

Abstract
Staphylococcus aureus is an opportunistic human pathogen, which can cause life-threatening disease. Proteome analyses of the bacterium can provide new insights into its pathophysiology and important facets of metabolic adaptation and, thus, aid the recognition of targets for intervention. However, the value of such proteome studies increases with their comprehensiveness. We present an MS–driven, proteome-wide characterization of the strain S. aureus HG001. Combining 144 high precision proteomic data sets, we identified 19 109 peptides from 2088 distinct S. aureus HG001 proteins, which account for 72% of the predicted ORFs. Peptides were further characterized concerning pI, GRAVY, and detectability scores in order to understand the low peptide coverage of 8.7% (19 109 out of 220 245 theoretical peptides). The high quality peptide-centric spectra have been organized into a comprehensive peptide fragmentation library (SpectraST) and used for identification of S. aureus-typic peptides in highly complex host–pathogen interaction experiments, which significantly improved the number of identified S. aureus proteins compared to a MASCOT search. This effort now allows the elucidation of crucial pathophysiological questions in S. aureus-specific host–pathogen interaction studies through comprehensive proteome analysis. The S. aureus-specific spectra resource developed here also represents an important spectral repository for SRM or for data-independent acquisition MS approaches. All MS data have been deposited in the ProteomeXchange with identifier PXD000702 (http://proteomecentral.proteomexchange.org/dataset/PXD000702).
Keywords: Host–pathogen interactions, Mass spectrometry (MS), Microbiology, Spectral library, Staphylococcus aureus
1 Introduction
Although substantial progress in hygiene and medical care has been achieved in recent decades, infectious diseases are still a significant risk factor for human health. Despite being a harmless commensal on the skin and in the anterior nares of about 20% (persistent carriers) to 60% (intermittent carriers) of the human population [1, 2], Staphylococcus aureus is an important pathogen causing a variety of diseases, not only mild to severe local infections, but also endocarditis and osteomyelitis, as well as systemic disease like sepsis and toxin-related diseases. The danger represented by S. aureus was aggravated by the spread of antibiotic resistance among S. aureus strains, especially when methicillin-resistant S. aureus strains (MRSA) appeared (reviewed in [3]).
The use of global-omics technologies provides important insight into cellular processes and helps to determine the pathophysiological reactions of S. aureus, which might someday reveal molecular targets for new antibiotic therapies and intervention strategies. With the invention and improvement of genomic sequencing techniques fast recording of complete genomes as well as comprehensive transcriptional profiling became routine procedures. However, these data allow only a selected and incomplete view on the cellular processes and the interaction with the human host. Genome-wide profiling on proteome level is more challenging because of difficulties in sample processing for analytical protein or peptide detection. Nevertheless, the comprehensive analysis of the staphylococcal proteome is especially important because it is the protein inventory of the bacterium that determines both the ability of S. aureus to react to its environment and its potential for causing disease.
Recently, MRSA and methicillin-sensitive S. aureus (MSSA) were compared for investigating the adaptation in the presence of sub-inhibitory concentrations of the beta-lactam antibiotic oxacillin using a spectral counting-based label-free quantitative proteomics approach [4]. The authors reported 1025 identified proteins in the individual samples and observed differentially regulated pathways after oxacillin treatment in MRSA and MSSA. A further study followed the fate of proteins during a shift from growth to a glucose-starvation induced stationary phase using a combination of pulse-chase in 13C/12C isotopically labeled medium and a 15N standard sample [5]. Protein amounts as well as changes in the solubility of proteins were determined from the quantitative data of approximately 900 proteins [5]. In order to improve the identification of low abundance proteins Muntel et al. employed exclusion lists during MS in combination with dedicated optimization of MS parameters and were able to quantify more than 990 S. aureus proteins without labeling techniques [6].
Recently, a study combining the results from six different analysis strategies has been published in order to make different cell compartments of S. aureus strain COL accessible for quantitative analysis [7]. This large effort – targeting exponential as well as stationary growth phase samples from cultures in BioExpress ® 1000 medium – led in total to the identification of about 1700 proteins when combining all approaches [7]. But still more than 1000 proteins expected from the genome sequence, which might comprise important physiological effectors, metabolic enzymes, and virulence factors and of which knowledge on protein abundance under different conditions might be important for understanding the virulence of S. aureus, were not detected. However, the analysis was confined to the strain COL, a MRSA strain isolated in the 1960s and sequenced in 2005 [8], but for broad applicability, data on other common laboratory S. aureus strains and clinical isolates are required.
In general, previous attempts to analyze the proteome of S. aureus have been limited by the necessity of preparing several samples from different bacterial cell compartments and the resulting long MS acquisition time in order to provide a more comprehensive coverage of the proteome. Technical advances in new MS instruments and applications have significantly improved the ability to provide comprehensive proteome coverage with higher degrees of quantitative accuracy [9]. A recent publication reports the identification of nearly 3000 yeast proteins and almost 5400 mammalian cell line proteins, both in triplicate single-run MS analyses [10], which demonstrates the significant improvement in proteome coverage. Thus, with adequate sample material, modern MS instruments, and sufficient MS measurement time a complete recording of a bacterial proteome like that of S. aureus with about 2800 to 2900 theoretically expected proteins is feasible today. Nevertheless, efficient proteome approaches will also include considerations on the effort, like work and measurement time, in relation to the gain of information. Here, a practical perspective will probably restrict extensive prefractionation and measurements to proof of principle projects targeting only few samples. Standard projects will probably be performed in settings constituting a compromise between reasonable effort and acceptable reduction of completeness in proteome recording. Most promising in this regard is the application of data-independent acquisition (DIA) approaches, where a comprehensive data library is recorded which is subsequently applied for the analysis of other experimental data sets.
The Human Proteome Project (HPP) is a very clear and relevant example for the need and the use of databases. In case of host–pathogen interaction studies, data from the HPP can be used to elucidate the response of the host when encountering the pathogen. The number of human proteins documented in neXtProt increased continuously and amounts to 15 646 in one of the latest publications from the HPP [11]. Although the HPP has already reported an overwhelming number of human proteins, still about 20% of predicted human proteins are not yet validated. Recent re-analysis of 16 857 LCMS/MS data sets, partly obtained from public databases, provided evidence for 18 097 human proteins [12]. Furthermore, tissue- and biofluid-specific proteome analyses can contribute to comprehensiveness [13]. The proteome coverage can also be furthermore extended by the use of proteogenomic technologies [14].
In order to map the expressed proteome of S. aureus, we combined three workflows for efficient proteome analysis of the model strain S. aureus HG001 [15]: (i) the standard method of reversed phase LC-MS/MS without prefractionation [16, 17]; (ii) off-gel IEF fractionation [18, 19]; (iii) two-dimensional strong cationic exchange chromatography (2D-SCX) [20] (Fig. 1) with the objectives (a) to detect a maximum of different peptides; (b) to apply published scores for the prediction of the detectability of peptides which could further improve the planning of targeted approaches like SRM assays, and to investigate whether such scores could be further adapted to and improved for our settings; and (c) to establish a spectral database as a prerequisite for the identification of proteins from highly complex mixtures and DIA or sequential window acquisition of all theoretical fragmentation spectra (SWATH) based approaches [21, 22]. Here, we present the so far most comprehensive S. aureus protein map comprising 90% of the expressed proteome [7]. This map allows confidential identification of S. aureus originated peptides in host–pathogen interaction studies. It further provides a promising data and spectral library repository for upcoming experiments.
Figure 1.
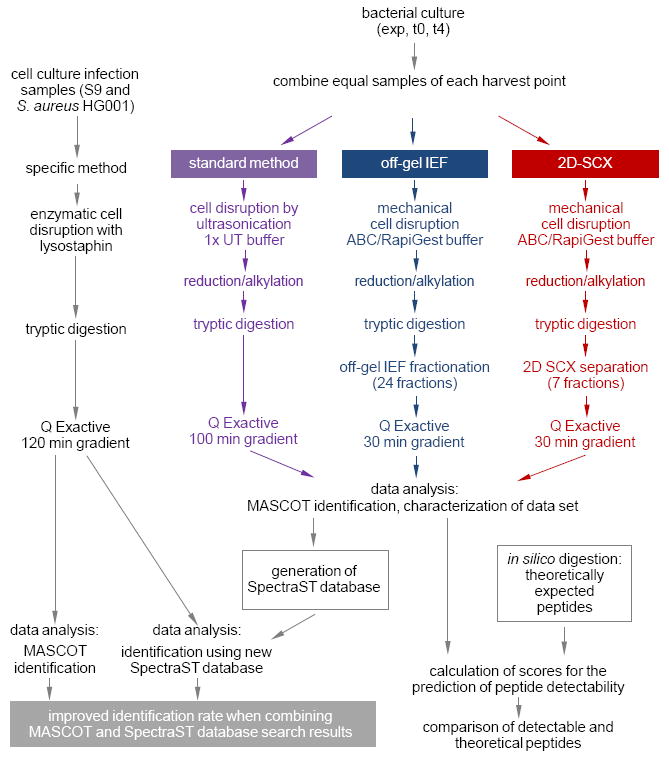
Experimental workflow. Equal samples, which were expected to cover many proteins of the theoretical proteome of S. aureus HG001, were subjected to three different methods of proteome analysis either with or without fractionation prior to mass spectrometric analysis. Peptides and proteins were identified from the resulting data sets of biological and technical replicates using MASCOT. All data were then combined in a new SpectraST database. Exemplary protein extracts from S9 host cell cultures infected with S. aureus HG001 were processed in a sample-specific way. On comparing the identification of peptides and proteins which resulted from a MASCOT search with those retrieved from a search using the newly generated SpectraST database, the new SpectraST database led to higher identification rates than the traditional MASCOT approach using a database of the theoretical proteins. An improvement of protein identification was especially observed when the identification results of both the MASCOT and the SpectraST database search were combined. exp – exponential growth phase; t0 – entry into stationary growth phase; t4 – 4 h after t0; ABC – ammonium bicarbonate; UT – urea/thiourea.
2 Material and methods
2.1 Bacterial cell culture
S. aureus HG001, a derivative of NCTC8325 in which the rsbU allele carrying a 11 bp deletion was replaced by its wild type copy [15] and which has been used in host–pathogen interaction studies before [17, 23-27], was cultivated in tryptic soy broth (TSB; Becton, Dickinson and Company, Franklin Lakes, NJ, USA). Cells were harvested at three different points in time during growth (Supporting Information Fig. S1): exponential growth phase (exp, optical density at 600 nm/OD600nm = 0.5), entry into stationary growth phase (t0, OD600nm ≈ 6.5), and stationary growth phase (t4, OD600nm ≈ 10). Each harvested sample comprised 40 OD 600nm-units. Three biological replicates (BR) were included. Subsequently, the three different samples (exp, t0, t4) were combined to a single sample for thorough analysis in order to cover a high fraction of the theoretical proteome. After washing with 50 mM ammonium bicarbonate (ABC), the cell mixture was divided into equal aliquots for use in the different analytical approaches (Fig. 1).
2.2 Cell disruption, protein extraction, and determination of protein concentration
For processing of samples intended to be analyzed with the standard method, pellets were washed once with TE buffer (10 mM Tris/HCl pH 8, 1 mM EDTA pH 8) and afterwards resuspended in 0.5 ml 1x UT buffer (8 M urea, 2 M thiourea). Subsequently, cells were disrupted in three cycles of 30 s ultrasonication and 30 s cooling on ice. After centrifugation (10 min, 4°C, 8600 × g) supernatants containing crude protein extract were used for MS analysis.
For samples, which were collected for IEF fractionation and two 2D-SCX, cell pellets were resuspended in lysis buffer (50 mM ABC, 0.1% RapiGest (Waters Corp., Milford, MA, USA)) and mechanically disintegrated in the presence of acid- washed glass beads (diameter 0.4–0.6 μm; Sigma-Aldrich, St. Louis, MO, USA) by vigorous shaking using a TurboMix Adapter and VortexGenie2 (Scientific Industries, Inc., Bohemia, NY, USA) for 10 min at 4°C. After centrifugation (10 min, 4°C, 15 000 × g), supernatants were collected and the extraction procedure was repeated three times.
Protein concentrations of all protein extracts were determined photometrically using a Bradford Assay (Bio-Rad Laboratories, Hercules, CA, USA).
2.3 Reduction, alkylation, and tryptic digestion of protein samples with subsequent C18-purification of the resulting peptides
Protein samples of 4 μg (standard method),1 mg (off-gel IEF fractionation), and 10 μg (2D-SCX) were reduced in 5 mM tris-2-carboxyethyl-phosphine (TCEP) [30 min, 37°C, shaking at 500 rpm, in the dark] and afterwards alkylated in 10 mM iodoacetamide (IAA) (1 h, 25°C, shaking at 500 rpm; protected from light). The excess of TCEP and IAA was removed by incubating the samples with 12.5 mM N-acetyl-cysteine (NAC) (10 min; 25°C, shaking at 500 rpm). Subsequently, proteins were digested with trypsin (Promega, Madison, WI, USA) at 37°C overnight using an enzyme:protein mass ratio of 1:25 (standard method) or 1:100 (off-gel IEF/2D-SCX). Digestion was stopped with a final concentration of 1% acetic acid and 1% TFA in the standard method and the off-gel IEF/2D-SCX method, respectively. All samples were centrifuged (10 min, 4°C, 8656 × g). The peptide-containing supernatants of 4 μg samples were subjected to C18 peptide purification using 2 μg-ZipTip μ-C18 pipette tips (Merck Millipore, Billerica, MA, USA). The pH of 1 mg samples was adjusted to about 3 with 0.5 M ABC, and afterwards peptides were purified using SepPak Vac tC18 1cc Cartridges (Waters Corp., Milford, MA, USA). The 10 μg samples were temporarily divided into two equal fractions and purified using 5 μg-ZipTipC18 pipette tips (Merck Millipore).
2.4 Off-gel IEF fractionation
C18-purified peptides resulting from a 1 mg protein sample were fractionated according to their pI using the Agilent OFFGEL 3100 Fractionator system (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s protocol. The rehydration was carried out for 1 h at a voltage of 500 V (current limit: 50 μA, power limit: 200 mW). Afterwards, the peptides were focused using IPG-strips (24 cm, pI range: 3–10; GE Healthcare Europe, Glattbrugg, Switzerland) at a voltage of 8000 V (current limit: 100 μA, power limit: 300 mW) until 50 kVh were reached (after about 16 h). Finally, the pH of each of the 24 collected fractions was adjusted to about 2 to 3 with 5% acetic acid, and peptides were purified using MicroSpin columns C18 Silica (The Nest Group, Southborough, MA, USA). In order to standardize the peptide concentration of samples prior to injecting a constant volume into the LC-MS instrument, 10% of the volume of each purified fraction was again subjected to a further purification using 2 μg-ZipTip μ-C18 pipette tips (Merck Millipore, Billerica, MA, USA). Since this ZipTip type has the capacity to bind 2 μg of peptides, this second purification step resulted in a comparable eluate amount and concentration for all samples after off-gel IEF fractionation.
2.5 2D-SCX
Peptides were separated by online SCX fractionation using 2D salt steps on a nano column (Poros 10S, 300 μm × 10 cm, nanoViper, SCX, 10 μm) via an Ultimate 3000 RSLC- nano (Thermo Scientific, former Dionex, Idstein, Germany). Five μg of trypsin-digested peptides were loaded and subsequently eluted in seven fractions by injection of 12 μl salt buffer using salt plug concentrations ranging from 2 to 500 mM NaCl. Further details are listed in Supporting Information S1.
2.6 LC-MS/MS mass spectrometric analysis
Reversed phase nano LC was done with an Ultimate 3000 RSLCnano (Dionex/Thermo Fisher Scientific, Idstein, Germany). Peptides were first loaded on a trap column (Acclaim PepMap 100, 100 µm × 2 cm nano Viper, C18, 5 μm, 100 A°; Thermo Fisher Scientific, Waltham, MA, USA) and then analyzed using an analytical column (Acclaim PepMap RSLC, 75 µm × 15 cm nano Viper, C18, 2 μm, 100 A°; Thermo Fisher Scientific, Waltham, MA, USA). Reversed phase chromatography was performed with a binary buffer system consisting of 0.1% acetic acid, 2% ACN (buffer A), and 0.1% acetic acid in 100% ACN (buffer B). The peptides were separated by a linear gradient of buffer B from 2% up to 25% for 30, 100, and 120 min for SCX or off-gel IEF pre-fractionated samples, for non-fractionated samples (standard method), and for samples from the cell culture infection model, respectively, with a flow rate of 300 nL/min. The columns were operated at a constant temperature of 40°C. The LC was coupled to a Q Exactive mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) via a TriVersa NanoMate (Advion Biosciences, Norwich, UK). The MS-instrument was operated in data-dependent acquisition mode. MS settings were as follow: survey full-scan spectra were acquired with a resolution R = 70 000, automated gain control (AGC) target was set to 3e6 ions, the maximum injection time was set to 250 ms. MS/MS scan events were repeated for top ten peaks using the higher energy collisional dissociation mode at normalized collision induced energy of 27.5%, underfill ratio (5%) with an intensity threshold of 8.3e4 ions was selected. Already targeted ions for MS/MS were dynamically excluded for 40 s with monoisotopic precursor selection enabled. Further details are listed in Supporting Information S1.
2.7 Data analysis
MS raw files were converted into mzXML and mgf formats using the Trans Proteomics Pipeline (TPP) version v4.6 OCCUPY rev 2, Build 201308090511. Each mgf file was searched against an S. aureus-specific database on a MASCOT Server by MASCOT Daemon version 2.3.2. The four most frequent modifications – deamidation, carbamidomethylation (C), oxidation (M), and conversion of Gln to pyroGlu (N-term Q), which were determined prior to analysis by an error-tolerant search were included as variable modifications. Further chemical processing modifications or biological PTMs were not considered or analyzed, respectively. Identification quality was further controlled via the Percolator score false discovery rate (FDR) [28]. Further details are listed in Supporting Information S1. All data are available at the PRIDE Archive [29] (http://www.ebi.ac.uk/pride/archive/) of the ProteomeXchange Consortium [30] through accession number PXD000702.
Data filtered for ion scores of at least 20 were exported to Microsoft Office Excel. For each file, an ion score >20 is higher than the threshold score which indicates identity or extensive homology with p < 0.05 according to the MASCOT algorithm [31]. The average ion score at the threshold of p < 0.05 was 16.5, and the corresponding FDR averaged out at 0.19. Individual ion scores at the threshold of p < 0.05 for the individual files are listed in Supporting Information S1. Thus, limiting data to results with an ion score >20 included only the most reliable data into the analysis.
Peptide and protein lists were compared and Venn diagrams were generated with the VENNY tool (Juan Carlos Oliveros; http://bioinfogp.cnb.csic.es/tools/venny/index.html; 2007). Area-proportional Venn diagrams displaying the area in proportion to the numbers of items were generated based on the eulerAPE v3 (Luana Micallef and Peter Rodgers, http://www.eulerdiagrams.org/eulerAPE/; 2013). ProteinCenter (Version 3.10.10016; Thermo Fisher Scientific Inc., Waltham, MA, USA) was utilized to calculate pI values and sequence coverage. Peptide data are available as Supporting Information Table S2. Density functions as well as histogram data were calculated and depicted using R scripts. Voronoi treemaps [32] were created using the Paver software (DECODON GmbH, Greifswald, Germany), which employed the assignment of proteins to functions from the TIGRFAMs protein family classification scheme [33] by using HMMER/HMMScan [34], an all protein sequences containing FASTA-file of S. aureus HG001, and the TIGRFAMs Hidden Markov models (HMMs; http://www.jcvi.org/cgibin/tigrfams/index.cgi; Current Release: 13.0, 4284 families, August 15 2012). Only the best sequence to HMM hit was considered as a “protein sequence to TIGRFAMs assignment”. Proteins without a TIGRFAMs assignment are not displayed. By definition, TIGRFAMs are quasi-hierarchically structured meaning that TIGRFAMs-distinguished proteins may be allocated to multiple functional groups, so-called sub-roles, which are classified in 16 main roles. For a clearer representation, we divided these 16 main roles into six meta-roles whose definition was inspired by KEGG Brite [35]. The six meta-roles are termed “metabolism”, “cell structure”, “cellular processes”, “signal transduction”, and “genetic information processing” as well as a class of non-allocated TIGRFAMs (“unmapped”). Newly TIGRFAMs-distinguished proteins were, wherever possible, allocated manually to a meta-role other than the “unmapped” one (data not shown). To avoid the occurrence of proteins in more than one meta-role, only primary functions were considered with multiple or secondary assignments being manually removed (data not shown). Finally, 1941 proteins were assigned to a functional group (including the “unmapped” group).
The areas of the Voronoi cells representing the proteins were used to encode the proteins’ sequence lengths. Subdivision of the protein Voronoi cells was performed in such a way that the length of a peptide resulting from a zero missed cleavage theoretical tryptic digestion determined the size of the peptide cell.
2.8 SpectraST database
Using TPP, a SpectraST database [36] was generated from 144 LC-MS/MS runs which resulted from the analysis of the three different methods described above: (i) off-gel IEF fractionation (72 runs of three BR with 24 fractions each); (ii) SCX fractionation (21 runs of three BR with 7 fractions each); and (iii) the standard method (three BR without fractionation). Furthermore, 48 files resulting from a technical replication of the IEF off-gel fractionation of two BR were integrated into the database, which extended the peptide data complexity. Briefly, the raw data were translated into mzXML and later to the MASCOT generic file format (mgf). These mgf-files were searched using MASCOT and the resulting dat-files were restricted to those spectra/peptide identifications with p < 0.05 according to the MASCOT algorithm (indicating identity or extensive homology) [31] as described above for data analysis.
In order to use only peptide matches of highest confidence, only data with ion scores > 20 were further converted to pep.XML-files, which were subsequently compiled into a SpectraST specific library (spec.lib library). This SpectraST library for the comprehensive S. aureus proteome is available online at the PRIDE Archive [29] (http://www.ebi.ac.uk/pride/archive/) of the ProteomeXchange Consortium [30] through accession number PXD000702. After the spec.lib library had been used for identification searches only results with an absolute precursor tolerance of less than 0.01 and a DOT score of at least 0.5 were considered as reliably identified. The dot score cutoff of 0.5 was determined after plotting the number of identified proteins depending on the dot score values (Supporting Information Fig. S2) where the total number of identified proteins was inversely correlated with the dot score only at dot score values of 0.5 or greater. Since dot scores between 0 and 0.5 always resulted in constant total numbers of identified proteins we concluded that dot scores in this range did not discriminate true identifications from unspecific false identifications (Supporting Information Fig. S2).
2.9 Cell culture infection experiments
Cell culture infection experiments were performed as described [37, 38]. Briefly, confluent cell culture dishes of S9 cells were infected with S. aureus HG001 pJL74, a green fluorescent protein (GFP)-expressing strain, which had been incubated in the presence of ferric-oxide core nanoparticles (FeOx-NP) for 24 h prior to infection. The GFP-expressing strain was only chosen for control purposes because it allowed determination of bacterial cell counts using a Guava EasyCyte flow cytometer (Millipore, Billerica, MA, USA). After an internalization phase of 1 h, non-internalized bacteria were lysed using lysostaphin. After washing with PBS, the infected host cells were lysed in 0.3% Triton X-100 in A. dest. 2.5 h and 4.5 h after infection, and intact, FeOx-NP-labeled bacteria were captured by a 2 Tesla magnet (HOKIMag, Hoock GmbH, Kiel, Germany). Captured bacteria were washed with PBS when still remaining in the magnetic field in order to remove contaminants of host cell debris. Bacteria were collected on a filter membrane, washed again, and digested with lysostaphin and trypsin without reduction and alkylation directly on the membrane. The resulting peptides were subjected to MS analysis with the standard method, except that the 15 cm analytical column was replaced with a 25 cm column and that the gradient time was increased to 120 min. MS raw data were transformed to mzXML and mgf formats and subjected to a MASCOT database search as well as a search based on the newly generated SpectraST database. MASCOT identifications were accepted only with ion scores of at least 20, thus, at the same cutoff as for the pure TSB-culture samples, while identifications from the SpectraST database search approach had to pass the criteria of a minimal DOT score 0.5 and an absolute precursor tolerance of less than 0.01 as described above.
2.10 In silico tryptic digestion and calculation of score values for the prediction of peptide detectability
The FASTA-database including the complete predicted proteome of S. aureus was subjected to an in silico tryptic digestion using R scripts with the following parameter settings: peptide mass 550–5500; minimal peptide length 5; maximal missed cleavages 2; without static or variable modifications. This in silico digestion resulted in a set of about 220 245 theoretical peptides. Approximately 99% of these peptides were unique for distinct proteins. For the calculation of score values, different data (sub) sets were employed: (i) all detected peptide sequences from the 144 LC-MS runs; peptides with missed cleavage(s) counted as individual peptides while modified peptides were not counted as separate peptides; (ii) all theoretical peptides from the in silico digestion excluding all peptides of proteins for which no peptide was detected; (iii) all theoretical peptides from the in silico digestion. CHEMscore values for peptides [39] were calculated using the ProteinProspector/MS-Digest tool (v 5.10.12; UCSF – University of California, San Francisco, CA, USA). The algorithm was modified for equal weighting of Arg- and Lys-tryptic peptides since the preference of Arg-peptides over Lys-peptides which occurs during MALDI measurements [40, 41] was not observed for our ESI data set. CONSeQuence score values [42] were calculated using the provided online-tool without modification of the algorithm. The DetectabilityPredictor score was calculated as indicated [43].
3 Results
3.1 Identification of peptides and proteins by three different methods
Three different methods for sample analysis were conducted in our study: (i) the standard method of reversed phase LC- MS/MS without pre-fractionation; (ii) off-gel IEF fractionation; (iii) 2D-SCX (Fig. 1). We aimed to identify as many different peptides as possible and to build a comprehensive S. aureus proteome catalogue in order to provide a data repository for further host–pathogen experiments.
The analysis of three independent BR by the three methods resulted in a set of 16 681 detected peptides and 1936 identified proteins. For each method, we observed a specific set of detected peptides and identified proteins (Fig. 2). The number of method-specific peptides totaled to 1585 (off-gel IEF), 1462 (2D-SCX), and 6575 (standard method) (Fig. 2A), while the number of method-specific proteins totaled to about 133 (off-gel IEF), 85 (2D-SCX), and 125 (standard method) (Fig. 2B). The summed results of all three methods covered about 67% of the theoretically predicted proteome of S. aureus HG001 (2891 predicted proteins). Including all technical LC-MS replicate measurements, the number of identified proteins was increased to 2088, which corresponded to >72% of the predicted S. aureus HG001 proteome. We further observed an overrepresentation of cytoplasmic membrane and extracellular proteins (PSort annotation) in the set of non-detected proteins (Supporting Information Fig. S3).
Figure 2.
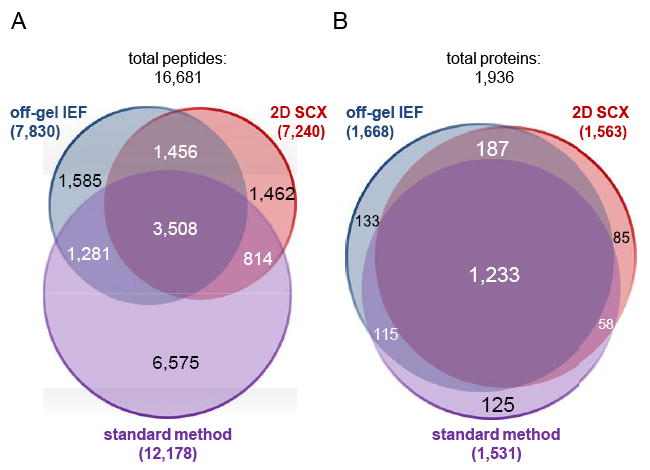
Detection of peptides and protein identification using three different approaches of proteome analysis. Comparison of detected peptides including peptides with missed cleavages (A) and identified proteins (B) for the three methods of proteome analysis under investigation (three biological replicates per method, without technical replicates). Some peptides were additionally detected in different charge states or in a modified form (e.g. oxidized), but the Venn diagram only shows unique peptide sequences independent of further parameters.
Assuming that based on transcriptional profiling data [7] only 80% of the predicted S. aureus HG001 proteome is in fact expressed in the selected sample set (i.e. that the expected number of proteins amounts to around 2300), our identified 2088 proteins cover 90% of the expected, sample-specific proteome.
In order to characterize the identified peptides and proteins, we displayed each peptide identified in individual proteins in a Voronoi treemap [32] created on the basis of the TIGRFAMs protein family classification scheme [33] by using HMMER/HMMScan [34] (Fig. 3). The identified proteins covered all displayed functional groups to a similar extent except for under-represented groups of transport and binding proteins, cellular processes, parts of the protein fate or amino acid biosynthesis (Fig. 3A). Abundant proteins belonging the TCA cycle or the tRNA aminoacylation were well-covered and showed high peptide coverage, and vice versa proteins involved in transport of cations and iron carrying compounds, prophage functions or toxin production and resistance were under-represented.
Figure 3.

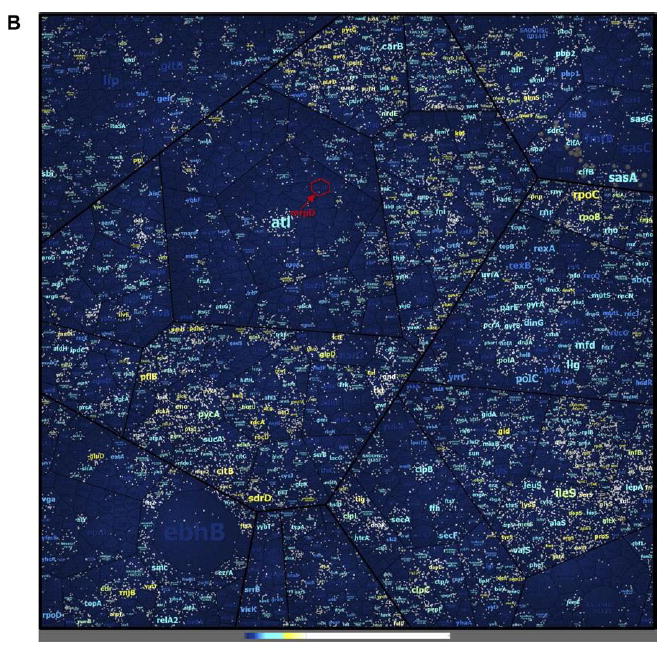
Peptide and protein coverage. The coverage of the annotated proteome of S. aureus HG001 using the three methods of proteome analysis under investigation (A).The Voronoi treemap was created on the basis the TIGRFAMs protein family classification scheme [33] by using HMMER/HMMScan [34]. The small graphs in the upper part display the included functional annotations. Peptides form the lowest level of area subdivision. The area per peptide represents the peptide length (number of amino acids). Therefore, the area per protein correlates with the protein size. Detected peptides of proteins identified by at least one of the three methods applied in this study (three biological replicates, without technical replicates) are colored in shades of orange. The color represents the sequence coverage of the proteins by the detected peptides. Peptides not detected in any set of three biological replicates per method are colored gray. Nevertheless, some of these proteins were identified when technical replicates were included to generate an even more comprehensive new database (SpectraST). Coverage of peptides from an in silico digestion in the real MS data sets (B). Included functional annotations are the same as depicted in Fig. 3A. Additionally, the protein label size correlates with the protein size. White dots indicate the detected peptides from the MS data sets. Coloring was applied to the protein labels: Dark blue labels indicate proteins not identified. Light blue, yellow, and white coloring indicates in this order increasing coverage of identified proteins. The Voronoi treemap contains about 220245 theoretically expected peptides from an in silico digestion. Of these, about 19109 peptides were detected in the MS data sets.
Interestingly, the high coverage of the proteome was possible albeit a much lower coverage of the theoretical peptides expected from an in silico digestion, where only 8.7% (19 109 of 220 245) of the expected peptides were detected (Fig. 3B). The identification of peptides from membrane- spanning proteins belonging to transport/binding protein groups was particularly scarce because these proteins were not well accessible with the methods applied here. For example, the 54 kDa protein monovalent cation/H+ antiporter subunit D (MrpD) contains 14 transmembrane regions and only a small proportion of the protein is solvent accessible. From about 20 theoretical tryptic peptides in MrpD, only one could be finally detected with the three proteome analysis methods applied here.
3.2 Distribution of peptide pI values, GRAVY scores, and monoisotopic mass for the different methods
As part of this study, we wanted to determine which parameters provided comprehensive sets of accessible peptides from the three different proteome analysis methods under investigation. Therefore, we analyzed the frequency of peptides along the observed pI range, especially since in one of the methods this physicochemical property is used to fractionate the peptides. First, we observed that the density of the pI was not evenly distributed along the accessed range. Three density maxima were observed at pI values of about 4, 7, and 10 (Supporting Information Fig. S4). Furthermore, peptides of acidic pI were observed most often, those with alkaline pI values least often, and those with neutral pI values were ranked in between (Supporting Information Fig. S4).
Since the distribution was highly similar for the detected peptides for each of the three methods (Supporting Information Fig. S4), we decided to employ only the combined data set for further comparisons.
We then extended the data set to all theoretical peptides retrieved from an in silico tryptic digestion of the complete S. aureus proteome but only for proteins identified in at least one of our 144 data sets. Interestingly, the three maxima were also observed when the pI values of this theoretical peptide set were plotted in histograms (Supporting Information Fig. S5A). But the frequency of acidic pI and alkaline pI peptides was clearly different in the theoretical dataset compared to the mass spectrometrically detected peptides, with an over-representation of acidic and neutral range peptides and an under-representation of alkaline peptides in the MS data set (Supporting Information Fig. S5A). In addition to pI values, we compared GRAVY score values – which indicate the hydropathy of peptides – between the detected and the theoretical set of peptides (Supporting Information Fig. S5B). Both sets possessed a highly similar distribution with slightly fewer peptides with higher GRAVY scores than the median in the set of theoretical peptides.
Further, the monoisotopic mass of the peptides was plotted as a control measure. The distributions of these values were similar for the detected and the theoretical set of peptides (Supporting Information Fig. S5C). However, there was a clear over-representation of peptides between the MH+ mass range 750–1500 Da. As expected, the lowest and highest values were not included in the set of detected peptides, which is due to the restricted MS detection window used for mgf file conversion (400 to 6000 Da).
3.3 Calculation of score values in order to predict the MS-detectability of peptides in a selected analytical setting
Having observed different densities in the pI values of MS- detected and theoretically computed peptides, but only slight differences in GRAVY score and monoisotopic mass, we investigated the question of whether we could use the already recorded data sets to predict in advance whether a specific peptide might be detectable in our selected analytical MS settings.
A score focusing on the rating of peptides before using them as QConCat peptides has been published recently [42]. When these so-called CONSeQuence score values were calculated and plotted for our detected and theoretical data sets, we observed a maximal frequency of score values around 0.21 to 0.22 for the group of theoretical peptides (Fig. 4A). The distribution of score values for the set of detected peptides was split into two subgroups, with a maximum of frequency around 0.21 to 0.22, as seen for the theoretical peptides, and another maximum around 0.59 to 0.64 (Fig. 4A).
Figure 4.
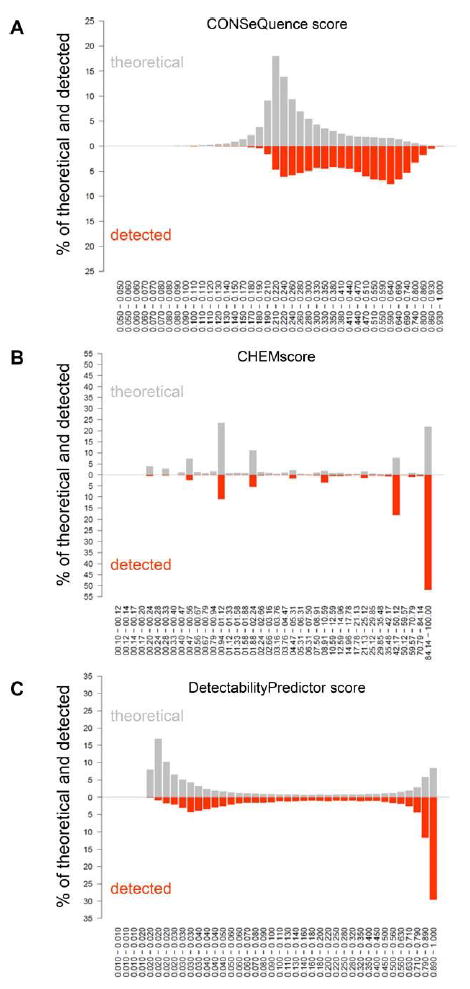
Characterization of the detected peptides and peptides retrieved from an in silico digestion by different scoring methods. The frequency of peptides is depicted in classes of CONSeQuence score [42] values (A), CHEMscore [39] values (B), and Detectability Predictor score [43] values (C). The upper part of the histograms refers to the set of theoretical peptides from the in silico digestion (gray), and the lower part of the histograms displays the data set of detected peptides (orange).
A second score, the CHEMscore [39], possessed a distribution which more clearly distinguished detected peptides from the theoretically possible peptides. Higher score values were clearly over-represented while lower score values were clearly under-represented in the detected set of peptides (Fig. 4B).
Another smooth distribution of score values was observed for a third score, called DetectabilityPredictor score, which ranks peptides provided in the PeptideAtlas repositories [43]. Similar to the results for the CHEMscore, higher score values were overrepresented while lower score values were under-represented in the detected set of peptides also for the DetectabilityPredictor score (Fig. 4C). Although a high fraction of detected peptides possessed a high DetectabilityPredictor score, we again observed a second maximum of frequency in the set of detected peptides with score values around 0.028 to 0.032, which was, however, not visible in the set of theoretically expected peptides. Peptides with lowest score values (0.016 to 0.02) were rarely detected, although they represented an important fraction in the set of peptides from the in silico digestion.
Analyzing the distribution of the three score values in the set of detected peptides two distinct peptide populations could be distinguished for all three scores: (i) peptides with higher score values which were detected in our combined data set; and (ii) peptides with lower score values, i.e. peptides not predicted to be present in the data set, which were nonetheless detected in our study.
We now investigated the question of whether we could identify a parameter whose value determined whether a peptide with a lower score value would be detectable or not. We hypothesized that the sets of detected peptides with the highest and the lowest possible score values might help to identify the dominant influence which determined detectability of peptides featuring low score values (Supporting Information Fig. S6).
We therefore compared the distribution of exponentially modified protein abundance index (emPAI) [44] of the proteins from which peptides originated with the score distribution but could not find any correlation between them (Supporting Information Fig. S6A). The comparison of pI or GRAVY score distribution between the set of peptides with highest and lowest score values indicated that both distributions differed between the low and the high score sets. The difference was especially visible in the subsets of peptides which possessed low or high values for all three scoring algorithms (Supporting Information Fig. S6B and C, each intersection abc). Peptides with lower score values tended to possess higher pI values (Supporting Information Fig. S6B) and smaller GRAVY scores values (Supporting Information Fig. S6C).
Using Voronoi treemaps with the full set of about 220 245 theoretical peptides (Supporting Information Fig. S7A), we noticed an influence of the GRAVY score values (Supporting Information Fig. S7B) on the CONSeQuence score and the DetectabilityPredictor score (Supporting Information Fig. S7C–E), while the CHEMscore did not consider the GRAVY score.
3.4 Spectral library enabling fast and sensitive identification of S. aureus proteins as shown in a cell culture infection model
Data sets collected in selected experimental and analytical settings can be transformed into usable spectral libraries. Such a database library can be used to improve protein identification in proteome analyses by matching true fragmentation abundances of peptides. Our goal was to take advantage of our extensive data set and to employ a spectral library to improve the proteome analysis in host-pathogen interaction studies. An exemplary data set of four samples from S. aureus HG001 internalized into human bronchial epithelial S9 cells was subjected to a MASCOT search, as well as to an identification search using a SpectraST database generated from the 144 data files of S. aureus HG001 obtained in the present study. These samples from the cell culture infection assay contained only a limited number of bacterial cells, and, therefore, protein identification rates lower than for the non-limited samples used before were expected. While MASCOT allowed between 335 and 625 protein identifications, the identifications obtained by SpectraST database searching were always significantly higher ranging from 542 to 714 (Fig. 5). The intersection of MASCOT and SpectraST results accounted for 60% to 82% of the single result sets from the MASCOT search. Thus, protein identifications specific for the MASCOT search (MASCOT score ≥ 20) contributed only to a small proportion to the total sum of identifications (from 102 to 135). Gain of protein information was strongly pronounced when the SpectraST database search was applied (DOT score ≥ 0.5 and absolute precursor tolerance ≤ 0.01), which yielded increases in protein identification of 194 to 342 proteins corresponding to 32% to 102% increase in relation to the results from the MASCOT search (Fig. 5). Thus, including SpectraST results improved protein identification significantly. The group of proteins which were specifically identified by SpectraST searches in all four samples and, therefore, not identified in any MASCOT search included for example regulators such as SarS [staphylococcal accessory regulator-like protein], GlpP [glycerol uptake operon antiterminator regulatory protein, putative], and CodY [transcriptional repressor CodY] (data not shown). We provide the SpectraST database generated in this study at the PRIDE Archive [29] (http://www.ebi.ac.uk/pride/archive/) of the ProteomeXchange Consortium [30] through accession number PXD000702 for use in further analyses.
Figure 5.
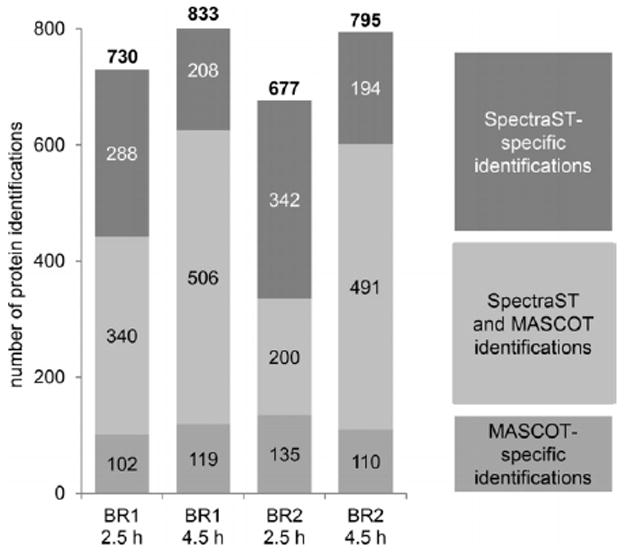
Protein identifications from four data sets of S. aureus HG001 cells after internalization into human bronchial epithelial S9 cells. Two biological replicates and two points in time after infection were analyzed: biological replicate 1 (BR1); biological replicate 2 (BR2); 2.5 h after infection; 4.5 h after infection, respectively. The numbers indicate identified proteins, including proteins with only one detected peptide.
4 Discussion
We compared three different approaches for proteome analysis, and our results clearly indicated that a majority of proteins could be identified with any of the three methods. We interpret this as a result of the improvements in mass spectrometers and search algorithms. Furthermore, for each method we observed a set of identified proteins specific for this method. When comparing, for example, off-gel IEF fractionation and the standard method, we found that the intensive fractionation approach led to the identification of 320 proteins not identified with the standard method. Overall, the power of the analytic strategies lay in their combination, which yielded a total of 1936 proteins identified from samples of the exponential and stationary phases during growth in a complex medium such as TSB. It was not surprising that the total number of identified proteins increased to 2088 proteins when technical replicates were included.
Our set of identified proteins was shown to cover all categories of functional annotations of the S. aureus proteome. A reduced coverage was only observed in the categories “amino acid biosynthesis” and “transport and binding proteins”. Coverage of these protein classes was expected to be low since S. aureus was grown in TSB, a rich medium providing amino acids for bacterial growth, and thus, amino acid synthesis genes were probably not expressed. The observed under- representation of cytoplasmic membrane proteins in the set of detected proteins may be explained by the fact that membrane proteins were less accessible to our analytical strategy. Membrane transporter and binding proteins contain large regions with mainly hydrophobic amino acids that are inserted into the membrane. Being less soluble in water-based buffer systems, many of them were probably retained in the fraction of cell membrane debris after cell disruption and, therefore, not included in the sample. Furthermore, these proteins are known to possess only few proteolytic sites accessible for tryptic cleavage as exemplified by MrpD. Therefore, several membrane transport proteins, although included in the sample, were probably not eligible for analysis with our approaches. Extracellular/secreted proteins (PSort annotation) were under-represented because they were mostly lost during sampling because the analysis was confined to pelleted bacteria and not extended to the culture supernatant.
In total, we identified 2088 staphylococcal proteins from samples of the exponential and stationary phases during growth in complex medium TSB. This number of identified proteins represents a further increase in the coverage of the bacterial proteome using MS analysis. A previous study reported the identification of about 1700 proteins in total from MS analyses of S. aureus COL [7]. That study included more than 230 MS runs when counting only tryptically digested samples, while we only performed 144 MS runs which furthermore took less than about one fourth of the MS measurement time compared to the S. aureus COL study. The increased number of protein identifications with a significant reduction in MS measurement time is a consequence of the tremendous advance in MS instruments. It has to be mentioned that we have not included bacterial culture supernatants in our analysis yet. Only 56 of 109 proteins annotated extracellularly (PSort) were identified in our investigation. The remaining 53 proteins might be identified on analyzing an extracellular protein extract enriched with those proteins. However, while these proteins are certainly important in infection, they will not be accessible by many in vivo proteomics approaches employing cell sorting or magnetic enrichment of bacteria, because proteins not adhering to the bacterial surface will be lost by these enrichment techniques. The data set presented here and the newly generated spectral library is complementary to targeted proteomics approaches. Our library is a helpful resource for defining SRM assays, which will allow addressing more specific pathophysiological questions in host-S. aureus interaction experiments. Our database will provide the discovery-driven information which is needed prior to setting up targeted proteomics experiments. Although we provide the so far most complete S. aureus protein map there is still a fraction of proteins not detected yet. Different options might be used to close this gap: (i) inclusion of different protein fractions with prior prefractionation, such as the secreted proteins or specifically enriched membrane or cell wall bound proteins. However, one has to consider the effort-gain-balance. (ii) Another option is the setup of specific targeted assays that would then address proteins of specific importance.
Our data set was characterized by a distribution of peptide pI values whose density was not congruent with that of a theoretical peptide data set generated by an in silico digestion. We preferably detected peptides with acidic and neutral pI values with our mass spectrometric setting. Given the differences between the MS-detected and the theoretically expected data sets, it was apparent that peptide properties determine the detectability of peptides for defined experimental and analytical settings in addition to biological effects which regulate the protein abundance and, in consequence, the detectability of proteins/peptides. We use the term “detectability” to describe a combination of the influence of sample, sample preparation, sample separation/elution by chromatography, peptide ionization, peptide MS detection, and computational peptide identification [42, 43]. Since other peptide properties such as the GRAVY score did not reveal similar differences between detected and theoretical data sets like the pI value distributions did, we applied different published score values [39, 42, 43] which combine several peptide properties and which are intended to allow prediction of the detectability of a selected peptide prior to MS measurement. For all three scores, we observed an over-representation of higher score values in our detected peptide set in comparison to the score values in the data set of peptides from the in silico digestion. But all three scores allowed only partial prediction of the detectability because a relevant set of detected peptides possessed low score values and, thus, was unlikely to be detectable. The emPAI score values had only a minor impact, and the distributions were comparable between the sets. The reason is probably related to the calculation of emPAI score values from the MS-detected data sets [44], since emPAI score values result from the amount of the specific protein in the analyzed sample. They are calculated based on the ratio of observed peptides to theoretically observable peptides in each protein. Thus, it is not possible to judge on the influence of protein abundance of the non-detected proteins because abundance data are obviously unavailable. We were able to identify an influence of the peptide properties characterized by pI and GRAVY score values on the mass spectrometric detection of a peptide in our analytical settings. This influence is probably not adequately represented in the scoring algorithms, at least for our analytical settings. A low GRAVY score, indicating a hydrophilic peptide, might lead to a reduced binding on a C18-column during reversed- phase liquid chromatography and consequently impede the detection in the MS. Contrarily, a higher GRAVY score, i.e. higher hydropathy, is not sufficient for detection. Since the GRAVY score value represents the averaged hydropathy of all amino acids of the corresponding sequence but does not take into account the three-dimensional and transmembrane domain structure, it characterizes only restricted properties of the peptide. Further characteristics like the transmembrane structure, e.g. of membrane proteins with inaccessible tryptic cleavage sites, influence the detectability.
In the future, it might be an option to train facility-specific score values on already existing data sets in order to further optimize and sharpen the predictive separation of peptides that are very likely from those that are highly unlikely to be detected. Such score was presented most recently by Qeli and colleagues [45]. Their score, called PeptideRank, uses a rank- based algorithm for the in silico prediction of the detectability of peptides, starting best with species-specific training and testing sets [45]. We can now provide a comprehensive peptide data set for S. aureus HG001 which is available at the PRIDE Archive [29] (http://www.ebi.ac.uk/pride/archive/) of the ProteomeXchange Consortium [30] through accession number PXD000702. Technically, our data indicate that methodological modifications to access hydrophobic proteins/peptides could have significant impact and increase the number of detected peptides further.
Today, such an MS data collection for a specific experimental and analytical setting provides a unique opportunity to further increase the options for protein identification in proteome studies performed with the same settings. When our data collection was combined into a spectral database and used for the identification search of four sample data sets from a cell culture infection assay, we were able to strongly increase the number of identified proteins as compared to the MASCOT search results. For example, SpectraST searches always identified proteins SarS, GlpP, and CodY, which fulfill regulatory functions at central nodes of virulence and metabolism. Their identification in all SpectraST searches but in none of the MASCOT searches illustrates the advantage of applying the SpectraST database from this study to proteomic data from host–pathogen interaction studies using S. aureus. Increased improvement of identification can be expected when further search tools are integrated into the analysis, as has been shown in a prior publication [46]. This is especially important for data sets of samples from host– pathogen interaction studies, which often contain only low cell numbers and which most often consist of a mixture of (enriched) pathogen material and host material contamination in significant amounts.
Our data will enable us and others to analyze future experiments with better coverage of the proteome. Additionally, existing data sets could be re-analyzed in order to capture cellular reactions in the protein patterns in more depth.
In conclusion, our data indicated the improvement of proteome analysis with respect to protein identification when the different pre-fractionation techniques were applied. It would be advisable to generate a broad data set which can be transformed into a spectral database since such a database, in combination with a classical sequence database search, will help to increase protein identification, even with samples that do not allow the application of pre-fractionation techniques. We now provide a complex and comprehensive protein repository of S. aureus HG001 (ProteomeXchange / PRIDE Archive accession PXD000702; http://www.ebi.ac.uk/pride/archive/) to the scientific community.
Supplementary Material
Significance of the study.
Global, comprehensive proteome recording of pathogens provide insights into important facets of virulence, pathophysiology, adaptation to their hosts, and, furthermore, can generate knowledge helping in recognition of intervention targets. Such proteome recording was applied to Staphylococcus aureus, an opportunistic human pathogen, which can cause life-threatening disease and of which multidrug- resistant strains are arising nowadays. We identified 2088 distinct S. aureus HG001 proteins, characterized the detected peptides of these proteins, and provided a comprehensive peptide tandem-MS library (SpectraST) from our measurements, which was shown to be applicable for identification of proteins from samples of highly complex host–pathogen interaction experiments. With our data set and spectral database we now provide a valuable tool to the scientific community which can facilitate elucidation of crucial pathophysiological questions in S. aureus-specific host pathogen interaction studies through comprehensive proteome analysis, which represents an important spectral repository for SRM or for DIA MS approaches, and whose high quality data can be used for validation of spectral data from subsequent MS analyses.
Acknowledgments
The authors thank Olga Schubert for alternative protein extraction protocols, Imke Meyer and Andrea Jacobs for their introduction to off-gel IEF fractionation protocols, Kirsten Bartels for technical assistance, and Peter Baker and Robert Chalkley for providing the ProteinProspector tool and for their help with CHEM- score calculations. Additionally, the authors are grateful to Anne Giese for her support in allocating functionally unassigned TIGR- FAMs to known functional roles and acknowledge the PRIDE team for data deposition to the ProteomeXchange Consortium. This work has been funded in part with US federal funds from National Science Foundation Major Research Instrumentation Grant 0923536, by funds from the American Recovery and Reinvestment Act through Grant RC2 HG005805 from the National Human Genome Research Institute, and Grant S10 RR027584, R01 GM087221, Center for Systems Biology/2P50 GM076547 from the National Institute of General Medical Sciences, by the BMBF/“Unternehmen Region” as part of the ZIK-FunGene, and by the DFG in the framework of the SFB Transregio 34.
Abbreviations
- 2D-SCX
two-dimensional strong cationic exchange chromatography
- ABC
ammonium bicarbonate
- DIA
data-independent acquisition
- emPAI
exponentially modified protein abundance index
- FeOx-NP
ferric-oxide core nanoparticles
- HPP
Human Proteome Project
- IAA
iodoacetamide
- NAC
N-acetyl-cysteine
- SpectraST
Spectra Search Tool
- TCEP
tris-2- carboxyethyl-phosphine
- TE
Tris/EDTA
- TSB
tryptic soy broth
- UT
urea/thiourea
Footnotes
The authors have declared no conflict of interest.
References
- 1.Kluytmans J, van Belkum A, Verbrugh H. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev. 1997;10:505–520. doi: 10.1128/cmr.10.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wertheim HF, Melles DC, Vos MC, van Leeuwen W, et al. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis. 2005;5:751–762. doi: 10.1016/S1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- 3.Watkins RR, David MZ, Salata RA. Current concepts on the virulence mechanisms of meticillin-resistant Staphylococcus aureus. J Med Microbiol. 2012;61:1179–1193. doi: 10.1099/jmm.0.043513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu X, Hu Y, Pai PJ, Chen D, Lam H. Label-free quantitative proteomics analysis of antibiotic response in Staphylococcus aureus to oxacillin. J Proteome Res. 201413:1223–1233. doi: 10.1021/pr400669d. [DOI] [PubMed] [Google Scholar]
- 5.Michalik S, Bernhardt J, Otto A, Moche M, et al. Life and death of proteins: a case study of glucose-starved Staphylococcus aureus. Mol Cell Proteomics. 2012;11:558–570. doi: 10.1074/mcp.M112.017004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muntel J, Hecker M, Becher D. An exclusion list based label-free proteome quantification approach using an LTQ Orbitrap. Rapid Commun. Mass Spectrom. 2012;26:701–709. doi: 10.1002/rcm.6147. [DOI] [PubMed] [Google Scholar]
- 7.Becher D, Hempel K, Sievers S, Zuhlke D, et al. A proteomic view of an important human pathogen–towards the quantification of the entire Staphylococcus aureus proteome. PloS One. 2009;4:e8176, 1–11. doi: 10.1371/journal.pone.0008176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gill SR, Fouts DE, Archer GL, Mongodin EF, et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol. 2005;187:2426–2438. doi: 10.1128/JB.187.7.2426-2438.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamond AI, Uhlen M, Horning S, Makarov A, et al. Advancing cell biology through proteomics in space and time (PROSPECTS) Mol Cell Proteomics. 2012;11:1–12. doi: 10.1074/mcp.O112.017731. O112 017731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thakur SS, Geiger T, Chatterjee B, Bandilla P, et al. Deep and highly sensitive proteome coverage by LC-MS/MS without prefractionation. Mol Cell Proteomics. 2011;10:1–9. doi: 10.1074/mcp.M110.003699. M110 003699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lane L, Bairoch A, Beavis RC, Deutsch EW, et al. Metrics for the Human Proteome Project 2013–2014 and strategies for finding missing proteins. J Proteome Res. 2014;13:15–20. doi: 10.1021/pr401144x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilhelm M, Schlegl J, Hahne H, Moghaddas Gholami A, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509:582–587. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 13.Farrah T, Deutsch EW, Omenn GS, Sun Z, et al. State of the human proteome in 2013 as viewed through PeptideAtlas: comparing the kidney, urine, and plasma proteomes for the biology- and disease-driven Human Proteome Project. J Proteome Res. 2014;13:60–75. doi: 10.1021/pr4010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim MS, Pinto SM, Getnet D, Nirujogi RS, et al. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herbert S, Ziebandt AK, Ohlsen K, Schafer T, et al. Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect Immun. 2010;78:2877–2889. doi: 10.1128/IAI.00088-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pfortner H, Burian MS, Michalik S, Depke M, et al. Activation of the alternative sigma factor SigB of Staphylococcus aureus following internalization by epithelial cells an in vivo proteomics perspective. Int J Med Microbiol. 2014;304:177–187. doi: 10.1016/j.ijmm.2013.11.014. [DOI] [PubMed] [Google Scholar]
- 17.Surmann K, Michalik S, Hildebrandt P, Gierok P, et al. Comparative proteome analysis reveals conserved and specific adaptation patterns of Staphylococcus aureus after internalization by different types of human non-professional phagocytic host cells. Front Microbiol. 2014;5:392, 1–14. doi: 10.3389/fmicb.2014.00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Michel PE, Reymond F, Arnaud IL, Josserand J, et al. Protein fractionation in a multicompartment device using Off-Gel isoelectric focusing. Electrophoresis. 2003;24:3–11. doi: 10.1002/elps.200390030. [DOI] [PubMed] [Google Scholar]
- 19.Horth P, Miller CA, Preckel T, Wenz C. Efficient fractionation and improved protein identification by peptide OFFGEL electrophoresis. Mol Cell Proteomics. 2006;5:1968–1974. doi: 10.1074/mcp.T600037-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Link AJ, Eng J, Schieltz DM, Carmack E, et al. Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17:676–682. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 21.Gillet LC, Navarro P, Tate S, Rost H, et al. Targeted data extraction of the MS/MS spectra generated by data- independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11:1–17. doi: 10.1074/mcp.O111.016717. O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Egertson JD, Kuehn A, Merrihew GE, Bateman NW, et al. Multiplexed MS/MS for improved data-independent acquisition. Nat Methods. 2013;10:744–746. doi: 10.1038/nmeth.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Depke M, Burian M, Schafer T, Broker BM, et al. The alternative sigma factor B modulates virulence gene expression in a murine Staphylococcus aureus infection model but does not influence kidney gene expression pattern of the host. Int J Med Microbiol. 2012;302:33–39. doi: 10.1016/j.ijmm.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 24.Geiger T, Francois P, Liebeke M, Fraunholz M, et al. The stringent response of Staphylococcus aureus and its impact on survival after phagocytosis through the induction of intracellular PSMs expression. PLoS Pathog. 2012;8:e1003016, 1–15. doi: 10.1371/journal.ppat.1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mauthe M, Yu W, Krut O, Kronke M, et al. WIPI-1 positive autophagosome-like vesicles entrap pathogenic Staphylococcus aureus for lysosomal degradation. Int J Cell Biol. 2012;2012:1–13. doi: 10.1155/2012/179207. 179207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blanchet C, Jouvion G, Fitting C, Cavaillon JM, AdibConquy M. Protective or deleterious role of scavenger receptors SRA and CD36 on host resistance to Staphylococcus aureus depends on the site of infection. PloS one. 2014;9:e87927, 1–9. doi: 10.1371/journal.pone.0087927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valour F, Trouillet-Assant S, Riffard N, Tasse J, et al. Antimicrobial activity against intraosteoblastic Staphylococcus aureus. Antimicrob Agents Chemother. 2015;59:2029–2036. doi: 10.1128/AAC.04359-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods. 2007;4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 29.Vizcaino JA, Cote RG, Csordas A, Dianes JA, et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucl Acids Res. 2013;41:D1063–D1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vizcaino JA, Deutsch EW, Wang R, Csordas A, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 32.Bernhardt J, Funke S, Hecker M, Siebourg J. Visualizing gene expression data via Voronoi treemaps. ISVD. 2009:233–241. [Google Scholar]
- 33.Haft DH, Loftus BJ, Richardson DL, Yang F, et al. TIGRFAMs: a protein family resource for the functional identification of proteins. Nucl Acids Res. 2001;29:41–43. doi: 10.1093/nar/29.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucl Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aoki KF, Kanehisa M. Using the KEGG database resource. Current protocols in bioinformatics. 2005;11:12–54. doi: 10.1002/0471250953.bi0112s11. [DOI] [PubMed] [Google Scholar]
- 36.Lam H, Deutsch EW, Eddes JS, Eng JK, et al. Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics. 2007;7:655–667. doi: 10.1002/pmic.200600625. [DOI] [PubMed] [Google Scholar]
- 37.Pfortner H, Wagner J, Surmann K, Hildebrandt P, et al. A proteomics workflow for quantitative and time-resolved analysis of adaptation reactions of internalized bacteria. Methods. 2013;61:244–250. doi: 10.1016/j.ymeth.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 38.Depke M, Surmann K, Hildebrandt P, Jehmlich N, et al. Labeling of the pathogenic bacterium Staphylococcus aureus with gold or ferric oxide-core nanoparticles highlights new capabilities for investigation of host-pathogen interactions. Cytometry A. 2014;85:140–150. doi: 10.1002/cyto.a.22425. [DOI] [PubMed] [Google Scholar]
- 39.Parker KC. Scoring methods in MALDI peptide mass fingerprinting: ChemScore, and the ChemApplex program. J Am Soc Mass Spectrom. 2002;13:22–39. doi: 10.1016/S1044-0305(01)00320-8. [DOI] [PubMed] [Google Scholar]
- 40.Krause E, Wenschuh H, Jungblut PR. The dominance of arginine-containing peptides in MALDI-derived tryptic mass fingerprints of proteins. Anal Chem. 1999;71:4160–4165. doi: 10.1021/ac990298f. [DOI] [PubMed] [Google Scholar]
- 41.Hessling B, Buttner K, Hecker M, Becher D. Global relative quantification with liquid chromatography-matrix- assisted laser desorption ionization time-of-flight (LC- MALDI-TOF)–cross-validation with LTQ-Orbitrap proves reliability and reveals complementary ionization preferences. Mol Cell Proteomics. 2013;12:2911–2920. doi: 10.1074/mcp.M112.023457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eyers CE, Lawless C, Wedge DC, Lau KW, et al. CONSeQuence: prediction of reference peptides for absolute quantitative proteomics using consensus machine learning approaches. Mol Cell Proteomics. 2011;10:1–12. doi: 10.1074/mcp.M110.003384. M110 003384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang H, Arnold RJ, Alves P, Xun Z, et al. A computational approach toward label-free protein quantification using predicted peptide detectability. Bioinformatics. 2006;22:e481–488. doi: 10.1093/bioinformatics/btl237. [DOI] [PubMed] [Google Scholar]
- 44.Ishihama Y, Oda Y, Tabata T, Sato T, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4:1265–1272. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- 45.Qeli E, Omasits U, Goetze S, Stekhoven DJ, et al. Improved prediction of peptide detectability for targeted proteomics using a rank-based algorithm and organism-specific data. J Proteomics. 2014;108C:269–283. doi: 10.1016/j.jprot.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 46.Shteynberg D, Nesvizhskii AI, Moritz RL, Deutsch EW. Combining results of multiple search engines in proteomics. Mol Cell Proteomics. 2013;12:2383–2393. doi: 10.1074/mcp.R113.027797. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.