

Abstract
Identifying bacterial strains in metagenome and microbiome samples using computational analyses of short-read sequence remains a difficult problem. Here, we present an analysis of a human gut microbiome using on Tru-seq synthetic long reads combined with new computational tools for metagenomic long-read assembly, variant-calling and haplotyping (Nanoscope and Lens). Our analysis identifies 178 bacterial species of which 51 were not found using short sequence reads alone. We recover bacterial contigs that comprise multiple operons, including 22 contigs of >1Mbp. Extensive intraspecies variation among microbial strains in the form of haplotypes that span up to hundreds of Kbp can be observed using our approach. Our method incorporates synthetic long-read sequencing technology with standard shotgun approaches to move towards rapid, precise and comprehensive analyses of metagenome and microbiome samples.
As yet, only a small fraction of the microbial world has been isolated and studied in the laboratory and little is known about species that cannot be cultured1. Metagenomics has begun to shed light on this unculturable ‘microbial dark matter’ by sequencing the DNA of microbial communities directly from the environment2. Metagenomic analyses of soil3, water4 and human microbiome5 samples have already increased our understanding of the microorganisms present in these environments.
Although short-read sequencing technologies have enabled high-throughput metagenomic studies, the limited read lengths combined with the complexity of microbial samples (hundreds to thousands of species) can make it difficult to accurately identify bacterial strains, recover whole genomes, catalog sample diversity and assess the abundance of species and strains.
Many approaches have been proposed to overcome these problems. Binning de novo assembled contigs using metrics such as tetranucleotide frequencies6, genomic coverage under different DNA extraction methods7 or abundance correlations between samples from multiple individuals8 have all been used to identify species and recover their genomes. However, each approach has limitations: tetranucleotide frequencies may vary within the same species6, closely related bacteria have similar DNA extraction efficiencies7, and establishing abundance correlations among individuals may require a large number of samples8. Metagenomic contigs can also be scaffolded using chromatin-level contact probability maps generated by the high-throughput chromosome conformation capture (Hi-C) technology9; however, Hi-C has high input-DNA requirements and the performance of this scaffolding method has not yet been assessed on bona fide high-complexity metagenomes. An alternative technology, single-molecule real-time sequencing10, has been used to sequence 16S rRNA amplicons11, but has seen limited application to whole metagenomes.
Recently, Tru-seq synthetic long-read sequencing has been developed to increase the effective read length available using the Illumina platform from hundreds to more than ten thousand base pairs. This method uses a modified library-preparation protocol, in which kilobase-long DNA fragments are extracted, diluted, amplified, and reassembled from regular short reads12. Synthetic long-read sequencing has been used in metagenomics for assembly validation13 and for studying environmental metagenomes14. Here, we report the first study of the human gut microbiome using synthetic long reads.
We present three improvements compared with previous technologies. First, we demonstrate that long reads, in conjunction with Lens, an algorithm developed for this study, reveal extensive haplotype diversity among individual bacterial strains of the same species. This level of resolution was inaccessible using previous technologies, which at best studied microorganisms at the strain level. Second, we use long reads to assemble de-novo hundreds of megabases of genomic sequence in contigs of complete operons and whole bacterial chromosomes. Finally, we determine microbial composition accurately.
Results
Sequencing the gut microbiome using synthetic long reads
We applied our long read sequencing approach to two metagenomic datasets: the human microbiome project staggered mock metagenomic community5 (mock metagenome), and a sample from the gut of a healthy male adult individual (human gut metagenome). The mock metagenome is a synthetic community of 20 organisms with known reference genomes (Supplementary Table 1) that is widely used for validation. We generated three Tru-seq synthetic long read libraries (2.9 Gbp of sequence, N50 read length of 9.2 kbp; Supplementary Figure 1) for this dataset, in addition to 3.1 Gbp of standard 101-bp paired-end Illumina short read libraries (Supplementary Table 2). For the human gut metagenome, we generated seven Tru-seq synthetic long read libraries (8.3 Gbp of sequence, N50 read length of 8.6 kbp; Supplementary Figure 2), and complemented this data with 8.1 Gbp of standard Illumina libraries (Supplementary Table 3). The similar sequencing amounts of short and long reads for both samples helped assess the benefits of using longer read lengths.
We maped the long reads to the known reference genomes of the mock community using the MUMmer aligner. Accuracy was high, with less than 0.5% of reads misassembled (Supplementary Table 4). However, we observed differences in coverage between short- and long-read technologies, with long reads covering ~ 15% fewer base pairs overall than short reads (Supplementary Tables 5, 6; Supplementary Figure 3). In particular, four organisms were highly covered (>98%) by short reads but long read coverage was substantially less thorough (<75%), suggesting that long reads have more sequence bias. Interestingly, six organisms had at least 10% of their genomes covered by long reads but not by short reads, suggesting that the two technologies can be complementary (Supplementary Table 7). We also found that abundance estimates were substantially different between the technologies (sometimes by more than an order of magnitude), indicating that long reads on their own may be insufficient for abundance estimation (Supplementary Figure 4). Differences in coverage have been previously linked to the long reads’ increased sensitivity to GC content during the PCR amplification step12,19. We suggest that for best results, both types of data should be used.
Assembly of bacterial operons and chromosomes
We assembled long reads from the human gut metagenome sample using Nanoscope, a new bioinformatics pipeline we created. Nanoscope automates metagenomic assembly, species identification, substrain analysis, and abundance estimation from a combination of short and long read data (Figure 1; Supplementary Code). It is available online as a free open-source tool (https://github.com/kuleshov/nanoscope).
Figure 1.
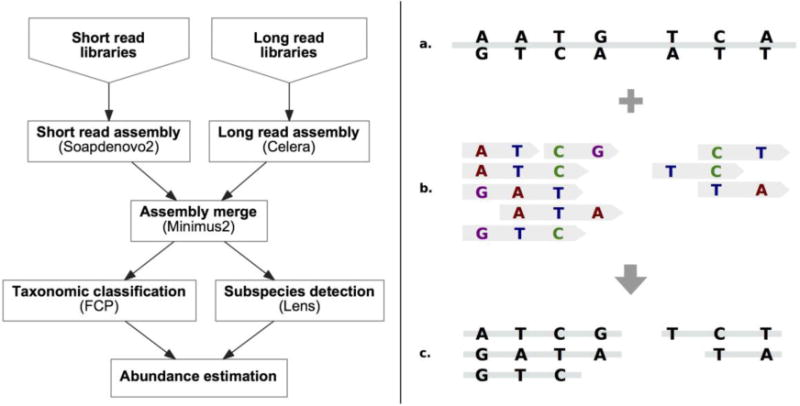
The Nanoscope pipeline and the Lens algorithm. Left: Nanoscope first assembles short and long reads using the Soapdenovo2 and Celera assemblers and merges the results with Minimus2; it then assigns taxonomic labels to contigs with the Fragment Classification Package (FCP) and identifies bacterial strains with Lens; finally, it estimates abundances of detected bacterial species by mapping short reads to contigs and by aggregating the coverage over all contigs assigned to the same species. Right: The Lens algorithm identifies heterozygous variants in the assembled genomic contigs (a); these variants are supported by long reads (b) aligned to the contigs. Each long read originates from a single organism; thus the variants it supports must belong to the same substrain. By connecting reads at their overlapping variants, Lens places the variants into multi-kilobase-long haplotypes (c) associated with bacterial strains. The number of haplotypes is a priori unknown and is inferred from the data.
Nanoscope starts by invoking the Soapdenovo20 and Celera21 assemblers to independently assemble the short and long read libraries, before merging the results using Minimus222. This method produced contigs for more than 650 Mbp of the human gut metagenome (N50 length of 49 kbp; Table 1); these were longer and more complete than ones assembled from either long reads (600 Mbp of sequence; N50 length of 38 kbp; Celera assembler) or short reads (232 Mbp of sequence; N50 length of 8.6 kbp; Soapdenovo2 assembler) alone. Twenty-two of the contigs we obtained were longer than 1 Mbp (Supplementary Figure 5), indicating that multiple organisms could be assembled completely or almost completely. The longest contig we recovered was 3.9 Mbp; its length and number of predicted ORFs23 were comparable to that of a closely related complete bacterial genome (see below). For comparison, long reads by themselves produced 19 contigs longer than 1 Mbp, whereas the longest contig from short reads alone was 410 Kbp. This indicates that synthetic long reads assemble a small number of complete chromosomes in addition to dozens of contigs that are almost an order of magnitude longer than ones obtained from short reads.
Table 1.
Assembly of the human gut metagenome. Short and long read libraries were assembled with the Soapdenovo2 and Celera assemblers, respectively. The results were merged using Minimus2 to produce a joint assembly. Long reads assemble into significantly longer contigs that contain about twice as many genes.
| Short | Long | Joint | |
|---|---|---|---|
| Number of contigs | 92,247 | 24,199 | 34,786 |
| Largest contig (Mbp) | 0.63 | 3.94 | 3.94 |
| Total length (Mbp) | 233 | 610 | 656 |
| N50 (Kbp) | 8.7 | 37.3 | 49.2 |
| Number of predicted genes | 274,600 | 523,358 | 552,680 |
| Average number of genes/contig | 2.98 | 21.62 | 15.88 |
We also assessed our assembly strategy using the mock metagenome. Our merged assembly of long- and short-reads recovered 42 Mbp of sequence (of 83 Mbp total) into contigs with an N50 length of 92 kbp (Table 1, Supplementary Table 8). This assembly was quite accurate, with approximately one misassembly per 400 kbp on average (Supplementary Table 9), a rate that compares favorably to accuracies reported by previous empirical studies of de-novo assembly algorithms24. Using only long reads resulted in much shorter contigs (33 Mbp of sequence; N50 length of 43 kbp), suggesting that for low-complexity metagenomes, combining short- and long-read technologies might substantially improve assembly quality. We assembled on this dataset one contig longer than 1Mbp and three contigs longer than 500 Kbp, all of these were assembled using long reads alone, suggesting that short reads mainly improved assembly completeness rather than contiguity.
One of the potential advantages of long-read sequencing is the recovery of complete bacterial operons (clusters of functionally related genes that are transcribed together). By using the known positions of operons in the reference genomes of the species in the mock metagenome25, we confirm that operon recovery is feasible with a combined assembly of short- and long- reads (Supplementary Table 10). Our combined assembly recovered 4,500 operons, which represents more than half of all the known operons in the mock metagenome and twice the number that can be obtained using short reads alone. Interestingly, long and short reads by themselves reconstructed only about 2,500 operons each, and many could be assembled from only one dataset (Supplementary Figures 6, 7). We attribute this discrepancy to differences in coverage between short and long reads.
In particular, long reads enabled us to recover long flagellar operons present in E. coli (Supplementary Table 11); these operons are clinically relevant as flagella contribute to pathogenicity. We assembled complete sequences of 11 flagellar operons from three bacterial species (E. coli, R. sphaeroides, P. aeruginosa; Supplementary Table 12), which comprised half of the known flagellar operons in the mock metagenome. We also recovered multiple flagellar operons from the gut metagenome (Supplementary Table 13). For example, a 2.3 Mbp contig belonging to the genus Acinetobacter was found to contain 10 flagellar operons, the longest of which contained 11 genes.
Identification of substrains in the gut microbiome
We assessed variation among the bacterial strains whose genomes we assembled using Lens, a new tool that we created. (Table 2). In brief, we mapped long reads to assembled contigs using the BWA aligner; at many positions, read and contig sequences differed, which we interpreted as variation among recently diverged strains of the same species. We used Lens to determine and phase single-nucleotide variants (SNVs) and short indels based on this alignment; Lens performed these tasks via new algorithms that do not make any assumptions on either the read length or the ploidy of the organism (see Supplementary Methods; Supplementary Code; https://github.com/kuleshov/lens).
Table 2.
Overview of the variation among bacterial strains identified by the Lens algorithm. The human gut contains hundreds of thousands of variants, which are distributed across 2,204 genomic regions of up to 112 kbp in length. A region is defined as a maximal set of variants that can be phased by Lens using long reads.
| Genomic variants | 202,574 |
| Genomic regions harboring haplotypes | 2,204 |
| Number of haplotypes | 5,024 |
| N50 region length (bp) | 18,985 |
| Max region length (bp) | 112,271 |
| Fraction of regions intersecting a gene | 95% |
| Fraction of genes intersecting a region | 4.4% |
Lens found extensive intraspecies variation in almost every bacterial species in the human gut metagenome (Figure 2 (a); Supplementary Figures 8, 9), which in total contained more than 200,000 variants (Supplementary Methods). Lens assembled these variants into 5,024 haplotypes distributed across about 2,204 genomic regions with an N50 length of 19 kbp (Supplementary Figure 10); each region contained on average 3.93 bacterial haplotypes. More than 95% of regions overlapped with ORFs, and the longest region we found spanned 112 Kbp or 242 variants and contained four distinct haplotypes.
Figure 2.
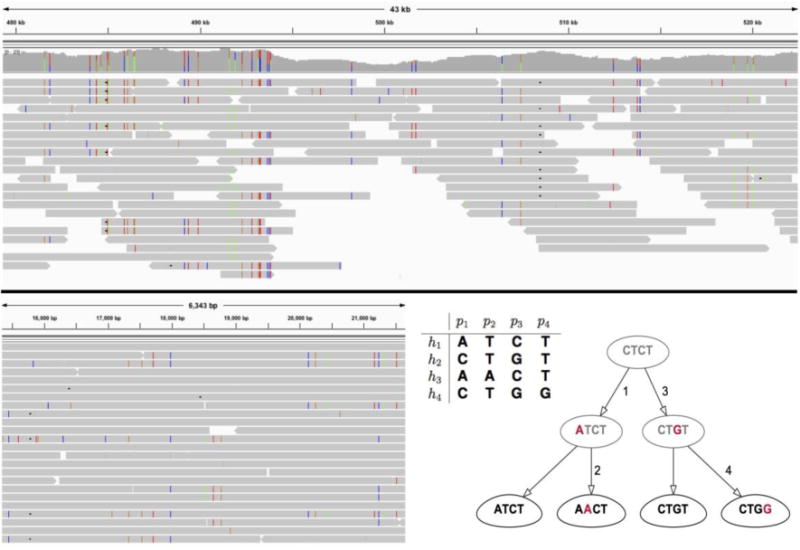
Long reads aligned to assembled metagenomic contigs reveal extensive variation among bacterial strains. Top: Fragment of a 110 kbp long region within a metagenomic contig belonging to the species Odoribacter splanchnicus; the region harbors numerous strain variants that can be assembled into bacterial haplotypes. Bottom left: Fragment of a bacterial region containing 32 genomic variants that assemble into four bacterial haplotypes. Bottom right: These haplotypes can be placed in an evolutionary tree satisfying perfect phylogeny; for simplicity, we visualize this tree over 4 of the 32 positions in the region (upper left corner).
We observed that significantly fewer variants were present in essential genes26 compared to non-essential genes, as expected from evolutionary pressure (Supplementary Table 14; p < 0.02). We repeated the same analysis on the mock metagenome and also uncovered a small number of genomic variants (few were expected, as the sample is synthetic; see Supplementary Table 15); as in the gut metagenome, significantly fewer variants were found in essential genes26 (Supplementary Table 14; p < 1e-3). We used the variant annotation package SNPEff27 to predict the deleteriousness of each mutation in the genome of E. coli (Supplementary Table 16), and found that most variants had low to moderate effects. Only six variants had high effect and were all found in non-essential genes rhsB, ydfK, icd, and perR. These observations suggest that the variants Lens uncovers are not attributable to noise.
To evaluate the correctness of the phased haplotypes, we determined whether they satisfy perfect phylogeny28. A tree over haplotypes satisfies perfect phylogeny if all strains evolved from a common ancestor, and during this process, each position mutated at most once. Although this criterion is not applicable to distantly diverged species, it is useful when organisms undergo short evolutionary distances, as in the case of bacterial subspecies. In the human gut metagenome, most (85%) of the genomic regions harboring at least four haplotypes satisfied perfect phylogeny (for three haplotypes or less, perfect phylogeny always holds). When the model is not met (such as when certain positions have mutated twice), it is possible to measure the extent to which it is violated by estimating the number of positions that can be excluded to make perfect phylogeny hold. We were able to place more than 92% of all gut metagenome regions in perfect phylogeny by excluding at most 5% of positions within each region (Supplementary Figures 10, 11). These observations support the hypothesis that the variants we find correspond to distinct bacterial strains that have evolved from one another. Our approach is the first, to our knowledge, to uncover substrain resolution and offers a snapshot of how strains evolve in vivo.
Assessing strain abundance with long reads
Nanoscope uses the FCP software package29 to assign taxonomic labels to assembled contigs. FCP determines labels using either a homology-based approach (based on the lowest common ancestor or LCA algorithm), or a composition-based30 approach (a Naïve Bayes or NB classifier trained on k-mer frequencies). In principle, longer contigs should be easier to label because they contain more species-specific sequences and they should map with less ambiguity to known reference genomes.
In the human gut metagenome, 61.4% of contigs assembled from long reads could be labeled using the LCA method, compared with 46.5% of contigs derived from short reads (Supplementary Table 17). Similarly, 89.8% of contigs assembled using long reads could be labeled by the NB method, compared to 11.0% of contigs assembled using short reads. To assess the accuracy of these assignments, we used the mock metagenome. LCA assigned contigs with 100% accuracy on both long and short reads, whereas NB had accuracies of 99% and 98% on contigs obtained from long and short reads respectively (Supplementary Tables 18–21).
By examining the taxonomic labels assigned to the longest contigs in the gut metagenome, we were able to identify which bacteria could be assembled completely or almost completely (Supplementary Table 22). Five of the ten longest contigs belonged to the genus Bacteroides, and the longest 3.9 Mbp contig was from the genus Odoribacter. Many contigs – including one measuring 2.3 Mbp – could not be assigned accurate labels; these contigs correspond to either unknown species, or to species whose genomes have not yet been fully assembled (Supplementary Tables 23, 24). Notably, the above 2.3 Mbp contig did not match any known reference genome by more than 3.2 Kbp; however, we found that contigs from a fragmented draft assembly of a species from the genus Acinetobacter mapped to the 2.3 Mbp contig completely (Supplementary Figure 12). This suggests that we were able to recover the genome of that bacterium at a higher level of quality than a previous study that used metagenomics samples from 396 different individuals8. We observed similar mapping results for other unclassified contigs as well.
Finally, we determined the abundance of each bacterium by mapping short reads to gut metagenome contigs and then using the above taxonomic labels to propagate the resulting coverage estimates to each identified bacterium (Supplementary Methods). We found 178 species in the human gut metagenome and these species greatly varied in their abundance: some comprised as much as 5% of the metagenome, and others as little as 0.02% (Figure 3; Supplementary Table 25). Interestingly, different species were recovered by short and by long reads: short reads helped finding two relatively high-abundance bacteria, whereas long reads uncovered 51 species (mostly of low abundance) that were missed by short reads (given the same amount of sequencing). Moreover, by combining both short and long reads, 58 additional low-abundance bacteria could be identified. Finally, we found that on the mock metagenome, abundance estimates were highly concordant with those obtained from mapping short reads directly to the 20 known reference genomes (rˆ2 = 0.97; Supplementary Figure 13; Supplementary Table 26). This serves as an indicator of the accuracy of our approach.
Figure 3.
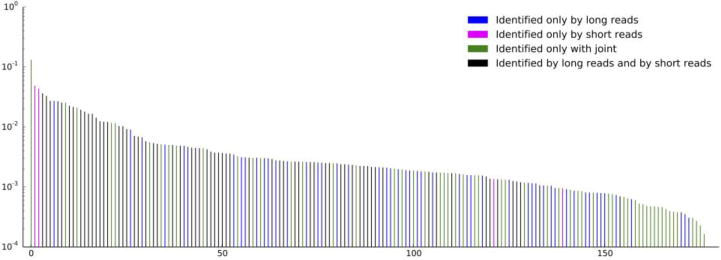
Bacterial strains identified only by long reads (blue), only by short reads (magenta), by both technologies (green), and only by a combination of the two (black), ordered by abundance. Long reads identify 51 species that short reads do not detect; combining short and long reads identifies 58 additional species, including ones having the lowest abundance. A total of 178 species are detected using all the methods.
Discussion
Including synthetic long reads in metagenomic analyses significantly improves the delineation of complex metagenomic samples relative to short-read sequencing. Although synthetic long reads have sequencing biases that affect coverage in specific genomic regions, these biases can be overcome using a small amount of additional shotgun sequencing. The resulting approach offers three main advantages over existing methods. First, we recover long bacterial contigs (up to megabases in length) that span operons that could not be recovered by short read sequencing alone. Second, analysis of long reads produces kilobase-long haplotypes that can reveal evolutionary trends in microbial communities. Finally, longer sequencing read lengths enable identification of bacteria at abundances as low as 0.02% that are undetected by short reads.
Our approach is of course not without limitations. Synthetic long read technologies rely on a dilution step that attempts to reduce the number of copies of each repeat to at most one per well; genomes of bacteria at high abundances of 10% or more may not be sufficiently diluted, and several repeat copies may remain in a single well, preventing the subassembly of their long reads. This may explain why some gut metagenome bacteria at ~5% abundance were not identified by long reads alone. A related issue is the presence of short tandem repeats, which may also prevent subassembly. A third shortcoming of our approach is the reliance on a PCR step, which could introduce bias (see Supplementary Figures 3–4) and error. However validation using the mock metagenome and comparison with short shotgun reads indicates that this error does not exceed 2–3% (Supplementary Methods).
Despite these limitations, our approach can more readily shed light on the configuration of complete operons, and facilitate the identification of pathogenic strains (especially in a mixed population). For instance, we assembled multiple kilobase-long flagellar operons that affect motility and thus play a role during infection. Furthermore, the substrain resolution enabled by our methods could assist in understanding how evolution shaped strains over time in situ. Finally, the ability to identify low-abundance bacteria will help reveal the complete composition of environmental samples and discover new species.
These results are comparable in many ways to previous methods that required hundreds of human subjects8, multiple DNA extraction methods7 or tetranucleotide binning with a mix of Sanger and mate-pair sequencing6 (Table 3). Our method, on the other hand, requires only a single metagenomic sample, does not involve binning, and is superior at identifying bacterial strains. It is also feasible that long reads might work in tandem with previous approaches, since longer contigs will increase the accuracy of their statistical components. Similarly, longer contigs might improve the accuracy of scaffolding techniques such as ones based on Hi-C34. Finally, our approach is most closely related to single-molecule real-time sequencing (SMRT), an alternative long-read technology10. To compare our two methods, we assembled a publically available dataset for the mock metagenome (Supplementary Materials) using the MHAP assembly strategy33. This produced long contigs with fewer misassemblies than ones we obtained from synthetic long reads; however, the SMRT contigs also had a 5x higher indel rate and only 88% of SNVs called using Lens in these contigs could be confirmed with short reads (compared to 99% for synthetic long reads). It thus appears that SMRT reads produce excellent draft metagenomic assemblies, but their high error rate makes it difficult to identify and phase variants in the metagenome.
Table 3.
Comparison to alternative technologies. We obtain similar results to alternative techniques that used hundreds of pooled samples (Nielsen et al.) or potentially inaccurate binning approaches (Albertsen et al., Iverson et al.). We also analyze strains at the resolution of individual variants and haplotypes rather than strains or species.
| Our method | Nielsen et al. | Albertsen et al. | Iverson et al. | Sharon et al. | |
|---|---|---|---|---|---|
| Sample type | Gut microbiome | Gut microbiome | Environmental | Environmental | Environmental |
| # of samples | 1 | 18–396 | 2 | 1 | 3 independent |
| Seq. platform | Tru-seq SLR | Illumina WGS | Illumina WGS | SOLiD mate-pairs Sanger sequencing |
Tru-seq SLR |
| Seq. amount | 8 Gbp (long reads) ×40 (subassembly) | 4.5 Gbp/sample | 86 Gbp | 59 Gbp | 1.5 Gbp (long reads) ×40 (subassembly) |
| Analysis type | De-novo assembly; Phasing |
Correlation across multiple samples | DNA extraction efficiency binning | Tetranucleotide binning | De-novo assembly |
| Resolution | Individual SNV | Strain | Species with diff. GC content | Family | Strain |
| Longest scaffold | 3.9 Mbp | 733 Kbp | 3.6 Mbp | 2.2 Mbp | <20 Kbp |
| Scaffold N50 | 49 Kbp | 39 Kbp1 | 4.1 Kbp overall ~100 Kbp for top species |
6.8 Kbp | 8.2 Kbp |
| Bases assembled | 656 Mbp | 45 Mbp (genes) 35 Gbp (total) |
423 Mbp | 300 Mbp | 500 Mbp/sample |
| # Variants | 200K | n/a | n/a | n/a | n/a |
| # Haplotypes | 5K | n/a | n/a | n/a | n/a |
Finally, synthetic long reads were recently used by Sharon et al. to analyze a soil metagenome sample14. By focusing on a set of marker genes, they showed that the community comprised a combination of closely related strains and rare species. Our work demonstrates that long reads can produce much longer contigs than ones described by Sharon et al. (Table 3); in addition, owing to the lack of need for a marker gene set, our analysis pipeline can find additional species, including a phylum that marker genes did not reveal on the Sharon et al. environmental dataset (see Supplementary Material for full discussion).
In conclusion we reveal that the human gut microbiome is more complex than previously thought, particularly in terms of subspecies diversity. The rapid evolution of bacterial strains at the subspecies level could affect human physiology16. Armed with more complete inventories of microbiomes, it might be possible to strengthen associations between human and bacterial phenotypes.
Online Methods
Metagenomic sample preparation
Mock microbioal DNA, HM-277D Staggered v5.2H, was obtained from BEIresources. Gut microbiome DNA was isolated from the frozen feces of a healthy subject using PowerSoil DNA Isolation Kit (MO BIO Laboratories, Inc.). Both DNA samples were sequenced using Illumina Tru-seq synthetic long reads technique (three and seven libraries for mock and gut microbiome samples, respectively) and the standard shortgun technique, with each library sequenced on one full lane of HiSeq. All libraries were prepared according the manufacturer’s standard protocol. Shotgun sequence reads for both the mock and the gut metagenomic samples were subsampled at random to produce subsampled libraries containing the same amount of base pairs as the in the Tru-seq synthetic long read libraries. The results were assembled on the Illumina Basespace platform, according to standard protocol.
We used Ovation Ultralow DR Multiplex Systems 1–8 (0330-32, NuGEN Technologies, Inc.) for whole genome library preparation. Briefly, 100 ng of intact gDNA was diluted into 120 μL of 1X low EDTA-TE buffer and transferred to Covaris snap cap microtube and Fragmented to 300 bp following Covaris recommended settings. Fragmented DNA was purified using Agencourt RNAClean XP bead, provided by Nugen Library preparation kit. The sheared DNA was then subjected to end repair and adaptor ligation. Adaptor ligated libraries were purified with Agencourt RNAClean XP bead and amplified using 18 PCR cycle of 94°C for 30 sec, 60°C –for 30 sec, and 72°C for 1 min. Agencourt RNAClean XP bead was used for amplified Library Purification and libraries Fragment distribution was validated on Bioanalyzer DNA Chip 1000.
Overview of the Nanoscope pipeline
In order to facilitate the analysis of synthetic long read data for in the context of metagenomics, we have developed a bioinformatics pipeline called Nanoscope (Figure 1). Nanoscope takes as input a set of long read libraries together with optional (but highly recommended) short read libraries. It then performs a four-stage analysis of this data that includes de-novo assembly, variant calling and haplotyping, taxonomic identification, and abundance estimation.
Nanoscope starts by invoking the Soapdenovo20 and Celera21 assemblers to independently assemble the short and long read libraries, before merging the results using Minimus222. In the next step, it invokes a variant calling and phasing algorithm called Lens to analyze the assembled contigs for strain variation. Lens reveals hundreds to thousands of sites where individual bacteria of the same strain differ from each other and then phases these variants into bacterial haplotypes. A typical contig might harbor more than a dozen different strain haplotypes, each of which may contain thousands of sequence variants. Variants and haplotypes are determined using a simple model (see the section on Lens below) that, unlike previous approaches35,36, does not make any assumptions on the length of sequencing reads or the ploidy of the organism; we have found that these factors may confuse existing bacterial variant callers and lead to suboptimal results.
Finally, Nanoscope invokes the FCP software package29 to assign taxonomic labels to assembled contigs and to estimate bacterial abundances. The latter task is done by mapping short reads to assembled contigs and by aggregating the coverage over all contigs assigned to the same species. Computing abundances from short reads avoids certain biases inherent to synthetic long reads; mapping reads to contigs enables estimation of abundances for bacteria whose genomes are not present in standard databases. At each stage, Nanoscope uses the popular Quast tool41 to assess its performance and to generate reports.
Nanoscope differs from existing metagenomic pipelines42,43 because it includes additional programs for dealing with synthetic long reads (most notably, the Celera and Minimus2 assemblers). We modified the source code of some of these packages to handle longer genomic sequences (see Supplementary Material); all programs used by Nanoscope have also been tuned for longer read lengths. The source code of Nanoscope is publically available in an open-source repository.
The Lens haplotyper and variant caller
Lens is a new variant calling and phasing tool specialized for metagenomes and synthetic long reads. It is based on algorithms that, unlike previous approaches35,36, do not make any assumptions on the length of sequencing reads or the ploidy of the organism; we have found that these factors may confuse existing bacterial variant callers and lead to suboptimal results (see Supplementary Material). At a high level, Lens does two things: starting from an alignment of long reads to assembled contigs (or to bacterial reference genomes), it first determines positions at which the reads and the reference differ; these positions are indicative of multiple closely related strains of the same bacterium. Then, Lens phases these variants into long haplotypes, each haplotype being defined in this context as a set of variants that co-occur within the same bacterial substrain.
The Lens haplotyper leverages the fact that each long read originates from a single organism, and therefore all variants within a read must belong to the same substrain. By connecting reads at their overlapping variants, Lens places the variants into multi-kilobase-long haplotypes in a process that is reminiscent of single-individual haplotyping (SIH) techniques37. In our setting, the number of true haplotypes is an unknown parameter that may be greater than two, making the phasing problem considerably more difficult. Although there exist well-known phasing algorithms for polyploid genomes (e.g. plants or cancer genomes), they all assume a fixed, known ploidy38,39, with the notable exception of some recent methods developed while this paper was under review 44,45; Lens on the other hand infers the ploidy directly from the data. More precisely, Lens assembles haplotypes using an approximate greedy procedure (see Supplementary Material); this choice is in part due to the fact that the SIH problem (of which ours is a generalization) is computationally intractable40. In brief, Lens sorts aligned reads from left to right and in turn uses each read to either extend an existing haplotype or to form a new one, depending on the read-haplotype overlap and on the cost of forming a new cluster (both are tunable parameters for the algorithm). Our high-level approach may in principle have applications outside metagenomics, such as in cancer genome phasing.
Supplementary Material
Acknowledgments
This work was supported by NIH/NHGRI grant T32 HG000044. V.K. was supported by an NSERC post-graduate fellowship. We thank Illumina, Inc. for their assistance in sample preparation.
Footnotes
Contributions
S.B. and M.S. conceived the study. W.Z. and F.J. performed library preparation. V.K. developed the Nanoscope pipeline and the Lens algorithm. V.K. and C.J. performed computational analyses. V.K., C.J., S.B. and M.S. wrote the paper. S.B. and M.S. supervised the study.
Competing interests
V.K. serves as a consultant for Illumina Inc. SB is a co-founder of DNAnexus and a member of the scientific advisory boards of 23andMe and Eve Biomedical. MS is a co-founder of Personalis and a member of the scientific advisory boards of Personalis, AxioMx and Genapsys.
Note that the contigs of Nielsen et al. are also clustered into unordered sets belonging to the same species.
References
- 1.Rinke C, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013 doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 2.Thomas T, Gilbert J, Meyer F. Metagenomics - a guide from sampling to data analysis. Microbial Informatics and Experimentation. 2012;2:3. doi: 10.1186/2042-5783-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daniel R. The metagenomics of soil. Nat Rev Micro. 2005;3:470–478. doi: 10.1038/nrmicro1160. [DOI] [PubMed] [Google Scholar]
- 4.Venter JC, et al. Environmental Genome Shotgun Sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- 5.Human Microbiome Project Consortium Structure function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iverson V, et al. Untangling Genomes from Metagenomes: Revealing an Uncultured Class of Marine Euryarchaeota. Science. 2012;335:587–590. doi: 10.1126/science.1212665. [DOI] [PubMed] [Google Scholar]
- 7.Albertsen M, et al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotech. 31:533–538. doi: 10.1038/nbt.2579. [DOI] [PubMed] [Google Scholar]
- 8.Nielsen HB, et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotech. 32:822–828. doi: 10.1038/nbt.2939. [DOI] [PubMed] [Google Scholar]
- 9.Burton JN, Liachko I, Dunham MJ, Shendure J. Species-Level Deconvolution of Metagenome Assemblies with Hi-C–Based Contact Probability Maps. G3: Genes|Genomes|Genetics. 2014;4:1339–1346. doi: 10.1534/g3.114.011825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eid J, et al. Real-Time DNA Sequencing from Single Polymerase Molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 11.Fichot E, Norman RS. Microbial phylogenetic profiling with the Pacific Biosciences sequencing platform. Microbiome. 2013;1:10. doi: 10.1186/2049-2618-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuleshov V, et al. Whole-genome haplotyping using long reads and statistical methods. Nat Biotechnol. 2014 doi: 10.1038/nbt.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Rienzi SC, et al. The human gut and groundwater harbor non-photosynthetic bacteria belonging to a new candidate phylum sibling to Cyanobacteria. Elife. 2:e01102. doi: 10.7554/eLife.01102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharon I, et al. Accurate, multi-kb reads resolve complex populations and detect rare microorganisms. Genome Res. 2015;25:534–543. doi: 10.1101/gr.183012.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castillo-Rodal AI, et al. Mycobacterium bovis BCG substrains confer different levels of protection against Mycobacterium tuberculosis infection in a BALB/c model of progressive pulmonary tuberculosis. Infect Immun. 2006;74:1718–1724. doi: 10.1128/IAI.74.3.1718-1724.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lieberman TD, et al. Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat Genet. 2011;43:1275–1280. doi: 10.1038/ng.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Welch RA, et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci USA. 2002;99:17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gire SK, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science. 2014;345:1369–1372. doi: 10.1126/science.1259657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCoy, et al. Illumina TruSeq Synthetic Long-Reads Empower De Novo Assembly and Resolve Complex, Highly-Repetitive Transposable Elements. PLoS ONE. 2014;9:e106689. doi: 10.1371/journal.pone.0106689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li R, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2009 doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Myers EW, et al. A Whole-Genome Assembly of Drosophila. Science. 2000;287:2196–2204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- 22.Sommer D, Delcher A, Salzberg S, Pop M. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics. 2007;8:64. doi: 10.1186/1471-2105-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu W, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010;38:e132. doi: 10.1093/nar/gkq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magoc T, et al. GAGE-B: an evaluation of genome assemblers for bacterial organisms. Bioinformatics. 29:1718–1725. doi: 10.1093/bioinformatics/btt273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mao F, Dam P, Chou J, Olman V, Xu Y. DOOR: a database for prokaryotic operons. Nucleic Acids Res. 37:D459–D463. doi: 10.1093/nar/gkn757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen W, Minguez P, Lercher MJ, Bork P. OGEE: an online gene essentiality database. Nucleic Acids Res. 40:D901–D906. doi: 10.1093/nar/gkr986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118); iso-2; iso-3. Fly. 6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gusfield D. Efficient algorithms for inferring evolutionary trees. Networks. 1991;21:19–28. [Google Scholar]
- 29.Parks D, MacDonald N, Beiko R. Classifying short genomic fragments from novel lineages using composition and homology. BMC Bioinformatics. 2011;12:328. doi: 10.1186/1471-2105-12-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alneberg J, et al. Binning metagenomic contigs by coverage and composition. Nat Meth. 11:1144–1146. doi: 10.1038/nmeth.3103. [DOI] [PubMed] [Google Scholar]
- 31.Schloissnig S, et al. Genomic variation landscape of the human gut microbiome. Nature. 2013;493:45–50. doi: 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greenblum S, Carr R, Borenstein E. Extensive strain-level copy-number variation across human gut microbiome species. Cell. 2015;160:583–594. doi: 10.1016/j.cell.2014.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berlin K, et al. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol. 2015;33:623–630. doi: 10.1038/nbt.3238. [DOI] [PubMed] [Google Scholar]
- 34.Burton JN, et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol. 2013;31:1119–1125. doi: 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walker BJ, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nijkamp JF, Pop M, Reinders MJT, de Ridder D. Exploring variation-aware contig graphs for (comparative) metagenomics using MaryGold. Bioinformatics. 2013;29:2826–2834. doi: 10.1093/bioinformatics/btt502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duitama J, et al. Fosmid-based whole genome haplotyping of a HapMap trio child: evaluation of Single Individual Haplotyping techniques. Nucleic Acids Res. 2012;40:2041–2053. doi: 10.1093/nar/gkr1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berger E, Yorukoglu D, Peng J, Berger B. HapTree: a novel Bayesian framework for single individual polyplotyping using NGS data. PLoS Comput Biol. 2014;10:e1003502. doi: 10.1371/journal.pcbi.1003502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aguiar D, Istrail S. Haplotype assembly in polyploid genomes and identical by descent shared tracts. Bioinformatics. 2013;29:i352–60. doi: 10.1093/bioinformatics/btt213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gusfield D. Inference of haplotypes from samples of diploid populations: complexity and algorithms. J Comput Biol. 2001;8:305–323. doi: 10.1089/10665270152530863. [DOI] [PubMed] [Google Scholar]
- 41.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Treangen T, et al. MetAMOS: a modular and open source metagenomic assembly and analysis pipeline. Genome Biology. 2013;14:R2. doi: 10.1186/gb-2013-14-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schloss PD, et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Applied and Environmental Microbiology. 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niklas N, et al. cFinder: definition and quantification of multiple haplotypes in a mixed sample. BMC Res Notes. 2015;8:422. doi: 10.1186/s13104-015-1382-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pulido-Tamayo S, et al. Frequency-based haplotype reconstruction from deep sequencing data of bacterial populations. Nucleic Acids Res. 2015;43:e105. doi: 10.1093/nar/gkv478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.