

Abstract
To realize the potential of human embryonic stem cells (hESCs) in regenerative medicine and drug discovery applications, large numbers of cells that accurately recapitulate cell and tissue function must be robustly produced. Previous studies have suggested that genetic instability and epigenetic changes occur as a consequence of enzymatic passaging. However, the potential impacts of such passaging methods on the metabolism of hESCs have not been described. Using stable isotope tracing and mass spectrometry-based metabolomics, we have explored how different passaging reagents impact hESC metabolism. Enzymatic passaging caused significant decreases in glucose utilization throughout central carbon metabolism along with attenuated de novo lipogenesis. In addition, we developed and validated a method for rapidly quantifying glycan abundance and isotopic labeling in hydrolyzed biomass. Enzymatic passaging reagents significantly altered levels of glycans immediately after digestion but surprisingly glucose contribution to glycans was not affected. These results demonstrate that there is an immediate effect on hESC metabolism after enzymatic passaging in both central carbon metabolism and biosynthesis. HESCs subjected to enzymatic passaging are routinely placed in a state requiring re-synthesis of biomass components, subtly influencing their metabolic needs in a manner that may impact cell performance in regenerative medicine applications.
Keywords: Glycans, Lipids, Pluripotent stem cells, Stable isotope tracing, Stem cell metabolism
1 Introduction
Human embryonic stem cells (hESCs) are characterized by their ability to differentiate into the three terminal germ layers and self-renew indefinitely. This makes them ideal cell types for regenerative medicine and drug discovery applications. However, the impact of in vitro culture conditions on cell performance and stability must be monitored and validated to ensure these cells recapitulate actual tissue functions. Since the initial isolation and expansion of hESC lines on feeder layer conditions [1], more recently developed, chemically-defined culture conditions have become commonly used for the isolation and culture of hESCs and induced pluripotent stem cells (iPSCs) [2]. Along with the engineering of defined culture conditions, a concomitant development of enzymatic passaging methods to replace laborious mechanical passaging methods also occurred. Analysis of cells cultivated using these enzymatic passaging techniques has suggested these approaches increase karyotypic instability of hESCs [3–5]. Another recent study also indicated that enzymatic passaging may cause increased genetic instability and differential DNA methylation [6]. While these studies have addressed the effects of enzymatic passaging on cellular genetics and epigenetic alterations, the impacts of passaging method on cellular metabolism have not been examined.
HESC metabolism is characterized by a greater reliance on glycolysis as compared to cells in the differentiated state [7–9]. Glycolytic flux is essential for maintenance of the stem cell phenotype, as evidenced by the impact of inhibitors of glycolysis on pluripotency marker expression and cellular reprogramming [10, 11]. While glucose metabolism provides cellular energy by producing adenosine triphosphate (ATP) and reducing equivalents, glucose is also the primary carbon source for a myriad of biosynthetic precursors, including ribose for nucleotides, non-essential amino acids, and acetyl-coenzyme A (AcCoA) for lipids. Though commonly overlooked in studies of intermediary metabolism, glucose (in addition to glutamine) provides the necessary building blocks for the synthesis of glycan moieties, which are essential for protein function and trafficking. Glycans are also the key components that comprise the glycocalyx, which surrounds the cell membrane of some cells [12]. Through the cleavage of extracellular proteins and their associated glycans, enzymatic digestion could affect the phenotype of hESCs by impacting their metabolic demands or ability to respond to extracellular signaling cues [13–17].
To understand the influence of enzymatic passaging on hESC metabolism we utilized stable isotope tracing and mass spectrometry-based metabolomics to characterize central carbon metabolism after passaging. We developed and applied a method for rapid quantitation of glycan, nucleotide, and amino acid pools to explore the impact of passaging reagents on hESC biomass. Finally, we determined how enzymatic passaging affects biosynthetic flux to lipids, nucleotides and carbohydrates. Our results demonstrate that enzymatic passaging alters hESC metabolism and the cell’s ability to synthesize biosynthetic intermediates while highlighting the quantitative importance of metabolic flux to glycan pools.
2 Materials and methods
2.1 Cell culture
WA09 hESCs (H9s) were maintained on Synthemax II-SC coated (Corning, Corning, NY) plates in mTESR1 (Stem Cell Technologies, Vancouver, BC). hESCs were passaged every five days by exposure to Versene (Gibco, Grand Island, NY) for 10 min at 37°C. Synthemax II-SC coating was performed by dispensing 2 mL of working dilution (25 μg/mL) to each well of a six-well of a tissue culture polystyrene plate and incubating for 2 h. For isotopic labeling experiments, cells were maintained in mTESR1 media with uniformly-labeled glucose(tracer mTESR) by adding 5× mTESR1 supplement to custom DMEM/F-12. Custom DMEM/F-12 (Hyclone Laboratories, Logan, UT) without amino acids, D-glucose, sodium pyruvate, sodium bicarbonate, and phenol red was supplemented with all amino acids, sodium pyruvate, sodium bicarbonate (14 mM; Sigma-Aldrich, St. Louis, MO), HEPES (15 mM; from 1 M stock, Gibco, Grand Island, NY), and [U-13C6] Glucose (99%; Cambridge Isotopes, Cambridge, MA) at DMEM/F-12 levels.
2.2 Enzymatic passaging experiments
H9s (p29–35) were grown on Synthemax II-SC coated plates to 50–70% confluency. Cells were rinsed with 1 mL PBS and then exposed at 37°C to either 1 mL Versene for 10 min, Accutase (Innovative Cell Technology, San Diego, CA) for 5 min, or Trypsin-EDTA (0.25%; Gibco, Grand Island, NY) for 5 min. Versene-treated cells were then split to three wells by aspirating Versene and resuspending in 6 mL mTESR. Accutase-treated cells were split to three wells by adding 9 mL PBS to Accutase solution, centrifuging at 300 ×g for 5 min, and resuspending in 6 mL mTESR after aspiration. Trypsin-treated cells were split to three wells by adding 9 mL PBS to Trypsin solution, centrifuging at 300 ×g for 5 min, and resuspendingpellet in 6 mL mTESR after aspiration. Cells traced immediately after passaging were resuspended in tracer mTESR. Cells traced 24 h after passaging were resuspended in mTESR1 immediately after passaging, rinsed with PBS 24 h later, and changed into tracer mTESR before extracting 4 h later. For experiments with ROCK inhibitor, 5 μM of Y-27632 (Tocris, Avon, UK) was added to media.
For quantitation of biomass abundances after passaging, cellsin triplicate were rinsed with 1 mL PBS and exposed at 37°C to 1 mL Versene for 10 min, TrypLE Express (Gibco, Grand Island, NY) for 5 min, Accutase for 5 min, or Trypsin-EDTA for 5 min. 1 mL of PBS was immediately added after incubation to quench enzymatic digestion and then transferred to 15 mL conical tube containing 7 mL PBS. Each well was then washed with 1 mL PBS and added to the respective conical tube. Cells were then centrifuged at 300 ×g for 5 min and supernatant was aspirated. Cells were then washed twice by resuspension of the pellet in 1 mL 0.9% w/v saline, centrifugation at 300 ×g for 5 min, and aspiration of supernatant. Pellets were then stored at −20°C for metabolite extraction.
2.3 Metabolite extraction and GC-MS analysis
Polar metabolites and fatty acids were extracted using methanol/water/chloroform as previously described [18]. Briefly, cells were rinsed with 0.9% w/v saline and 250 μL of −80°C MeOH was added to quench metabolic reactions. 100 μL of ice-cold water supplemented with 10 μg/mL norvaline was then added to each well and cells were collected by scraping. The lysate was moved to a fresh 1.5 mL Eppendorf tube and 250 μL of −20°C chloroform supplemented with 10 μg/mL heptadecanoate was added. After vortexing and centrifugation, the top aqueous layer and bottom organic layer were collected and dried under airflow. The remaining “interface” layer containing biomass was washed twice by addition of −80°C 500 μL of MeOH, centrifugation at 21 000 ×g, and decanting of supernatant. Interface layers were then dried by ambient air overnight and stored at −20°C. For cell pellets, a similar procedure was performed as previously described, except the cell pellet was resuspended in ice cold MeOH/water solution with norvaline by pipetting and then cells were lysed by vortexing for 1 min. Chloroform was then added and polar/non-polar fractions were collected. To prepare biomass components for relative quantitation and isotopomer analysis, acid hydrolysis of interface layer was performed by first drying the rinsed interface under airflow, then incubating in 500 μL of 6 M HCl at 80°C for 2 h. Hydrolyzed biomass solution was split to five aliquots and dried by airflow overnight for subsequent GC/MS analysis.
Fatty acids and polar metabolites were derivatized as previously described [19]. For fatty acids, dried non-polar fraction was saponified and esterified to form fatty acid methyl esters (FAMEs) by addition of 500 μL of 2% w/v H2SO4 in MeOH and incubated at 50°C for 120 min. FAMEs were then extracted by addition of saturated NaCl and hexane before collection and drying of the inorganic layer. For polar metabolites, methoxime-tBDMS derivatives were formed by addition of 15 μL 2% w/v methoxylamine hydrochloride (MP Biomedicals, Solon, OH) in pyridine and incubated at 45°C for 60 min. Samples were then silylated by addition of 15 μL of N-tert-butyldimethylsily-N-methyltrifluoroacetamide (MTBSTFA) with 1% tert-butyldimethylchlorosilane (tBDMS) (Regis Technologies, Morton Grove, IL) and incubated at 45°C for 30 min. For biomass analysis of sugars and glycan residues that were too large for tBDMS derivatization, methoxime-trimethylsilyl (TMS) derivatives were formed by addition of 15 μL 2% w/v methoxylamine hydrochloride (MP Biomedicals, Solon, OH) in pyridine and incubated at 37°C for 60 min. Samples were then silylated by addition of 15 μL of N-methyltrimethylsilyltrifluoroacetamide (MSTFA; Regis Technologies, Morton Grove, IL) and incubated at 45°C for 30 min.
Polar and interface samples were analyzed by GC-MS using a DB-35MS column (30 m × 0.25 mm i.d. × 0.25 μm, Agilent J&W Scientific, Santa Clara, CA) in an Agilent 7890B gas chromatograph (GC) interfaced with a 5977C mass spectrometer (MS). Electron impact ionization was performed with the MS scanning over the range of 100–650 m/z for polar metabolites and 70–850 m/z for biomass metabolites. For separation of polar metabolites the GC oven was held at 100°C for 1 min after injection, increased to 255°C at 3.5°C/min, and finally increased to 320°C at 15°C/min and held for 3 min. For separation of the biomass metabolites the GC oven was held at 80°C for 6 min after injection, increased to 300°C at 6°C/min and held for 10 min, and finally increased to 325°C at 10°C/min and held for 4 min.
Derivatized fatty acids were analyzed by GC-MS using a select FAME column (100 m × 0.25 mm i.d. × 0.25 μm; Agilent J&W Scientific, Santa Clara, CA) as above, with the MS scanning over the range 120–400 m/z. For separation the GC oven was held at 80°C for 1 min after injection, increased to 160°C at 20°C/min, increased to 198°C at 1°C/min, and finally increased to 250°C at 5°C/min and held for 15 min.
2.4 Mass isotopomer distributions, isotopomer spectral analysis (ISA), and flux analysis
Mass isotopomer distributions and total abundances were determined by integration of mass fragments (Table 1) and correcting for natural abundances using in-house algorithms [19]. Total abundances were normalized by counts of adenine and guanine. Isotopomer spectral analysis (ISA) was performed as previously described [19]. ISA compares experimental labeling of fatty acids to simulated labeling using a reaction network where C14:0 is condensation of 7 AcCoAs, C16:0 is condensation of 8 AcCoAs, C16:1c is condensation of 8 AcCoAs, C18:0 is condensation of 9 AcCoAs, and C18:1n9c is condensation of 9 AcCoAs. Parameters for contribution of glucose to lipogenic AcCoA (D value) and percentage of newly synthesized fatty acid (g(t) value) and their 95% confidence intervals are then calculated using best-fit model from INCA MFA software [20]. Per biomass molar quantitation of glucose, galactose, glucosamine, mannosamine, serine, ribose, and leucine was accomplished by determining the ratio of each and comparing to the molar amount of leucine in mammalian cells per gram dry weight [21]. Amino acid fragments were taken from previously described work and validated [22].
Table 1.
Metabolite fragments used for GC/MS analysis.
| Metabolite | Derivatization | Fragments for integration | m/z | Positions of labeled carbons | Retention Time (min) |
|---|---|---|---|---|---|
| Alanine | tBDMS | C11H26O2NSi2 | 260 | 123 | 13.1 |
| Lactate | tBDMS | C11H25O3Si2 | 261 | 123 | 11.9 |
| Citrate | tBDMS | C26H55O7Si4 | 591 | 123456 | 41.2 |
| Adenine | TMS | C11H21N5Si2 | 279 | 27.4 | |
| TMS | C10H18N5Si2 | 264 | |||
| Galactose | TMS | C13H31O3Si3 | 319 | 3456 | 23.8 |
| Glucose | TMS | C13H31O3Si3 | 319 | 3456 | 23.9 |
| Glucosamine | TMS | C13H31O3Si3 | 319 | 3456 | 24.7 |
| Guanine | TMS | C14H29ON5Si3 | 367 | 31.5 | |
| TMS | C13H26ON5Si3 | 352 | |||
| Mannosamine | TMS | C13H31O3Si3 | 319 | 3456 | 24.5 |
| Ribose | TMS | C12H31O3Si3 | 307 | 345 | 20.3 |
| Serine | TMS | C11H28O3NSi3 | 306 | 123 | 14.4 |
| Leucine | TMS | C8H20NSi | 158 | 12.1 |
2.5 Statistical analyses
All results shown as averages of triplicates presented as mean ± standard deviation (SD). p-values were calculated using a Student’s two-tailed t-test; *, p-value between 0.01 and 0.05; **, p-value between 0.001 and 0.01; ***, p-value < 0.001. All errors associated with ISA are 95% confidence intervals determined via confidence interval analysis [23].
3 Results
3.1 Enzymatic passaging decreases glucose oxidation and fatty acid synthesis in hESCs
To investigate the effects of enzymatic passaging on hESC metabolism we used stable isotope tracing with [U-13C6]glucose and GC/MS analysis to probe intermediary metabolism and lipid synthesis (Fig. 1A). By quantifying the extent of metabolite labeling and pool sizes in hESCs after various treatments we were able to quantify relative changes in flux through each pathway. Accutase treatment induced a significant decrease in glucose contribution to both lactate and alanine as compared to clumped passaging (Versene treatment) (Fig. 1B). Additionally, a significantly lower contribution of glucose to the TCA-intermediate citrate was seen in Accutase treated cells (Fig. 1B; Supporting information, Fig. S1A). Importantly, the pool sizes of each metabolite 4 h after enzyme treatment were similar to or lower than that observed with clumped passaging (Supporting information, Fig. S1B). Therefore, the differential labeling we observed indicated that flux through glycolysis and into mitochondria were significantly decreased in Accutase treated cells. Similar changes in glucose-derived labeling were observed when cells were passaged with Trypsin, indicating the impact on central carbon metabolism is not specific to Accutase (Supporting information, Fig. S1D). Previous work has demonstrated that addition of a Rho-associated kinase (ROCK) inhibitor can prevent single cell dissociation-induced apoptosis [24]. To account for these effects we also investigated whether the addition of Y-27632 could rescue defects in glucose metabolism. While addition of ROCK inhibitor rescued labeling in lactate and citrate, flux to alanine only increased slightly (Figure 1B; Supporting information, Fig. S1A). Taken together, these results suggest that enzymatic passaging lowered flux through glycolysis and into the TCA cycle, and addition of ROCK inhibitor only partially rescued this metabolic phenotype.
Figure 1.
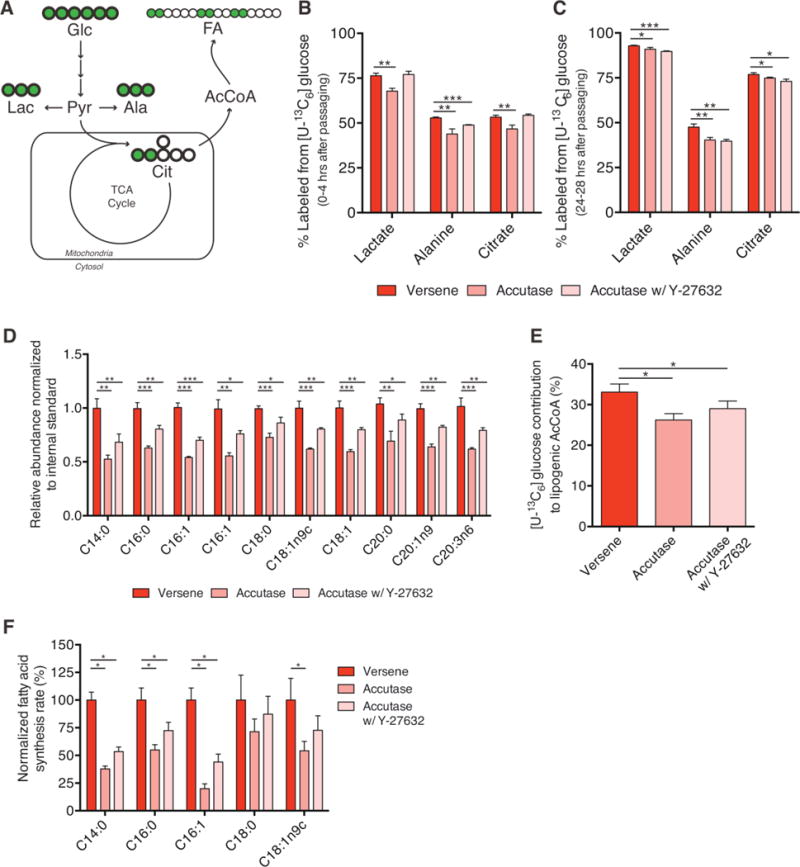
Enzymatic passaging alters central carbon metabolism. (A) Atom-transition map depicting flow of [U-13C6]glucose (UGlc) carbon through central carbon metabolism and lipid biosynthesis. Green circles depict 13C atoms and open circles depict 12C atoms. (B) Percentage of labeled metabolites from UGlc 4 h after non-enzymatic or enzymatic passaging. Higher labeling indicates greater glucose usage for glycolysis, non-essential amino acid synthesis, and TCA metabolism. (C) Percentage of labeled metabolites from UGlc one day after non-enzymatic or enzymatic passaging (i.e. labeled from 24–28 h after passaging). Defects in glucose catabolism mediated through enzymatic passaging are still present. (D) Relative abundance of fatty acid species after enzymatic or non-enzymatic passaging. (E) Contribution of UGlc to lipogenic AcCoA as determined by ISA model. Decrease in contribution is consistent with decreased labeling in the lipogenic metabolite citrate. (F) Normalized fatty acid flux for synthesized fatty acid species calculated using total pool size and fractional synthesis from ISA model. Error bars represent SD (B–D) or 95% CI (E–F) for three replicates. *, p-value between 0.01 and 0.05; **, p-value between 0.001 and 0.01; ***, p-value < 0.001 by Student’s two-tailed t-test; or, * indicates significance by nonoverlapping 95% confidence intervals
To determine whether enzymatic passaging elicited sustained effects on intermediary metabolism we quantified abundance and labeling of the same metabolite pools when applying tracer 24 h after re-plating. Interestingly, even after 24 h we still observed significant, though slight, decreases in lactate, alanine, and citrate abundances (Supporting information, Fig. S1C) and labeling (Fig. 1C). These data suggest that enzymatic passaging may impact cell metabolism last longer than the immediate period after passaging.
Having observed differences in flux to the lipogenic intermediate citrate, we then explored whether enzymatic passaging impacted lipid metabolism. Specifically, we quantified the relative abundance and isotopic labeling in various fatty acid species from saponified lipid fractions of hESCs. Measurement of total cellular fatty acid abundances showed a clear decrease in all measured species upon enzymatic passaging, and addition of ROCK inhibitor failed to rescue any defect (Fig. 1D). Decreases in abundance were observed in saturated, unsaturated, and polyunsaturated fatty acids, implicating pan defects in lipid metabolism. Indeed, since decreases were observed in both non-essential (e.g. C16:0) and essential (e.g. C20:3n6) fatty acids, these data indicate that lipid synthesis and uptake were compromised in cells passaged using enzymatic reagents. To quantify biosynthetic fluxes in greater detail we then applied isotopomer spectral analysis (ISA) to determine the relative contribution of glucose to lipogenic AcCoA pools as well as the extent of de novo lipogenesis for each fatty acid measured [25]. Consistent with the above effects on glucose flux to citrate (Fig. 1B), enzymatic passaging with and without ROCK inhibitor supplementation significantly decreased the extent of glucose conversion to lipogenic AcCoA as compared to clumped passaging (Fig. 1E). Using both pool size (Fig. 1D) and fractional synthesis/turnover data obtained from ISA, we observed that enzymatic passaging significantly decreased the synthesis rates of saturated myristic acid (C14:0), saturated palmitic acid (C16:0), and unsaturated palmitoleic acid (C16:1) (Fig. 1F). In nearly all cases these effects were not rescued by addition of a ROCK inhibitor. These data therefore indicate that enzymatic passaging lowers the ability of hESCs to utilize glucose for biosynthesis in central carbon metabolism and lipid synthesis.
3.2 Rapid quantitation of total glycan pools and synthesis in hESCs
Rapidly dividing cells have considerable biosynthetic demands for structural components as well as bioenergetic demands for maintenance and division [26]. Glucose metabolism has been demonstrated as an essential source of carbon and ATP generation for hESC proliferation [27, 28]. At the nexus of glucose metabolism and biosynthesis, post-translational modifications (PTM) have also been demonstrated to be essential to maintain stem cell pluripotency and function [29–31]. Large classes of PTMs that also involve glucose metabolism are N-linked and O-linked glycosylation moieties [32, 33]. Indeed, N-linked glycosylation is the major structural component of the glycocalyx that surrounds cell membranes [33]. Given the significant abundance of these intermediates at the cell surface, we hypothesized that glycan pools and synthesis might be significantly affected by enzymatic passaging of hESCs.
To better quantify flux through the hexosamine biosynthesis pathway we developed a method for measuring relative glycan pool sizes and isotopic labeling from tracers in hESCs or other cultured cell types. While enzyme-mediated dissociation is commonly used for glycomic-platform analyses, such methods can be costly and time-consuming. Rather than conducting whole-glycan analyses we instead collected the biomass fraction of hESC extracts and performed acid hydrolysis to release individual glycan residues, nucleobases, sugars from nucleotides or glycogen, and proteinogenic amino acids. Acid hydrolysis is commonly used in metabolic flux analysis (MFA) applications, but the extent of labeling in glycans is not commonly measured [34]. This is particularly true in MFA applied to mammalian cells [35–37]. To validate our approach we analyzed standards for specific glycan residues and compared the mass isotopomer distributions (MIDs) of specific fragments to those measured in cells cultured in the presence of [U-13C6]glucose. Glucose labeling was readily incorporated into glycan pools through the hexosamine biosynthesis pathway (Fig. 2A). Acid hydrolysis of cellular biomass in turn releases proteinogenic amino acids, ribose, sugars, and aminosugars for GC/MS analysis (Fig. 2B). However, since several glycan species are labile in the conditions used for release, in some cases (e.g. acetylated glycan moieties) we relied on the measurement of proxy molecules (Fig. 2C). The MIDs of specific glycan sugars from standards and hydrolyzed hESCs are depicted in Fig. 2D and 2E, tabulated in Supporting information, Tables S1 and S2; all of which were corrected for natural isotope abundance using in-house algorithms and calculated fragment formulae (Table 1). In each case the corrected MID matched that of the standards. Notably, some glycan sugars were not present at detectable levels to include here (e.g. fucose, xylose, mannose, galactosamine), and the relatively low abundance of mannosamine caused some deviation from unity in the measured and corrected MID. Furthermore, labeling from [U-13C6]glucose indicated the number of carbons present from the parent molecule (Fig. 2F). Although free metabolites were removed from the biomass interface prior to hydrolysis and derivatization, we conducted parallel treatments and quantitation on the free, polar metabolites present in our extract to the quantities in each subcellular pool. While serine, ribose, glucose, and glucosamine were five to ten-fold more abundant in biomass compared to free metabolites (including those present as nucleotide-sugars or phosphorylated intermediates), the abundance of galactose and mannosamine from biomass hydrolysates was only two-fold higher than that quantified from free metabolites (Supporting information, Fig. S2A). Therefore, this method allowed for the measurement of relative glycan residue abundance and labeling from cellular biomass pools (Fig. 2B). Previous methods focusing on biomass pools have relied on targeted digestion of nucleotides, proteins, and glycans individually [38–40]. Our method instead allows profiling of all three classes of biosynthetic intermediates simultaneously.
Figure 2.
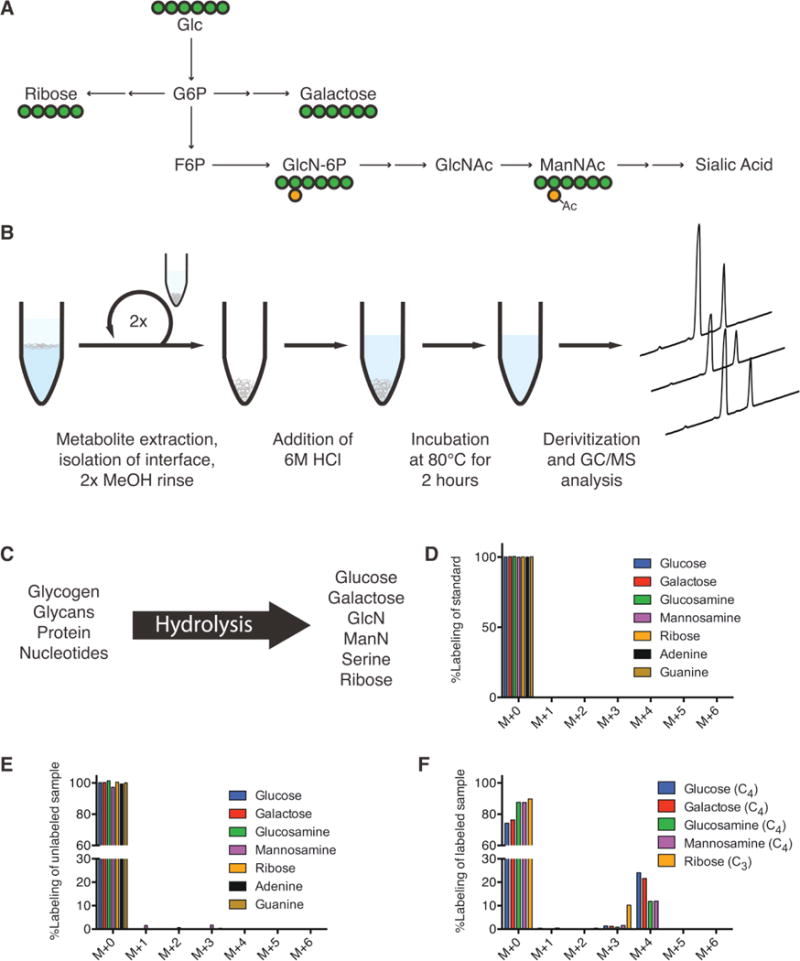
Quantitation of glycan residue abundance and labeling in cellular biomass. (A) Atom-transition map depicting flow of [U-13C6]glucose (UGlc) into ribose, galactose, and hexosamines. Green circles depict carbon atoms and orange circles depict nitrogen atoms. (B) Schematic of biomass hydrolysis method. Insoluble interface layer is isolated from initial methanol/water/chloroform quench/extraction, rinsed twice with methanol, and acid hydrolyzed. (C) Diagram of detectable metabolites after acid hydrolysis. Major macromolecules (nucleotides, protein, glycans) are broken down into primary components (ribose/nucleobases, amino acids, sugars/amino-sugars, respectively), which can be measured on GC/MS. (D) Corrected mass isotopomer distribution (MID) of each metabolite standard. Corrected M + 0 peak equal to unity ensures accuracy of MIDs. (E) Corrected MID of metabolites from unlabeled cell hydrolysates. Corrected M + 0 peak deviation from unity is informative of MID accuracy and potential contaminating fragments in hydrolysates. (F) Corrected MID of metabolites measured in hydrolysates from hESCs labeled using UGlc. Glucose, galactose, glucosamine, and mannosamine fragments have four carbons labeled from glucose. Ribose has three carbons labeled from glucose. Error bars represent SD (E–F) for three independent hydrolysates.
3.3 Glycan and carbohydrate pools are significantly depleted upon enzymatic passaging
To test the effect of enzymatic-treatment on glycan and macromolecule abundance immediately after dissociation, non-enzymatic (Versene) and enzymatic methods (TrypLE, Accutase, and Trypsin) of increasing dissociation strength were used to dissociate cells. All enzymatic reagents significantly altered carbohydrate abundances in biomass as compared to non-enzymatic control treatment (Fig. 3A and 3B). However, while galactose abundance was significantly reduced with enzymatic treatment (Fig. 3A), glucose abundance significantly increased (Fig. 3B). Given the presence of glucose in cells as both glycosylation intermediate [32] and component of glycogen, the differential catabolism of glycogen presumably caused such changes. Indeed, this result would be expected given the decreased flux through glycolysis observed in Fig. 1. Galactose, on the other hand, is primarily present in cells as the glycan residue proximate to terminal sialylation [41].
Figure 3.
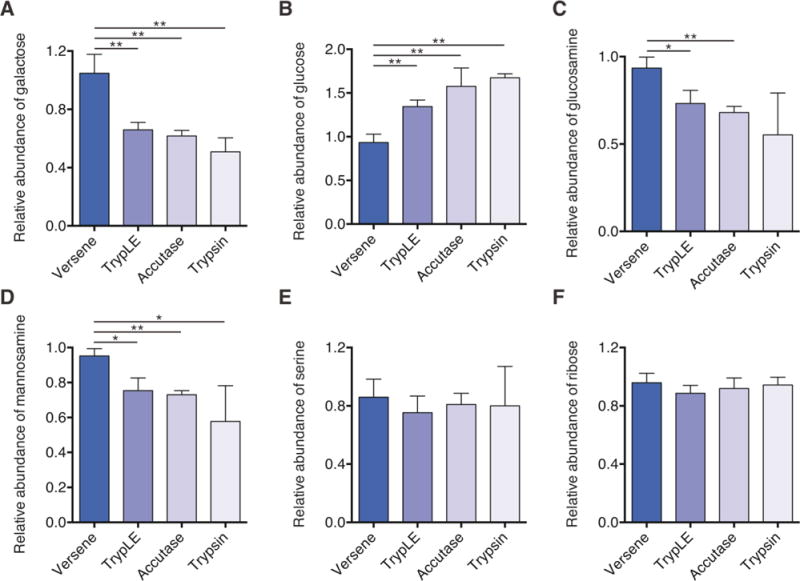
Enzymatic passaging alters glycan abundance of hESCs. (A–F) Relative abundance of biomass-derived galactose (A), glucose (B), glucosamine (C), mannosamine (D), serine (E), and ribose (F) immediately after passaging. All data are reported relative to Versene. Decreases in hexose (galactose) and hexosamine (mannosamine and glucosamine) abundances suggest that glycans are impacted by enzymatic passaging. This change in abundance is not observed in protein-derived amino acids (serine) or nucleotide/cofactor-derived ribose. Error bars represent SD (A–F) for three replicates. *, p-value between 0.01 and 0.05; **, p-value between 0.001 and 0.01; ***, p-value < 0.001 by Student’s two-tailed t-test.
Similar to our results quantifying galactose, the abundance of glucosamine and mannosamine also decreased with increasing strength of passaging reagents used (Fig. 3C and 3D). Importantly, even milder reagents like TrypLE and Accutase showed a significant reduction in abundance of both amino sugars as compared to non-enzymatic control (Fig. 3C and 3D). As expected, intracellular serine and ribose levels were unaffected by extracellular enzymatic digestion (Fig. 3E and 3F). These results suggest that enzymatic passaging significantly affects biomass composition directly after dissociation.
3.4 Biosynthetic fluxes to nucleotides and glycans are similar in cultured hESCs
Since the total pools of specific glycans as well as the flux to various fatty acids in biomass were significantly altered after enzymatic passaging, we then hypothesized that fluxes to these and other (e.g. nucleotides, protein) biomass pools of hESCs cells would be affected in a similar manner. We again employed stable isotope tracing with [U-13C6]glucose and GC/MS analysis to quantify labeling of hydrolyzed hESC biomass, focusing on components that are representative of protein (serine), nucleotide (ribose), and hexosamine synthesis (Fig. 2A and 2C). Cells treated with Accutase and ROCK inhibitor exhibited a slight decrease in labeling of proteinogenic serine (Fig. 4A), although this measurement is impacted by changes in serine synthesis and uptake from the culture medium. Ribose labeling indicated that enzymatic passaging also decreased the extent of ribose labeling with or without ROCK inhibitor (Fig. 4A).
Figure 4.
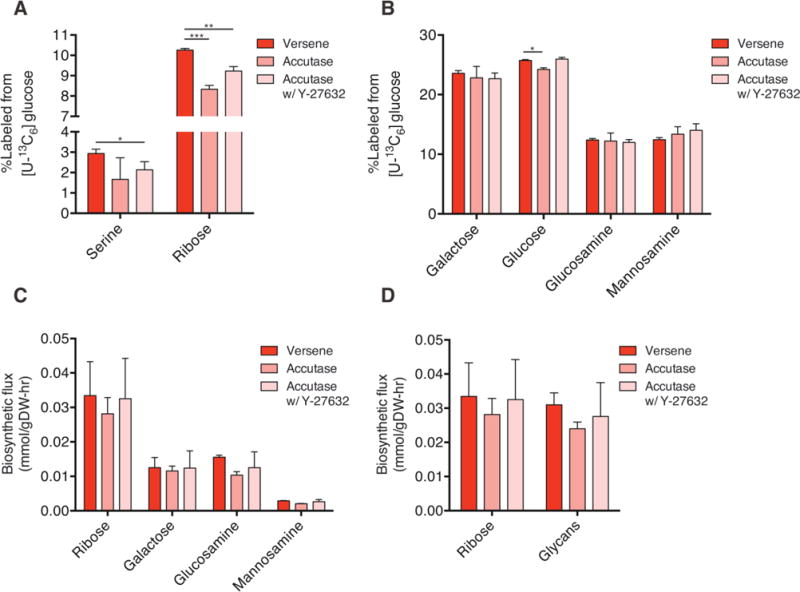
Biosynthetic fluxes to glycans and nucleotides are similar in cultured hESCs. (A) Percentage of labeled serine and ribose in cells cultured for 4 h after passaging in the presence of [U-13C6]glucose (UGlc). (B) Percentage of labeled glycan moieties from biomass in cells treated as in (A). (C) Quantitation of biosynthetic flux to different metabolites calculated using MIDs and molar pool sizes. (D) Comparison of fluxes to ribose versus glycans demonstrates similar biosynthetic needs in hESCs. Error bars represent SD (A–D) for three replicates. *, p-value between 0.01 and 0.05; **, p-value between 0.001 and 0.01; ***, p-value < 0.001 by Student’s two-tailed t-test.
Surprisingly, upon examining labeling from glucose in glycan moieties we observed minimal effects when comparing non-enzymatic to enzymatic passaging (Fig. 4B). While a slight decrease in labeling of biomass-derived glucose was noted upon Accutase treatment, this result was likely due to the increased pool size maintained in enzyme treated cells after passaging (Fig. 3B). Since routine passaging using enzymatic reagents is an extremely common and frequent insult experienced by hESCs cultivated in vitro, we calculated the flux to each biomass compartments using pool size and labeling information. Glucose and serine were not included in these calculations to avoid convoluting effects of glycogen turnover and serine uptake. While molar fluxes associated with galactose, glucosamine, and mannosamine synthesis were significantly lower than that observed for ribose in hESCs (Fig. 4C), flux to glycans in aggregate were similar to that observed for nucleotides (Fig. 4D). These results highlight the significance of glucose flux to galactose and through the hexosamine biosynthesis pathway (Fig. 4D). Notably, the flux of glucose to glycans was unaffected by enzymatic digestion (Fig. 4C) due to the rapid recovery of pool sizes after the initial 4 h of growth (Supporting information, Fig. S2B). This finding is perhaps not surprising due to long term selection experienced by hESCs in standard culture. On the other hand, these calculations demonstrate how high the flux to glycan moieties is in standard culture conditions. Although nucleotides are routinely considered a large biosynthetic pool, our measurements indicate that flux to glycans is approximately the same as that of ribose, which is a component of RNA and various cofactors (e.g. ATP, NAD+) (Fig. 4D).
Taking these results together, although abundance of glycan moieties is significantly altered after enzymatic digestion, the flux through these pathways is high enough to recover from these cleavage events. However, the contribution of glucose to fatty acids, proteinogenic amino acids, and nucleotides remains diminished, suggesting that such passaging methods impact metabolism for at least several hours after hESC subculture.
4 Discussion
We have demonstrated that the use of enzymatic reagents of hESCs has an immediate and significant impact on metabolic activity after passaging. Through the use of 13C MFA we have demonstrated that glucose flux through glycolysis and the TCA cycle as well as lipid biosynthesis are decreased after splitting cells using enzyme-based passaging methods. Using a method that can rapidly probe the abundance and labeling of glycans in hydrolyzed biomass, we observed that enzymatic passaging significantly impacts the abundance of glycan moieties in hESCs.
4.1 Potential pitfalls in advanced hESC culture methods
For the past decade efforts in stem cell bioprocess engineering have focused on the development of well-defined but less laborious methods of cell expansion. While there is a clear need to streamline processes and assimilate current good manufacturing practices (cGMPs) [42], these advances come at the expense of compromising the stem cell niche. Widely used stem cells lines (H1, H9, etc.) were isolated in fully undefined conditions derived from mESC engineering from the early 1980s [1, 43, 44]. Here we demonstrate that the transition to more “modern” passaging methods has direct consequences on cell function and behavior.
4.2 Potential selective pressure of enzymatic passaging through altered metabolism
Presumably, treatment with enzymatic reagents leads to proteolytic cleavage of various receptors and other proteoglycans at the cell surface. In turn, the decreased receptor abundance mitigates the responsiveness of cells to endogenous signaling factors and exogenous growth factors in hESC media. A wide range of cell surface proteins may drive this phenotype, including solute carriers, glucose transporters, and receptor tyrosine kinases. Indeed, the increased abundance of glucose within enzyme-passaged cells indicates that cells may even be compromised with respect to their ability to access glycogen pools. Energetic stress has previously been associated with cells cultured for 24 h under non-adherent conditions (i.e. detachment from the matrix) [45, 46], but the immediate impacts on metabolism after dissociation were not previously appreciated. Although cells presumably recover rapidly to replenish glycan and biosynthetic intermediate pools, even a temporary selective pressure like that observed here will have lasting and significant effects on cell populations. Such effects may impact cells from the time of isolation (i.e. blastocyst or primary cell isolation) throughout passaging in vitro. Indeed, any functional application that makes use of hESCs or their derivatives requires that they accurately represent the metabolic activity of the somatic tissues that one attempts to model. For example, the metabolic behavior of hPSC-derived cardiomyocytes is known to significantly differ compared to adult heart cells with respect to their capacity for fatty acid oxidation [47, 48]. The extent of developmental maturation and selective pressures due to in vitro culture on such phenomena must both be considered.
4.3 Glycocalyx is a significant biomass pool in cultured hESCs
We also developed an analytical method for quantifying the overall abundance and isotopic labeling of glycan residues, proteinogenic amino acids, and ribose moieties from nucleotides and cofactors in cell cultures. This approach highlighted the profound impact of enzyme passaging on carbohydrate and glycan abundances in cells. While this method contrasts traditional methods of enzyme-mediated digestion of glycans from their protein cores and direct analysis of their structures (i.e. glycomics), the rapid nature of our methods makes it attractive for studying general effects on hexosamine metabolism. Furthermore, analysis of glycan biomass affords reliable quantitation of overall synthesis rates compared to measurements of sugar nucleotides. The glycocalyx and glycosylation profile is particularly important for cell signaling and protein function [49, 50]. Recent studies also suggest that modulation of flux through the hexosamine biosynthesis pathway directly impacts the glycoprofile of cells [51]. Consistent with this concept, we demonstrate that glucose flux to glycans is similar to that observed for flux to ribose, which contributes to nucleotide synthesis and maintenance of cellular redox levels [52, 53]. As such, hexosamine biosynthesis is an underappreciated biomass sink in metabolic studies. Indeed, studies on the metabolism of cancer cells and stem cells commonly ignore the importance of glycan production while focusing primarily on the importance of nucleotide, lipid, and amino acid metabolism [18, 54–57]. Far fewer studies address or attempt to quantify or modulate flux to glycans [13, 58].
4.4 Concluding remarks
These results and other recent studies [6, 59] are beginning to illustrate how the in vitro culture environment influences hESC phenotype. Cells are routinely subjected to periods of starvation during incubation with passaging reagents as well as cleavage of their glycocalyx and cell surface proteins. As cultures age, changes to gene expression and epigenetic markers will be selected for to deal with these stresses. In this context, upregulation of flux through the hexosamine biosynthesis pathway is to be expected. Future engineering strategies must identify and address sources of cellular stresses at the genomic, transcriptional, signaling, and metabolic levels in order to mitigate the deleterious effects of in vitro culture in regenerative medicine applications.
Supplementary Material
Acknowledgments
This research was supported by the California Institute of Regenerative Medicine (RB5-07356), NIH grant (5 R01 CA188652-02), and a Searle Scholar Award to C.M.M. M.G.B. is supported by a NSF Graduate Research Fellowship (DGE-1144086).
Abbreviations
- AcCoA
acetyl coenzyme A
- GC/MS
gas chromatography/mass spectrometry
- MID
mass isotopomer distribution
- hESC
human embryonic stem cell
- ISA
isotopomer spectral analysis
- MFA
metabolic flux analysis
- UGlc
[U-13C6]glucose
- TCA
tricarboxylic acid
Footnotes
Supporting information available online
The authors declare no financial or commercial conflict of interest.
References
- 1.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 2.Ludwig TE, Levenstein ME, Jones JM, Berggren WT, et al. Derivation of human embryonic stem cells in defined conditions. Nat Biotechnol. 2006;24:185–187. doi: 10.1038/nbt1177. [DOI] [PubMed] [Google Scholar]
- 3.Draper JS, Smith K, Gokhale P, Moore HD, et al. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat Biotechnol. 2004;22:53–54. doi: 10.1038/nbt922. [DOI] [PubMed] [Google Scholar]
- 4.Buzzard JJ, Gough NM, Crook JM, Colman A. Karyotype of human ES cells during extended culture. Nat Biotechnol. 2004;22:381–382. doi: 10.1038/nbt0404-381. author reply 382. [DOI] [PubMed] [Google Scholar]
- 5.Mitalipova MM, Rao RR, Hoyer DM, Johnson JA, et al. Preserving the genetic integrity of human embryonic stem cells. Nat Biotechnol. 2005;23:19–20. doi: 10.1038/nbt0105-19. [DOI] [PubMed] [Google Scholar]
- 6.Garitaonandia I, Amir H, Boscolo FS, Wambua GK, et al. Increased risk of genetic and epigenetic instability in human embryonic stem cells associated with specific culture conditions. PLoS One. 2015;10:e0118307. doi: 10.1371/journal.pone.0118307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shyh-Chang N, Daley GQ, Cantley LC. Stem cell metabolism in tissue development and aging. Development. 2013;140:2535–2547. doi: 10.1242/dev.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Panopoulos AD, Yanes O, Ruiz S, Kida YS, et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012;22:168–177. doi: 10.1038/cr.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J, Khvorostov I, Hong JS, Oktay Y, et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011;30:4860–4873. doi: 10.1038/emboj.2011.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Nuebel E, Daley GQ, Koehler CM, Teitell MA. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell. 2012;11:589–595. doi: 10.1016/j.stem.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, et al. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011;14:264–271. doi: 10.1016/j.cmet.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: Composition, functions, and visualization. Pfluegers Arch. 2007;454:345–359. doi: 10.1007/s00424-007-0212-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wellen KE, Lu C, Mancuso A, Lemons JM, et al. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 2010;24:2784–2799. doi: 10.1101/gad.1985910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang M, Shen Z, Huang S, Zhao L, et al. The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell. 2010;143:711–724. doi: 10.1016/j.cell.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 15.Metallo CM, Vander Heiden MG. Metabolism strikes back: Metabolic flux regulates cell signaling. Genes Dev. 2010;24:2717–2722. doi: 10.1101/gad.2010510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lauzier B, Vaillant F, Merlen C, Gelinas R, et al. Metabolic effects of glutamine on the heart: Anaplerosis versus the hexosamine biosynthetic pathway. J Mol Cell Cardiol. 2013;55:92–100. doi: 10.1016/j.yjmcc.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lau KS, Partridge EA, Grigorian A, Silvescu CI, et al. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell. 2007;129:123–134. doi: 10.1016/j.cell.2007.01.049. [DOI] [PubMed] [Google Scholar]
- 18.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014;74:3317–3331. doi: 10.1158/0008-5472.CAN-14-0772-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young JD. INCA: A computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics. 2014;30:1333–1335. doi: 10.1093/bioinformatics/btu015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheikh K, Forster J, Nielsen LK. Modeling hybridoma cell metabolism using a generic genome-scale metabolic model of Mus musculus. Biotechnol Progr. 2005;21:112–121. doi: 10.1021/bp0498138. [DOI] [PubMed] [Google Scholar]
- 22.Wegner A, Weindl D, Jager C, Sapcariu SC, et al. Fragment formula calculator (FFC): Determination of chemical formulas for fragment ions in mass spectrometric data. Anal Chem. 2014;86:2221–2228. doi: 10.1021/ac403879d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antoniewicz MR, Kelleher JK, Stephanopoulos G. Determination of confidence intervals of metabolic fluxes estimated from stable isotope measurements. Metab Eng. 2006;8:324–337. doi: 10.1016/j.ymben.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe K, Ueno M, Kamiya D, Nishiyama A, et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol. 2007;25:681–686. doi: 10.1038/nbt1310. [DOI] [PubMed] [Google Scholar]
- 25.Kharroubi AT, Masterson TM, Aldaghlas TA, Kennedy KA, Kelleher JK. Isotopomer spectral analysis of triglyceride fatty acid synthesis in 3T3-L1 cells. Am J Physiol. 1992;263:E667–E675. doi: 10.1152/ajpendo.1992.263.4.E667. [DOI] [PubMed] [Google Scholar]
- 26.Metallo CM, Vander Heiden MG. Understanding metabolic regulation and its influence on cell physiology. Mol Cell. 2013;49:388–398. doi: 10.1016/j.molcel.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X, Duan S, Yi F, Ocampo A, et al. Mitochondrial regulation in pluripotent stem cells. Cell Metab. 2013;18:325–332. doi: 10.1016/j.cmet.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Vacanti NM, Metallo CM. Exploring metabolic pathways that contribute to the stem cell phenotype. Biochim Biophys Acta. 2013;1830:2361–2369. doi: 10.1016/j.bbagen.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 29.Wang YC, Peterson SE, Loring JF. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014;24:143–160. doi: 10.1038/cr.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang YC, Lin V, Loring JF, Peterson SE. The ‘sweet’ spot of cellular pluripotency: Protein glycosylation in human pluripotent stem cells and its applications in regenerative medicine. Expert Opin Biol Ther. 2015:1–9. doi: 10.1517/14712598.2015.1021329. [DOI] [PubMed] [Google Scholar]
- 31.Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11:596–606. doi: 10.1016/j.stem.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat Rev Mol Cell Biol. 2012;13:448–462. doi: 10.1038/nrm3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dennis JW, Nabi IR, Demetriou M. Metabolism cell surface organization, and disease. Cell. 2009;139:1229–1241. doi: 10.1016/j.cell.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Long CP, Antoniewicz MR. Quantifying biomass composition by gas chromatography/mass spectrometry. Anal Chem. 2014;86:9423–9427. doi: 10.1021/ac502734e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murphy TA, Dang CV, Young JD. Isotopically nonstationary 13C flux analysis of Myc-induced metabolic reprogramming in B-cells. Metab Eng. 2013;15:206–217. doi: 10.1016/j.ymben.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munger J, Bennett BD, Parikh A, Feng XJ, et al. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nature Biotechnology. 2008;26:1179–1186. doi: 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hofmann U, Maier K, Nicbel A, Vacun G, et al. Identification of metabolic fluxes in hepatic cells from transient C-13-labeling experiments: Part I. Experimental observations. Biotechnol Bioeng. 2008;100:344–354. doi: 10.1002/bit.21747. [DOI] [PubMed] [Google Scholar]
- 38.Miranda-Santos I, Gramacho S, Pineiro M, Martinez-Gomez K, et al. Mass isotopomer analysis of nucleosides isolated from RNA and DNA using GC/MS. Anal Chem. 2015;87:617–623. doi: 10.1021/ac503305w. [DOI] [PubMed] [Google Scholar]
- 39.Jang-Lee J, North SJ, Sutton-Smith M, Goldberg D, et al. Glycomic profiling of cells and tissues by mass spectrometry: Fingerprinting and sequencing methodologies. Methods Enzymol. 2006;415:59–86. doi: 10.1016/S0076-6879(06)15005-3. [DOI] [PubMed] [Google Scholar]
- 40.Fountoulakis M, Lahm HW. Hydrolysis and amino acid composition of proteins. J Chromatogr A. 1998;826:109–134. doi: 10.1016/s0021-9673(98)00721-3. [DOI] [PubMed] [Google Scholar]
- 41.Tomiya N, Narang S, Lee YC, Betenbaugh MJ. Comparing N-glycan processing in mammalian cell lines to native and engineered lepidopteran insect cell lines. Glycoconjugate J. 2004;21:343–360. doi: 10.1023/B:GLYC.0000046275.28315.87. [DOI] [PubMed] [Google Scholar]
- 42.Unger C, Skottman H, Blomberg P, Dilber MS, Hovatta O. Good manufacturing practice and clinical-grade human embryonic stem cell lines. Hum Mol Genet. 2008;17:R48–R53. doi: 10.1093/hmg/ddn079. [DOI] [PubMed] [Google Scholar]
- 43.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 44.Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci USA. 1981;78:7634–7638. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schafer ZT, Grassian AR, Song L, Jiang Z, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grassian AR, Metallo CM, Coloff JL, Stephanopoulos G, Brugge JS. Erk regulation of pyruvate dehydrogenase flux through PDK4 modulates cell proliferation. Genes Dev. 2011;25:1716–1733. doi: 10.1101/gad.16771811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lopaschuk GD, Jaswal JS. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol. 2010;56:130–140. doi: 10.1097/FJC.0b013e3181e74a14. [DOI] [PubMed] [Google Scholar]
- 48.Kim C, Wong J, Wen J, Wang S, et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;494:105–110. doi: 10.1038/nature11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoosdally SJ, Andress EJ, Wooding C, Martin CA, Linton KJ. The Human Scavenger Receptor CD36: Glycosylation status and its role in trafficking and function. The J Biol Chem. 2009;284:16277–16288. doi: 10.1074/jbc.M109.007849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmid-Schonbein GW. An emerging role of degrading proteinases in hypertension and the metabolic syndrome: Autodigestion and receptor cleavage. Curr Hypertens Rep. 2012;14:88–96. doi: 10.1007/s11906-011-0240-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Almaraz RT, Tian Y, Bhattarcharya R, Tan E, et al. Metabolic flux increases glycoprotein sialylation: Implications for cell adhesion and cancer metastasis. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.M112.017558. M112.017558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lewis CA, Parker SJ, Fiske BP, McCloskey D, et al. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol Cell. 2014;55:253–263. doi: 10.1016/j.molcel.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fan J, Ye J, Kamphorst JJ, Shlomi T, et al. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benjamin DI, Li DS, Lowe W, Heuer T, et al. Diacylglycerol metabolism and signaling is a driving force underlying FASN inhibitor sensitivity in cancer cells. ACS Chem Biol. 2015;10:1616–1623. doi: 10.1021/acschembio.5b00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shiraki N, Shiraki Y, Tsuyama T, Obata F, et al. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 2014;19:780–794. doi: 10.1016/j.cmet.2014.03.017. [DOI] [PubMed] [Google Scholar]
- 56.Moussaieff A, Rouleau M, Kitsberg D, Cohen M, et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015;21:392–402. doi: 10.1016/j.cmet.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 57.Jain M, Nilsson R, Sharma S, Madhusudhan N, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Almaraz RT, Aich U, Khanna HS, Tan E, et al. Metabolic oligosaccharide engineering with N-Acyl functionalized ManNAc analogs: Cytotoxicity, metabolic flux, and glycan-display considerations. Biotechnol Bioeng. 2012;109:992–1006. doi: 10.1002/bit.24363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laurent LC, Ulitsky I, Slavin I, Tran H, et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell. 2011;8:106–118. doi: 10.1016/j.stem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.