

Abstract
Adrenocortical carcinoma (ACC) generally has poor prognosis. Existing treatments provide limited benefit for most patients with locally advanced or metastatic tumors. We investigated the mechanisms for the cytotoxicity, xenograft suppression and adrenalytic activity of ATR-101 (PD132301-02), a prospective agent for ACC treatment. Oral ATR-101 administration inhibited the establishment and impeded the growth of ACC-derived H295R cell xenografts in mice. ATR-101 induced H295R cell apoptosis in culture and in xenografts. ATR-101 caused mitochondrial hyperpolarization, reactive oxygen release and ATP depletion within hours after exposure, followed by cytochrome c release, caspase-3 activation, and membrane permeabilization. The increase in mitochondrial membrane potential occurred concurrently with the decrease in cellular ATP levels. When combined with ATR-101, lipophilic free radical scavengers suppressed the reactive oxygen release, and glycolytic precursors prevented the ATP depletion, abrogating ATR-101 cytotoxicity. ATR-101 directly inhibited F1F0-ATPase activity and suppressed ATP synthesis in mitochondrial fractions. ATR-101 administration to guinea pigs caused oxidized lipofuscin accumulation in the zona fasciculata layer of the adrenal cortex, implicating reactive oxygen release in the adrenalytic effect of ATR-101. These results support the development of ATR-101 and other adrenalytic compounds for the treatment of ACC.
Keywords: Apoptosis of adrenocortical carcinoma cells in culture and in xenografts, Hyperpolarization of mitochondrial membranes and reactive oxygen production, Suppression of cytotoxicity by lipophilic antioxidants, Inhibition of F1F0-ATPase activity and oxidative phosphorylation, Roles of reactive oxygen in adrenalytic activity
Introduction
A classical approach for the development of anti-cancer drugs is based on the identification of compounds that have selective toxicity in specific normal cells or tissues (Bergenstal DH 1959; Goodman, et al. 1946). In order to develop such compounds for therapeutic use, it is essential to determine their efficacy in cell and animal models of cancer and to characterize their molecular mechanisms of action.
Adrenocortical carcinoma (ACC) is an aggressive disease with poor prognosis and few treatment options (Creemers, et al. 2015; Else, et al. 2014). The adrenalytic compound mitotane is frequently used for ACC treatment (Kerkhofs, et al. 2015). Mitotane has unfavorable pharmacokinetics, complex drug interactions due to CYP3A4 activation, unclear mechanisms of action, and significant adverse effects in most patients (Hahner and Fassnacht 2005; Terzolo, et al. 2014; van Erp, et al. 2011). Due to the limitations of existing targeted agents, the current standard of care for patients with locally advanced or metastatic disease is combination chemotherapy. The most effective chemotherapy regimen produces a tumor response in one quarter to one third of ACC patients with locally advanced disease (Fassnacht, et al. 2012). We sought to identify a new agent for ACC treatment and to characterize its mechanisms of action in cultured cells and in animals.
To identify agents that could be used for ACC treatment, we focused on compounds that cause selective damage to the adrenal cortex (adrenalytic activity). Some inhibitors of acyl-CoA:cholesterol acyltransferase (ACAT; sterol O-acyl transferase; SOAT), that were originally developed for atherosclerosis, have adrenalytic activity (Sliskovic and White 1991). In particular, ATR-101 has adrenalytic activity in guinea pigs, beagle dogs and cynomolgus monkeys (Dominick, et al. 1993a; Dominick, et al. 1993b; Reindel, et al. 1994). ATR-101 reduces the ATP level in the adrenal cortex of guinea pigs and in cultured adrenocortical cells (Vernetti, et al. 1993; Wolfgang, et al. 1995). ATR-101 cytotoxicity correlates with the inhibition of respiration in primary adrenocortical cells and in mitochondrial fractions (Vernetti et al. 1993). The observed toxicity of ATR-101 is limited to the adrenal cortex, and to the ovaries and sebaceous glands in cynomolgus monkeys (Reindel et al. 1994). The selective adrenalytic activity of ATR-101 suggests that it could be used to treat diseases that are caused by excess adrenocortical cell growth or activity.
A subset of other ACAT inhibitors can affect adrenocortical functions (Nishimura, et al. 2013; Sliskovic, et al. 1998). However, many potent ACAT inhibitors, including avasimibe (CI-1011), eflucimibe (F 12511), and pactimibe sulfate (CS-505) do not have detectable adrenalytic activity (Junquero, et al. 2001; Kotsuma, et al. 2008; Robertson, et al. 2001). Targeted deletion of the mouse gene that encodes the adrenocortial ACAT1 enzyme (Acact) depletes cholesterol esters in adrenals, but it does not cause corticosteroid insufficiency (Meiner, et al. 1996). It is therefore likely that the adrenalytic activity of ATR-101 involves molecular mechanisms unrelated to ACAT inhibition.
To evaluate the efficacy and mechanisms of ATR-101 action in cell and animal models of ACC, we investigated the effects of ATR-101 on ACC-derived cells in culture and in mouse xenografts. We compared the mechanisms of ATR-101 in vitro with its adrenalytic activity in vivo.
Materials and Methods
Cell culture and pharmacological reagents
NCI H295R cells were obtained from American Type Culture Collection (ATCC) and were maintained in DMEM:F-12 Medium with 10% FCS at 37 °C in an atmosphere with 5% CO2. The cells were validated by determining the transcription of genes and synthesis of corticosteroids that are characteristic for the NCI 295R cell line. The NCI H295R cell line is the most extensively characterized ACC-derived cell line and carries a mutation in the CTNNB1 gene, which is the most frequently observed mutation in ACC tumors (Assie, et al. 2014; Juhlin, et al. 2015; Pinto, et al. 2015). To investigate the effects of ATR-101, the cells were transferred into DMEM with 5% FCS, 10mM Galactose, 2mM Glutamine, 1mM Na-Pyruvate at least 24 hours before each experiment.
ATR-101 was synthesized by AAPharmasyn and by PharmAgra Labs and was purified to >97% homogeneity using protocols similar to those that have been described (Trivedi, et al. 1994).
Experimental animals, analysis of ATR-101 effects on xenografts and adrenalytic activity
CB17-SCID mice with H295R cell xenografts were produced and the effects of ATR-101 administration were analyzed using protocols adapted from previous studies (Barlaskar, et al. 2009). To measure the effects of ATR-101 on xenograft growth, the mice were randomized into groups that had similar distributions of xenograft sizes with an average of 100 mm3. To measure the effects of ATR-101 on xenograft establishment, the sizes of swelling produced by cell implantation were measured twice during the week after cell injection, and the mice were randomized into groups that had similar distributions of the sizes of the swellings. Urinary free cortisol was measured using EIA kits (DetectX, Arbor Assays). ATR-101 was administered in 0.5% CMC-saline by oral gavage.
Guinea pigs (Dunkin-Hartley, 500–600 g) and rats (SD 2–3 and 24 months) were administered ATR-101 and the adrenals were examined using protocols modified from previous studies (Dominick et al. 1993a). Autofluorescence was visualized in freshly mounted cryosections and adjacent sections were stained with Sudan Black B or H&E.
All animal experiments and care followed policies established by the University of Michigan Committee for the Use and Care of Animals.
Cell and tissue imaging and analysis of cell viability
To measure cell proliferation in xenografts, mice were injected intraperitoneally with 2 mg BrdU and euthanized 6 hours after injection. BrdU incorporation was detected by immunostaining. Apoptotic cells in xenografts were detected using the Cell Death Detection kit (TUNEL staining, Roche). Apoptosis of cultured cells was detected using the Annexin V Apoptosis Detection Kit I (Becton, Dickinson and Company). Cytochrome c release from mitochondria was analyzed by separating mitochondria from cytosol by differential centrifugation and immunoblotting analysis. Caspase 3/7 activities were measured using the Caspase-GloW 3/7 kit (Promega North America, Madison, WI).
Cellular reducing activities were measured using MTS (Promega) and PrestoBlue (Molecular Probes) reagents. ATP levels were measured using the CellTiter-Glo Assay kit (Promega). Cell membrane permeability was measured by incubating the cells with 50 nM SYTOX green (Invitrogen). Cells and tissue sections were incubated with the labeling reagents indicated or immunostained with the indicated antibodies after fixation using protocols modified from previous studies (Cheng, et al. 2009). Cells and cell extracts were analyzed by using indicators of cell viability and protocols modified from previous studies (Marroquin, et al. 2007). The fluorescence was visualized by microscopy and the fluorescence or luminescence intensities or absorabance were measured using a microwell plate reader.
Analysis of oxidative phosphorylation and reactive oxygen in cells and in mitochondrial fractions
Mitochondrial morphology, membrane potential, and the levels of reactive oxygen species in cells were analyzed using protocols modified from previous studies (Frezza, et al. 2007a). Mitochondrial membrane potential was measured by incubating the cells with 250 nM JC-1 or 100 nM TMRM for 30 minutes. Mitochondrial morphology was analyzed by preloading cells with 250 nM Mitotracker Red for 2 hours followed by fixation and immunostaining using anti-cytochrome c antibody. The levels of reactive oxygen species were measured by incubation with 2.5 μM DCFH, 2.5 μM MitoSOX™ Red or 2.5 μM DHE for 60 minutes.
Rat liver mitochondrial fractions were prepared and the activities of complexes required for oxidative phosphorylation were measured as described (Frezza, et al. 2007b). ATP synthesis was measured using a coupled luciferase assay. Complex I activity was determined by measuring NADH absorbance. Complex III and complex IV activities were determined by measuring cytochrome c absorbance. F1F0-ATPase activity was determined by measuring phosphate release using the ATPase Assay Kit (Novus Biologicals).
Detailed protocols and the sources of reagents for all experimental procedures are provided in supplemental material and methods.
Results
Effects of ATR-101 on ACC xenograft establishment and growth
We examined the effect of ATR-101 on the growth of xenografts formed by human adrenocortical carcinoma H295R cells. Mice with H295R cell xenografts of equivalent sizes were randomized into ATR-101 and vehicle cohorts. ATR-101 administration (1 mg/g/day po) was begun when the mean size of the xenografts reached 100 mm3. The growth of the xenografts was slower in the mice that were administered ATR-101 than in the mice that were administered vehicle (Fig. 1A). The weight of the xenografts excised from mice that were administered ATR-101 was lower than that of xenografts from control mice. ATR-101 administration had no significant effect on the mean body weight of the mice and did not cause overt symptoms of weakness or fatigue in the mice (Fig. 1A, data not shown).
Figure 1. Inhibition of ACC cell xenograft establishment and growth by ATR-101.
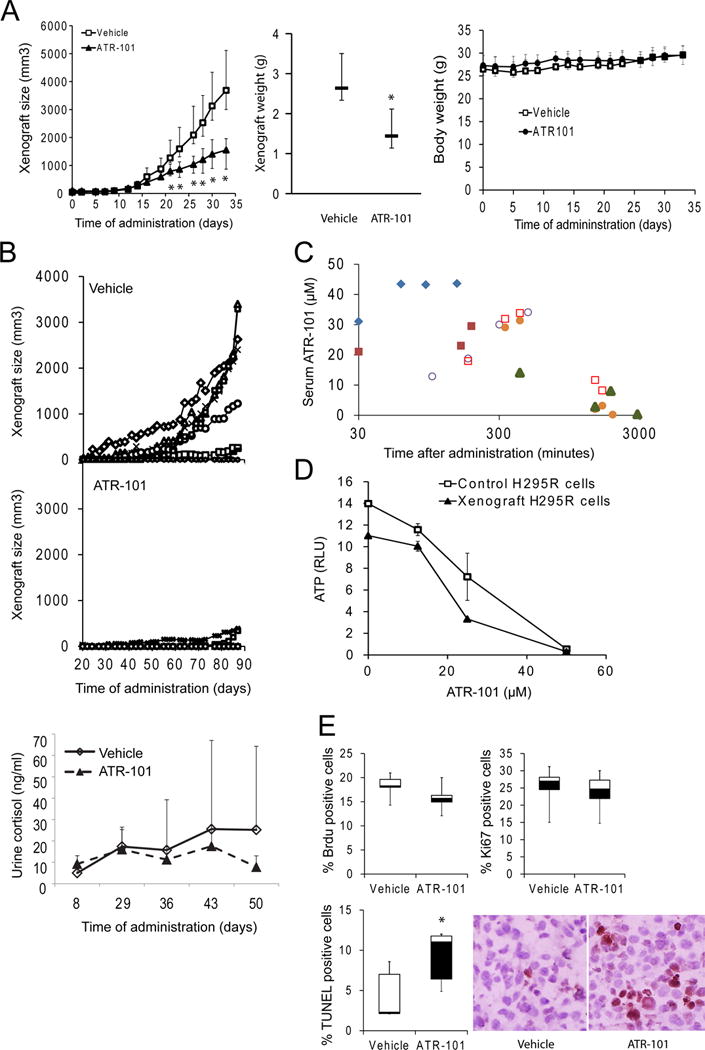
(A) Xenograft growth in mice that were administered ATR-101 (1 mg/g/day po) or vehicle starting when the mean size of the xenografts reached 100 mm3. The median and middle quartiles of the xenograft sizes and weights are plotted for each cohort. The median body weight for each cohort is shown in in the graph on the right.
(B) Xenograft establishment and growth, and urinary free cortisol levels in mice that were administered ATR-101 (0.7 mg/kg/day po) or vehicle starting two weeks after cell implantation. The sizes of the xenografts in individual mice that were administered vehicle (upper panel) or ATR-101 (middle panel) are plotted as a function of the time after beginning administration. The levels of urinary free cortisol were measured in the mice from each cohort that urinated during a 2 hour collection period and the mean and standard deviation are plotted as a function of the time after beginning administration (bottom panel).
(C) Pharmacokinetic analysis of the minimum serum concentration of ATR-101 that affected xenograft growth in mice. The concentration of ATR-101 in serum was measured using LC-MS at the times indicated after the administration of a single dose (0.3 mg/g po) of ATR-101. Each symbol indicates serum samples that were collected from the same mouse.
(D) Effects of long-term ATR-101 administration to mice on the sensitivity of cells reisolated from the xenografts to ATR-101. The ATP levels of H295R cells and cells that were reisolated from the xenografts of mice that that had been administered ATR-101 for 110 days were compared after 16 h culture in the presence of the indicated concentrations of ATR-101 or vehicle. The graph shows the means and standard deviations of three parallel cultures and is representative of two independent experiments.
(E) Effects of ATR-101 administration on the proportions of cortical xenograft cells that were stained by anti-BrdU antibodies (left graph), by anti-Ki67 antibodies (middle graph), and by TUNEL reagents (right graph and image to the right of the graphs). The median, middle quartiles and range are plotted for each cohort. The results shown are representative of two or more independent experiments. (*: p<0.05, Mann–Whitney non-parametric U-test, n=6)
We determined the effect of ATR-101 on xenograft establishment by starting ATR-101 (0.7 mg/g/day po) or vehicle administration two weeks after cell implantation. This protocol is designed to mimic the use of adjuvant chemotherapy in conjunction with surgery to counteract ACC tumor recurrence (Lindhe and Skogseid 2010). The mice were randomized into ATR-101 and control cohorts that had similar distributions of the sizes of the swellings produced by cell injection, which was taken to indicate that comparable numbers of H295R cells were implanted into the mice. The proportion of the mice that developed xenografts and the rates of xenograft growth were lower for the mice that were administered ATR-101 than for the control mice (Fig. 1B, Table 1). The cortisol levels of individual mice varied over a wide range. Nevertheless, the cortisol levels of mice that were administered ATR-101 were lower on average than the cortisol levels of control mice with H295R cell implants (Fig. 1B). No new xenografts were observed when ATR-101 administration was discontinued after 88 days, suggesting that ATR-101 administration eradicated the implanted cells in most mice. Re-implantation of H295R cells into the contra-lateral flank in the absence of ATR-101 administration produced xenografts in all of the mice.
Table 1.
Effect of ATR-101 administration on xenograft establishment.
| Cohort | Proportion (percentage) of mice with detectable xenografts |
|---|---|
| Vehicle | 7/10 (70%) |
| ATR-101 | 2/10 (20%)* |
| Control 1** | 25/49 (51%) |
| Control 2** | 3/5 (60%) |
| Control 3** | 15/22 (68%) |
| Control 4** | 16/18 (89%) |
| Control 5** | 12/12 (100%) |
p<0.05 vs Vehicle (chi-square test)
Controls included both cohorts of mice that were administered vehicle as well as mice that were not administered test compounds or vehicle
To determine the minimum serum concentration of ATR-101 that affected xenograft growth in mice, we evaluated the effects of different doses of ATR-101. An ATR-101 dose of 0.3 mg/g/day po had a transient effect on xenograft growth in a subset of the mice. At this dose, the serum ATR-101 concentration reached a maximum of 40 μM at 60–150 minutes after initial administration, and then decreased with a t½ of about 6 hours (Fig. 1C). The doses that were used to evaluate the effects of long-term ATR-101 administration on xenograft growth were 2–3 times higher than the minimal dose that had a detectable effect.
Cells that were re-isolated from the few xenografts that formed in mice during ATR-101 administration had the same susceptibility to ATR-101 as the cells that were implanted into the mice (Fig. 1D). ATR-101 administration therefore did not give rise to ATR-101 resistance in the xenografts of these mice.
We examined cell proliferation and apoptosis in the cortex of the xenografts to determine how ATR-101 suppressed xenograft growth. ATR-101 administration did not alter the proportions of cells that incorporated BrdU or those that reacted with anti-Ki67 antibodies (Fig. 1E). In contrast, the proportion of cells that produced TUNEL signal was higher in the xenografts from mice that were administered ATR-101 than in xenografts from control mice. ATR-101 administration therefore increased the proportion of apoptotic cells in mouse xenografts.
Effects of ATR-101 and related compounds on cultured ACC-derived cells
We compared the effects of culturing the ACC-derived H295R cell line with different concentrations of ATR-101 on several independent characteristics that are used as indicators of cell viability. ATR-101 caused rapid and opposite changes in the rates of formazan (MTS) versus resorufin (Prestoblue) reduction in H295R cells (Fig. 2A). The balance of cellular reducing compounds shifted within tens of minutes after ATR-101 addition to cultured cells. The same ATR-101 concentrations that shifted the balance of reducing compounds depleted the ATP in H295R cells (Fig. 2B). The ATP level was reduced within 2 hours after ATR-101 addition and was depleted within 12 hours (Fig. 4C). After 16 hours of culture with ATR-101, the cell membrane became permeable to SYTOX (Fig. 2C). When the cells were washed 4 h after ATR-101 addition, a majority of the cells recovered and resumed growth (Fig. 2D). The cells did not recover when ATR-101 was removed after 20 hours.
Figure 2. Mechanisms of ATR-101 cytotoxicity in cultured ACC-derived cells.
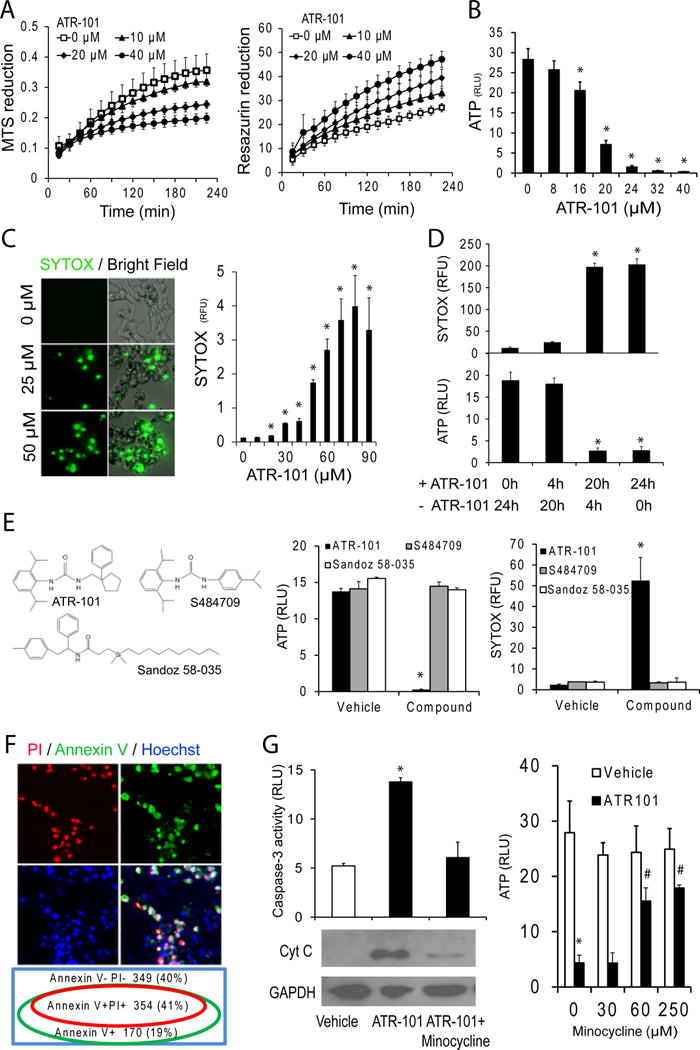
(A) Suppression of MTS reduction and enhancement of resazurin reduction in cells cultured with ATR-101. The MTS absorbance (left graph) and resazurin fluorescence (right graph) of H295R cells are plotted as a function of time after addition of the indicated concentrations of ATR-101.
(B) ATP depletion in H295R cells cultured with ATR-101. The ATP levels of H295R cells that were cultured in the presence of the indicated concentrations of ATR-101 for 20 h are plotted
(C) Membrane permeabilization in cells cultured with ATR-101. The SYTOX fluorescence in H295R cells that were cultured in the presence of the indicated concentrations of ATR-101 for 20 h is shown.
(D) Reversibility of membrane permeability and ATP depletion following ATR-101 removal at different times after exposure. The SYTOX fluorescence and ATP levels of cells that were cultured with 30 μM ATR-101 for the time indicated below the graphs (+ ATR-101), followed by culture without ATR-101 for the time indicated below the graphs (−ATR-101) are plotted. The graphs show the means and standard deviations from three replicate cell cultures and are representative of two independent experiments (*: p<0.05 vs 0h ATR-101; two-tailed unpaired Student’s t test).
(E) Comparison of the effects of ATR-101, a structurally related compound (Sigma S484709), and an ACAT inhibitor (Sandoz 58-035) on ATP levels and membrane permeability. The ATP levels and SYTOX fluorescence of H295R cells that were cultured with 40 μM of the indicated compounds for 24 h are plotted.
(F) Apoptotic markers in cells cultured with ATR-101. The Annexin V (green), Propidium Iodide (PI, red), and Hoechst (blue) fluorescence of H295R cells that were cultured in the presence of 30 μM ATR-101 for 20 h are shown. The Venn diagram indicates the percentage of cells labeled by each combination of fluorophores.
(G) Effects of the inhibition of cytochrome c release on ATR-101 cytotoxicity. The levels of cytochrome c and GAPDH in cytoplasmic extracts (lower images), caspase-3/7 activities (left graph) and ATP (right graph) in H295R cells that were cultured in the presence of vehicle, ATR-101 alone or ATR-101 in combination with minocycline for 16–20 h are shown.
All graphs plot the means and standard deviations for two or more replicate cultures. Each graph is representative of two or more independent experiments. (*: p<0.05 vs vehicle, #: p<0.05 vs ATR-101; two-tailed unpaired Student’s t test).
Figure 4. Release of reactive oxygen in cells cultured with ATR-101.
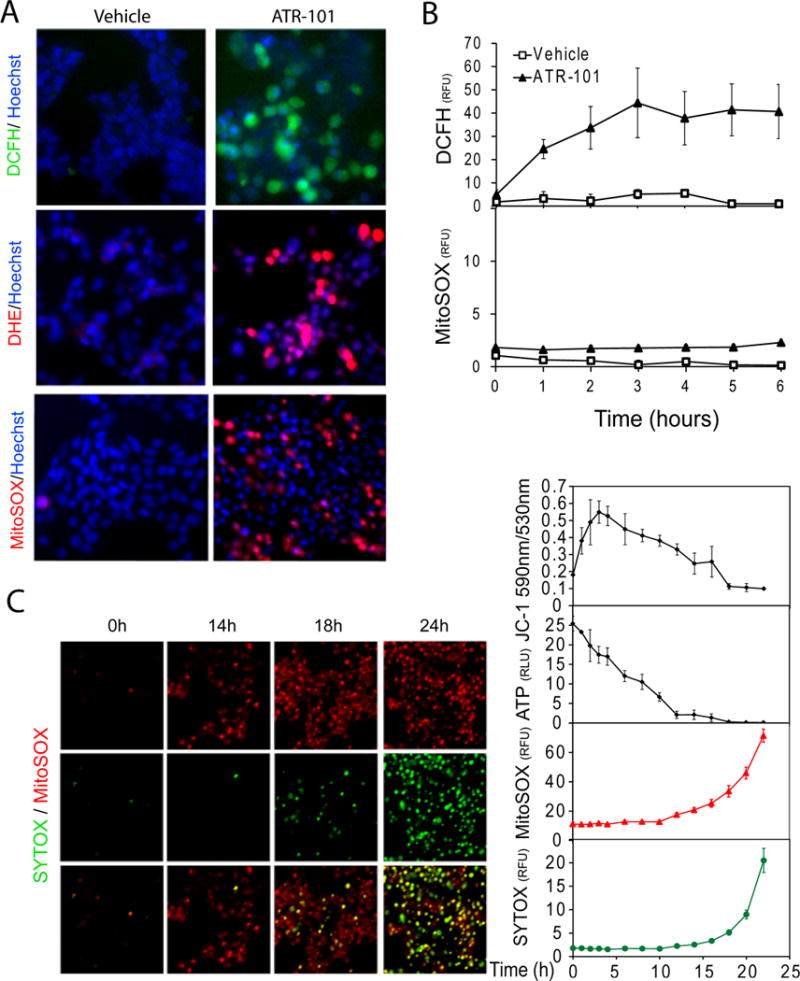
(A) Increases in reactive oxygen and superoxide levels in cells cultured with ATR-101. The DCFH (top - green), DHE (middle – red), and MitoSOX (bottom - red) fluorescence are shown in H295R cells that were cultured with vehicle (left images) or with 40 μM ATR-101 (right images) for 5 h (DCFH), 6 h (DHE) and 16 h (MitoSOX), respectively. The nuclei are counterstained with Hoechst (blue).
(B) Timing of the changes in the levels of different reactive oxygen species in cells cultured with ATR-101. The graphs show the fluorescence of DCFH and MitoSOX in H295R cells that were cultured with 40 μM ATR-101 for the times indicated.
(C) Succession of mitochondrial membrane hyperpolarization, ATP depletion, superoxide production and cell membrane permeabilization after ATR-101 addition. The MitoSOX (upper row - red) and SYTOX (middle row - green) fluorescence and their overlap (bottom row – merged) are shown in H295R cells that were cultured with 40 μM ATR-101 for the times indicated above the images on the left. The levels of ATP, and of JC-1, MitoSOX and SYTOX fluorescence are plotted for H295R cells that were cultured with 30 μM ATR-101 for the times indicated below the graphs on the right. The graphs plot the means and standard deviations of three replicate cultures. Each graph is representative of data from at least two independent experiments.
The images are representative of the large majority of cells in each population and of two or more independent experiments.
We compared the cytotoxicity of ATR-101 with that of another ACAT inhibitor (Sandoz 58-035) as well as a compound that is structurally related to ATR-101 (S484709). No ATP depletion or membrane permeabilization was observed in H295R cells that were cultured with either of these compounds (Fig. 2E). Moreover, the concentration of ATR-101 that was required for cytotoxicity was orders of magnitude higher than the concentrations that inhibits ACAT activity in vitro (Trivedi et al. 1994). Likewise, the doses of ATR-101 that were required to suppress xenograft establishment and growth in mice were more than an order of magnitude larger than those that are required to reduce plasma cholesterol levels in rats, rabbits, guinea pigs and dogs (Krause, et al. 1993). The cytotoxicity of and xenograft suppression by ATR-101 are therefore likely to require activities unrelated to ACAT inhibition.
We investigated if ATR-101 caused the apoptosis of cultured H295R cells. Annexin V labeled a majority of the cells that were cultured with ATR-101, and all of the cells that were labeled by propidium iodide (Fig. 2F). Cytochrome c was released into the cytoplasm and the caspase-3 activity increased in cells cultured with ATR-101 (Fig. 2G). Culture of cells with minocycline (Zhu, et al. 2002) suppressed cytochrome c release, caspase-3 activation and ATP depletion by ATR-101. ATR-101 therefore caused H295R cell apoptosis both in culture and in xenografts.
Effects of ATR-101 on mitochondrial membrane potential and on mitochondrial morphology
To investigate the causes for the rapid depletion of ATP in the presence of ATR-101, we examined the mitochondria of H295R cells that were cultured with ATR-101. ATR-101 caused an increase in the mitochondrial membrane potential as detected by JC-1 as well as by TMRM fluorescence (Fig. 3A). This increase occurred within hours after ATR-101 addition to cultured cells (Fig. 3B). Thus, ATR-101 had simultaneous and opposite effects on the mitochondrial membrane potential and on the ATP level at early times after addition (Fig. 4C). After 3 hours, the mitochondrial membrane potential gradually declined and reached a level below that of untreated cells 16 hours after ATR-101 addition at the same time as the cellular energy charge was depleted.
Figure 3. Mitochondrial membrane hyperpolarization and fragmentation in cells cultured with ATR-101.
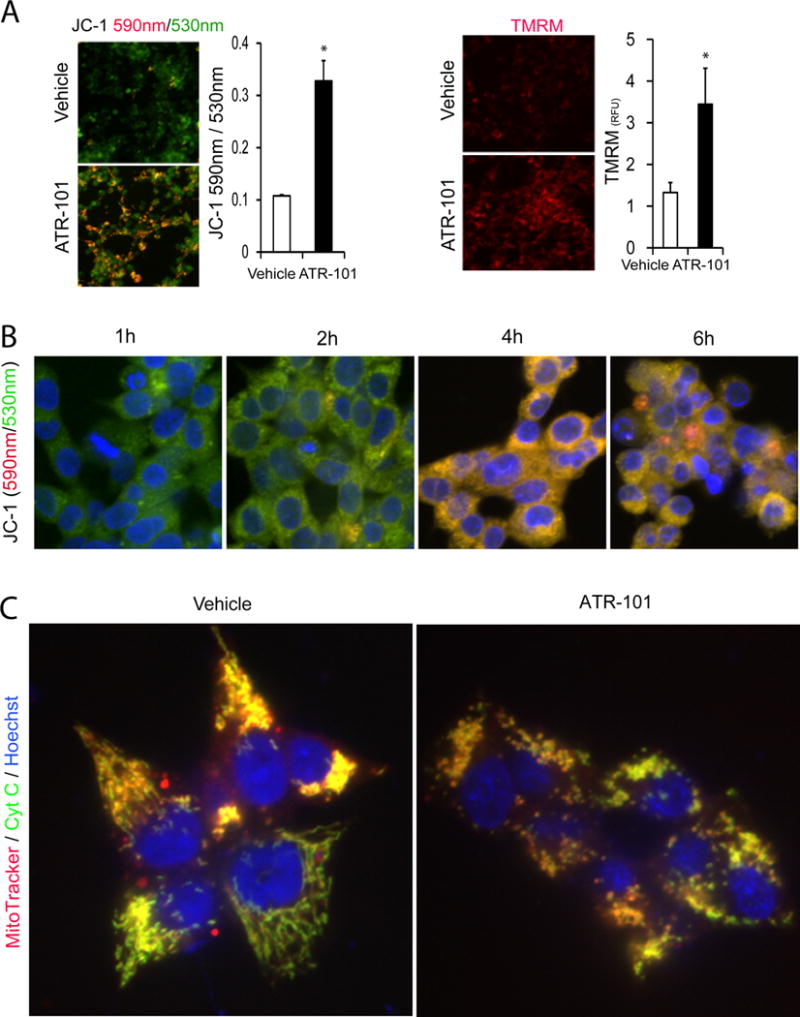
(A) Mitochondrial membrane hyperpolarization in cells cultured with ATR-101. The images show JC-1 [left panels; 530 nm (green), 590 nm (red)] and TMRM (right panels; 590 nm (red)) fluorescence in H295R cells that were cultured with vehicle (upper images) or with 30 μM ATR-101 (lower images) for 4 h. The graph on the left plots the JC-1 fluorescence emissions ratio (590 nm/530 nm) and the graph on the right plots the TMRM fluorescence intensity. The graphs plot the means and standard deviations of three replicate cultures, and are representative of data from two or more independent experiments (*: p<0.05 vs vehicle; two-tailed unpaired Student’s t test).
(B) Timing of mitochondrial membrane hyperpolarization in cells cultured with ATR-101. The images show JC-1 fluorescence [530 nm (green), 590 nm (red)] of H295R cells that were cultured with 40 μM ATR-101 for the times indicated, followed by incubation with JC-1.
(C) Mitochondrial fragmentation in cells cultured with ATR-101. The MitoTracker (red) and anti-cytochrome c (green) fluorescence of H295R cells that were cultured with vehicle (left image) or with 50 μM ATR-101 (right image) for 6 h are shown.
The images are representative of the large majority of cells in each population and of data from two independent experiments.
We compared the morphologies of mitochondria in cells that were cultured with vehicle and with ATR-101. Mitochondria that were visualized by MitoTracker fluorescence and anti-cytochrome c immunofluorescence appeared as small round puncta in cells cultured with ATR-101, whereas mitochondria in cells cultured with vehicle appeared as long reticular strands (Fig. 3C). ATR-101 therefore caused mitochondrial fragmentation in parallel with its effects on mitochondrial membrane potential and ATP depletion.
Effects of ATR-101 on reactive oxygen levels
We tested the hypothesis that the increase in mitochondrial membrane potential caused the release of reactive oxygen from the electron transport chain. Cells that were cultured with ATR-101 had elevated levels of reactive oxygen species detected by 2′,7′-dichlorodihydrofluorescin diacetate (DCFH), dihydroethidium (DHE) and MitoSOX (hydroxyethidine) fluorescence (Fig. 4A). DCFH can be oxidized by many different reactive oxygen species, whereas DHE and MitoSOX react primarily with superoxide (Dikalov, et al. 2007; Robinson, et al. 2006). MitoSOX is enriched in mitochondria, and binds nucleic acids upon oxidation, resulting in nuclear fluorescence. The DCFH fluorescence increased in parallel with the mitochondrial membrane potential, whereas the MitoSOX fluorescence increased only after ATP depletion (Fig. 4B, 4C).
Effects of lipophilic antioxidants on ATR-101 cytotoxicity
We tested if the release of reactive oxygen mediated ATR-101 cytotoxicity by determining if antioxidants protected the cells from the effects of ATR-101. Addition of α-tocopherol (vitamin E) to the cells in combination with ATR-101 suppressed the increase in superoxide levels, caspase activation and membrane permeabilization (Fig. 5A, 5B). By contrast, ATR-101 had the same effect on JC-1 fluorescence when added together with α-tocopherol, indicating that α-tocopherol did not block ATR-101 entry into cells or its effect on the mitochondrial membrane potential (Fig. 5A, 5B). Higher α-tocopherol concentrations suppressed the effects of proportionately higher ATR-101 concentrations on the ATP levels of cells (Fig. 5C). The counterbalancing effects of ATR-101 and α-tocopherol on the ATP level suggest that they had directly opposing effects on processes that were critical for the maintenance of cellular ATP.
Figure 5. Suppression of ATR-101 cytotoxicity by α-tocopherol.
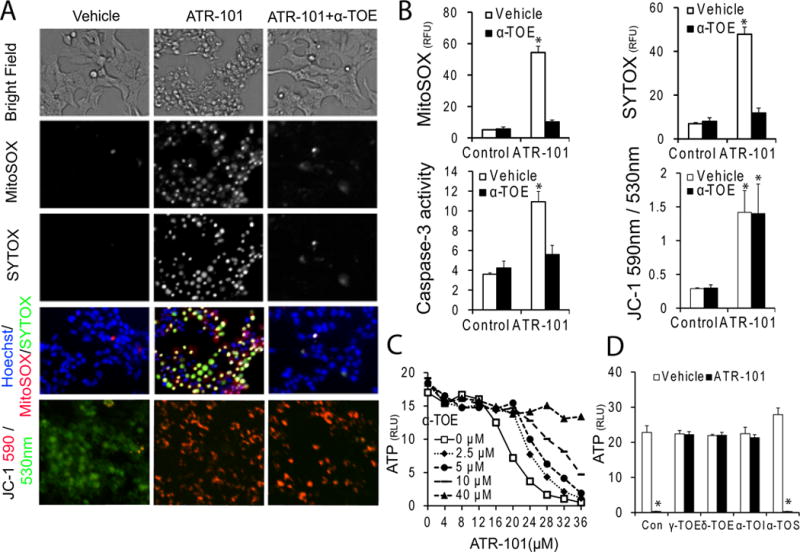
(A) α-tocopherol suppresses the effects of ATR-101 on superoxide levels and on mebrane permeability, but not on mitochondrial hyperpolarization. The left column shows cells cultured with vehicle, the middle column shown cells cultured with 30 μM ATR-101 alone, and the right column shows cell cultured with 30 μM ATR-101 and 40 μM α-tocopherol (α-TOE). The upper 4 rows show phase contrast (top row), MitoSOX fluorescence (second row), SYTOX fluorescence (third row) and merged MitoSOX (red), SYTOX (green) and Hoechst (blue) fluorescence (fourth row) for the same field of H295R cells after 20 hour culture with the indicated compounds. The bottom row shows JC-1 fluorescence [530 nm (green); 590 nm (red)] of H295R cells after 6 hour culture with the indicated compounds.
(B) Differential effects of α-tocopherol on the superoxide levels, membrane permeability and caspase-3/7 activities versus mitochondrial membrane potential of cells cultured with ATR-101. The MitoSOX (upper left), SYTOX (upper right) and JC-1 (lower right) fluorescence, as well as the caspase-3 activity (lower left) are plotted for H295R cells that were cultured with 30 μM ATR-101 and 40 μM α-tocopherol separately and in combination for 20 hours (MitoSOX, SYTOX and caspase-3) or for 3 hours (JC-1).
(C) Counterbalancing effects of α-tocopherol and ATR-101 on cell viability. The ATP levels of H295R cells that were cultured with the indicated concentrations of α-tocopherol and ATR-101 for 20 hours are plotted.
(D) Selective effects of different tocopherol derivatives on ATR-101 cytotoxicity. The ATP levels of H295R cells that were cultured with 40 μM of the indicated tocopherols together with 25 μM ATR-101 or vehicle for 20 hours are plotted (α-TOI: α-tocotrienol, α-TOS: α-tocopherol-succinate).
Each graph plots the means and standard deviations of three replicate cultures and is representative of data from two independent experiments (*: p<0.05 vs vehicle, #: p<0.05 vs ATR-101; two-tailed unpaired Student’s t tests). The images are representative of the large majority of cells in each population and of two or more independent experiments.
Several different tocopherols suppressed ATP depletion by ATR-101 (Fig. 5D). These tocopherols also have antioxidant activity. In contrast, α-tocopherol-succinate did not prevent ATP depletion by ATR-101. α-tocopherol-succinate does not have antioxidant activity, but retains many functions of α-tocopherol that are independent of its antioxidant activity (Birringer, et al. 2003; Galli, et al. 2004; Neuzil, et al. 2001). The protection from ATR-101 cytotoxicity therefore correlated with antioxidant activity.
Structurally unrelated lipophilic antioxidants, including 3-phytylmenadione (Vitamin K) and probucol also protected cells from the release of reactive oxygen and ATP depletion in the presence of ATR-101 (Fig. 6A, 6B). None of these antioxidants prevented the change in JC-1 fluorescence caused by ATR-101, indicating that mitochondrial membrane hyperpolarization preceded the release of reactive oxygen and its cytotoxic effects (Fig. 6C). Nevertheless, lipophilic antioxidants restored the ATP level in cells cultured with ATR-101, suggesting that the elevation of reactive oxygen levels contributed to ATP depletion. None of the many hydrophilic antioxidants tested prevented ATP depletion by ATR-101, indicating that protection from ATR-101 cytotoxicity required antioxidant activity in a lipophilic environment (Fig. 6D).
Figure 6. Suppression of ATR-101 cytotoxicity by structurally dissimilar lipophilic antioxidants.
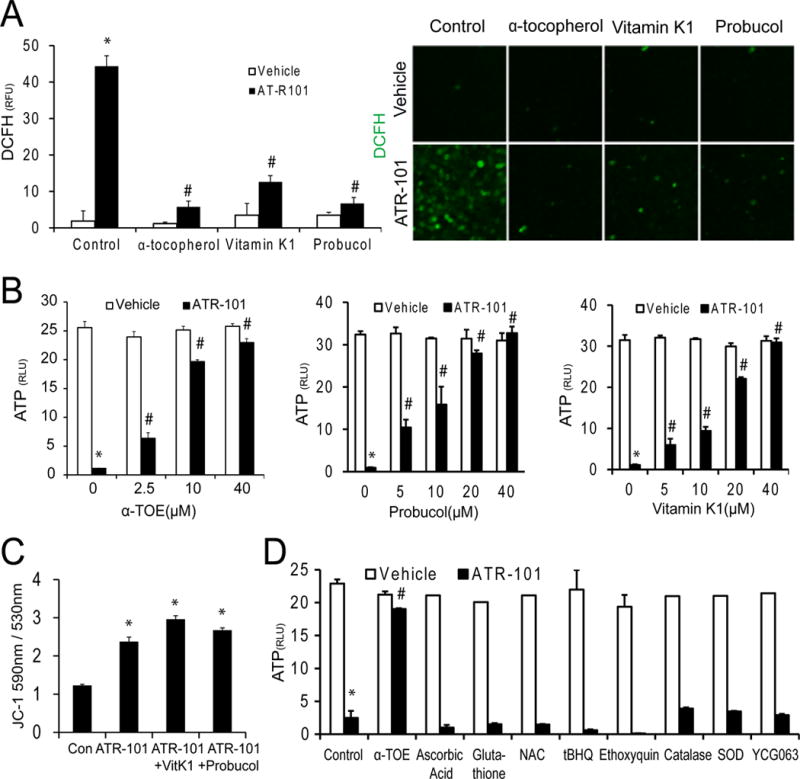
(A) Effects of structurally dissimilar antioxidants on the levels of reactive oxygen species in cells cultured with ATR-101. The images and the graph show the DCFH fluorescence of H295R cells that were cultured with vehicle or with 40 μM ATR-101 together with or without 40 μM α-tocopherol, 3-phytylmenadione (Vitamin K), or probucol for 6 hours, followed by incubation with DCFH (green).
(B) Effects of α-tocopherol, probucol and Vitamin K on the changes in ATP levels caused by ATR-101. The graphs show the ATP levels of H295R cells that were cultured with the indicated concentrations of α-tocopherol (left graph), probucol (middle graph) or Vitamin K (right graph) in combination with 25 μM ATR-101 or vehicle for 20 hours.
(C) Effects of 3-phytylmenadione (Vitamin K) and probucol on the mitochondrial membrane potentials of cells cultured with ATR-101. The graph shows the JC-1 fluorescence emissions ratio (590 nm/530 nm) of H295R cells that were cultured with vehicle or 40 μM ATR-101 alone or in combination with 40 μM 3-phytylmenadione or probucol or for 3 hours.
(D) Effects of various hydrophilic antioxidants on the ATP levels of cells cultured with ATR-101. The graph shows the ATP levels of H295R cells that were cultured with 25 μM ATR-101 or vehicle in combination with 40 μM α-tocopherol, 1 mM Ascorbic Acid, 10 mM Glutathione, 5 mM NAC, 10 μM tBHQ, 20 μM Ethoxyquin, 500 U/ml Catalase, 500 U/ml SOD, or 5 μM YCG063 for 20 hours.
Each graph plots the means and standard deviations of three replicate cultures and is representative of data from two independent experiments (*: p<0.05 vs vehicle, #: p<0.05 vs ATR-101; two-tailed unpaired Student’s t tests). The images are representative of the large majority of cells in each population and of two or more independent experiments.
Effects of glycolysis on ATR-101 cytotoxicity
H295R cells that were cultured with glycolytic substrates maintained their ATP levels and cell membrane integrity in the presence of ATR-101 (Fig. 7A). Conversely, culture of H295R cells with 2-deoxy-D-glucose aggravated the ATP depletion and membrane permeabilization caused by ATR-101 (Fig. 7B). ATR-101 cytotoxicity therefore correlated with a reliance on respiration for ATP synthesis. Glycolytic substrates did not eliminate the rapid increase in mitochondrial membrane potential and DCFH reactive oxygen caused by ATR-101, but they suppressed the slow increase in MitoSOX fluorescence (Fig. 7C, 7D). The increases in mitochondrial membrane potential and reactive oxygen levels over the short term were therefore not sufficient for ATR-101 cytotoxicity, but additional factors over the long term, potentially including ATP depletion or elevated superoxide levels, were required for ATR-101 cytotoxicity.
Figure 7. Suppression of ATR-101 cytotoxicity by glycolytic substrates.
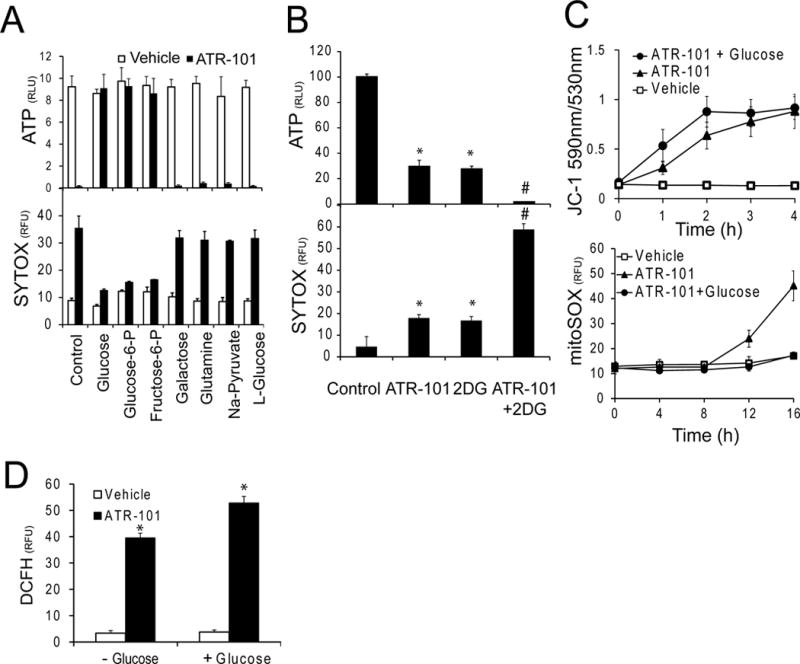
(A) Effects of glycolytic substrates on ATR-101 cytotoxicity. The ATP levels (upper graph) and SYTOX fluorescence (lower graph) of H295R cells that were cultured in the presence of the energy sources indicated below the bars together with vehicle or 30 μM ATR-101 for 20 hours are plotted.
(B) Combined effects of 2-deoxy-D-glucose and ATR-101 on cytotoxicity. The ATP levels (upper graph) and the SYTOX fluorescence (lower graph) of H295R cells that were cultured with 20 μM ATR-101 and 10 mM 2-deoxy-D-glucose separately or in combination for 20 hours are plotted.
(C) Glucose selectively alleviates the effect of ATR-101 on superoxide accumulation, but not on the mitochondrial membrane potential. The JC-1 (upper graph) and MitoSOX (lower graph) fluorescence are plotted for H295R cells that were cultured with vehicle or with 30 μM ATR-101 alone or in combination with 10 mM D-Glucose for the times indicated.
(D) Glucose does not suppress short-term elevation of reactive oxygen levels by ATR-101. The graph shows the DCFH fluorescence of H295R cells that were cultured with vehicle or with 30 μM ATR-101 in combination with or without 10 mM D-Glucose for 4 hours.
Each graph plots the means and standard deviations of three replicate cultures and is representative of data from two or more independent experiments (*: p<0.05 vs vehicle, #: p<0.05 vs ATR-101; two-tailed unpaired Student’s t test).
Relationships between ATR-101 cytotoxicity and oxidative phosphorylation
To identify steps in oxidative phosphorylation that were affected by ATR-101 in cells, we measured the combined effects of ATR-101 and inhibitors of individual steps of oxidative phosphorylation on the ATP level and on cell membrane permeability. ATR-101 and inhibitors of complex I (rotenone) and of complex III (antimycin A) had synergistic effects on the ATP level and on SYTOX fluorescence (Fig. 8A). ATR-101 and the complex IV inhibitor sodium azide as well as the uncoupler CCCP had additive effects on the ATP level and moderately synergistic effects on SYTOX fluoresence. By contrast, ATR-101 and the F1F0-ATP synthase inhibitor oligomycin had additive effects both on the ATP level and on SYTOX fluorescence. Thus, ATR-101 had synergistic effects in combination with compounds that inhibit early steps in the electron transport pathway, additive effects in combination with compounds that inhibit ATP synthase, and different effects on the ATP level and on cell membrane permeability in combination with compounds that inhibit late steps in the electron transport pathway or that dissipate the proton motive force.
Figure 8. Synergistic versus additive effects of ATR-101 in combination with inhibitors of oxidative phosphorylation.
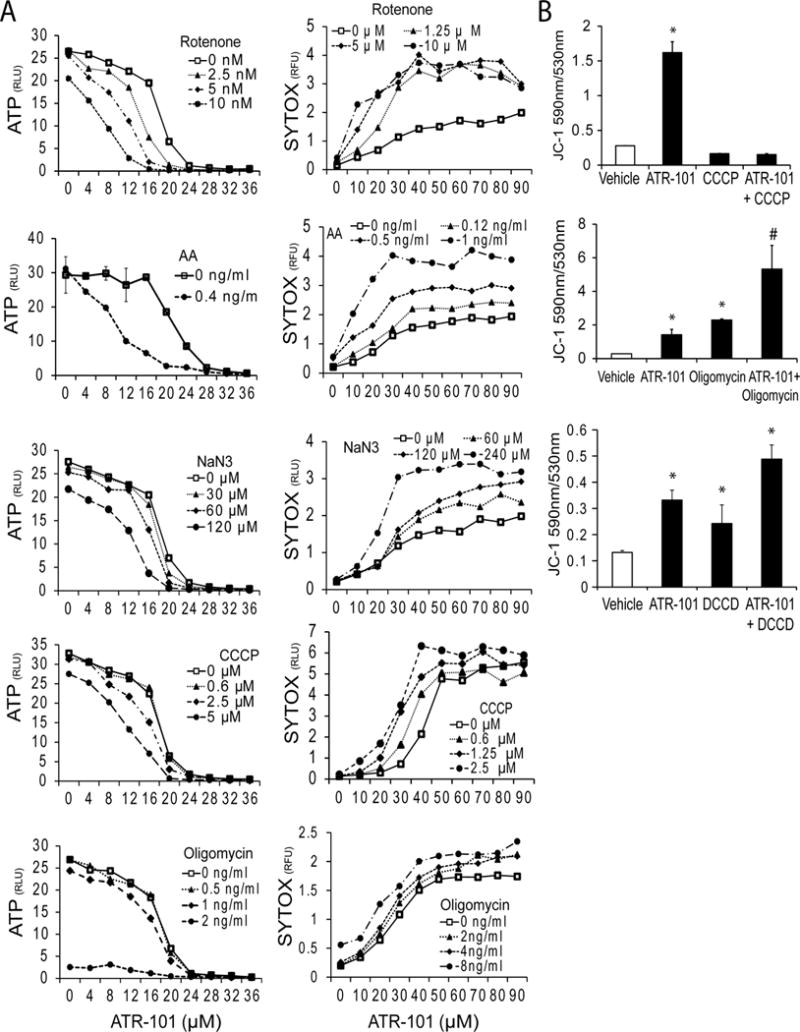
(A) ATR-101 has distinct effects in combination with different inhibitors of electron transport versus ATP synthase on cellular ATP levels. The ATP levels and SYTOX fluorescence of H295R cells that were cultured with the indicated concentrations of H295R cells were cultured with the indicated concentrations of rotenone, antimycin A, sodium azide, CCCP and oligomycin (from top to bottom) in combination with different concentrations of ATR-101 for 20 hours are plotted.
(B) ATR-101 has distinct effects in combination with uncouplers versus inhibitors of F1F0-ATPase on the mitochondrial membrane potential. The JC-1 fluorescence of H295R cells that were cultured with 20 μM ATR-101 and 10 μM CCCP, 100 ng/ml oligomycin or 3 μM DCCD (from top to bottom) separately and in combination for 3 hours are plotted. The graphs show the means and standard deviations of three replicate cultures and are representative of two independent experiments (*: p<0.05 vs vehicle, #: p<0.05 vs ATR-101; two-tailed unpaired Student’s t test).
We determined the combined effects of ATR-101 with CCCP, oligomycin as well as DCCD on the mitochondrial membrane potential. CCCP reversed the increase in the JC-1 fluorescence ratio caused by ATR-101 (Fig. 8B). In contrast, both oligomycin and DCCD increased the JC-1 fluorescence ratio further in combination with ATR-101. Taken together, these results are consistent with the interpretation that ATR-101 blocks ATP synthase activity, and does not inhibit electron transport directly.
Effects of ATR-101 on the membrane potential and on oxidative phosphorylation in mitochondrial fractions
To establish if ATR-101 had direct effects on mitochondria, we investigated the effects of ATR-101 in mitochondrial fractions that were prepared by differential centrifugation (Fig. 9A). Mitochondria that respired in the presence of ATR-101 produced higher JC-1 fluorescence than mitochondria that respired in the absence of ATR-101 (Fig. 9B). ATR-101 therefore had a direct effect on the membrane potential generated by mitochondrial respiration. The increase in JC-1 fluorescence required an electron donor and was prevented by CCCP.
Figure 9. ATR-101 inhibits oxidative phosphorylation and F1F0-ATPase in mitochondrial fractions.
(A) Enrichment of mitochondria by differential centrifugation. Equal proportions of the mitochondrial pellet (mito) and cytoplasmic supernatant (Cyto) fractions were separated by SDS-gel electrophoresis and analyzed by immunoblotting blot using anti-cytochrome c and anti-GAPDH antibodies. The data shown are representative of two independent mitochondrial preparations.
(B) Membrane hyperpolarization in mitochondria respiring in the presence of ATR-101. The JC-1 fluorescence of mitochondria that respired in the presence of the indicated concentrations of ATR-101 alone or in combination with 5 μM CCCP for 30 minutes is plotted.
(C) Reduced ATP synthesis in mitochondria respiring in the presence of ATR-101. The ATP levels synthesized in mitochondria that respired in the presence of the indicated concentrations of ATR-101 for 30 minutes are plotted.
(D) ATR-101 inhibits ATP synthesis by Complex I and Complex II electron acceptors, but not electron transfer by Complex III or Complex IV. The amounts of ATP synthesized by mitochondria that respired in the absence or in the presence of ATR-101 using either Complex I (glutamate and malate; upper graph) or Complex II (succinate; lower graph) are plotted on the left. The rates of electron transfer by Complex III (cytochrome c reduction; upper graph) and by Complex IV (cytochrome c oxidation; lower graph) in mitochondria are plotted on the right.
(E) ATR-101 does not inhibit NADH oxidation by complex I. The graph shows the change in NADH absorbance that was produced by the respiration of mitochondria that were pre-incubated with vehicle, 50 μM ATR-101, or 10 μM rotenone
(F) ATR-101 inhibits F1F0-ATPase activity in isolated mitochondria. The graph shows the phosphate released by freeze-thawed rat liver mitochondria that were pre-incubated with vehicle, 50 μM ATR-101, 4 mM sodium azide (NaN3) or 5 μg/ml oligomycin for 30 minutes, followed by incubation with ATP for 10 minutes.
(G) Model for the mechanisms of ATR-101 cytotoxicity in cultured ACC-derived cells.
The graphs in panels C, D, E and F show the means and standard deviations of three replicate assays and are representative of at least two independent experiments (*: p<0.05 vs vehicle; two-tailed unpaired Student’s t test). The graph in panel B shows multiple reactions stopped at different times and is representative of two independent experiments.
ATR-101 reduced the amount of ATP that was produced by respiring mitochondria (Fig. 9C). ATR-101 inhibited ATP synthesis when glutamate and malate, which are utilized by complex I, or succinate, which is utilized by complex II, were used as electron donors (Fig. 9D). Similar ATR-101 concentrations increased the mitochondrial membrane potential and inhibited ATP synthesis in mitochondrial fractions as well as in cultured cells.
We examined the effects of ATR-101 on individual steps of oxidative phosphorylation. ATR-101 had no detectable effect on uncoupled NADH oxidation by complex I, on cytochrome c reduction by complex III, or on cytochrome c oxidation by complex IV (Fig. 9D, 9E). In contrast, ATR-101 inhibited F1F0-ATPase activity in mitochondrial fractions (Fig. 9F). Similar ATR-101 concentrations reduced F1F0-ATPase activity, increased JC-1 fluorescence and reduced ATP synthesis. These results indicate that ATR-101 increased the mitochondrial membrane potential and inhibited oxidative phosphorylation by direct inhibition of F1F0-ATPase.
Role of reactive oxygen in ATR-101 adrenalytic activity in vivo
We investigated the significance of the molecular mechanisms of ATR-101 action identified in vitro by studying the adrenalytic effect of ATR-101 in guinea pigs. The adrenal cortex of guinea pigs that were administered ATR-101 for two weeks contained a layer of autofluorescent foci (Fig. 10A). The fluorescence intensities of these foci were up to 5-fold higher than the fluorescence of other layers of the adrenal cortex and of the corresponding layer of the adrenal cortex from control animals. The autofluorescent layer contained brown granules and the autofluorescence was quenched by Sudan Black (Fig. 10B, 10C).
Figure 10. Lipofuscin fluorescence in the zona fasciculata of guinea pigs fed ATR-101.
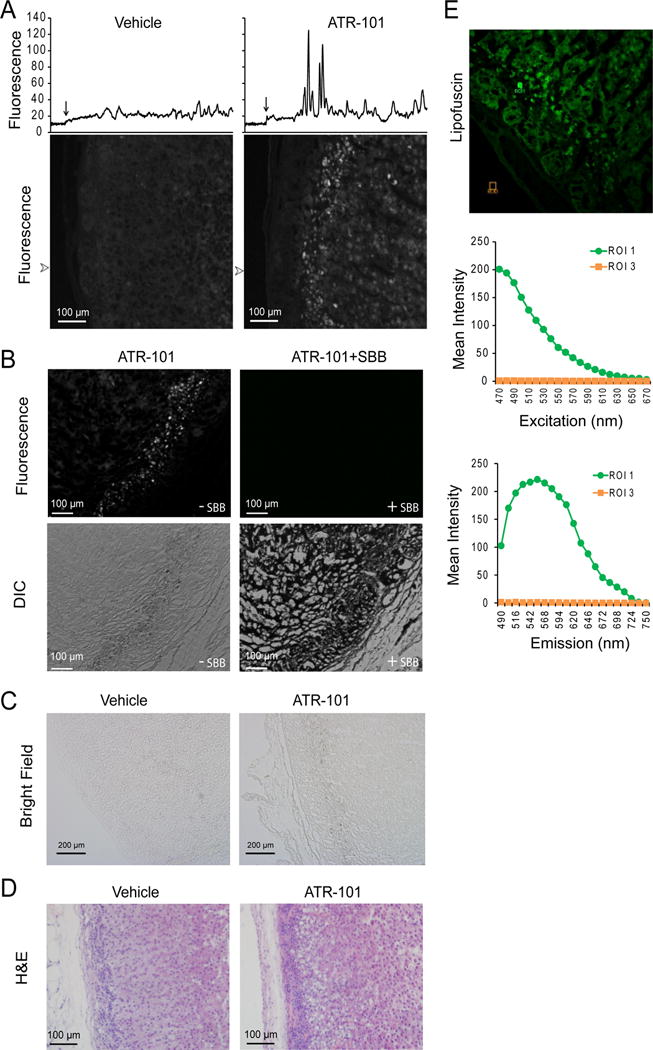
(A) ATR-101 administration to guinea pigs causes autofluorescence of the zona fasciculata. The panels show autofluorescence images of sections of the adrenal cortex from guinea pigs that were administered vehicle or ATR-101 for two weeks. The traces above the images show the fluorescence intensities along lines traversing from the capsule through the zona reticularis at the arrowheads to the left of the images. The location of the edge of the capsule is shown by an arrow above each trace.
(B) The zona fasciculata layer of guinea pigs administered ATR-101 contains granules and its autofluorescence is quenched by Sudan Black staining. The upper panels show autofluorescence and the lower panels show differential interference contrast images of adjacent sections of the adrenal cortex from a guinea pig that was administered ATR-101 for two weeks. The sections on the right were stained using Sudan Black, and the sections on the left were unstained.
(C) The granules in the zona fasciculata layer of guinea pigs administered ATR-101 are pigmented. The panels show transmitted light images of sections of the adrenal cortex from guinea pigs that were administered vehicle or ATR-101 for two weeks.
(D) The autofluorescent zona fasciculata layer of guinea pigs that were administered ATR-101 overlaps a region of adrenalytic vacuolation. The panels show images of H&E stained sections of the adrenal cortex from guinea pigs that were administered ATR-101 or vehicle for two weeks.
(E) The spectrum of the autofluorescence in adrenals from guinea pigs that were administered ATR-101. The autofluorescence excitation and emission spectra of the regions indicated in the image on the left were analyzed using a Leica SP5X Inverted 2-Photon FLIM Confocal microscope.
The images shown in each panel are representative of adrenacortical sections from 6 guinea pigs that were administered ATR-101 (0.1 g/kg/day po) and from 5 control guinea pigs that were administered vehicle.
Tissue autofluorescence with pigmented granules that is quenched by Sudan Black is characteristic of oxidized lipids known as lipofuscin. The spectrum of the autofluorescent foci in the adrenals from guinea pigs that were administered ATR-101 was characteristic of lipofuscins (Fig. 10E) (Georgakopoulou, et al. 2013). No autofluorescence or granules were observed in the adrenal cortex from guinea pigs sacrificed 6–8 h after the administration of single dose (500 mg/kg po) of ATR-101, suggesting that lipofuscin accumulation required an extended period of ATR-101 exposure. The layer of autofluorescent foci overlapped with the region of the zona fasciculata that exhibited adrenalytic damage in guinea pigs that were administered ATR-101 (Fig. 10D). The overlap between these regions implicates reactive oxygen species as a cause of the adrenalytic activity of ATR-101.
ATR-101 administration had no detectable effect on the low level of autofluorescence or on the integrity of the adrenal cortex in young rats (data not shown). ATR-101 also had no detectable effect on the autofluorescence of the adrenal cortex of old rats, which had scattered autofluorescent cells in the zona reticularis. Changes in the morphology of the adrenal cortex were observed in some of the old rats that were administered ATR-101. These changes were not as extensive or reproducible as those that were observed in guinea pigs. Also, whereas adrenalytic effects of ATR-101 in guinea pigs were observed consistently at a dose of 0.1 g/kg/day po, as reported previously (Dominick et al. 1993a), the variable effects of ATR-101 on the adrenal cortex of rats were observed at a dose of 1 g/kg/day po. The difference between the effects of ATR-101 on lipofuscin accumulation in the adrenal cortex of guinea pigs versus rats correlated with the difference between the adrenalytic effects of ATR-101 in these species (Reindel et al. 1994).
Discussion
The genetic and epigenetic causes of adrenocortical carcinoma are complex (Assie et al. 2014; Fonseca, et al. 2012; Juhlin et al. 2015; Pinto et al. 2015). Clinical trials of molecularly targeted agents have not demonstrated therapeutic benefit for a majority of the patients (Fassnacht, et al. 2015; O’Sullivan, et al. 2014; Quinkler, et al. 2008; Saif, et al. 2012).
ATR-101 counteracted the establishment and growth of xenografts by ACC-derived H295R cells in SCID mice, but it did not shrink established xenografts. The lower efficacy of ATR-101 against established xenografts could be due to changes in ATR-101 access to, or the metabolism of, H295R cells at different stages of xenograft formation. Compounds with a higher efficacy and potency against established xenografts could have better prospects of therapeutic benefit.
Multiple independent lines of evidence implicate the inhibition of oxidative phosphorylation in the molecular mechanism for ATR-101 cytotoxicity. ATR-101 inhibited F1F0-ATP synthase and ATP synthesis in isolated mitochondria and elevated the mitochondrial membrane potential and depleted ATP concurrently in cells. This mitochondrial dysfunction caused the release of reactive oxygen and triggered cytochrome c release, resulting in caspase-mediated apoptosis. The cytotoxicity of ATR-101 was suppressed by antioxidants as well as by glycolytic precursors. Both ATP depletion and the release of reactive oxygen species are likely to contribute to ATR-101 cytotoxicity (Fig. 9G).
Similar ATR-101 concentrations were required for the inhibition of oxidative phosphorylation in isolated mitochondria and for the elevation of mitochondrial membrane potential and release of reactive oxygen in cells. The concentration of ATR-101 in serum that was required for xenograft suppression in mice was similar to the concentration of ATR-101 that inhibited mitochondrial functions in cells and in vitro. The effects of ATR-101 on mitochondrial fragmentation and their functions in cultured cells corresponded with its effects on mitochondrial morphology and the ATP levels in the adrenal cortex (Dominick et al. 1993a). ATR-101 increased the reactive oxygen levels in both cultured cells and in the zona fasciculata. It is likely that the same mechanisms mediated these effects of ATR-101 in cultured cells and in vivo.
Many inhibitors of electron transport as well as of F1F0-ATPase cause cell death under conditions that require respiration, but not under glycolytic conditions (Marroquin et al. 2007; Poli, et al. 2013; Tettamanti, et al. 2008). Numerous inhibitors of oxidative phosphorylation have been investigated as potential anti-cancer agents (Rohlena, et al. 2013). A majority of these agents have adverse effects that are likely to be due to mitochondrial dysfunctions in normal tissues. The inhibition of mitochondrial functions and the release of reactive oxygen have many effects on carcinogenesis and could have both beneficial and adverse effects for ACC patients (Kuznetsov, et al. 2011; Ralph, et al. 2010).
Many compounds that are in clinical use or in trials for the treatment ACC cause mitochondrial dysfunctions in cancer cell lines (Andre, et al. 2002; Bull, et al. 2012; Hescot, et al. 2013; Marullo, et al. 2013). The mitochondria of the adrenal cortex have unique functions, including corticosteroid biosynthesis, making it plausible that the inhibition of mitochondrial functions can selectively kill cells of adrenocortical origin. Based on the selective oxidative damage caused by ATR-101 in the adrenal cortex of guinea pigs and its apoptotic effects on ACC-derived cells in culture and in xenografts, we speculate that this compound can disrupt mitochondrial functions in tumors of adrenocortical origin.
The close correspondence between the layer of the adrenal cortex that accumulates autofluorescent lipofuscin and that which is subject to adrenalytic damage in guinea pigs that are administered ATR-101 suggests that the adrenalytic activity of ATR-101 involves lipid oxidation. The ATR-101 dependent accumulation of lipofuscin in the guinea pig adrenal cortex, but not in the rat adrenal cortex, is consistent with the species-specific differences in the adrenalytic activity of ATR-101 (Reindel et al. 1994). The mechanisms that cause the species-specific differences in the adrenalytic activity of ATR-101 remain unknown.
The difference in lipofuscin accumulation and adrenalytic damage between guinea pigs and rats that were administered ATR-101 correlates with the difference in the α-tocopherol concentrations of rat and guinea pig adrenals (Burczynski, et al. 1999). Mice that are fed a tocopherol-deficient diet have higher lipofuscin levels in the adrenal cortex, but not in the brain (Davies, et al. 1987). Microsomes from the outer zone of the adrenal cortex of guinea pigs that are fed a tocopherol-deficient diet have higher levels of lipid peroxidation (Staats, et al. 1988). Chemical depletion of tocopherols from microsomes isolated from the human adrenal cortex also increases lipid peroxidation (Takayanagi, et al. 1986). It is therefore likely that lipid oxidation in the adrenal cortex is a general consequence of conditions that increase the level of reactive oxygen in this tissue.
Supplementary Material
Acknowledgments
We thank Wendell Wierenga, Robert Sliskovic and members of the Parke-Davis Pharmaceuticals team that developed PD132301-02 for their insights that were instrumental for initiation of the project; Gary Hammer for assistance with xenografts; Duxin Sun for pharmacokinetic analysis; Veronica Burns and other members of the Kerppola laboratory for critical discussions.
Funding
The research was supported by the National Institute on Drug Abuse (DA030339) and by the National Center for Advancing Translational Sciences (UL1RR024986).
Footnotes
Declaration of interest
The Raili Emilia Kerppola Estate owned shares in Atterocor, Inc, now Millendo, Inc. Tom K. Kerppola was a co-inventor of the use of ATR-101 for the treatment of adrenocortical carcinoma, which was licensed for development by Atterocor, Inc., now Millendo, Inc.
Author contributions
Y. Cheng designed and performed most of the experiments, analyzed the data, produced the figures and drafted the manuscript.
R. E. Kerppola proposed the investigation of PD132301-02 for the treatment of ACC.
T. K. Kerppola designed the study, performed some of the experiments, interpreted the results and wrote the paper.
References
- Andre N, Carre M, Brasseur G, Pourroy B, Kovacic H, Briand C, Braguer D. Paclitaxel targets mitochondria upstream of caspase activation in intact human neuroblastoma cells. FEBS Lett. 2002;532:256–260. doi: 10.1016/s0014-5793(02)03691-8. [DOI] [PubMed] [Google Scholar]
- Assie G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, Rene-Corail F, et al. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet. 2014;46:607–612. doi: 10.1038/ng.2953. [DOI] [PubMed] [Google Scholar]
- Barlaskar FM, Spalding AC, Heaton JH, Kuick R, Kim AC, Thomas DG, Giordano TJ, Ben-Josef E, Hammer GD. Preclinical targeting of the type I insulin-like growth factor receptor in adrenocortical carcinoma. J Clin Endocrinol Metab. 2009;94:204–212. doi: 10.1210/jc.2008-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergenstal DH, L M, Moy RH, et al. Regression of adrenal cancer and suppression of adrenal function in man by o, p’ DDD. Transactions of the Association of American Physicians. 1959;72:10. [Google Scholar]
- Birringer M, EyTina JH, Salvatore BA, Neuzil J. Vitamin E analogues as inducers of apoptosis: structure-function relation. Br J Cancer. 2003;88:1948–1955. doi: 10.1038/sj.bjc.6600981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull VH, Rajalingam K, Thiede B. Sorafenib-induced mitochondrial complex I inactivation and cell death in human neuroblastoma cells. J Proteome Res. 2012;11:1609–1620. doi: 10.1021/pr200790e. [DOI] [PubMed] [Google Scholar]
- Burczynski JM, Hayes JR, Longhurst PA, Colby HD. Species differences in adrenal lipid peroxidation: role of alpha-tocopherol. Free Radic Biol Med. 1999;26:987–991. doi: 10.1016/s0891-5849(98)00289-5. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Liu X, Zhang S, Lin Y, Yang J, Zhang C. MicroRNA-21 protects against the H(2)O(2)-induced injury on cardiac myocytes via its target gene PDCD4. J Mol Cell Cardiol. 2009;47:5–14. doi: 10.1016/j.yjmcc.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creemers SG, Hofland L, Korpershoek E, Franssen GJ, van Kemenade FJ, de Herder WW, Feelders RA. Future directions in the diagnosis and medical treatment of adrenocortical carcinoma. Endocr Relat Cancer. 2015 doi: 10.1530/ERC-15-0452. [DOI] [PubMed] [Google Scholar]
- Davies I, Davidson Y, Fotheringham AP. The effect of vitamin E deficiency on the induction of age pigment in various tissues of the mouse. Exp Gerontol. 1987;22:127–137. doi: 10.1016/0531-5565(87)90047-7. [DOI] [PubMed] [Google Scholar]
- Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49:717–727. doi: 10.1161/01.HYP.0000258594.87211.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominick MA, Bobrowski WA, MacDonald JR, Gough AW. Morphogenesis of a zone-specific adrenocortical cytotoxicity in guinea pigs administered PD 132301-2, an inhibitor of acyl-CoA:cholesterol acyltransferase. Toxicol Pathol. 1993a;21:54–62. doi: 10.1177/019262339302100107. [DOI] [PubMed] [Google Scholar]
- Dominick MA, McGuire EJ, Reindel JF, Bobrowski WF, Bocan TM, Gough AW. Subacute toxicity of a novel inhibitor of acyl-CoA: cholesterol acyltransferase in beagle dogs. Fundam Appl Toxicol. 1993b;20:217–224. doi: 10.1006/faat.1993.1029. [DOI] [PubMed] [Google Scholar]
- Else T, Kim AC, Sabolch A, Raymond VM, Kandathil A, Caoili EM, Jolly S, Miller BS, Giordano TJ, Hammer GD. Adrenocortical carcinoma. Endocr Rev. 2014;35:282–326. doi: 10.1210/er.2013-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassnacht M, Berruti A, Baudin E, Demeure MJ, Gilbert J, Haak H, Kroiss M, Quinn DI, Hesseltine E, Ronchi CL, et al. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: a double-blind, randomised, phase 3 study. Lancet Oncol. 2015;16:426–435. doi: 10.1016/S1470-2045(15)70081-1. [DOI] [PubMed] [Google Scholar]
- Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, Welin S, Schade-Brittinger C, Lacroix A, Jarzab B, et al. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med. 2012;366:2189–2197. doi: 10.1056/NEJMoa1200966. [DOI] [PubMed] [Google Scholar]
- Fonseca AL, Kugelberg J, Starker LF, Scholl U, Choi M, Hellman P, Akerstrom G, Westin G, Lifton RP, Bjorklund P, et al. Comprehensive DNA methylation analysis of benign and malignant adrenocortical tumors. Genes Chromosomes Cancer. 2012;51:949–960. doi: 10.1002/gcc.21978. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Scorrano L. Measuring mitochondrial shape changes and their consequences on mitochondrial involvement during apoptosis. Methods Mol Biol. 2007a;372:405–420. doi: 10.1007/978-1-59745-365-3_29. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007b;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- Galli F, Stabile AM, Betti M, Conte C, Pistilli A, Rende M, Floridi A, Azzi A. The effect of alpha- and gamma-tocopherol and their carboxyethyl hydroxychroman metabolites on prostate cancer cell proliferation. Arch Biochem Biophys. 2004;423:97–102. doi: 10.1016/j.abb.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Georgakopoulou EA, Tsimaratou K, Evangelou K, Fernandez Marcos PJ, Zoumpourlis V, Trougakos IP, Kletsas D, Bartek J, Serrano M, Gorgoulis VG. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging (Albany NY) 2013;5:37–50. doi: 10.18632/aging.100527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman LS, Wintrobe MM, et al. Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. J Am Med Assoc. 1946;132:126–132. doi: 10.1001/jama.1946.02870380008004. [DOI] [PubMed] [Google Scholar]
- Hahner S, Fassnacht M. Mitotane for adrenocortical carcinoma treatment. Curr Opin Investig Drugs. 2005;6:386–394. [PubMed] [Google Scholar]
- Hescot S, Slama A, Lombes A, Paci A, Remy H, Leboulleux S, Chadarevian R, Trabado S, Amazit L, Young J, et al. Mitotane alters mitochondrial respiratory chain activity by inducing cytochrome c oxidase defect in human adrenocortical cells. Endocr Relat Cancer. 2013;20:371–381. doi: 10.1530/ERC-12-0368. [DOI] [PubMed] [Google Scholar]
- Juhlin CC, Goh G, Healy JM, Fonseca AL, Scholl UI, Stenman A, Kunstman JW, Brown TC, Overton JD, Mane SM, et al. Whole-exome sequencing characterizes the landscape of somatic mutations and copy number alterations in adrenocortical carcinoma. J Clin Endocrinol Metab. 2015;100:E493–502. doi: 10.1210/jc.2014-3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junquero D, Bruniquel F, N’Guyen X, Autin JM, Patoiseau JF, Degryse AD, Colpaert FC, Delhon A. F 12511, a novel ACAT inhibitor, and atorvastatin regulate endogenous hypercholesterolemia in a synergistic manner in New Zealand rabbits fed a casein-enriched diet. Atherosclerosis. 2001;155:131–142. doi: 10.1016/s0021-9150(00)00559-1. [DOI] [PubMed] [Google Scholar]
- Kerkhofs TM, Ettaieb MH, Hermsen IG, Haak HR. Developing treatment for adrenocortical carcinoma. Endocr Relat Cancer. 2015;22:R325–338. doi: 10.1530/ERC-15-0318. [DOI] [PubMed] [Google Scholar]
- Kotsuma M, Tokui T, Freudenthaler S, Nishimura K. Effects of ketoconazole and quinidine on pharmacokinetics of pactimibe and its plasma metabolite, R-125528, in humans. Drug Metab Dispos. 2008;36:1505–1511. doi: 10.1124/dmd.108.021394. [DOI] [PubMed] [Google Scholar]
- Krause BR, Black A, Bousley R, Essenburg A, Cornicelli J, Holmes A, Homan R, Kieft K, Sekerke C, Shaw-Hes MK, et al. Divergent pharmacologic activities of PD 132301-2 and CL 277,082, urea inhibitors of acyl-CoA:cholesterol acyltransferase. J Pharmacol Exp Ther. 1993;267:734–743. [PubMed] [Google Scholar]
- Kuznetsov AV, Margreiter R, Amberger A, Saks V, Grimm M. Changes in mitochondrial redox state, membrane potential and calcium precede mitochondrial dysfunction in doxorubicin-induced cell death. Biochim Biophys Acta. 2011;1813:1144–1152. doi: 10.1016/j.bbamcr.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Lindhe O, Skogseid B. Mitotane effects in a H295R xenograft model of adjuvant treatment of adrenocortical cancer. Horm Metab Res. 2010;42:725–730. doi: 10.1055/s-0030-1261923. [DOI] [PubMed] [Google Scholar]
- Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol Sci. 2007;97:539–547. doi: 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS, Doetsch PW. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS One. 2013;8:e81162. doi: 10.1371/journal.pone.0081162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiner VL, Cases S, Myers HM, Sande ER, Bellosta S, Schambelan M, Pitas RE, McGuire J, Herz J, Farese RV., Jr Disruption of the acyl-CoA:cholesterol acyltransferase gene in mice: evidence suggesting multiple cholesterol esterification enzymes in mammals. Proc Natl Acad Sci U S A. 1996;93:14041–14046. doi: 10.1073/pnas.93.24.14041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuzil J, Weber T, Schroder A, Lu M, Ostermann G, Gellert N, Mayne GC, Olejnicka B, Negre-Salvayre A, Sticha M, et al. Induction of cancer cell apoptosis by alpha-tocopheryl succinate: molecular pathways and structural requirements. FASEB J. 2001;15:403–415. doi: 10.1096/fj.00-0251com. [DOI] [PubMed] [Google Scholar]
- Nishimura J, Ohmichi K, Wato E, Saito T, Takashima K, Tanaka T, Hiwatashi Y, Kobayashi K, Tsujimoto T, Asahiyama M, et al. Effects of compound X, a novel potent inhibitor of acyl-coenzyme A:cholesterol O-acyltransferase, on the adrenal gland of rats. Exp Toxicol Pathol. 2013;65:961–971. doi: 10.1016/j.etp.2013.01.010. [DOI] [PubMed] [Google Scholar]
- O’Sullivan C, Edgerly M, Velarde M, Wilkerson J, Venkatesan AM, Pittaluga S, Yang SX, Nguyen D, Balasubramaniam S, Fojo T. The VEGF inhibitor axitinib has limited effectiveness as a therapy for adrenocortical cancer. J Clin Endocrinol Metab. 2014;99:1291–1297. doi: 10.1210/jc.2013-2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto EM, Chen X, Easton J, Finkelstein D, Liu Z, Pounds S, Rodriguez-Galindo C, Lund TC, Mardis ER, Wilson RK, et al. Genomic landscape of paediatric adrenocortical tumours. Nat Commun. 2015;6:6302. doi: 10.1038/ncomms7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli G, Guasti D, Rapizzi E, Fucci R, Canu L, Bandini A, Cini N, Bani D, Mannelli M, Luconi M. Morphofunctional effects of mitotane on mitochondria in human adrenocortical cancer cells. Endocr Relat Cancer. 2013;20:537–550. doi: 10.1530/ERC-13-0150. [DOI] [PubMed] [Google Scholar]
- Quinkler M, Hahner S, Wortmann S, Johanssen S, Adam P, Ritter C, Strasburger C, Allolio B, Fassnacht M. Treatment of advanced adrenocortical carcinoma with erlotinib plus gemcitabine. J Clin Endocrinol Metab. 2008;93:2057–2062. doi: 10.1210/jc.2007-2564. [DOI] [PubMed] [Google Scholar]
- Ralph SJ, Rodriguez-Enriquez S, Neuzil J, Saavedra E, Moreno-Sanchez R. The causes of cancer revisited: “mitochondrial malignancy” and ROS-induced oncogenic transformation - why mitochondria are targets for cancer therapy. Mol Aspects Med. 2010;31:145–170. doi: 10.1016/j.mam.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Reindel JF, Dominick MA, Bocan TM, Gough AW, McGuire EJ. Toxicologic effects of a novel acyl-CoA:cholesterol acyltransferase inhibitor in cynomolgus monkeys. Toxicol Pathol. 1994;22:510–518. doi: 10.1177/019262339402200505. [DOI] [PubMed] [Google Scholar]
- Robertson DG, Breider MA, Milad MA. Preclinical safety evaluation of avasimibe in beagle dogs: an ACAT inhibitor with minimal adrenal effects. Toxicol Sci. 2001;59:324–334. doi: 10.1093/toxsci/59.2.324. [DOI] [PubMed] [Google Scholar]
- Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci U S A. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlena J, Dong LF, Neuzil J. Targeting the mitochondrial electron transport chain complexes for the induction of apoptosis and cancer treatment. Curr Pharm Biotechnol. 2013;14:377–389. doi: 10.2174/1389201011314030011. [DOI] [PubMed] [Google Scholar]
- Saif MW, Fallon B, Syrigos KN. Is there a role of targeted agents in the management of adrenocortical cancers? Case Rep Endocrinol. 2012;2012:875764. doi: 10.1155/2012/875764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliskovic DR, Picard JA, O’Brien PM, Liao P, Roark WH, Roth BD, Anderson MA, Mueller SB, Bocan TM, Bousley RF, et al. alpha-Substituted malonester amides: tools to define the relationship between ACAT inhibition and adrenal toxicity. J Med Chem. 1998;41:682–690. doi: 10.1021/jm970560h. [DOI] [PubMed] [Google Scholar]
- Sliskovic DR, White AD. Therapeutic potential of ACAT inhibitors as lipid lowering and anti-atherosclerotic agents. Trends Pharmacol Sci. 1991;12:194–199. doi: 10.1016/0165-6147(91)90546-5. [DOI] [PubMed] [Google Scholar]
- Staats DA, Lohr DP, Colby HD. Effects of tocopherol depletion on the regional differences in adrenal microsomal lipid peroxidation and steroid metabolism. Endocrinology. 1988;123:975–980. doi: 10.1210/endo-123-2-975. [DOI] [PubMed] [Google Scholar]
- Takayanagi R, Kato K, Ibayashi H. Relative inactivation of steroidogenic enzyme activities of in vitro vitamin E-depleted human adrenal microsomes by lipid peroxidation. Endocrinology. 1986;119:464–469. doi: 10.1210/endo-119-2-464. [DOI] [PubMed] [Google Scholar]
- Terzolo M, Zaggia B, Allasino B, De Francia S. Practical treatment using mitotane for adrenocortical carcinoma. Curr Opin Endocrinol Diabetes Obes. 2014;21:159–165. doi: 10.1097/MED.0000000000000056. [DOI] [PubMed] [Google Scholar]
- Tettamanti G, Malagoli D, Ottaviani E, de Eguileor M. Oligomycin A and the IPLB-LdFB insect cell line: actin and mitochondrial responses. Cell Biol Int. 2008;32:287–292. doi: 10.1016/j.cellbi.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Trivedi BK, Purchase TS, Holmes A, Augelli-Szafran CE, Essenburg AD, Hamelehle KL, Stanfield RL, Bousley RF, Krause BR. Inhibitors of acyl-CoA:cholesterol acyltransferase (ACAT). 7. Development of a series of substituted N-phenyl-N′-[(1-phenylcyclopentyl)methyl]ureas with enhanced hypocholesterolemic activity. J Med Chem. 1994;37:1652–1659. doi: 10.1021/jm00037a016. [DOI] [PubMed] [Google Scholar]
- van Erp NP, Guchelaar HJ, Ploeger BA, Romijn JA, Hartigh J, Gelderblom H. Mitotane has a strong and a durable inducing effect on CYP3A4 activity. Eur J Endocrinol. 2011;164:621–626. doi: 10.1530/EJE-10-0956. [DOI] [PubMed] [Google Scholar]
- Vernetti LA, MacDonald JR, Wolfgang GH, Dominick MA, Pegg DG. ATP depletion is associated with cytotoxicity of a novel lipid regulator in guinea pig adrenocortical cells. Toxicol Appl Pharmacol. 1993;118:30–38. doi: 10.1006/taap.1993.1005. [DOI] [PubMed] [Google Scholar]
- Wolfgang GH, MacDonald JR, Vernetti LA, Pegg DG, Robertson DG. Biochemical alterations in guinea pig adrenal cortex following administration of PD 132301-2, an inhibitor of acyl-CoA:cholesterol acyltransferase. Life Sci. 1995;56:1089–1093. doi: 10.1016/0024-3205(95)00045-8. [DOI] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.