

Abstract
A eubiotic microbiota influences many physiological processes in the metazoan host, including development and intestinal homeostasis. Here, we have shown that the intestinal microbiota modulates inflammatory responses caused by sex steroid deficiency, leading to trabecular bone loss. In murine models, sex steroid deficiency increased gut permeability, expanded Th17 cells, and upregulated the osteoclastogenic cytokines TNFα (TNF), RANKL, and IL-17 in the small intestine and the BM. In germ-free (GF) mice, sex steroid deficiency failed to increase osteoclastogenic cytokine production, stimulate bone resorption, and cause trabecular bone loss, demonstrating that the gut microbiota is central in sex steroid deficiency–induced trabecular bone loss. Furthermore, we demonstrated that twice-weekly treatment of sex steroid–deficient mice with the probiotics Lactobacillus rhamnosus GG (LGG) or the commercially available probiotic supplement VSL#3 reduces gut permeability, dampens intestinal and BM inflammation, and completely protects against bone loss. In contrast, supplementation with a nonprobiotic strain of E. coli or a mutant LGG was not protective. Together, these data highlight the role that the gut luminal microbiota and increased gut permeability play in triggering inflammatory pathways that are critical for inducing bone loss in sex steroid–deficient mice. Our data further suggest that probiotics that decrease gut permeability have potential as a therapeutic strategy for postmenopausal osteoporosis.
Introduction
The exposed surfaces of metazoans are colonized with trillions of bacteria, fungi, and viruses, creating a diverse ecosystem known as the microbiota (1, 2). Of these surfaces, the gastrointestinal tract harbors the largest and most diverse population of microorganisms, which have been shown to elicit positive influences on health. For example, the intestinal microbiota induces the generation of immunomodulatory factors that regulate intestinal inflammatory responses (3), cytoprotection (4), barrier integrity, and homeostasis (5), as well as wound restitution (6). It is now clear that the gut microbiota influences not only the local immune response, but also contributes to immune responses distant from mucosal surfaces, including the CNS, joints, and lungs (3, 7–9). Surprisingly, the intestinal microbiota has also been found to influence bone homeostasis. Indeed, GF mice have increased bone mass, fewer CD4+ T cells and osteoclast precursors in the BM, and lower levels of osteoclastogenic cytokines (10). In addition, antibiotic administration increases bone density in young mice (11, 12), while select probiotics blunt the bone loss that normally ensues following ovariectomy (ovx) (13–15). However, the molecular mechanisms that mediate and function in probiotic-induced host responses have not yet been fully characterized.
Postmenopausal osteoporosis is a common skeletal disease that leads to bone fractures and disability. The disease stems mainly from the cessation of ovarian function, where declining estrogen levels result in the stimulation of bone resorption and — to a lesser extent — bone formation, leading a period of rapid bone loss (16). In addition, many genetic and nongenetic factors intensify the negative impact of estrogen deficiency on the skeleton (17, 18). In mice, the effects of estrogen depletion are modeled by ovx or by treatment with gonadotropin-releasing hormone (GnRH) agonists (19, 20). At the cellular level, the central mechanism by which sex steroid deficiency induces bone loss is via an increase in osteoclast formation (21, 22) and osteoclast lifespan (23, 24). The primary driver of increased osteoclastogenesis is the enhanced production of the immune factors RANKL and TNF (25, 26). Many cell types have been shown to be generators of RANKL, including hemopoietic cells, T cells, B cells, osteoblastic cells, and osteocytes (27–29). In the mouse, osteocytes are probably the most relevant source of RANKL in estrogen-deficient mice (29). In humans, estrogen deficiency is associated with an expansion of RANKL- and TNF-expressing T cells and B cells (27, 28, 30, 31). The contributing influences of IL-1 and TNF in humans was underscored by reports showing that menopause increases the levels of both of these immune factors (32–35), while treatment with TNF and IL-1 inhibitors prevents the increase in bone resorption that results from estrogen deficiency (36). Indeed, the causal role of TNF in ovx-induced bone loss in mice has been demonstrated in multiple models (37–39), where the key mechanisms by which TNF stimulates bone resorption were identified as the potentiation of RANKL activity (40, 41) and induction of Th17 cells (42–44). Th17 cells are an osteoclastogenic population of CD4+ T cells (45, 46) defined by the capacity to produce IL-17 (47). Th17 cells potently induce osteoclastogenesis by secreting IL-17A, RANKL, TNF, IL-1, and IL-6, along with low levels of IFNγ (48–50). IL-17A stimulates the release of RANKL by all osteoblastic cells including osteocytes (42, 51) and increases the osteoclastogenic activity of RANKL by upregulating RANK (52).
We proposed that T cell–produced TNF is required for ovx-induced bone loss based on the findings that no bone loss occurred and no increased TNF production was detected in T cell–null mice or mice depleted of T cells (40, 53). Furthermore bone loss did not occur in mice lacking T cell TNF production (37, 40, 54, 55) or lacking the costimulatory molecule CD40L (53) or in mice treated with CTLA4-Ig, an agent that transmits an inhibitory signal to T cells (56). Clinical evidence corroborates the role of T cell–produced TNF in postmenopausal bone loss (30, 31). Mechanistically, ovx induces proliferation of conventional T cells via an antigen-dependent process (56, 57), thereby increasing the number of CD4+ and CD8+ T cells in the BM and enhancing their production of TNF (37, 53). This process is driven by enhanced antigen presentation by macrophages and DCs (56, 57), although the nature of the involved antigen remains unknown. Because the T cells of ovx mice have similar features to T cells exposed to bacteria (58), we hypothesized that increased exposure to microorganisms provides the antigens required for T cell activation, and ensuing systemic immune responses required for sex steroid deficiency induced bone loss.
Herein, we show that sex steroid–depleted mice raised in GF conditions are protected against trabecular bone loss and that microbial recolonization of GF mice restores the capacity of sex steroid deficiency to induce bone loss. Moreover, we show that sex steroid deficiency resulted in increased epithelial barrier permeability and decreased expression of gut epithelial tight junction proteins. Strikingly, supplementation of the normal flora of sex steroid–depleted mice with the commonly used probiotics Lactobacillus rhamnosus GG (LGG) or VSL#3 significantly tightened intestinal barrier integrity and completely protected mice against sex steroid depletion–induced bone loss.
Results
Germ-free mice are protected against the bone loss induced by sex steroid deficiency.
To investigate the role of the microbiota in sex steroid deficiency–induced bone loss, female C57BL/6J mice were raised in either conventional conditions (Conv.R mice), germ-free conditions (GF mice), or a GF environment until 4 weeks of age and then recolonized with conventional microbiota (Col.GF mice). Since ovx is not technically feasible in GF conditions, sex steroid deficiency was induced pharmacologically. All mice were administered the GnRH agonists Lupron Depot (375 μg every 4 weeks) or control vehicle for 10 weeks, starting at 10 weeks of age. Therefore, Col.GF mice were conventionalized with bacteria 6 weeks before starting the 10-week leuprolide treatment. Leuprolide causes a 4-week stimulation of sex steroid production due to the GnRH agonistic activity of this compound. This is followed by long-term blockade of ovarian sex steroid production due to blunted secretion of follicle-stimulating hormone (FSH) and leuteinizing hormone (LH) secondary to desensitization of GnRH receptors on the pituitary gland. At the end of the treatment period, leuprolide-induced hypogonadism was verified by the finding of a significant decrease in uterus weight compared with mice injected with vehicle only (Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/JCI86062DS1). Assessment of femoral bone structure by in vitro microcomputed tomography (μCT) revealed that trabecular bone volume fraction (BV/TV) was similar in all groups of vehicle-treated mice, although a trend toward higher BV/TV was observed in GF mice (Figure 1, A and B). Importantly, leuprolide failed to induce a loss of BV/TV in GF mice, while it caused a significant decrease in BV/TV in Conv.R mice and Col.GF mice, indicating that sex steroid deprivation induces cortical bone loss via a microbiota-dependent mechanism. By contrast, vehicle-treated GF mice had significantly more cortical volume (Ct.V) and cortical thickness (Ct.Th) compared with vehicle-treated conventionally raised or gnotobiotically colonized mice (Figure 1, C and D). However, leuprolide treatment resulted in a similar loss of Ct.V and Ct.Th in all groups of mice. Assessment of parameters of trabecular structure revealed that vehicle-treated GF mice had higher trabecular number (Tb.N) (Figure 1E), lower trabecular space (Tb.Sp) (Figure 1F), and similar trabecular thickness (Tb.Th) (Figure 1G) as compared with vehicle-treated Conv.R mice and Col.GF mice. Leuprolide treatment did not alter Tb.N and Tb.Sp in any group of mice (Figure 1, E and F) and induced similar changes to Tb.Th in the 3 groups of mice (Figure 1G). Together, these data reveal that the microbiota has specific effects on cortical and trabecular bone volume and structure following sex steroid deprivation.
Figure 1. GF mice are protected against the loss of trabecular bone induced by sex steroid deficiency.

Assessment of femoral bone structure by in vitro μCT in Conv.R mice, GF mice, and Col.GF mice following either leuprolide (375 μg/month) or vehicle control treatment for 10 weeks. (A) Images of representative 3-dimensional μCT reconstructions of examined femurs from each group. (B) BV/TV. (C) Ct.V. (D) Ct.Th. (E) Tb.N. (F) Tb.Sp. (G) Tb.Th. n = 10 mice per group in all panels. Data are expressed as mean ± SEM. All data were normally distributed according to the Shapiro-Wilk normality test and analyzed by 2-way ANOVA and post hoc tests applying the Bonferroni correction for multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 compared with the indicated groups.
GF mice are protected against the increase in bone turnover induced by sex steroid deprivation.
Analysis of femoral cancellous bone by quantitative histomorphometry revealed that the 3 groups of vehicle-treated mice had a similar number of osteoclasts (OCs) per bone surface (N.Oc/BS) and percentage of surfaces covered by OCs (Oc.S/BS), which are 2 indices of bone resorption (Table 1 and Figure 2A). However, leuprolide induced a significant increase in indices of bone resorption in Conv.R and Col.GF mice, but not in GF mice, compared with their respective vehicle controls (Table 1 and Figure 2A), indicating that the microbiota is required for leuprolide to increase bone resorption.
Table 1. Histomorphometric indices of bone turnover in Conv.R mice, GF mice, and Col.GF mice treated with leuprolide (Leu) at 375 μg/month or vehicle (veh).
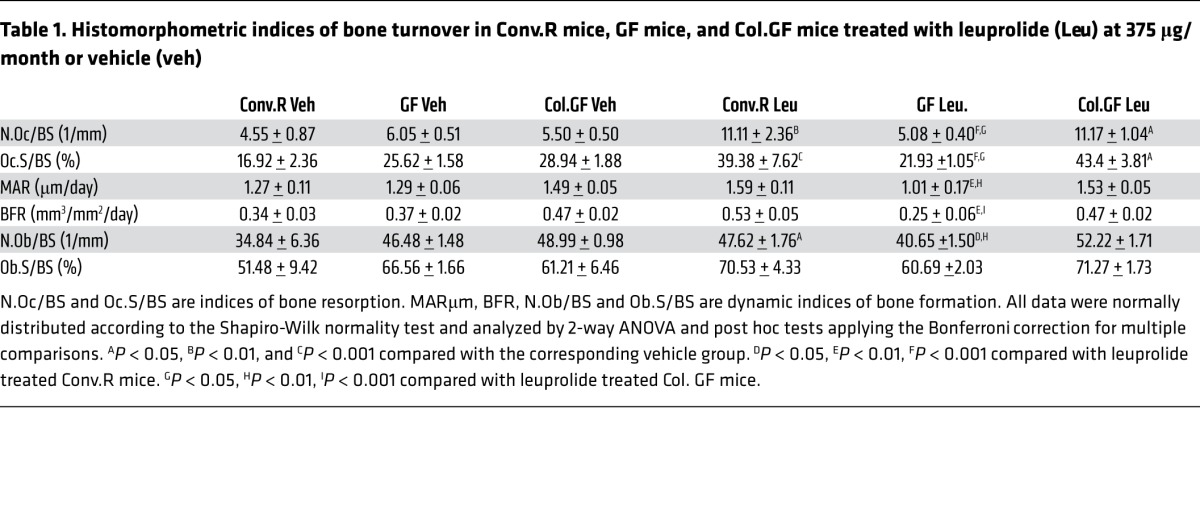
Figure 2. GF mice are protected against the increase in bone turnover induced by sex steroid deprivation.

(A) The images show tartrate-resistant acid phosphatase–stained (TRAP-stained) sections of the distal femur. Original magnification ×40. (B) Images are representative sections displaying the calcein double-fluorescence labeling. Original magnification ×20. (C) Serum levels of type 1 cross-linked CTX. (D) Serum levels of osteocalcin. n = 10 mice per group in all panels. Data are expressed as mean ±SEM. All data were normally distributed according to the Shapiro-Wilk normality test and analyzed by 2-way ANOVA and post hoc tests applying the Bonferroni correction for multiple comparisons. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the corresponding vehicle group.
Determination of dynamic indices of bone formation revealed that the 3 experimental groups of vehicle-treated mice had similar mineral apposition rate (MAR) and bone formation rate (BFR) and that leuprolide treatment did not increase MAR and BFR (Table 1 and Figure 2B). However, leuprolide-treated GF mice exhibited significantly lower MAR and BFR than leuprolide-treated Conv.R and Col.GF mice. Analysis of static indices of bone formation revealed similar findings, as all vehicle-treated mice exhibited no difference in the number of osteoblasts per BS (N.Ob/BS) or in the percentage of surfaces covered by osteoblasts (Ob.S/BS), while leuprolide-treated GF mice exhibited lower N.Ob/BS than leuprolide-treated conventional and colonized mice (Table 1 and Figure 2B). However, leuprolide treatment increased N.Ob/BS in conventional mice but not in GF and colonized mice, while GF mice and control mice had similar Ob.S/BS. Together, the data suggest that sex steroid–depleted mice lacking the microbiota have lower rates of compensatory bone formation than control sex steroid–depleted mice. Measurements of the serum level of C-terminal telopeptide of collagen (CTX), a marker for bone resorption, and of osteocalcin, a marker for bone formation, revealed that the 3 strains of vehicle-treated mice had similar rates of bone turnover (Figure 2, C and D). Leuprolide increased both CTX and osteocalcin levels in conventional and colonized mice but not in GF mice. As a result, leuprolide-treated control mice had higher levels of markers of bone turnover compared with similarly treated GF mice.
The BM and the intestine of germ-free mice are protected against the increase in osteoclastogenic cytokines induced by sex steroid deprivation.
Measurements of BM cytokines reveal that only in conventional and colonized mice did leuprolide increase the expression of the osteoclastogenic cytokines TNF, RANKL, and IL-17, whereas no significant increase in the expression of these cytokines were detected in leuprolide-treated GF mice (Figure 3, A–C). By contrast, leuprolide did not increase IFNγ and IL-4 in all groups of mice (Supplemental Figure 2, A and B). Similarly, leuprolide increased the mRNA levels of Rankl, Tnf, and Il17 in purified BM T cells of Conv.R and Col.GF mice but not in GF mice (Supplemental Figure 3).
Figure 3. The BM and the intestine of GF mice are protected against the increase in osteoclastogenic cytokines induced by sex steroid deprivation.

(A–C) Levels of the osteoclastogenic cytokines TNF, RANKL, and IL-17 in the BM of Conv.R, GF, and Col.GF mice following either leuprolide (375 μg/month) or vehicle control treatment for 10 weeks. (D–F) Relative frequency of BM CD4+TNF+ cells, of CD8+TNF+ cells, and of Th17 cells in mice from experimental groups described in A. (G and H) qPCR analysis measuring transcript levels of the Th17 cell–inducing transcription factors Rora and Rorgt in CD4+ T cells from the BM of mice in experimental groups described in A. (I–K) qPCR analysis measuring transcript levels of TNF, Rankl, and Il17 in the SI of mice in experimental groups described in A. (L–N) Flow cytometry analysis to measure the relative abundance of cytokine-producing CD4+ T cells in the SILP of ovx and sham-operated mice at 4 weeks following surgery. n = 10 mice per group in all panels. Data are expressed as mean ±SEM. All data were normally distributed according to the Shapiro-Wilk normality test. A–K were analyzed by 2-way ANOVA and post hoc tests applying the Bonferroni correction for multiple comparisons. L–N were analyzed by unpaired t tests. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 compared with the indicated group.
In addition, leuprolide treatment upregulated the number of CD4+TNF+ cells, CD8+TNF+ cells, and Th17 cells (CD4+IL-17A+ cells) in Conv.R and Col.GF mice but not in GF mice (Figure 3, D–F). Furthermore, transcript numbers of the IL-17A–inducing transcription factors Rora and Rorgt in BM CD4+ T cells (Figure 3, G and H) were all elevated in conventional and colonized mice — though not in GF mice — following leuprolide treatment. Thus, because the signature inflammatory profile that occurs in response to sex steroid depletion was not observed in GF mice, our data support the hypothesis that gut microbiota–dependent bone loss is caused by microbiota-driven inflammatory signaling.
To support the concept that sex steroid deprivation enhances inflammatory responses initiated by intestinal microbiota, we measured the levels of osteoclastogenic cytokines within tissues of the small intestine (SI). Indeed, similar to responses in the BM, leuprolide treatment increased transcript levels of Tnf, Rankl, and Il17 in the SI of conventional and colonized mice but had no effects in the SI of GF mice (Figure 3, I–K), while it did not increase the mRNA levels of Ifng and Il4 in all groups (Supplemental Figure 2, C and D).
Since little information is available about the effect of sex steroid deprivation on intestinal cytokine production, in addition to inducing sex steroid deficiency by the administration of Leuprolide, a method feasible in a GF isolator, we also assessed the influence of ovx on intestinal cytokine production in Conv.R mice. Consistent with our findings following the dosing of leuprolide, analysis 4 weeks after surgery revealed that ovx resulted in higher number of TNF-, RANKL-, and IL-17–producing CD4+ T cells in the SI lamina propria (SILP) compared with sham-operated mice (Figure 3, L–N). Thus, we report compelling evidence showing the proinflammatory effects of sex steroid depletion within tissues of the BM and SI.
Sex steroid deprivation increases intestinal permeability.
We investigated the extent to which sex steroid depletion augments inflammation in the intestine by increasing the antigenic load traversing the intestinal barrier. To this end, we first measured transcript levels of a number of the gap junction proteins of the claudin family and of JAM3, which have all been shown to modulate intestinal barrier integrity (59, 60). As compared with Conv.R and Col.GF conditions, vehicle-treated GF mice had lower mRNA levels of claudin 2, 3, and 15, and of Jam3 (Figure 4, A–D), a finding consistent with the capacity of the microbiota to induce gap junction protein gene expression (61). Leuprolide treatment resulted in reduced transcript levels of claudin 2, 3, and 15, and of Jam3 in Conv.R and Col.GF mice but, importantly, did not alter expression levels of these genes in GF mice. In addition, and as expected, serum endotoxin levels — which correlate with gut permeability — were undetectable in all GF mice but were measurable in Conv.R and Col.GF mice (Figure 4E). Leuprolide-treated Conv.R and Col.GF mice displayed higher serum endotoxin levels as compared with those treated with vehicle, confirming that sex steroid deprivation increases gut permeability. To further explore the effects of sex steroid withdrawal on gut permeability, Conv.R ovx and sham-operated mice were gavaged with FITC-dextran 4 weeks after surgery, and 4 hours later, the serum levels of FITC-dextran — an index of gut permeability — were measured (62). Ovx mice had higher serum levels of FITC-dextran than sham-operated mice (Figure 4F), altogether showing that the intestinal barrier permeability is compromised following sex steroid depletion.
Figure 4. Sex steroid deprivation promotes intestinal permeability.

(A–D) qPCR analysis measuring transcript levels of the tight junction proteins claudin 2, claudin 3, claudin 15, and Jam3 in the SI of Conv.R, and GF mice, and in Col.GF mice, following either leuprolide (375 μg/month) or vehicle control treatment for 10 weeks. (E) Measurement of serum endotoxin levels in mice from experimental groups described in A. (F) Measurement of gut permeability in conventional ovx and sham-operated mice. At 4 weeks after surgery, FITC-dextran was administered orally to each experimental subject. Serum FITC-dextran levels were measured 4 hours later. n = 10 mice per group in all panels. Data are expressed as mean ±SEM. All data were normally distributed according to the Shapiro-Wilk normality test. A–E were analyzed by 2-way ANOVA and post hoc tests applying the Bonferroni correction for multiple comparisons. F was analyzed by unpaired t tests. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with the corresponding vehicle or sham group. ND, not detectable. EU, endotoxin units.
Supplementation of the indigenous microbiota with probiotics prevents sex steroid–induced bone loss.
To determine the extent to which supplementation of the normal diet with symbiotic bacteria tightens barrier integrity and influences the rate of bone loss following sex steroid depletion, 10-week-old conventionally raised mice were ovx or sham operated, treated with probiotics or control vehicle for the first 4 weeks after surgery, and then sacrificed. Mice were supplemented by oral gavage with vehicle or purified cultures of the extensively studied probiotic LGG, or with commercially available VSL#3 preparation, which contains 8 strains of live bacteria — Bifidobacterium breve, Bifidobacterium longum, Bifidobacterium infantis, Lactobacillus acidophilus, Lactobacillus plantarum, Lactobacillus paracasei, Lactobacillus bulgaricus, and Streptococcus thermophilus — over a 4-week period. Additional control groups of sham-operated and ovx mice were treated with purified cultures of laboratory E. coli DH5alpha, a strain that does not exert a probiotic effect, and the LGG pili mutant LGG(ΔSpaC), herein referred to as LGGM — a strain with defective epithelial adhesion — which is required for LGG-induced cellular responses (63). Mice were fed 1 × 109 total bacteria at 3.5-day intervals for 4 weeks. No change in murine body weight was detected over this period (Supplemental Figure 1B); however, the expected ovx-associated decrease in uterus weight was confirmed (Supplemental Figure 1C). In vivo prospective μCT measurements of indices of spinal trabecular bone structure revealed that, in sham-operated mice, feeding of LGG or VSL#3 — but not LGGM or E. coli — caused a marked increase in BV/TV compared with baseline and vehicle at both 2 and 4 weeks from surgery (Figure 5A). Strikingly, whereas ovx mice fed with vehicle or E. coli exhibited a significant loss of BV/TV at 2 and 4 weeks, treatment with LGGM resulted in a partial bone sparing effect, and administration of LGG or VSL#3 completely prevented the loss of BV/TV induced by ovx. In fact, at the end of the study, ovx mice treated with LGG or VSL#3 had BV/TV values similar to baseline and to those of vehicle-treated sham mice and significantly higher than those of vehicle-treated ovx mice. By contrast, ovx mice treated with E. coli or LGGM had lower BV/TV values compared with baseline and vehicle-treated sham mice but were not statistically different than those of vehicle-treated ovx mice (Figure 5A). Similarly, the changes in spinal Tb.Th, Tb.N, and Tb.Sp induced by ovx were completely prevented by LGG or VSL#3, partially prevented by LGGM, and not prevented at all by E. coli (Supplemental Figure 4, A–C). Since in vivo measurements of the femur were beyond the capability of our scanner, the distal femur was analyzed by in vitro μCT using samples harvested at sacrifice. This analysis confirmed that LGG and VSL#3 completely prevented the loss of BV/TV induced by ovx, while LGGM and E. coli had no bone sparing activity (Figure 5B). In addition, LGG and VSL#3 had a partial protective effect against the loss of Ct.V induced by ovx (Figure 5C). The changes in Tb.Th, Tb.N, Tb.Sp, and Ct.Th induced by ovx in vehicle-treated mice were also prevented by LGG and VSL#3, partially prevented by LGGM treatment, and not prevented at all by E. coli (Supplemental Figure 4, D–G).
Figure 5. Supplementation of the indigenous microbiota with probiotics prevents sex steroid–induced bone loss.

Conventional ovx and sham-operated mice were supplemented twice a week with 1 × 109 CFU of either VSL#3, LGG, LGG-M, E. coli, or vehicle. Data are expressed as mean ±SEM. (A) In vivo prospective measurements of spine BV/TV by μCT scanning at baseline and 2 and 4 weeks after surgery. n = 10–14 mice per group. Data were normally distributed according to the Shapiro-Wilk normality test and analyzed by ANOVA for repeated measures and post hoc tests applying the Bonferroni correction for multiple comparisons. (B and C) In vitro analysis of femoral BV/TV and Ct.V by μCT scanning. n = 10–14 mice per group. (D and E) Serum levels of type 1 cross-linked CTX and serum levels of osteocalcin. n = 10 mice per group randomly selected from a total of 10–14 mice per group. Data in B–E were normally distributed according to the Shapiro-Wilk normality test and analyzed by 2-way ANOVA and post hoc tests applying the Bonferroni correction for multiple comparisons. †P < 0.05, ††P < 0.01 †††P < 0.001, and ††††P < 0.0001 compared with baseline. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 compared with sham vehicle. #P < 0.05, ##P < 0.01, ###P < 0.001, and ####P < 0.0001 compared with ovx vehicle.
In our studies, markers of bone turnover proved to be more faithful and sensitive predictors to assess the effects of sex steroid deficiency on bone turnover compared with bone histomorphometry. Due to these observations, we assessed the effects of probiotic feeding on bone turnover by measuring the serum levels of CTX and osteocalcin. We found that ovx increased serum CTX levels in mice treated with vehicle, LGGM, and E. coli but not in those fed with LGG and VSL#3 (Figure 5D). As expected, ovx increased the serum levels of osteocalcin in vehicle-treated mice (Figure 5E). However, feeding of LGG or VSL#3 further increased bone formation in ovx mice, while treatment with LGGM or E. coli did not. Importantly, treatment with LGG or VSL#3 also increased osteocalcin levels in sham-operate mice (Figure 5E), demonstrating that probiotics induce a net anabolic effect independent of sex steroid deprivation.
Mechanistic studies revealed that feeding of LGG or VSL#3, but not LGG-M or E. coli, tightened intestinal barrier integrity, thus preventing the increase in gut permeability induced by ovx. This was demonstrated by the finding that ovx mice fed LGG or VSL#3, but not LGGM or E. coli, had lower levels of orally administered FITC-dextran (Figure 6A) and of serum endotoxin (Figure 6B), as well as increased expression of the tight junction proteins claudin 2, 3, and 15 and Jam3 compared with ovx mice treated with vehicle (Figure 6, C–F). In accordance with their effects on barrier function, treatment with LGG or VSL#3 prevented an ovx-induced increase in intestinal mRNA levels of Rankl, Tnf, and Il17 mRNAs, (Figure 7, A–C), as well as BM mRNA levels of Rankl, Tnf, and Il17 (Figure 7, D–F). By contrast, LGGM or E. coli did not prevent the increase in the mRNA levels of cytokines in the SI and the BM. Because of the key role of BM T cells in inducing bone loss in sex steroid–deprived mice, we also assessed the BM T cell production of inflammatory cytokines. This analysis revealed that treatment with LGG or VSL#3, but not E. coli and LGGM, prevented the increase in mRNA levels of Rankl, Tnf, and Il17 mRNAs in purified BM T cells (Supplemental Figure 5).
Figure 6. Supplementation of the indigenous microbiota with probiotics prevents the increase in gut permeability induced by sex steroid depletion.

(A) Measurement of gut permeability in conventional ovx and sham-operated mice. At 4 weeks after surgery, FITC-dextran was administered orally to each experimental subject. Serum FITC-dextran levels were measured 4 hours later. Mice were supplemented twice a week with 1 × 109 CFU total of either VSL#3, LGG, LGG-M, E. coli, or vehicle. (B) Endotoxin levels in the serum of mice from experimental groups described in A. (C–F) qPCR analysis measuring transcript levels of the tight junction proteins claudin 2, claudin 3, claudin 15, and Jam3 in the SI of mice from experimental groups described in A. n = 10 mice per group in all panels randomly selected from a total of 10–14 mice per group. Data are expressed as mean ±SEM. All data were normally distributed according to the Shapiro-Wilk normality test and analyzed by 2-way ANOVA and post hoc tests applying the Bonferroni correction for multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 compared with sham vehicle. ##P < 0.01, ###P < 0.001, and ####P < 0.0001 compared with ovx vehicle.
Figure 7. Supplementation of the indigenous microbiota with probiotics modulates the generation of SI and BM osteoclastogenic cytokines following sex steroid depletion.

(A–C) qPCR analysis measuring transcript levels of Tnf, Rankl, and Il17 in the SI tissue of conventional ovx and sham-operated mice at 4 weeks after surgery. Mice were supplemented twice a week with 1 × 109 CFU total of either VSL#3, LGG, LGG-M, E. coli, or vehicle. (D–F) qPCR analysis measuring transcript levels of Tnf, Rankl, and Il17 in the BM of mice treated as described in A. n = 10 mice per group in all panels randomly selected from a total of 10–14 mice per group. Data are expressed as mean ±SEM. All data were normally distributed according to the Shapiro-Wilk normality test and analyzed by 2-way ANOVA and post hoc tests applying the Bonferroni correction for multiple comparisons. **P < 0.01, ***P < 0.001, and ****P < 0.0001 compared with sham vehicle. #P < 0.05, ##P < 0.01, ###P < 0.001, and ####P < 0.0001 compared with ovx vehicle.
Discussion
We report that sex steroid depletion increases gut permeability and the production of osteoclastogenic cytokines in the intestine and the BM. Mice housed in GF conditions are protected against intestinal and BM inflammatory responses and the loss of trabecular bone induced by sex steroid deficiency. We also show that treatment with probiotics prevents the increase in gut permeability, the production of intestinal and BM cytokines, and the bone loss induced by sex steroid depletion.
We utilized mice raised in GF conditions, an established model for investigating the role of the microbiota in pathologic processes. However, GF mice have intestinal and immunological abnormalities, including fewer T cells and effector T cells skewed toward a Th2 phenotype (3). Because these deviations resolve within 2–3 weeks following colonization with normal flora (3), GF mice that were colonized with the microbiota of conventional mice were included in the study to control for phenotypic aberrations of GF mice not reversed by microbial colonization. In addition, because surgical procedures such as ovx are not technically feasible within a GF isolator, we utilized the GnRH agonist leuprolide to alter the production of ovarian sex steroids; leuprolide has been used previously to induce bone loss in rodents (19, 20).
Since the microbiota could affect the response to sex steroid deprivation of either bone resorption or bone formation, analysis of biochemical and histomorphometric indices of bone turnover was undertaken at the end of the 10-week treatment period. Our data revealed that leuprolide increased bone resorption in conventional and colonized mice but not in GF mice. Serum osteocalcin, a biochemical index of the overall rate of bone formation in the entire skeleton, was differentially affected by leuprolide in GF mice and control mice. By contrast, histomorphometric indices of bone formation, which reflect only the status of the sampled region, were scarcely affected by leuprolide. However, both methods revealed that GF sex steroid–depleted mice had lower bone formation as compared with sex steroid–depleted mice with intestinal microbiota. Therefore, the microbiota is required for sex steroid deficiency to increase bone resorption and some aspects of bone formation.
Postmenopausal bone loss is regarded as an inflammatory condition because of the causal role of the immune system (21) and the presence of increased BM levels of inflammatory cytokines (26). Among the mechanisms responsible for the bone loss induced by surgical menopause in mice and humans is an expansion in the BM of activated T cells that produce TNF (30, 31, 37, 40). In the mouse, this phenomenon results from enhanced thymic-dependent differentiation of BM-derived progenitors and IFNγ-driven peripheral expansion of mature T cells (57, 64). Ovx also increases the population of premature senescent CD4+CD28– T cells (65), a lineage that produces high levels of TNF. Another factor responsible for the bone loss induced by estrogen deficiency is IL-17, the signature cytokine of Th17 cells (47), which are an osteoclastogenic subset of CD4+ T cells (45, 46). Intriguingly, postmenopausal women with osteoporosis exhibit elevated levels of serum IL-17 (66–68). In mice, ovx expands Th17 cells by promoting the differentiation of naive CD4+ T cells into mature Th17 cells (69), a phenomenon driven by cytokines such as TGFβ, IL-6, IL-1β, and TNF (43, 44, 47, 70) — factors that are all disregulated by estrogen deficiency (25). In addition, Th17 differentiation is inhibited by estrogen via a direct effect on CD4+ T cells mediated by estrogen receptor α (ERα) (71). The importance of IL-17 in bone loss is highlighted by the fact that silencing of IL-17R (72) or treatment with an anti–IL-17 antibody (73) prevent ovx-induced bone loss. In our study, we report that, in GF mice, sex steroid deficiency does not expand TNF-producing T cells and does not increase the production of TNF in the BM and the gut. Moreover, in the absence of microbiota, no increase in IL-17 production occurred in the BM and tissues of the SI following sex steroid deficiency. Taken together, the present findings indicate that the microbiota is required for sex steroid deprivation to induce bone loss because it drives the expansion of conventional T cells and Th17 cells, thereby increasing their production of TNF and IL-17.
Importantly, we found sex steroid deficiency to alter cytokine levels not only in the BM, but also within tissues of the SI. These tissues are in intimate contact with the gut luminal microbiota and constantly sample and respond to the antigenic load within the intestinal lumen (74). The detected intestinal osteoclastogenic cytokines are produced by immune cells that reside in intestinal subepithelial compartments, presumably in response to live microorganisms residing within the gut luminal content. Still unclear is whether bone loss is induced by gut cytokines that reach the BM, by immune cells activated in the gut that home to the BM, or by BM cells that are activated by foreign antigens of intestinal origin. Irrespective of the specific mechanism involved, our data indicate that the intestinal microbiota is critical for inducing osteoclastogenic cytokine and the resulting bone loss.
The intestinal epithelium forms a tight physiological barrier to separate systemic tissue compartments from the resident flora of the gut lumen. A breakdown in barrier integrity leads to increased bacterial translocation, often resulting in chronic immunogenic disease states. In response to sex steroid deprivation, we detected an increased gut permeability. Our proposed model is that increased permeability allows an expanded range of molecules and potential antigenic load to enter epithelial submucosa, initiating aberrant intestinal and systemic immune responses (75–78). Taken together, the present findings support the proposed role for sex steroids in maintaining a tight physiological barrier between the systemic tissue compartments from the resident flora of the gut lumen. Depletion of sex steroid levels leads to increased gut permeability and offers a positive link between a weakened intestinal barrier and the signature osteoclastogenic cytokine profile associated with osteoporosis.
One of the most prominent features of our studies is the observation that supplementation of the microbiota with known probiotics entirely protected ovx mice against bone loss and caused a net gain in bone mass in sex steroid–replete mice. Consistent with these physiological findings, probiotics also tightened gut epithelial barrier integrity and dampened the levels of osteoclastogenic cytokines following sex steroid depletion. These profound effects were elicited by oral administration of either LGG or VSL#3 to ovx mice with a conventional microflora. These data indicate that both LGG and VSL#3 have dynamic interactions with the epithelial layer of the intestine and actively induce the expression of proteins involved with tightening the gut epithelial barrier. In addition, it is also possible that probiotics function by displacing bacterial species that downregulate tight junctional proteins in the intestinal epithelium.
Control assays in our studies showed that protection against bone loss is bacterial-strain specific. Here, treatment of ovx mice with an attenuated laboratory strain of E. coli DH5alpha exerted no detectable modulatory influence on bone loss or on cytokine production. Intriguingly, treatment of ovx mice with the LGGM partially protected ovx mice from bone loss. The SpaC protein was shown by our research group to be necessary for LGG to bind to the intestinal mucus layer and for LGG to exert its positive influence on gut tissue health (63). The fact that LGGM maintained a degree of protection from bone loss suggests that direct contact with the mucus layer may not be the only mode by which LGG exerts probiotic effects and that other properties of LGG — such as the production of short chain fatty acids, which have been shown to modulate cytokine levels in the intestine — is also a mechanism to consider (79).
LGG has been shown to facilitate stem cell turnover (5), to facilitate wound restitution in the gut (6), to mediate cytoprotective effects in the gut (4), and to facilitate cell proliferation and migration (80, 81). It is also known that permeability along paracellular spaces in the epithelium is controlled by intercellular tight junctional proteins (82, 83) and regulated, at least in part, by the MAPK and PI3 kinase signaling pathways (84). This may suggest that contact of epithelium cells with LGG and VSL#3 is able to compensate for the loss of MAPK activity following sex steroid depletion (85). Because gut epithelial cells express estrogen receptors (86, 87) — and estrogen receptor signaling activates MAPK (85) — one may speculate that sex steroids binding to their receptors in the gut epithelia induce MAPK expression that maintain and/or tighten gut permeability. Sex steroid deficiency would then result in weakening the MAPK pathway expression and barrier integrity, as detected in ovx or leuprolide-treated mice.
A relationship between decreased taxonomic diversity and disease state has been found in many inflammatory (88, 89) and metabolic conditions (90). As a result, decreased diversity is regarded as an indicator of disease state, reflecting a deteriorating habitat and/or depleted resources (91). Interestingly, estrogen depletion has also been associated with decreased α-diversity in the gut microbiome (92, 93), while microbial diversity in the gut transiently increases with administration of probiotics (94). Therefore, we hypothesize that sex steroid deficiency leads to decreased microbiota diversity and that LGG and VSL#3 probiotics restore a greater diversity. Further studies will be required to investigate this hypothesis and to determine whether decreased diversity contributes to the bone loss induced by sex steroid deprivation.
Consistent with our data, reports have shown beneficial effects of probiotics for bone health. Probiotics have been shown to alter the composition or the metabolic activity of the gut microbiota (95), regulating the immune response of the host (96, 97) and enhancing epithelial barrier function (98, 99). Others have reported on the phenomenology that probiotics prevented trabecular bone loss as effectively as the bisphosphonate alendronate (13) and that lactobacilli can prevent the loss of cortical but not trabecular bone and reverses the decrease in Tregs induced by ovx (14). In addition, Lactobacillus reuteri prevented the loss of trabecular bone induced by ovx (15). This probiotic prevented the increase in CD4+ T cells induced by ovx, secreted anti-TNF factors, and modified microbial communities in the ovx mouse gut (15).
In summary, the dysregulation in the interactions between the gut microbiota and the intestinal wall play a pivotal role in inducing inflammation and bone loss in sex steroid–deficient mice. Direct clinical applications of these findings arise because of the availability of pre- and probiotic agents that may prevent bone loss by tightening the gut barrier integrity, thus limiting the production of osteoclastogenic cytokines.
Methods
Animals.
Female C57BL6/J mice were purchased from Taconic. Conv.R mice were maintained under specific pathogen-free conditions and fed sterilized food and autoclaved water ad libitum. GF mice were maintained in a semiflexible isolators (Park Bioservices) under a strict 12-hour light cycle and fed with autoclaved chow diet ad libitum at the GF facility of Georgia State University. GF status was confirmed at the end of the treatment period by fecal DNA extraction using QIAamp DNA Stool Mini Kit (QIAGEN) and subsequent quantitative PCR (qPCR) using 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) primers. Conv.R mice and water were used as positive and negative controls, respectively.
Colonization of GF mice.
The colon and cecal contents of 10-week-old Conv.R mice were homogenized and suspended in 10 ml of sterile PBS. Microbiota reconstitutions were performed by oral gavage with 0.1 ml of suspension and by spreading 2 ml aliquots of suspension on the fur of 4-week-old GF recipients as described (100).
Leuprolide administration.
Lupron Depot (Takeda) or vehicle was injected at doses of 375 μg/mouse s.c. once per month for 3 months starting with 10-week-old mice. Mice were sacrificed after 10 weeks of treatment.
Intestinal permeability assays.
Barrier function was evaluated by measuring in vivo paracellular permeability to fluorescent-labeled dextran as described (101). Briefly, mice were administered 0.5 ml of 22 mg/ml fluorescein isothiocyanate–labeled 4.4-kDa dextran (FD4; Sigma-Aldrich) by oral gavage. Serum was obtained 4 hours after administration by terminal cardiac puncture under isoflurane anesthesia. The serum dextran concentration was calculated by comparing samples with serial dilutions of known standards using a Synergy HT fluorimeter (BioTek) with excitation at 485 nm and emission at 530 nm. Serum endotoxin levels were determined using the Limulus Amebocyte Lysate kit from Associates of Cape Cod Inc. according to the manufacturer’s instructions.
μCT measurements.
μCT scanning and analysis was performed as reported previously (42, 102, 103) using Scanco μCT-40 and Scanco vivaCT 40 scanners. Voxel size was 10.5 μm3 for the in vitro measurements of femurs from Conv.R, GF, and Col.GF mice. For measurements of mice in the probiotics study, voxel sizes were 12 μm3 for the in vitro measurements of femurs and 21.5 μm3 for the in vivo measurements of the spine. For the femoral trabecular region, we analyzed 140 slices, beginning 50 slices below the distal growth plate. Femoral cortical bone was assessed using 80 continuous CT slides located at the femoral midshaft. In vivo measurements of spinal trabecular bone contours along the periosteal surfaces were drawn encompassing 50 slices of the L4 vertebra, starting at the beginning of trabecular bone within the spinal body, as described (39). X-ray tube potential was 70 kVp, and integration time was 350 ms for the in vivo measurements and 200 ms for the in vitro measurements.
Quantitative bone histomorphometry.
This analysis was performed as previously described (42). The measurements, terminology, and units used for histomorphometric analysis were those recommended by the Nomenclature Committee of the American Society for Bone and Mineral Research (104).
IL-17, TNF, RANKL, IFNγ, and IL-4 ELISA.
BM was collected at sacrifice and cultured in RPMI-1640 medium (Invitrogen) supplemented with 10% FBS in the presence of phorbol 12-myristate 13-acetate (20 ng/ml, Sigma-Aldrich) and ionomycin (1 μg/ml, Sigma-Aldrich) at 37°C for 48 hours. Supernatants were collected and assayed for IL-17, TNF, RANKL, IFNγ, and IL-4 by ELISA kits (R&D Systems) according to the manufacturer’s directions.
Markers of bone turnover.
Serum CTX and serum osteocalcin were measured by rodent-specific ELISAs (Immunodiagnostic Systems).
SILP lymphocyte isolation.
Lamina propria lymphocyte isolation was performed as described (105). Briefly, the SI was removed and flushed of fecal content, and intestinal segments containing Peyer’s patches were excised. The intestine was opened longitudinally and cut into 5-mm pieces. Tissues were transferred into a 50-ml conical tube and shaken at 2 g for 20 minutes at 37°C in HBSS medium (Invitrogen) supplemented with 5% FBS (Mediatech Inc., Corning) containing 2 mM EDTA. This process was repeated 2 additional times. The tissue suspension was passed through a strainer, and the remaining intestinal tissue was washed and then minced, transferred into a fresh 50-ml conical tube, and shaken for 20 minutes at 37°C in HBSS plus 5% FBS containing type VIII collagenase (Sigma-Aldrich) at 1.5 mg/ml. The tissue suspension was collected, passed through a strainer, and pelleted by centrifugation at 40 g for 3 minutes. The pellet was suspended in 10 ml HBSS medium and passed through glass wool halfway packed in a 10-ml syringe to remove undigested tissue pieces. The cell suspension was collected in a 50-ml tube and centrifuged at 2 g for 3 minutes. The pellet was suspended in 5 ml HBSS and 5 ml 90% isotonic Percoll and then transferred into a 15-ml tube and mixed by tilting back and forth. The cell content was layered onto 2 ml of 70% isotonic Percoll. The gradient was centrifuged at 112 g for 20 minutes. Cells were collected from the interface area and washed by centrifugation in 10 ml supplemented RPMI-1640 medium supplemented with 5% FBS. The cell pellet was resuspended in 10 ml supplemented RPMI-1640, and cell number was counted.
Flow cytometry.
Single cell suspension of BM, spleen, and SILP cells were prepared in RPMI 1640 culture medium. For intracellular cytokine staining, cells were incubated with phorbol 12-myristate 13-acetate (50 ng/ml, Sigma-Aldrich) and ionomycin (1 μg/ml, Sigma-Aldrich) in the presence of GolgiStop (1 μg/ml, BioLegend) at 37°C for 4 hours. Cells were then stained with FITC anti-TCRβ (clone H57-597, catalog 109206, BioLegend), PerCp/Cy5.5 anti-CD4 (clone RM4-5, catalog 100434, BioLegend), and PE/Cy7 anti-CD8 (clone 53-6.7, catalog 100722, BioLegend) followed by intracellular staining with PE anti–IL-17 (clone TC11-18H10.1, catalog 506904, BioLegend), APC anti-TNF (clone MP6-XT22, catalog 506308, BioLegend), or PE anti-RANKL (clone IK22-5, catalog 510006, BioLegend). All antibodies were obtained from BioLegend. Cells were analyzed by flow cytometry using a LSRII flow cytometer (BD Biosciences) and FlowJo software. Results are expressed as cell frequency (%). The percentage of IL-17+CD4+ T cells, TNF+CD4+ T cells, and TNF+CD8+ cells gated in TCRβ+ cells was quantified in all the groups.
RNA isolation from SI.
A 10-mm intestinal piece of each mouse was collected at sacrifice and completely submerged into 0.5 ml RNAlater (QIAGEN) in a 1.5-ml tube. The tissue was kept at 4°C overnight and then transferred to –80°C for storage. RNA was extracted using the QIAshredder kit and the RNeasy Mini kit (QIAGEN) according to the manufacturer’s directions.
qPCR and murine primers.
RNA levels were quantified by qPCR. RNA was isolated using RNeasy kit (QIAGEN) and cDNA synthesized using Superscript II (Invitrogen) and random hexamers according to the manufacturer’s instructions. Relative abundance of cDNAs was determined by qPCR analysis using the ABI StepOnePlus Real-Time PCR system (Applied Biosystems). The expression levels of murine Rora and Rorgt mRNAs levels were measured in purified BM CD4+ T cells. The expression levels of murine Il17, Tnf, and Rankl were measured in whole BM, purified BM T cells, and SI cells. For the survey of gap junction protein expression, the mRNA levels of claudin 2, claudin 3, claudin 15, and Jam3 were measured in SI. All the primers used were designed by Primer Express Software v3.0.1 (Applied Biosystems). Changes in relative gene expression between vehicle and leuprolide groups were calculated using the 2–ΔCT method with normalization to 18S rRNA. The primers used were as follows: for 18S rRNA, 5′-ATTCGAACGTCTGCCCTATCA-3′ (forward) and 5′-GTCACCCGTGGTCACCATG-3′ (reverse); for murine Il17a, 5′-TGACGCCCACCTACAACATC-3′ (forward) and 5′-CATCATGCAGTTCCGTCAGC-3′ (reverse); for Rora, 5′-GCCTCCTGCCACCTTGAGTA-3′ (forward) and 5′-TCTGCCTTCAGCTTTGCCTC -3′ (reverse); for Rorgt, 5′-TCAGCGTGTCCAAACACTGAG-3′ (forward) and 5′-CGCCAAGGGAGTTAAAGACTT-3′ (reverse); for Rankl, 5′-CCTGATGAAAGGAGGGAGCA-3′ (forward) and 5′-TGGAATTCAGAATTGCCCGA-3′ (reverse); for Tnfa, 5′-AACTCCAGGCGGTGCCTAT-3′ (forward) and 5′-TGCCACAAGCAGGAATGAGA-3′ (reverse); for Ifng, 5′-GGTCCAGCGCCAAGCAT-3′ (forward) and 5′-GCTGGATTCCGGCAACAG-3′ (reverse); for Il4, 5′-GCAGAGACTCTTTCGGGCTTT-3′ (forward) and 5′-CATTCATGGTGCAGCTTATCGA-3′ (reverse); for Cldn2, 5′-TCTCAGCCCTGTTTTCTTTGG-3′ (forward) and 5′-GGCGAGCAGGAAAAGCAA-3′ (reverse); for Cldn3, 5′-TCATCACGGCGCAGATCA-3′ (forward) and 5′-CTCTGCACCACGCAGTTCA-3′ (reverse); for Cldn15, 5′-GGCGGCATCTGTGTCTTCTC-3′ (forward) and 5′-TGGTGGCTGGTTCCTCCTT-3′ (reverse); and for Jam3, 5′-CACTACAGCTGGTACCGCAATG-3′ (forward) and 5′-CTGGGATTGGCTCTGGAATC-3′ (reverse).
Statistics.
Murine data were normally distributed according to the Shapiro-Wilk normality test and analyzed by unpaired 2-tailed t tests, 2-way ANOVA, or ANOVA for repeated measures as appropriate, which included the main effects for animal strain and treatment plus the statistical interaction between animal strain and treatment. When the statistical interaction between animal strain and treatment group was not statistically significant (P > 0.05) nor suggestive of an important interaction (P > 0.10), P values for the main effects tests were reported. When the statistical interaction was statistically significant (P < 0.05) or suggestive of an important interaction, then t tests were used to compare the differences between the treatment means for each animal strain, applying the Bonferroni correction for multiple comparisons. P < 0.05 was considered significant.
Study approval.
All animal procedures were approved by the Institutional Animal Care and Use Committee of Emory University and Georgia State University.
Author contributions
JYL, MNW, JGM, ATG, RMJ, and RP designed the studies. JYL, BC, AMT, CV, TL, JA, and TMD performed the research and analyzed the data. RP and RMJ wrote the manuscript.
Supplementary Material
Acknowledgments
This study was supported by grants from the NIH (R. Pacifici: R01AR54625, R01DK091780, DK007298, and RR028009; J.Y. Li: AR061453; R.M. Jones: R01DK098391; M.N. Weitzmann: R01AR059364, R01AG040013, R01AR068157, and R01AR068157). B. Chassaing is a recipient of the Career Development award from the Crohn’s and Colitis Foundation of America (CCFA). M.N. Weitzmann was also supported by a grant from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development (5I01BX000105).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(6):2049–2063. doi:10.1172/JCI86062.
See the related Commentary beginning on page 2045.
Contributor Information
Jau-Yi Li, Email: jli56@emory.edu.
Benoit Chassaing, Email: bchassaing@gsu.edu.
Chiara Vaccaro, Email: chiara.vaccaro24@gmail.com.
Tao Luo, Email: taoluocn@gmail.com.
Jonathan Adams, Email: jadam12@emory.edu.
M. Neale Weitzmann, Email: mweitzm@emory.edu.
Jennifer G. Mulle, Email: jmulle@emory.edu.
Roberto Pacifici, Email: roberto.pacifici@emory.edu.
References
- 1.Sommer F, Backhed F. The gut microbiota--masters of host development and physiology. Nat Rev Microbiol. 2013;11(4):227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- 2.Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489(7415):242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- 3.Surana NK, Kasper DL. Deciphering the tete-a-tete between the microbiota and the immune system. J Clin Invest. 2014;124(10):4197–4203. doi: 10.1172/JCI72332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones RM, et al. Lactobacilli modulate epithelial cytoprotection through the Nrf2 pathway. Cell Rep. 2015;12(8):1217–1225. doi: 10.1016/j.celrep.2015.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones RM, et al. Symbiotic lactobacilli stimulate gut epithelial proliferation via Nox-mediated generation of reactive oxygen species. EMBO J. 2013;32(23):3017–3028. doi: 10.1038/emboj.2013.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alam A, et al. Redox signaling regulates commensal-mediated mucosal homeostasis and restitution and requires formyl peptide receptor 1. Mucosal Immunol. 2014;7(3):645–655. doi: 10.1038/mi.2013.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2011;108(suppl 1):4615–4622. doi: 10.1073/pnas.1000082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noverr MC, Falkowski NR, McDonald RA, McKenzie AN, Huffnagle GB. Development of allergic airway disease in mice following antibiotic therapy and fungal microbiota increase: role of host genetics, antigen, and interleukin-13. Infect Immun. 2005;73(1):30–38. doi: 10.1128/IAI.73.1.30-38.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu HJ, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32(6):815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sjogren K, et al. The gut microbiota regulates bone mass in mice. J Bone Miner Res. 2012;27(6):1357–1367. doi: 10.1002/jbmr.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho I, et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012;488(7413):621–626. doi: 10.1038/nature11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pytlik M, Folwarczna J, Janiec W. Effects of doxycycline on mechanical properties of bones in rats with ovariectomy-induced osteopenia. Calcif Tissue Int. 2004;75(3):225–230. doi: 10.1007/s00223-004-0097-x. [DOI] [PubMed] [Google Scholar]
- 13.Chiang SS, Pan TM. Antiosteoporotic effects of Lactobacillus -fermented soy skim milk on bone mineral density and the microstructure of femoral bone in ovariectomized mice. J Agric Food Chem. 2011;59(14):7734–7742. doi: 10.1021/jf2013716. [DOI] [PubMed] [Google Scholar]
- 14.Ohlsson C, et al. Probiotics protect mice from ovariectomy-induced cortical bone loss. PLoS One. 2014;9(3): doi: 10.1371/journal.pone.0092368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Britton RA, et al. Probiotic L. reuteri treatment prevents bone loss in a menopausal ovariectomized mouse model. J Cell Physiol. 2014;229(11):1822–1830. doi: 10.1002/jcp.24636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13(7):791–801. doi: 10.1038/nm1593. [DOI] [PubMed] [Google Scholar]
- 17.Riggs BL, Melton LJ., 3rd Evidence for two distinct syndromes of involutional osteoporosis. Am J Med. 1983;75(6):899–901. doi: 10.1016/0002-9343(83)90860-4. [DOI] [PubMed] [Google Scholar]
- 18.Riggs BL, Melton LJ., 3rd Involutional osteoporosis. N Engl J Med. 1986;314(26):1676–1686. doi: 10.1056/NEJM198606263142605. [DOI] [PubMed] [Google Scholar]
- 19.Kurabayashi T, Fujimaki T, Yasuda M, Yamamoto Y, Tanaka K. Time-course of vertebral and femoral bone loss in rats administered gonadotrophin-releasing hormone agonist. J Endocrinol. 1993;138(1):115–125. doi: 10.1677/joe.0.1380115. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, et al. Comparison of the effects of add-back therapy with various natural oestrogens on bone metabolism in rats administered a long-acting gonadotrophin-releasing hormone agonist. J Endocrinol. 2000;165(2):467–473. doi: 10.1677/joe.0.1650467. [DOI] [PubMed] [Google Scholar]
- 21.Weitzmann MN, Pacifici R. Estrogen deficiency and bone loss: an inflammatory tale. J Clin Invest. 2006;116(5):1186–1194. doi: 10.1172/JCI28550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun L, et al. FSH directly regulates bone mass. Cell. 2006;125(2):247–260. doi: 10.1016/j.cell.2006.01.051. [DOI] [PubMed] [Google Scholar]
- 23.Nakamura T, et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130(5):811–823. doi: 10.1016/j.cell.2007.07.025. [DOI] [PubMed] [Google Scholar]
- 24.Krum SA, et al. Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO J. 2008;27(3):535–545. doi: 10.1038/sj.emboj.7601984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pacifici R. Role of T cells in ovariectomy induced bone loss-revisited. J Bone Miner Res. 2012;27(2):231–239. doi: 10.1002/jbmr.1500. [DOI] [PubMed] [Google Scholar]
- 26.Khosla S, Pacifici R. Osteoporosis. Vol. 1. Waltham, Massachusetts, USA: Academic Press; 2013. Estrogen deficiency, postmenopausal osteoporosis, and age-related bone loss. In: Marcus R, Feldman D, Dempster DW, Luckey M, Cauley JA, eds; pp. 1113–1138. 4th ed. [Google Scholar]
- 27.Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Invest. 2003;111(8):1221–1230. doi: 10.1172/JCI17215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taxel P, Kaneko H, Lee SK, Aguila HL, Raisz LG, Lorenzo JA. Estradiol rapidly inhibits osteoclastogenesis and RANKL expression in bone marrow cultures in postmenopausal women: a pilot study. Osteoporos Int. 2008;19(2):193–199. doi: 10.1007/s00198-007-0452-7. [DOI] [PubMed] [Google Scholar]
- 29.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17(10):1235–1241. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D’Amelio P, et al. Estrogen deficiency increases osteoclastogenesis up-regulating T cells activity: a key mechanism in osteoporosis. Bone. 2008;43(1):92–100. doi: 10.1016/j.bone.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 31.Adeel S, et al. Bone loss in surgically ovariectomized premenopausal women is associated with T lymphocyte activation and thymic hypertrophy. J Investig Med. 2013;61(8):1178–1183. doi: 10.231/JIM.0000000000000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pacifici R, et al. Ovarian steroid treatment blocks a postmenopausal increase in blood monocyte interleukin 1 release. Proc Natl Acad Sci U S A. 1989;86(7):2398–2402. doi: 10.1073/pnas.86.7.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pacifici R, et al. Effect of surgical menopause and estrogen replacement on cytokine release from human blood mononuclear cells. Proc Natl Acad Sci U S A. 1991;88(12):5134–5138. doi: 10.1073/pnas.88.12.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cohen-Solal ME, Graulet AM, Denne MA, Gueris J, Baylink D, de Vernejoul MC. Peripheral monocyte culture supernatants of menopausal women can induce bone resorption: involvement of cytokines. J Clin Endocrinol Metab. 1993;77(6):1648–1653. doi: 10.1210/jcem.77.6.8263153. [DOI] [PubMed] [Google Scholar]
- 35.Bernard-Poenaru O, Roux C, Blanque R, Gardner C, de Vemejoul MC, Cohen-Solal ME. Bone-resorbing cytokines from peripheral blood mononuclear cells after hormone replacement therapy: a longitudinal study. Osteoporos Int. 2001;12(9):769–776. doi: 10.1007/s001980170054. [DOI] [PubMed] [Google Scholar]
- 36.Charatcharoenwitthaya N, Khosla S, Atkinson EJ, McCready LK, Riggs BL. Effect of blockade of TNF-α and interleukin-1 action on bone resorption in early postmenopausal women. J Bone Miner Res. 2007;22(5):724–729. doi: 10.1359/jbmr.070207. [DOI] [PubMed] [Google Scholar]
- 37.Roggia C, et al. Up-regulation of TNF-producing T cells in the bone marrow: A key mechanism by which estrogen deficiency induces bone loss in vivo. Proc Natl Acad Sci U S A. 2001;98(24):13960–13965. doi: 10.1073/pnas.251534698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ammann P, et al. Transgenic mice expressing soluble tumor necrosis factor-receptor are protected against bone loss caused by estrogen deficiency. J Clin Invest. 1997;99(7):1699–1703. doi: 10.1172/JCI119333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kimble RB, Bain S, Pacifici R. The functional block of TNF but not of IL-6 prevents bone loss in ovariectomized mice. J Bone Miner Res. 1997;12(6):935–941. doi: 10.1359/jbmr.1997.12.6.935. [DOI] [PubMed] [Google Scholar]
- 40.Cenci S, et al. Estrogen deficiency induces bone loss by enhancing T-cell production of TNF-α. J Clin Invest. 2000;106(10):1229–1237. doi: 10.1172/JCI11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106(12):1481–1488. doi: 10.1172/JCI11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li JY, et al. IL-17A is increased in humans with primary hyperparathyroidism and mediates PTH-induced bone loss in mice. Cell Metab. 2015;22(5):799–810. doi: 10.1016/j.cmet.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen DY, Chen YM, Chen HH, Hsieh CW, Lin CC, Lan JL. Increasing levels of circulating Th17 cells and interleukin-17 in rheumatoid arthritis patients with an inadequate response to anti-TNF-α therapy. Arthritis Res Ther. 2011;13(4): doi: 10.1186/ar3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugita S, Kawazoe Y, Imai A, Yamada Y, Horie S, Mochizuki M. Inhibition of Th17 differentiation by anti-TNF-alpha therapy in uveitis patients with Behcet’s disease. Arthritis Res Ther. 2012;14(3): doi: 10.1186/ar3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sato K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203(12):2673–2682. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361(9):888–898. doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- 47.Basu R, Hatton RD, Weaver CT. The Th17 family: flexibility follows function. Immunol Rev. 2013;252(1):89–103. doi: 10.1111/imr.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Komatsu N, Takayanagi H. Autoimmune arthritis: the interface between the immune system and joints. Adv Immunol. 2012;115:45–71. doi: 10.1016/B978-0-12-394299-9.00002-3. [DOI] [PubMed] [Google Scholar]
- 49.Waisman A. T helper cell populations: as flexible as the skin? Eur J Immunol. 2011;41(9):2539–2543. doi: 10.1002/eji.201141938. [DOI] [PubMed] [Google Scholar]
- 50.Jovanovic DV, et al. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-β and TNF-α, by human macrophages. J Immunol. 1998;160(7):3513–3521. [PubMed] [Google Scholar]
- 51.Kotake S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103(9):1345–1352. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adamopoulos IE, et al. Interleukin-17A upregulates receptor activator of NF-κB on osteoclast precursors. Arthritis Res Ther. 2010;12(1): doi: 10.1186/ar2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li JY, et al. Ovariectomy disregulates osteoblast and osteoclast formation through the T-cell receptor CD40 ligand. Proc Natl Acad Sci U S A. 2011;108(2):768–773. doi: 10.1073/pnas.1013492108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao Y, et al. Estrogen prevents bone loss through transforming growth factor beta signaling in T cells. Proc Natl Acad Sci U S A. 2004;101(47):16618–16623. doi: 10.1073/pnas.0404888101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gao Y, et al. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest. 2007;117(1):122–132. doi: 10.1172/JCI30074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grassi F, et al. Oxidative stress causes bone loss in estrogen-deficient mice through enhanced bone marrow dendritic cell activation. Proc Natl Acad Sci U S A. 2007;104(38):15087–15092. doi: 10.1073/pnas.0703610104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cenci S, et al. Estrogen deficiency induces bone loss by increasing T cell proliferation and lifespan through IFN-gamma-induced class II transactivator. Proc Natl Acad Sci U S A. 2003;100(18):10405–10410. doi: 10.1073/pnas.1533207100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29(6):848–862. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 59.Zeissig S, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut. 2007;56(1):61–72. doi: 10.1136/gut.2006.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grootjans J, Thuijls G, Verdam F, Derikx JP, Lenaerts K, Buurman WA. Non-invasive assessment of barrier integrity and function of the human gut. World J Gastrointest Surg. 2010;2(3):61–69. doi: 10.4240/wjgs.v2.i3.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ulluwishewa D, Anderson RC, McNabb WC, Moughan PJ, Wells JM, Roy NC. Regulation of tight junction permeability by intestinal bacteria and dietary components. J Nutr. 2011;141(5):769–776. doi: 10.3945/jn.110.135657. [DOI] [PubMed] [Google Scholar]
- 62.Wang Q, Pantzar N, Jeppsson B, Westrom BR, Karlsson BW. Increased intestinal marker absorption due to regional permeability changes and decreased intestinal transit during sepsis in the rat. Scand J Gastroenterol. 1994;29(11):1001–1008. doi: 10.3109/00365529409094877. [DOI] [PubMed] [Google Scholar]
- 63.Ardita CS, et al. Epithelial adhesion mediated by pilin SpaC is required for Lactobacillus rhamnosus GG-induced cellular responses. Appl Environ Microbiol. 2014;80(16):5068–5077. doi: 10.1128/AEM.01039-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ryan MR, et al. An IL-7-dependent rebound in thymic T cell output contributes to the bone loss induced by estrogen deficiency. Proc Natl Acad Sci U S A. 2005;102(46):16735–16740. doi: 10.1073/pnas.0505168102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tyagi AM, et al. Premature T cell senescence in Ovx mice is inhibited by repletion of estrogen and medicarpin: a possible mechanism for alleviating bone loss. Osteoporos Int. 2012;23(3):1151–1161. doi: 10.1007/s00198-011-1650-x. [DOI] [PubMed] [Google Scholar]
- 66.Molnar I, Bohaty I, Somogyine-Vari E. IL-17A-mediated sRANK ligand elevation involved in postmenopausal osteoporosis. Osteoporos Int. 2014;25(2):783–786. doi: 10.1007/s00198-013-2548-6. [DOI] [PubMed] [Google Scholar]
- 67.Zhang J, Fu Q, Ren Z, Wang Y, Wang C, Shen T, Wang G, Wu L. Changes of serum cytokines-related Th1/Th2/Th17 concentration in patients with postmenopausal osteoporosis. Gynecol Endocrinol. 2015;31(3):183–190. doi: 10.3109/09513590.2014.975683. [DOI] [PubMed] [Google Scholar]
- 68.Molnar I, Bohaty I, Somogyine-Vari E. High prevalence of increased interleukin-17A serum levels in postmenopausal estrogen deficiency. Menopause. 2014;21(7):749–752. doi: 10.1097/GME.0000000000000125. [DOI] [PubMed] [Google Scholar]
- 69.Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: a new candidate in the pathogenesis of osteoporosis. PLoS One. 2012;7(9): doi: 10.1371/journal.pone.0044552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 71.Lelu K, et al. Estrogen receptor alpha signaling in T lymphocytes is required for estradiol-mediated inhibition of Th1 and Th17 cell differentiation and protection against experimental autoimmune encephalomyelitis. J Immunol. 2011;187(5):2386–2393. doi: 10.4049/jimmunol.1101578. [DOI] [PubMed] [Google Scholar]
- 72.DeSelm CJ, et al. IL-17 mediates estrogen-deficient osteoporosis in an Act1-dependent manner. J Cell Biochem. 2012;113(9):2895–2902. doi: 10.1002/jcb.24165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tyagi AM, et al. Enhanced immunoprotective effects by anti-IL-17 antibody translates to improved skeletal parameters under estrogen deficiency compared with anti-RANKL and anti-TNF-α antibodies. J Bone Miner Res. 2014;29(9):1981–1992. doi: 10.1002/jbmr.2228. [DOI] [PubMed] [Google Scholar]
- 74.McDole JR, et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature. 2012;483(7389):345–349. doi: 10.1038/nature10863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fasano A. Leaky gut and autoimmune diseases. Clin Rev Allergy Immunol. 2012;42(1):71–78. doi: 10.1007/s12016-011-8291-x. [DOI] [PubMed] [Google Scholar]
- 76.Heyman M, Abed J, Lebreton C, Cerf-Bensussan N. Intestinal permeability in coeliac disease: insight into mechanisms and relevance to pathogenesis. Gut. 2012;61(9):1355–1364. doi: 10.1136/gutjnl-2011-300327. [DOI] [PubMed] [Google Scholar]
- 77.Hijazi Z, Molla AM, Al-Habashi H, Muawad WMRA, Molla AM, Sharma PN. Intestinal permeability is increased in bronchial asthma. Arch Dis Child. 2004;89(3):227–229. doi: 10.1136/adc.2003.027680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Teixeira TFS, Collado MC, Ferreira CLLF, Bressan J, Peluzio MDG. Potential mechanisms for the emerging link between obesity and increased intestinal permeability. Nutr Res. 2012;32(9):637–647. doi: 10.1016/j.nutres.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 79.Asarat M, Apostolopoulos V, Vasiljevic T, Donkor O. Short-chain fatty acids produced by synbiotic mixtures in skim milk differentially regulate proliferation and cytokine production in peripheral blood mononuclear cells. Int J Food Sci Nutr. 2015;66(7):755–765. doi: 10.3109/09637486.2015.1088935. [DOI] [PubMed] [Google Scholar]
- 80.Wentworth CC, Alam A, Jones RM, Nusrat A, Neish AS. Enteric commensal bacteria induce ERK pathway signaling via formyl peptide receptor (FPR)-dependent redox modulation of Dual specific phosphatase 3 (DUSP3) J Biol Chem. 2011;286(44):38448–38455. doi: 10.1074/jbc.M111.268938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wentworth CC, Jones RM, Kwon YM, Nusrat A, Neish AS. Commensal-epithelial signaling mediated via formyl peptide receptors. Am J Pathol. 2010;177(6):2782–2790. doi: 10.2353/ajpath.2010.100529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ma TY, Anderson JM. Physiology of the Gastrointestinal Tract. Burlington, Massachusetts, USA: Elsevier Academic Press; 2006. Tight junctions and intestinal barrier. In: Johnson LR, ed. pp. 1559–1594. 4th ed. [Google Scholar]
- 83.Yu QH, Yang Q. Diversity of tight junctions (TJs) between gastrointestinal epithelial cells and their function in maintaining the mucosal barrier. Cell Biol Int. 2009;33(1):78–82. doi: 10.1016/j.cellbi.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 84.Wang Y, Zhang J, Yi XJ, Yu FSX. Activation of ERK1/2 MAP kinase pathway induces tight junction disruption in human corneal epithelial cells. Exp Eye Res. 2004;78(1):125–136. doi: 10.1016/j.exer.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 85.Filardo EJ, Quinn JA, Bland KI, Frackelton AR. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14(10):1649–1660. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- 86.Armstrong CM, Billimek AR, Allred KF, Sturino JM, Weeks BR, Allred CD. A novel shift in estrogen receptor expression occurs as estradiol suppresses inflammation-associated colon tumor formation. Endocr Relat Cancer. 2013;20(4):515–525. doi: 10.1530/ERC-12-0308. [DOI] [PubMed] [Google Scholar]
- 87.Qin B, et al. Expression of G protein-coupled estrogen receptor in irritable bowel syndrome and its clinical significance. Int J Clin Exp Pathol. 2014;7(5):2238–2246. [PMC free article] [PubMed] [Google Scholar]
- 88.Gevers D, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15(3):382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Scher JU, et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol. 2015;67(1):128–139. doi: 10.1002/art.38892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Knip M, Siljander H. The role of the intestinal microbiota in type 1 diabetes mellitus. Nat Rev Endocrinol. 2016;12(3):154–167. doi: 10.1038/nrendo.2015.218. [DOI] [PubMed] [Google Scholar]
- 91.Cardinale BJ, et al. Biodiversity loss and its impact on humanity. Nature. 2012;486(7401):59–67. doi: 10.1038/nature11148. [DOI] [PubMed] [Google Scholar]
- 92.Flores R, et al. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: a cross-sectional study. J Transl Med. 2012;10: doi: 10.1186/1479-5876-10-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fuhrman BJ, et al. Associations of the fecal microbiome with urinary estrogens and estrogen metabolites in postmenopausal women. J Clin Endocrinol Metab. 2014;99(12):4632–4640. doi: 10.1210/jc.2014-2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Preidis GA, et al. Probiotics stimulate enterocyte migration and microbial diversity in the neonatal mouse intestine. FASEB J. 2012;26(5):1960–1969. doi: 10.1096/fj.10-177980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bron PA, van Baarlen P, Kleerebezem M. Emerging molecular insights into the interaction between probiotics and the host intestinal mucosa. Nat Rev Microbiol. 2012;10(1):66–78. doi: 10.1038/nrmicro2690. [DOI] [PubMed] [Google Scholar]
- 96.Scholz-Ahrens KE, et al. Prebiotics, probiotics, and synbiotics affect mineral absorption, bone mineral content, and bone structure. J Nutr. 2007;137(3 suppl 2):838s–846s. doi: 10.1093/jn/137.3.838S. [DOI] [PubMed] [Google Scholar]
- 97.Yan F, Polk DB. Probiotics and immune health. Curr Opin Gastroenterol. 2011;27(6):496–501. doi: 10.1097/MOG.0b013e32834baa4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Anderson RC, et al. Lactobacillus plantarum MB452 enhances the function of the intestinal barrier by increasing the expression levels of genes involved in tight junction formation. BMC Microbiol. 2010;10: doi: 10.1186/1471-2180-10-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Seth A, Yan F, Polk DB, Rao RK. Probiotics ameliorate the hydrogen peroxide-induced epithelial barrier disruption by a PKC- and MAP kinase-dependent mechanism. Am J Physiol Gastrointest Liver Physiol. 2008;294(4):G1060–G1069. doi: 10.1152/ajpgi.00202.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bäckhed F, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101(44):15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Laukoetter MG, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204(13):3067–3076. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Robinson JW, et al. T cell-expressed CD40L potentiates the bone anabolic activity of intermittent PTH treatment. J Bone Miner Res. 2015;30(4):695–705. doi: 10.1002/jbmr.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li JY, Walker LD, Tyagi AM, Adams J, Weitzmann MN, Pacifici R. The sclerostin-independent bone anabolic activity of intermittent PTH treatment is mediated by T-cell-produced Wnt10b. J Bone Miner Res. 2014;29(1):43–54. doi: 10.1002/jbmr.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dempster DW, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 2013;28(1):2–17. doi: 10.1002/jbmr.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Denning TL, et al. Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J Immunol. 2011;187(2):733–747. doi: 10.4049/jimmunol.1002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.