceb.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Lew DJ, Burke DJ. The spindle assembly and spindle position checkpoints. Annu Rev Genet. 2003;37:251–282. doi: 10.1146/annurev.genet.37.042203.120656. [DOI] [PubMed] [Google Scholar]
- Lew DJ, Reed SI. A cell cycle checkpoint monitors cell morphogenesis in budding yeast. J Cell Biol. 1995;129:739–749. doi: 10.1083/jcb.129.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HH, Loy CJ, Zaman S, Surana U. Dephosphorylation of threonine 169 of Cdc28 is not required for exit from mitosis but may be necessary for start in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:4573–4583. doi: 10.1128/mcb.16.8.4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, III, DeMarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Longtine MS, Theesfeld CL, McMillan JN, Weaver E, Pringle JR, Lew DJ. Septin-dependent assembly of a cell-cycle-regulatory module in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20:4049–4061. doi: 10.1128/mcb.20.11.4049-4061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan JN, Longtine MS, Sia RAL, Theesfeld CL, Bardes ESG, Pringle JR, Lew DJ. The morphogenesis checkpoint in Saccharomyces cerevisiae: cell cycle control of Swe1p degradation by Hsl1p and Hsl7p. Mol Cell Biol. 1999a;19:6929–6939. doi: 10.1128/mcb.19.10.6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan JN, Sia RAL, Bardes ESG, Lew DJ. Phosphorylation-independent inhibition of Cdc28p by the tyrosine kinase Swe1p in the morphogenesis checkpoint. Mol Cell Biol. 1999b;19:5981–5990. doi: 10.1128/mcb.19.9.5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan JN, Sia RAL, Lew DJ. A morphogenesis checkpoint monitors the actin cytoskeleton in yeast. J Cell Biol. 1998;142:1487–1499. doi: 10.1083/jcb.142.6.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty JJ, Lew DJ. Swe1p responds to cytoskeletal perturbation, not bud size, in S. cerevisiae. Curr Biol. 2005;15:2190–2198. doi: 10.1016/j.cub.2005.11.039. [DOI] [PubMed] [Google Scholar]
- Moore JK, Chudalayandi P, Heil-Chapdelaine RA, Cooper JA. The spindle position checkpoint is coordinated by the Elm1 kinase. J Cell Biol. 2010;191:493–503. doi: 10.1083/jcb.201006092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moravcevic K, Mendrola JM, Schmitz KR, Wang YH, Slochower D, Janmey PA, Lemmon MA. Kinase associated-1 domains drive MARK/PAR1 kinases to membrane targets by binding acidic phospholipids. Cell. 2010;143:966–977. doi: 10.1016/j.cell.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997;13:261–291. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- Ong K, Wloka C, Okada S, Svitkina T, Bi E. Architecture and dynamic remodelling of the septin cytoskeleton during the cell cycle. Nat Commun. 2014;5:5698. doi: 10.1038/ncomms6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raspelli E, Cassani C, Lucchini G, Fraschini R. Budding yeast Dma1 and Dma2 participate in regulation of Swe1 levels and localization. Mol Biol Cell. 2011;22:2185–2197. doi: 10.1091/mbc.E11-02-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson HE, Wittenberg C, Cross F, Reed SI. An essential G1 function for cyclin-like proteins in yeast. Cell. 1989;59:1127–1133. doi: 10.1016/0092-8674(89)90768-x. [DOI] [PubMed] [Google Scholar]
- Russell P, Moreno S, Reed SI. Conservation of mitotic controls in fission and budding yeast. Cell. 1989;57:295–303. doi: 10.1016/0092-8674(89)90967-7. [DOI] [PubMed] [Google Scholar]
- Sakchaisri K, Asano S, Yu LR, Shulewitz MJ, Park CJ, Park JE, Cho YW, Veenstra TD, Thorner J, Lee KS. Coupling morphogenesis to mitotic entry. Proc Natl Acad Sci USA. 2004;101:4124–4129. doi: 10.1073/pnas.0400641101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulewitz MJ, Inouye CJ, Thorner J. Hsl7 localizes to a septin ring and serves as an adapter in a regulatory pathway that relieves tyrosine phosphorylation of Cdc28 protein kinase in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:7123–7137. doi: 10.1128/mcb.19.10.7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia RAL, Bardes ESG, Lew DJ. Control of Swe1p degradation by the morphogenesis checkpoint. EMBO J. 1998;17:6678–6688. doi: 10.1093/emboj/17.22.6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia RAL, Herald HA, Lew DJ. Cdc28 tyrosine phosphorylation and the morphogenesis checkpoint in budding yeast. Mol Biol Cell. 1996;7:1657–1666. doi: 10.1091/mbc.7.11.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szkotnicki L, Crutchley JM, Zyla TR, Bardes ES, Lew DJ. The checkpoint kinase Hsl1p is activated by Elm1p-dependent phosphorylation. Mol Biol Cell. 2008;19:4675–4686. doi: 10.1091/mbc.E08-06-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Pryciak PM. Membrane localization of scaffold proteins promotes graded signaling in the yeast MAP kinase cascade. Curr Biol. 2008;18:1184–1191. doi: 10.1016/j.cub.2008.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theesfeld CL, Zyla TR, Bardes EG, Lew DJ. A monitor for bud emergence in the yeast morphogenesis checkpoint. Mol Biol Cell. 2003;14:3280–3291. doi: 10.1091/mbc.E03-03-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Z, Gao XD, Howell AS, Bose I, Lew DJ, Bi E. Adjacent positioning of cellular structures enabled by a Cdc42 GTPase-activating protein-mediated zone of inhibition. J Cell Biol. 2007;179:1375–1384. doi: 10.1083/jcb.200705160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uesono Y, Ashe MP, Toh EA. Simultaneous yet independent regulation of actin cytoskeletal organization and translation initiation by glucose in Saccharomyces cerevisiae. Mol Biol Cell. 2004;15:1544–1556. doi: 10.1091/mbc.E03-12-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.