

Abstract
The glycerophosphodiesterase (GpdQ) from Enterobacter aerogenes is a promiscuous binuclear metallohydrolase that catalyzes the hydrolysis of mono-, di- and triester substrates, including some organophosphate pesticides and products of the degradation of nerve agents. GpdQ has attracted recent attention as a promising enzymatic bioremediator. Here, we have investigated the catalytic mechanism of this versatile enzyme using a range of techniques. An improved crystal structure (1.9 Å resolution) illustrates the presence of (i) an extended hydrogen bond network in the active site and (ii) two possible nucleophiles, i.e. water/hydroxide ligands coordinated to one or both metal ions. While it is at present not possible to unambiguously distinguish between these two possibilities a reaction mechanism is proposed whereby the terminally bound H2O/OH acts as the nucleophile, activated via hydrogen bonding by the bridging water molecule. Furthermore, the presence of substrate promotes the formation of a catalytically competent binuclear center by significantly enhancing the binding affinity of one of the metal ions in the active site. Asn80 appears to display coordination flexibility that may modulate enzyme activity. Kinetic data suggest that the rate-limiting step occurs after hydrolysis, i.e. the release of the phosphate moiety and the concomitant dissociation of one of the metal ions and/or associated conformational changes. Thus, it is proposed that GpdQ employs an intricate regulatory mechanism for catalysis, where coordination flexibility in one of the two metal binding sites is essential for optimal activity.
Keywords: Binuclear metallohydrolases, glycerophosphodiesterase, metal ion binding, enzyme regulation, catalysis
Graphical Abstract
1. INTRODUCTION
Binuclear metallophosphatases are important in a wide array of biochemical processes that involve the hydrolysis of phosphate ester bonds.1–5 These enzymes typically belong to one of three different classes, i.e. (i) mono-, (ii) di- and (iii) triesterases. Purple acid phosphatases (PAPs)1,3,6–9 and serine/threonine protein phosphatases2,10,11 are examples of phosphate monoester-cleaving enzymes. Phosphatases with activity towards diesters include 3′–5′ exonucleases,12,13 and 5′-nucleotidase,14 and recently PAPs have also been shown to exhibit some diesterase activity.15 Phosphate triesters do not occur naturally, however due to the widespread use of organophosphate pesticides, enzymes have evolved that are capable of hydrolyzing these compounds. The phosphotriesterase from Pseudomonas diminuta (PTE)16–19 and Agrobacterium radiobacter (OpdA)20–25 are examples of these and have gained increasing attention for their potential application in degrading phosphotriester nerve agents.
The current study focuses on the characterization of a promiscuous glycerophosphodiesterase (GpdQ; E.C. 3.1.4.46) from Enterobacter aerogenes.22,26–28 GpdQ was initially noted for its remarkable activity towards stable aliphatic diesters such as dimethyl phosphate (DMP) and ethyl methylphosphonate,27,28 and is at present the only known enzyme that is capable of hydrolyzing all three types of phosphate esters. Its biological function and that of other members of the glycerophosphodiesterase (GDPD) family is the hydrolysis of the 3′–5′ phosphodiester bond of glycerophosphodiesters such as glycerol-3-phosphoethanolamine.29 Other known substrates for GpdQ include p-nitrophenyl phosphate (pNPP), bis(p-nitrophenyl) phosphate (bpNPP) and, notably, EA 2192, the toxic hydrolysis product of the nerve agent VX.22,28,30
The crystal structures of ZnII- and CoII-substituted GpdQ have previously been solved to 2.9 Å and 3.0 Å, respectively.26 The oligomeric structure of the protein is hexameric, forming a trimer of dimers.26 Each subunit contains a binuclear metal center, and each metal ion is coordinated by four amino acid side chains, two aspartates and two histidines for the metal in the M1 or α site, and two histidines, one aspartate and one asparagine for the metal in the M2 or β site. A schematic illustration of the active site is shown in Fig. 1. One of the aspartates acts as a bridging ligand (Asp50). The active site structure of GpdQ is remarkably similar to that of a number of binuclear phosphomonoesterases, including PAPs,9,31–35 5′-nucleotidase36 and Mre11 nuclease.37
Figure 1.

Schematic representation of the active site of GpdQ based on the improved crystallographic data presented here. In two of the six active sites an additional water molecule is also seen bound terminally to the β metal and further hydrogen bonded to the amine nitrogen on N80.
The metal ion compositions amongst binuclear metallohydrolases is diverse, including homonuclear centers of the FeFe, ZnZn, MnMn and NiNi type and heteronuclear combinations suchs as the FeZn or FeMn centers in some plant PAPs.1,3 The in vivo metal content for GpdQ is currently unknown, but catalytic activity can be reconstituted in the presence of a range of divalent metal ions including CoII or ZnII.26 Analysis of Zn K-edge data indicate a lower metal ion occupancy in the β site, suggesting that GpdQ contains one metal center with high binding affinity (α site) and one with low affinity (β site) (Fig. 1).26 Differential metal ion binding affinities are not uncommon in binuclear enzymes and may be of regulatory significance. Examples include the 5′-nucleotidase,38 bacteriophage λ protein phosphatase39,40 and OpdA.41
In this work, the formation of a catalytically competent binuclear CoII metal center in GpdQ is monitored using a range of physicochemical techniques, and the catalytic reaction is probed using steady-state kinetics and the measurement of kinetic isotope effects (KIEs). The combined data are used to develop a model for the relevant molecular mechanism of hydrolysis employed by this enzyme.
2. MATERIALS and METHODS
Materials
The plasmid containing the gene for E. aerogenes GpdQ (GpdQ/pCY76) was previously described.22 p-NPP (bis(cyclohexylammonium)) salt and ethyl p-nitrophenyl phosphate (EtpNPP) sodium salt were synthesized by known methods.42,43 All other chemicals were purchased from Sigma-Aldrich, unless stated otherwise. bpNPP was purchased in the sodium salt form due to its higher solubility in water.
Site directed mutagenesis, expression and purification of GpdQ
Residue Asn80 (Fig. 1) in wild-type GpdQ was replaced by an alanine or an aspartate by site-directed mutagenesis. Wild-type and mutant GpdQ were expressed and purified using a modified version of a previously published procedure.22 In brief, DH5α E. coli cells were transformed with the GpdQ/pCY76 plasmid. Cells were grown in 4 L of TB medium containing 50 μg/mL ampicillin and 0.1 mM CoCl2.6H2O at 30°C for 30 hours. The plasmid used expresses GpdQ in a constitutive manner and no induction is required. The following steps were performed at 4 °C. Cells were harvested by centrifugation and then resuspended in 40 mL 20 mM Tris-HCl buffer, pH 8.0. Following lysis using a French Press at 10,000 lb/in2, the cell debris was pelleted by centrifugation. The supernatant was filtered using a 0.22 μm filter (Millipore) and loaded onto a HiPrep 16/10 DEAE FF column (GE Healthcare). A linear NaCl gradient (0 to 1 M) was applied to elute the protein over 10 column volumes, and the eluate was collected in 8 mL fractions. 5 μL of each fraction were assayed with 2 mM bpNPP, and the fractions with phosphodiesterase activity were combined. Dialysis against 1.5 M (NH4)2SO4, 20 mM HEPES, pH 8.0 was performed overnight. The protein solution was loaded onto a HiLoad 26/10 Phenyl Sepharose HP column (GE Healthcare). The proteins were eluted using an (NH4)2SO4 gradient (1.5 to 0 M) over 10 column volumes and collected again in 8 mL fractions. Fractions with phosphodiesterase activity were combined and the proteins were concentrated to approximately 4 mL. The solution was loaded onto a HiPrep 16/10 Sephacryl S-200 HR gel filtration column and eluted with 20 mM HEPES, 0.15 M NaCl, pH 8.0. Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) of the protein generally shows a single band migrating at ~31 kDa, corresponding to GpdQ at ≥ 95 % purity. The final yield was 70 mg/L. Protein concentrations were calculated using an ε280 = 39880 M−1 cm−1 (in monomer units).26 The metal ion content of the purified enzyme was determined by atomic absorption spectroscopy and resulted in 1.35(5) Fe, 0.25(5) Zn and 0.15(4) Mn atoms per active site.
Preparation of apoenzyme
3 mg of GpdQ were incubated at 4°C in a 3 mL solution containing 5 mM EDTA, 5 mM 1,10-phenanthroline, 5 mM 2,6-pyridine dicarboxylic acid, 5 mM 8-hydroxyquinoline-5-sulfonic acid, 5 mM 2-mercaptoethanol, 20 mM HEPES, pH 7.0 for 48 hours. The mixture was then loaded onto an Econo-Pac 10DG (Bio-Rad) gel filtration column and the apoenzyme was collected. The column was washed with the above chelating solution and equilibrated with Chelex-treated buffer prior to use.
X-ray data collection, model building and refinement
X-ray diffraction data were collected on beamline 3BM1 at the Australian Synchrotron, Melbourne.44 Data were collected to a resolution of 1.9 Å, using X-rays of 13.0008 keV energy, from a crystal formed in the space group P212121 from a solution of 60% Tacsimate, buffered with 100mM bis-tris pH 7.0. Three dimers were present in the asymmetric unit. The crystal was rotated in 0.5° intervals over a total rotation range of 120°. Data were integrated using MOSFLM45 and scaled using SCALA.46 The structure was readily solved by molecular replacement using the 2.9 Å structure crystallized in the cubic space group P213 (2DXL26), which contained one dimer in the asymmetric unit, as a search model. Rounds of manual rebuilding using the program COOT47 were interspersed with restrained refinement using REFMAC48 as implemented in the CCP4 suite of programs.46 In the final rounds of refinement TLS parameters49 were refined using the rigid body group assignments previously determined,26 i.e. 1–195, 195–256 and 257–274. Coordinates and structure factors have been deposited into the protein databank with accession number 3D03.
Determination of metal binding affinity by activity measurements
The effect of the metal ion concentration on activity was determined by varying the amount of added CoII (in the form of CoSO4.6H2O) in the activity assays (see below). The data were fit to Eq. 1, where r is the binding function, n is the number of sites associated with Kd, and [M]free is the free metal ion concentration. [M]free was calculated from Eq. 2, where [M]total is the total concentration of metal ions added and [E] is the concentration of the enzyme.
| Equation 1 |
| Equation 2 |
Enzyme activity assays and analysis
Phosphatase activity with bpNPP, EtpNPP and pNPP was measured spectrophotometrically by monitoring the formation of the product p-nitrophenolate at 405 nm. Exogenous [CoII] equal to the amount required for maximum activity (determined to be 40 μM) was maintained for all kinetic assays. Reactions were measured to less than 5 % of total substrate hydrolysis and were performed as a continuous assay for pH 5.5 and above, and as a discontinuous assay below pH 5.5. For the discontinuous assays, the reactions were quenched by addition of 5 μL of the reaction mixture into 3 mL of 0.1 M NaOH (ε400 = 18,300 M−1 cm−1 for p-nitrophenolate). For the continuous assays the extinction coefficient of the product p-nitrophenolate was determined at each relevant pH by using a standard solution of p-nitrophenol in the same buffer used for the enzymatic assays. Buffer systems used were 0.1 M sodium formate (pH 3.2 to pH 4.0), sodium acetate (pH 4.0 to pH 5.5), MES (pH 5.5 to pH 6.7), HEPES (pH 6.8 to pH 8.2), CHES (pH 8.6 to pH 9.7) or CAPS (pH 9.7 to 10.1). Adjustments for ionic strength were not required after activity assays conducted in a range of buffers and NaCl concentrations (0 to 1 M) indicated no dependence. Substrate concentrations ranged from 10 μM to 7.0 mM. The data were treated by the initial rate method and fit to the Michaelis-Menten equation (Eq. 3).
| Equation 3 |
The pH dependence of kcat and kcat/KM for the hydrolysis of pNPP and EtpNPP was determined in the range between pH 3 and pH 10 and the data were analyzed, where appropriate, with the following equations:
| Equation 4 |
| Equation 5 |
Eq. 4 was derived following an approach described by Sykes and co-workers,50 and employing a model involving three relevant protonation equilibria (Kes1 – Kes3).51 In Eq. 4 Kesn represents the nth protonation equilibrium of the enzyme-substrate complex and kcatn corresponds to the activity of the associated protonation state. Both kcat1 and kcat4 (corresponding to the fully protonated and fully deprotonated forms, respectively) were initially set to zero and then allowed to float as described previously.51
In Eq. 5 Ken represents the protonation equilibria associated with the free enzyme or free substrate.52 Eq. 5 takes into account that the catalytic efficiency (kcat/KM) for the hydrolysis of EtpNPP is pH-independent at high pH (see below). For the data measured with pNPP the (kcat/KM)highpH was omitted.
Inhibition of enzymatic activity
The inhibition of the hydrolysis of pNPP by inorganic phosphate (KH2PO4) was measured with inhibitor concentrations ranging from 0.1 to 5 mM. The inhibition data were evaluated by the general inhibition equation (Eq. 6). Kiuc and Kic represent the inhibition constants for uncompetitive and competitive inhibition, respectively.
| Equation 6 |
Kinetic isotope effects
Isotope effect experiments were conducted at pH 5.5 and pH 9.0 using 100 mM acetate (pH 5.5) or 100 mM CHES (pH 9.0). Buffer solutions contained 1 mM CoCl2.6H2O. Reactions of 100 μmol of substrate (pNPP and EtpNPP) were allowed to proceed to partial completion, typically from 40 to 60% of total hydrolysis, then stopped by addition of HCl to pH 2.0. Background hydrolysis was negligible under the KIE conditions, and reactions were performed in triplicate. Isotope ratio mass spectrometry was used to analyze the product and the residual substrate after partial reaction. The reaction product p-nitrophenol was isolated from each reaction mixture and prepared for analysis by previously published methods,43,53 The residual substrate was then subjected to complete hydrolysis. This was accomplished using alkaline phosphatase in the case of pNPP, and for EtpNPP the residual substrate was hydrolyzed by treatment at pH 14.0 at 370 K for 48 hours. KIEs were determined from the isotopic ratio of p-nitrophenol, in both the product (Rp) at fractional completion (f), the residual substrate (Rs) and in the starting material (Ro). Eqs 7 and 8 were used to calculate the KIEs.54
| Equation 7 |
| Equation 8 |
These equations give the 15N KIEs directly. The 18O isotope effects were determined by the remote label method where the nitrogen atom in the nitro group of the substrate is used as a reporter for both the bridging and non-bridging oxygen atoms.55 The observed isotope effects were corrected for the 15N effect of the remote label and for incomplete isotopic incorporation in the starting material.56
The nonbridge 18O KIEs for pNPP were corrected for the assumption that the monoanion is the catalytically active species. This correction was made using Eq. 9 where f is the fraction present as the dianion, and 0.9848 is the equilibrium isotope effect (EIE) for protonation of a phosphate ester.57
| Equation 9 |
KIEs for the hydrolysis of the analogous triester, diethyl 4-nitrophenyl phosphate (paraoxon) could not be measured due the large amount of enzyme required to turnover ~ 40 % of substrate.
Electronic absorption and magnetic circular dichroism
Electronic absorption spectra were recorded with a Varian Cary 6000i instrument using a 1 mm path length cell. Magnetic circular dichroism (MCD) spectra were recorded at 2 nm bandwidth with a Jasco J-600 spectropolarimeter equipped with an Oxford SM-4 magnet/cryostat and an Oxford ITC-4 temperature controller. Protein samples were diluted with glycerol to a ratio of 3 parts glycerol to 2 parts protein solution and stirred for at least one hour. The final concentration of protein was in the range of 0.38 mM to 0.78 mM. Protein solutions were buffered at pH 7.0 with 100 mM HEPES. The samples were added into a brass cell with quartz windows and 0.62 cm path length. Spectra were collected in the region between 280 nm to 800 nm.
3. RESULTS
Overall and active site structure of GpdQ
GpdQ crystallized in a different space group than in a previously reported study,26 resulting in the asymmetric unit to expand from a dimer to the entire (physiological) hexamer. Good electron density was obtained for all protein residues in chains A and F, while in chains B, C, D and E the three C-terminal residues (Glu272, Glu273 and Arg274) were poorly resolved and thus omitted from the model (Fig. S1). The final model has Rwork and Rfree values of 0.18 and 0.22, respectively. The refinement statistics are listed in Table 1. While the overall fold is essentially identical to that reported previously,26 the greatly increased resolution allowed the identification of numerous ordered solvent molecules, in particular those present in the active sites (vide infra). The six protein chains superimpose with excellent agreement (illustrated by a root mean square displacement (rmsd) of all main chain atoms of 0.19Å). Only the side chains of solvent exposed surface residues show some conformational flexibility. The stereochemical correctness of the model was satisfactorily checked using COOT,47 PROCHECK58 and SFCHECK.59 One residue in each protein chain, His195, was found in the disallowed region of the Ramachandran plot (ϕ,φ = 80°, −45°). His195 forms a hydrogen bond with its carbonyl oxygen atom to the metal ion bridging hydroxide in the active site, and its side chain imidazole group is a ligand to the metal ion in the β-site (Fig. 1). His195 forms part of a γ-turn. Histidine residues at an equivalent position and with similarly disallowed main chain torsion angles are also observed in pig PAP (1ute),31 and the serine/threonine protein phosphatases 1 (1fjm)60 and 2B (calcineurin; 1aui).61 A further residue, Arg205, was observed in the generously allowed region. This is a surface residue remote from the active site and it is located in well-defined electron density. It is unclear why this residue requires a strained main chain conformation. However, attempts to flip the peptide bond and refit to the electron density in order to achieve more conventional torsion angles were unsuccessful, resulting in a poor fit to density. All other residues were in allowed regions.
Table 1.
X-ray data collection and refinement statistics. Values shown in parenthesis in the data collection section refer to the highest resolution shell of data.
| Data Collection: | |
| Space Group | P212121 |
| Unit Cell Parameters (Å) | a,b,c = 94.97,133.84,168.94 |
| Temperature (K) | 100 |
| Wavelength/Energy (Å/keV) | 0.95361/13.0008 |
| Resolution (Å) | 43.3 - 1.9 (2.0 - 1.9) |
| Measurements | 691088 |
| Unique reflections | 163812 |
| Multiplicity | 4.2 (3.7) |
| Mean I/σ(I) | 11.9 (1.9) |
| Completeness (%) | 96.7 (91.0) |
| Rmerge | 0.098 (0.613) |
| Wilson B-factor (Å2) | 16.2 |
| Refinement: | |
| Resolution range (Å) | 43.3 - 1.9 |
| Number of Reflections (working set) | 155501 |
| Number of Reflections (test set) | 8219 (5%) |
| R (work + free) | 0.186 |
| Rwork | 0.185 |
| Rfree | 0.224 |
| Number of protein atoms | 12878 |
| Number of solvent molecules | 1036 |
| Number of metal ions | 12 |
| Ramachandran Plot: | |
| Favoured regions | 99.2% |
| Generously allowed regions | 0.4% (Arg 205) |
| Disallowed regions | 0.4% (His195) |
| Rms deviation from ideality: | |
| Bond lengths (Å) | 0.02 |
| Bond angles (°) | 1.57 |
| Estimated coordinate error: | |
| From Luzzati plot (Å) | 0.23 |
| From Cruickshank DPI (Å) | 0.12 |
All protein residues were modelled at full occupancy. The occupancies of the metal ions were fixed at 0.75 for the α site and 0.45 for the β site, supporting the hypothesis that the α site has a higher affinity for metal ions than the β site.26 This was based on a comparison of the temperature factors of coordinating atoms in refinements carried out with differing metal occupancies (note that the total metal content per subunit is 1.75, indicating that some metal ions are adventitiously bound). A schematic illustration of the active site based on the improved crystallographic data is shown in Fig. 1; the major difference in comparison to the previously reported structures of GpdQ is the observation of a water ligand, terminally bound to the metal ion in the α site. A network of hydrogen bonds connects this water ligand via two additional water molecules to His217, Tyr19 and Asn53, thus associating these residues with a role in catalysis and/or substrate binding (Fig. 1). An additional hydrogen bonding interaction was apparent in two of the six subunits, connecting the metal ion in the β site via a water molecule to the amino group of the ligand Asn80.
Magnetic Circular Dichroism
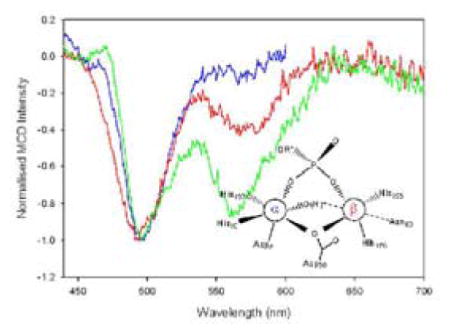
The visible region of the sample of apo-GpdQ was scanned for MCD active transitions (400–700 nm) and no features were observed. The addition of two equivalents of CoII to the apoenzyme gave rise to one negative C-term band at 495 nm (Fig. 2), characteristic of a six-coordinate mononuclear CoII center.62–65 This observation suggests that only one of the two metal ion binding sites in the active site, the α site (Fig. 1), is occupied. In order to populate the β-site, at least partially, an additional 48 equivalents of CoII were added, giving rise to a second C-term band at 574 nm (Fig. 2). The position of this d-d band indicates that a five-coordinate CoII ion binds to the β-site.62,63,65
Figure 2.

MCD spectra of wild-type GpdQ at 1.45 K, 3.5 T depicting a mononuclear CoII center, dinuclear CoII center and a dinuclear CoII center in the presence of phosphate.
The effect of inorganic phosphate on the metal ion binding affinities of the two metal sites was assessed by mixing a sample of GpdQ with two equivalents of CoII and ten equivalents of K2HPO4. Again, two C-term MCD bands were observed at 495 nm and 564 nm (Fig. 2), corresponding to a six- (at 495 nm) and five-coordinate (at 564 nm) CoII species. The position of the higher energy band is identical to that of the phosphate-free form, but the lower energy band is shifted by 10 nm, suggesting a binding interaction between the metal ion in the β site and the phosphate group. Importantly, the addition of phosphate greatly increases the metal ion affinity of the β site; only two equivalents of CoII are required to fully populate both metal ion centers.
Kinetic parameters of GpdQ
The pH dependence of kcat and kcat/KM for the hydrolysis of pNPP and EtpNPP are shown in Fig. 3. For both substrates, Michaelis-Menten behavior was observed at each pH. Above pH 7.5 the KM for pNPP was too large to be measured accurately, but kcat appears to be pH independent across the entire pH range investigated. This implies that the rate limiting step is either the release of the product or a conformational change. The pH dependence of kcat for the hydrolysis of EtpNPP was more complex and was analyzed using an equation derived for a model that includes four protonation states for the enzyme-substrate complex (Eq. 4). 51 Table 2 lists the obtained acid dissociation constants.66 The pH profiles for the catalytic efficiency ratio (kcat/KM) are bell-shaped for both substrates and were fit to Eq. 5 (Table 2; note that the deprotonated state retains some catalytic efficiency for the hydrolysis of EtpNPP at pH >8).52
Figure 3.

pH dependence of the activity (kcat) and catalytic efficiency (kcat/KM) of the hydrolysis of pNPP and EtpNPP by CoII-substituted GpdQ. The arrows indicate the pH values used for KIE measurements (see below).
Table 2.
Acid dissociation constants for the hydrolysis of pNPP and EtpNPP
| pKes1 | pKes2 | pKes3 | pKe1 | pKe2 | |
|---|---|---|---|---|---|
| pNPP | - | - | - | 3.8(4) | 5.1(3) |
| EtpNPP | < 3.2 | 6.2(3) | 10.2(4) | 3.7(3) | 6.8(3) |
The hydrolysis of bpNPP by CoII-GpdQ deviated from typical Michaelis-Menten behavior, as previously noted by Gerlt and collaborators.28 Illustrative examples are shown in Fig. S2, where especially at lower pH values the catalytic rate tends to saturate at low substrate concentrations, but a further increase in [bpNPP] leads to rate enhancements. Using sufficiently low substrate concentrations approximate fits to the Michaelis-Menten equation (Eq. 3) indicate that the profile of kcat is virtually pH independent between pH 5.5 to pH 10 (Fig. S3). The likely reason for the observed deviation from Michaelis-Menten behavior is the fact that the first reaction product of bpNPP hydrolysis (pNPP) is itself a substrate for GpdQ, at least at pH ≤8. At higher pH the KM value of pNPP is very large (Fig. 3) making this reagent a very poor substrate for GpdQ.
The effects of Asn80 mutations on metal ion binding and reactivity
Asn80 is a ligand of the metal ion in the β site (Fig. 1). It is anticipated that both the activity of the enzyme and the metal ion affinity of this site may be affected by mutations of that residue. The substitution by an alanine led to a considerable increase in reactivity albeit under the loss of substrate binding affinity (Table 3). The substrate affinity is affected in a similar manner in the Asn80Asp mutant, but the reactivity is greatly reduced in comparison to the wild-type enzyme.
Table 3.
Comparison of the kinetic parameters for the hydrolysis of bpNPP by wild type and variants of GpdQ. The assays were conducted at pH 7.00 (0.10 M HEPES buffer) and with 1 mM CoCl2.6H2O
| Variant | kcat (s−1) | KM (mM) | kcat/KM (s−1 mM−1) |
|---|---|---|---|
| Wild type | 1.89(1) | 0.12(3) | 16.4(3) |
| N80D | 0.24(2) | 39(6) | 0.006 |
| N80A | 6.4(6) | 49(6) | 0.13 |
CoII was added gradually to Asn80Ala-GpdQ and MCD spectra were recorded to monitor the binding of the metal ions to the active site. The transition at 495 nm, characteristic of a six-coordinate CoII species was observed after the addition of one equivalent, but no transition due to a five-coordinate species was detected, even after the addition of 100 equivalents of CoII (Fig. 4). However, after the addition of ten equivalents of phosphate, transitions at both 495 nm and 555 nm were observed after the addition of only two equivalents of CoII (Fig. 4), indicating the formation of a binuclear active site as discussed above.
Figure 4.

MCD spectra of CoII-substituted N80A and N80D mutants of GpdQ. Note that for N80A-GpdQ in the absence of phosphate up to 100 equivalents of CoII were added, but only the transition at 495 nm was observed.
In contrast to the Asn80Ala mutant, a fully occupied binuclear center was formed in Asn80Asp-GpdQ after the addition of two equivalents of CoII to the apoenzyme, even in the absence of phosphate (Fig. 4). Addition of more CoII did not alter the intensity of the band at 555 nm. Thus, in comparison to both the wild-type and Asp80Ala mutant the substitution of the asparagine by an aspartate greatly increases the metal ion affinity of the β site.
Kinetic Isotope Effects
Isotope effects were measured by the competitive method, and are thus effects on (V/K).67 The 15(V/K), 18(V/K)bridge and 18(V/K)nonbridge KIEs (Fig. 5) for the hydrolysis of pNPP and EtpNPP by wild-type GpdQ were measured at pH 5.5 and pH 9.0, and the data are listed in Table 4. Since the pH dependence of kcat/KM for pNPP (Fig. 3) suggests that the monoanion is the reactive form of this substrate, the observed nonbridge KIEs for this substrate were corrected as described in the Experimental section. No correction was made for the diester substrate, since the low pKa of EtpNPP ensured that only the deprotonated form is present under the experimental conditions (Fig. 3).
Figure 5.

The structure of pNPP and EtpNPP, and the isotope effects determined.
Table 4.
KIEs for the substrates pNPP and EtpNPP at pH 5.5 and pH 9.0. The nonbridge KIEs for pNPP have been corrected assuming the monoanion form of pNPP is the active form of the substrate.
| KIE | pNPP | EtpNPP | ||
|---|---|---|---|---|
| pH 5.5 | pH 9.0 | pH 5.5 | pH 9.0 | |
| 15(kcat/KM) | 1.0001(1) | 1.0009(3) | 1.0001(2) | 1.0003(2) |
| 18(kcat/KM)bridge | 1.0039(1) | 1.0109(6) | 1.0060(9) | 1.0031(2) |
| 18(kcat/KM)nonbridge | 0.9912(3) | 0.9923(1) | 0.9957(5) | 0.9947(8) |
Effect of Phosphate on Enzyme Activity
The inhibitory effect of phosphate (KH2PO4) towards the hydrolysis of pNPP was determined at pH 7.00. Phosphate acts as a competitive inhibitor (Fig. S4) with Kic = 78 ± 6 μM.
4. DISCUSSION
Binuclear metallohydrolases form a large group of enzymes that include numerous phosphatases, peptidases and lactamases.1–5,68 Common to all members of this family is the presence of two closely spaced metal ion binding sites in their active sites, but the metal-ion affinities of these sites may vary considerably. Consequently, the functional roles of the metal ions may be diverse, and catalytic mechanisms have been proposed that require only one or both metal ions.4 Here, the structure of the active site of GpdQ, its interaction with CoII and its mode of action were investigated using structural, spectroscopic and kinetic techniques.
Active Site Structure
The active site of GpdQ (Fig. 1) is very similar to that of other members of the family of binuclear metallohydrolases. Six of its seven metal ion ligands are identical to those of PAPs; the remaining ligand is a histidine in GpdQ (His10) and a tyrosine in PAPs.1,6,7,26 In this respect GpdQ resembles Ser/Thr protein phosphatases such as calcineurin61,69 or the bacteriophage λ protein phosphatase.70 In comparison to the previously reported structures of GpdQ the higher resolution model presented here provides strong evidence for the presence of at least two water ligands in the coordination spheres of the metal ions, i.e. a terminal H2O bound to the metal ion in the α site and a metal ion bridging water/hydroxide (Fig. 1). In addition, in two of six protein subunits a third water molecule was also bound terminally to the metal ion in the β site. Since at least one of these water molecules is expected to play a major functional role as nucleophile (see below) their presence was also investigated under catalytically relevant solution conditions using MCD. Two peaks were observed in the spectrum of CoII-reconstituted GpdQ, characteristic of a six- (at 495 nm) and five-coordinate CoII (574 nm), respectively (Fig. 2).62–65 Thus, the combined structural and spectroscopic data support a model with a six-coordinate metal ion in the α site and a five-coordinate species in the β site, as shown in Fig. 1.
The five-coordinate species only emerges upon the addition of excess metal ion, indicating weak binding of this metal ion. The binding affinities of different metal ion sites may vary significantly within the catalytic centers of an enzyme, and these affinities may be influenced, amongst other factors, by the presence or absence of substrates and products.1,4,5 MCD was employed to qualitatively assess the effect of the reaction product and possible substrate mimic phosphate on the metal ion affinity of GpdQ. In the presence of phosphate the addition of only two equivalents of CoII leads to a saturation of both the high and low energy transitions, indicating that both metal ion sites are fully occupied (Fig. 2). Thus, phosphate leads to a significant increase in the metal ion affinity of the β site, an estimate of which can be obtained by monitoring the catalytic activity as a function of added metal ion (Kd = 29 μM; Fig. S5). By analogy, we propose that the formation of a catalytically competent binuclear center in GpdQ is assisted by the presence of the substrate, hence assigning a mechanistic/regulatory role to the latter. Substrate-induced formation of a catalytically active binuclear center is not without precedence, and has been reported for related metallohydrolases such as the exonuclease activity of E. coli DNA polymerase I.1,71
Asn80 is a ligand of the metal ion in the β site (Fig. 1). In two of the six subunits of GpdQ the amine group of its side chain forms a hydrogen bond with a water molecule that is terminally bound to the metal ion in the β site. Since the position occupied by this water ligand is a probable binding site for the substrate molecule it is likely that Asn80 may play a crucial role in the modulation of both metal ion and substrate binding, and thus in the regulation of reactivity. To probe this residue’s role it was replaced by an alanine or aspartate. The alanine mutant displays greatly diminished metal ion binding affinity in the β site, whereas the the aspartate shows the opposite effect (Fig. 4). This is not an unusual observation since aspartate (but not alanine) is expected to be a good ligand for the metal ion. However, in Asn80Ala-GpdQ a binuclear center is formed readily in the presence of phosphate, similar to the wild-type enzyme (Fig. 4). Importantly, although the alanine residue is not able to coordinate the metal ion in the β site a five-coordinate species similar to that observed in the wild-type enzyme is observed. This observation suggests that (i) the fifth ligand of CoII in the β site is provided by the phosphate, and, by analogy, that (ii) in wild-type GpdQ the coordination bond between this metal ion and Asn80 is broken upon binding of substrate/product (thus yielding a five-coordinate species). Furthermore, the activity of Asn80Ala-GpdQ is ~four-fold higher than that of the wild type at pH 7.0, while the activity of the Asn80Asp mutant is greatly reduced (Table 3). Both mutants have greatly diminished affinity for the substrate with KM values ~400 times larger than that of the wild type enzyme. These findings demonstrate that Asn80 contributes to reactivity by (i) assisting in substrate binding, and (ii) facilitating the easy exchange of the metal ion in the β site.
Kinetic Properties of CoII-GpdQ
GpdQ has been shown to hydrolyze a range of substrates including both diesters and monoesters.22,30,72 Here, the catalytic properties of the enzyme were investigated using the diester EtpNPP and the monoester pNPP. For the monoesterase activity kcat does not depend on pH (Fig. 3), suggesting that the rate-limiting step is either product release or a conformational change associated with the protein. In contrast, kcat/KM displays a distinct bell-shaped dependence on pH (Fig. 3), indicating the involvement of at least two relevant protonation equilibria of the free enzyme and/or substrate in the catalytic cycle up to, and including, the first irreversible step, which is presumably hydrolysis of the P-O bond (pKe1 and pKe2; Table 2). At present it is unclear which protonation equilibrium pKe1 = 3.8 is attributable to, but it is likely to be associated with the free enzyme since its magnitude seems substrate-independent (Table 2). A possible candidate is a water ligand bound terminally to the metal in the α site (the pKa may be so low due to hydrogen bonding interactions in the active site) (Fig. 1). The assignment of pKe2 (5.1) to pNPP (i) is consistent with the second pKa of this substrate, (ii) agrees with previous kinetic studies that utilized pNPP,51,53,73,74 and (iii) suggests that the substrate binds and undergoes catalysis preferentially in its monoanionic form. The nonbridge KIE with pNPP listed in Table 4 was accordingly corrected assuming a monoanionic form of this substrate.
Previous enzymatic and nonenzymatic KIE measurements with pNPP give a framework in which to interpret KIEs. In a loose transition state with extensive P-O fission, the primary 18(V/K)bridge KIE can reach a maximum of ~1.03 and the secondary 15(V/K) KIE a maximum of ~1.003, reflecting a full negative charge on the departing nitrophenolate. If the leaving group is protonated by a general acid in the transition state, the primary 18(V/K)bridge KIE is reduced to the range 1.012 to 1.015, and 15(V/K) KIE is abolished to unity.75 Furthermore, the nonbridge KIE will be small and inverse in a metaphosphate-like transition state; however, in reactions involving a monoanion, a significant normal nonbridge KIE is observed if a proton transfer from the phosphoryl group occurs in the reaction. Modest inverse nonbridge KIEs have been observed for alkaline phosphatase-catalyzed reactions of phosphate monoester dianions, which have been attributed to effects from binding to the metal center.76 In general, the interpretation of KIEs of phosphorolytic reactions in terms of transition state structure also depends upon the extent to which the chemical step of phosphoryl transfer is rate-limiting. If other steps are fully or partially rate-limiting, the KIEs will be suppressed from their intrinsic values, thus inaccurately reflecting bonding changes in the transition state.
Because they are measured by the competitive method, the KIEs for the GpdQ-catalyzed reactions are those for kcat/KM, and thus reflect all steps from free substrate to the first irreversible step, i.e. P-O bond fission. For the reaction with the monoester substrate pNPP the flatness of the kcat vs pH profile (Fig. 3) suggests a nonchemical step is rate limiting in the overall mechanism, and several findings suggest that chemistry may not be fully rate-limiting for kcat/KM. These are (i) the fact that the magnitudes of the KIEs are significantly larger away from the pH optimum (9.0) than they are at the optimum (5.5), and (ii) the modest magnitudes of the primary bridge KIEs. With this caution in mind, the KIE data do permit some mechanistic conclusions to be drawn. Below, we first interpret the KIEs measured for for pNPP, and then those determined for the diester substrate.
The 15(V/K) KIE of 1.0009 at pH 9 indicates the presence of about a third of a charge on the leaving group, while at pH 5.5 no charge appears present (15(V/K) ~ unity; Table 4). Similarly, the 18(V/K)bridge KIE at pH 9 is about a third of its maximum value, while at pH 5.5 it is considerably smaller. These observations may indicate that a general acid protonates the leaving group at low pH, but which is no longer in the correct protonation state at high pH to perform its function. A possible candidate general acid is His81, an amino acid that lines the substrate binding pocket,26 and which is in a position equivalent to histidine residues in the second coordination sphere of other binuclear metallohydrolases, including PAPs (e.g. His295 in sweet potato PAP9) and bacteriophage λ protein phosphatase (His7677). Since the monoanion is the likely substrate, an alternative proton donor to the leaving group is the phosphoryl group of the substrate. For example, in the uncatalyzed hydrolysis of monoanions of phosphate monoesters, the proton is transferred from the phosphoryl group to the leaving group in the transition state, resulting in a normal nonbridge KIE for pNPP of 1.0184 at 95°C, and 1.0199 at 30°C.43,78 However, the inverse nonbridge KIE measured at pH 5.5 and pH 9.0 (Table 4) rules out the possibility that the phosphoryl group is a proton donor to the leaving group in the GpdQ-catalyzed reaction.
For the GpdQ-catalyzed hydrolysis of the diester EtpNPP, the modest pH dependence of both kcat and kcat/KM indicates that the chemical step is probably not fully rate-limiting (Fig. 3). pKe1 is virtually identical to that obtained when pNPP was used as substrate (Table 2). The pKa of EtpNPP is expected to be similar to that of 3,3-dimethylbutyl p-nitrophenyl phosphate, which is −0.36,79 and thus this substrate will also interact in its monoanionic form with GpdQ. pKe2 (6.8) can thus only be assigned to the free enzyme. We tentatively ascribe this pKa to a histidine residue in the vicinity of the active site. Likely candidates are His217 (Fig. 1) and His81, a residue in the substrate binding pocket (see preceding paragraph).26,80 His217 is involved in an extensive hydrogen bond network in the active site of GpdQ, and its deprotonation is expected to perturb this network, which may affect substrate binding and possibly the structure of the active site. However, crystallographic data and docking studies suggest that His81 is more likely to be associated with pKe2 since it is positioned ideally to interact with the substrate via hydrogen bonds.26,80 It is also important to note that the corresponding histidine residue in PAPs has been shown by site-directed mutagenesis to be important in substrate binding and orientation (see also the preceding paragraph).81,82 While the 18(V/K)bridge is in the range reported for uncatalyzed hydrolysis reactions of diesters, the 15(V/K) KIE at pH 5.5 is smaller (Table 5), consistent with the hypothesis of general acid catalysis at least at low pH. At higher pH the general acid may not be in the correct protonation state to function, but may not be required since the pKa of the leaving group pNP is low (~7). In support of this hypothesis kcat/KM does level off at high pH (Fig. 3). Alternatively, the very small leaving group KIEs at pH 9 may reflect rate-limitation by a nonchemical step, possibly substrate binding. We thus propose that His81 may have a dual function, (i) acting as a general acid in the hydrolysis of EtpNPP at low pH and (ii) orienting the substrate within the active site. Interestingly, the pKa of His81 is not observed in the hydrolysis of the monoester pNPP (Fig. 3), suggesting a mechanistic model whereby the rate-limiting steps depend on the nature of the substrate. For the bulkier diester substrate the formation of a catalytically competent enzyme-substrate complex is at least partially rate-limiting, whereas for the smaller monoester substrate product release or a conformational change may be rate-limiting.
The 18(V/K)nonbridge values for both substrates are more inverse than for uncatalyzed hydrolysis reactions, which probably reflects coordination to the metal center. Similar inverse 18(V/K)nonbridge KIEs are observed in alkaline phosphatase-catalyzed reactions, attributed to binding effects.76
A minimum of three protonation equilibria contribute to reactivity for the hydrolysis of EtpNPP (Fig. 3; Table 2), two of which in their deprotonated form (characterized by pKes1 and pKes2), and one (pKes3) in the protonated state.83 The latter may correspond to a residue that stabilizes and/or positions via H-bond interactions the substrate in the active site. With a magnitude of 10.2 a likely candidate for pKes3 is a tyrosine residue. In the vicinity of the active site three tyrosines are present, Tyr229, Tyr230 and Tyr263. According to a previous study in which the substrate glycerophosphoethanolamine was docked into the active site of GpdQ Tyr263 is close to the phosphate group where it may be involved in stabilizing the bound substrate. Again a correlation to a PAP from sweet potato is observed, where a tyrosine residue in a position similar to that of Tyr263 is believed to be involved substrate binding and orientation.9 pKes1 and pKes2 are likely to be associated with coordinating water/hydroxide ligands (Fig. 1), i.e. the terminal H2O bound to the metal in the α site, and the metal ion-bridging water molecule. The doubly Lewis-activated μ-aqua is anticipated to be more acidic than the terminal CoII-bound water molecule. Accordingly, pKes1 and pKes2 are assigned to the deprotonations of the μ-aqua and terminal water ligand, respectively. These are unusually low pKa values for bridging and terminal water molecules in CoII systems. The pKa value for Co(H2O)62+ is estimated to ~9.6,84 and for doubly-activated water ligands in cobalt complexes they range from 5 to 7,85–87 but can be as low as 4.4 in complexes of other divalent first row transitions metal ions.88 The likely reason for the lower pKas in GpdQ may be the presence of an extensive hydrogen bond network that connects the carbonyl oxygen of His195 to residues in the substrate binding pocket via the bridging and terminal water ligands (Fig. 1). Thus, in principle two possible nucleophiles are present in the active site of GpdQ, a situation similar to that recently reported for PAPs.51
Proposed Reaction Mechanism
The accumulated structural, spectroscopic and kinetic data facilitate the proposal of a plausible reaction mechanism for GpdQ-catalyzed hydrolysis (Scheme 1). In the resting state the enzyme is predominantly in a mononuclear form with a metal ion bound to the α site (Scheme 1a). In presence of a substrate the metal ion affinity of the β site is enhanced which leads to the formation of a catalytically competent binuclear active site. A similar behavior has recently been observed for the distantly related E. coli methionine aminopeptidase.89 The binding of the second metal ion also facilitates the formation of a μ-hydroxide bridge (Scheme 1b). From the MCD data (Fig. 2) it is apparent that substrate/product binding does not affect the coordination number of the two metal ions (i.e. six- and five-coordinate for the metal ions in the α and β sites, respectively). Hence, it is likely that in the resting mononuclear state the CoII in the α site has two terminal water ligands (Scheme 1a), one of which becomes bridging once the second metal ion binds to the β site (Scheme 1b). The subsequent interaction between a substrate oxygen atom and CoII in the β site (i) facilitates an attack by a nucleophile bound to the CoII in the α site, and (ii) leads to the disruption of the Asn80-CoII bond (Scheme 1c). The unusual pH profile observed for the kcat of EtpNPP hydrolysis (Fig. 3) is consistent with the presence of two ionisable groups involved in catalysis, one terminally bound to the metal in the α site, the other bridging the metal ions. It is at present not possible to unambiguously identify the reaction-initiating nucleophile. In the related binuclear metallohydrolase PAP both a terminal and a metal-bridging hydroxide have been proposed as candidates,1,3,4,5,9,15,35,51,73,74,82 and it has emerged that experimental conditions such as pH, metal ion composition and substrate used in the study may affect the identity of the nucleophile.15,51 The recent crystal structure of GpdQ with malonate bound in the active site indicates that the terminally bound hydroxide may be better aligned for a nucleophilic attack than the bridging one.80 We thus suggest a mechanism whereby the terminal bound water molecule attacks the phosphorus of the substrate, leading to the formation of μ-1,3 phosphate complex (Scheme 1c). The bridging hydroxide may then function as an activator of the nucleophile via a hydrogen bonding or electrostatic interaction (Fig. 1).90 Following the nucleophilic attack the leaving group alcohol is released. Upon the release of the phosphate moity the metal ion in the β site also dissociates, returning the active site into its mononuclear resting state (Scheme 1d). Based on the kinetic and KIE measurements it appears likely that this last step is rate-limiting.
Scheme 1.

Scheme for the proposed reaction mechanism for GpdQ-catalyzed hydrolysis.
The catalytic cycle that emerged from this study suggests a regulatory mechanism for reactivity. In the absence of substrate the enzyme is predominantly in an enzymatically inactive mononuclear state. Addition of substrate facilitates the formation of a catalytically competent binuclear center, presumably by increasing the binding affinity of the second metal ion (β site). According to the crystal structure (Fig. 1) Asn80 acts as a ligand to this metal ion, at least when no substrate analogue or product are present. However, in order to reconcile the spectroscopic data obtained for both wild-type and Asn80Ala-mutant GpdQ the coordination bond between Asn80 and the metal ion is broken when phosphate is present. This observation implies that the perturbation of this coordination interaction, possibly mediated via the hydrogen bonding network in the active site, plays an important role in the regulation of reactivity. This hypothesis is supported by the fact that the Asn80Asp mutant of GpdQ has a much lower activity than both the wild-type form and the Asn80Ala mutant. This type of regulation has been observed in the mononuclear non-heme iron enzyme lipoxygenase, where the coordination flexibility of an asparagine ligand was shown to be crucial for optimal reactivity.91
Supplementary Material
Acknowledgments
This work was funded by the Australian Research Council (DP0664039). JAL acknowledges support from the National Sciences Foundation, grant NSU/RUI CHE0554083. ACH acknowledges support from the National Institutes of Health (GM47297). The authors also thank Dr Tri Le for his assistance with the collection of NMR data. We are grateful to Dr Tom Caradoc-Davies and support staff at the Australian Synchrotron for their valuable assistance during X-ray data collection.
Abbreviations
- bpNPP
bis(para-nitrophenyl) phosphate
- EtpNPP
ethyl para-nitrophenyl phosphate
- KIE
kinetic isotope effect
- MCD
magnetic circular dichroism
- pNPP
para-nitrophenyl phosphate
Footnotes
Supplementary Information Available: Overall structure and oligomeric organization of GpdQ (Fig. S1); examples of the effect of substrate concentration on activity at pH 6.00 and pH 11.25 for bpNPP (Fig. S2); pH profile for bpNPP (Fig. S3); effect of inhibition of inorganic phosphate towards pNPP hydrolysis (Fig. S4); determination of metal binding to GpdQ by kinetic assay (Fig. S5); pH profile for EtpNPP fitted to an equation for two protonation equilibria (Fig. S6). This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mitić N, Smith SJ, Neves A, Guddat LW, Gahan LR, Schenk G. Chem Rev. 2006;106:3338–3363. doi: 10.1021/cr050318f. [DOI] [PubMed] [Google Scholar]
- 2.Jackson MD, Denu JM. Chem Rev. 2001;101:2313–2340. doi: 10.1021/cr000247e. [DOI] [PubMed] [Google Scholar]
- 3.Wilcox DE. Chem Rev. 1996;96:2435–2458. doi: 10.1021/cr950043b. [DOI] [PubMed] [Google Scholar]
- 4.Crowder MW, Spencer J, Vila AJ. Acc Chem Res. 2006;39:721–728. doi: 10.1021/ar0400241. [DOI] [PubMed] [Google Scholar]
- 5.Lowther WT, Matthews BW. Chem Rev. 2002;102:4581–4607. doi: 10.1021/cr0101757. [DOI] [PubMed] [Google Scholar]
- 6.Klabunde T, Krebs B. Struct Bonding. 1997;89:177–198. [Google Scholar]
- 7.Twitchett MB, Sykes AG. Eur J Inorg Chem. 1999;12:2105–2115. [Google Scholar]
- 8.Oddie GW, Schenk G, Angel NZ, Walsh N, Guddat LW, de Jersey J, CassadyAIHamilton SE, Hume DA. Bone. 2000;27:575–584. doi: 10.1016/s8756-3282(00)00368-9. [DOI] [PubMed] [Google Scholar]
- 9.Schenk G, Gahan LR, Carrington LE, Mitić N, Valizadeh M, Hamilton SE, de Jersey J, Guddat LW. Proc Natl Acad Sci USA. 2005;102:273–278. doi: 10.1073/pnas.0407239102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barford D, Das AK, Egloff MP. Annu Rev Biophys Biomol Struct. 1998;27:133–164. doi: 10.1146/annurev.biophys.27.1.133. [DOI] [PubMed] [Google Scholar]
- 11.Rusnak F, Mertz P. Physiol Rev. 2000;80:1483–1521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- 12.Scheuermann RH, Echols H. Proc Natl Acad Sci USA. 1984;81:7747–7751. doi: 10.1073/pnas.81.24.7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Catalano CE, Allen DJ, Benkovic SJ. Biochemistry. 1990;29:3612–3621. doi: 10.1021/bi00467a004. [DOI] [PubMed] [Google Scholar]
- 14.Sträter N. Handbook of Metalloproteins. 2004;3:199–207. [Google Scholar]
- 15.Cox RS, Schenk G, Mitić N, Gahan LR, Hengge AC. J Am Chem Soc. 2007;129:9550–9551. doi: 10.1021/ja072647q. [DOI] [PubMed] [Google Scholar]
- 16.Aubert SD, Li Y, Raushel FM. Biochemistry. 2004;43:5707–5715. doi: 10.1021/bi0497805. [DOI] [PubMed] [Google Scholar]
- 17.Shim H, Raushel FM. Biochemistry. 2000;39:7357–7364. doi: 10.1021/bi000291o. [DOI] [PubMed] [Google Scholar]
- 18.Dumas DP, Durst H, Landis WG, Raushel FM, Wild JR. Arch Biochem Biophys. 1990;277:155–159. doi: 10.1016/0003-9861(90)90564-f. [DOI] [PubMed] [Google Scholar]
- 19.Shim H, Hong SB, Raushel FM. J Biol Chem. 1998;273:17445–17450. doi: 10.1074/jbc.273.28.17445. [DOI] [PubMed] [Google Scholar]
- 20.Jackson CJ, Liu JW, Coote ML, Ollis DL. Org Biomol Chem. 2005;3:4343–4350. doi: 10.1039/b512399b. [DOI] [PubMed] [Google Scholar]
- 21.Jackson C, Kim HK, Carr PD, Liu JW, Ollis DL. Biochim Biophys Acta. 2005;1752:56–64. doi: 10.1016/j.bbapap.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 22.McLoughlin SY, Jackson C, Liu JW, Ollis DL. Appl Environ Microbiol. 2004;70:404–412. doi: 10.1128/AEM.70.1.404-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang H, Carr PD, McLoughlin SY, Liu JW, Horne I, Qiu X, Jeffries CMJ, Russell RJ, Oakeshott JG, Ollis DL. Protein Eng. 2003;16:135–145. doi: 10.1093/proeng/gzg013. [DOI] [PubMed] [Google Scholar]
- 24.McLoughlin SY, Jackson C, Liu JW, Ollis D. Protein Expression Purif. 2005;41:433–440. doi: 10.1016/j.pep.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 25.Ely F, Foo JL, Jackson CJ, Gahan LR, Ollis D, Schenk G. Curr Top Biochem Res. 2007;9:63–78. [Google Scholar]
- 26.Jackson CJ, Carr PD, Liu JW, Watt SJ, Beck JL, Ollis DL. J Mol Biol. 2007;367:1047–1062. doi: 10.1016/j.jmb.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 27.Gerlt JA, Westheimer FH. J Am Chem Soc. 1973;95:8166–8168. doi: 10.1021/ja00805a035. [DOI] [PubMed] [Google Scholar]
- 28.Gerlt JA, Whitman GJR. J Biol Chem. 1975;250:5053–5058. [PubMed] [Google Scholar]
- 29.Larson TJ, Ehrmann M, Boos W. J Biol Chem. 1983;258:5428–5432. [PubMed] [Google Scholar]
- 30.Ghanem E, Li Y, Xu C, Raushel FM. Biochemistry. 2007;46:9032–9040. doi: 10.1021/bi700561k. [DOI] [PubMed] [Google Scholar]
- 31.Guddat LW, McAlpine AS, Hume D, Hamilton S, De Jersey J, Martin JL. Structure. 1999;7:757–767. doi: 10.1016/s0969-2126(99)80100-2. [DOI] [PubMed] [Google Scholar]
- 32.Klabunde T, Sträter N, Fröhlich R, Witzel H, Krebs B. J Mol Biol. 1996;259:737–748. doi: 10.1006/jmbi.1996.0354. [DOI] [PubMed] [Google Scholar]
- 33.Lindqvist Y, Johansson E, Kaija H, Vihko P, Schneider G. J Mol Biol. 1999;291:135–147. doi: 10.1006/jmbi.1999.2962. [DOI] [PubMed] [Google Scholar]
- 34.Sträter N, Beate J, Scholte M, Krebs B, Duff AP, Langley DB, Han R, Averill BA, Freeman HC, Guss JM. J Mol Biol. 2005;351:233–246. doi: 10.1016/j.jmb.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 35.Schenk G, Elliott TW, Leung E, Carrington LE, Mitić N, Gahan LR, Guddat LW. BMC Struct Biol. 2008;8:6. doi: 10.1186/1472-6807-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knöfel T, Sträter N. Nat Struct Mol Biol. 1999;6:448–453. doi: 10.1038/8253. [DOI] [PubMed] [Google Scholar]
- 37.Hopfner KP, Karcher A, Craig L, Woo TT, Carney JP, Tainer JA. Cell. 2001;105:473–485. doi: 10.1016/s0092-8674(01)00335-x. [DOI] [PubMed] [Google Scholar]
- 38.McMillen L, Beacham IR, Burns DM. Biochem J. 2003;372:625–630. doi: 10.1042/BJ20021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.White DJ, Reiter NJ, Sikkink RA, Yu L, Rusnak F. Biochemistry. 2001;40:8918–8929. doi: 10.1021/bi010637a. [DOI] [PubMed] [Google Scholar]
- 40.Rusnak F, Yu L, Todorovic S, Mertz P. Biochemistry. 1999;38:6943–6952. doi: 10.1021/bi982606u. [DOI] [PubMed] [Google Scholar]
- 41.Jackson CJ, Carr PD, Kim HK, Liu JW, Herrald P, Mitić N, Schenk G, Clyde A, Ollis DL. Biochem J. 2006;397:501–508. doi: 10.1042/BJ20060276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hendry P, Sargeson AM. J Am Chem Soc. 1989;111:2521–2527. [Google Scholar]
- 43.Hengge AC, Edens WA, Elsing H. J Am Chem Soc. 1994;116:5045–5049. [Google Scholar]
- 44.Boldeman JW, Einfeld D. Nucl Instrum Methods Phys Res. 2004;A521:306–317. [Google Scholar]
- 45.Leslie AGW. Acta Crystallogr. 2006;D62:48–57. [Google Scholar]
- 46.Bailey S. Acta Crystallogr. 1994;D50:760–763. [Google Scholar]
- 47.Emsley P, Cowtan K. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 48.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 49.Winn MD, Isupov MN, Murshudov GN. Acta Crystallogr. 2001;D57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 50.Aquino MAS, Lim JS, Sykes AG. J Chem Soc, Dalton Trans. 1994:429–436. [Google Scholar]
- 51.Smith SJ, Casellato A, Hadler KS, Mitić N, Riley MJ, Bortoluzzi AJ, Szpoganicz B, Schenk G, Neves A, Gahan LR. J Biol Inorg Chem. 2007;12:1207–1220. doi: 10.1007/s00775-007-0286-y. [DOI] [PubMed] [Google Scholar]
- 52.Cleland WW. Adv Enzymol Relat Areas Mol Biol. 1977;45:273–387. doi: 10.1002/9780470122907.ch4. [DOI] [PubMed] [Google Scholar]
- 53.McCain DF, Catrina IE, Hengge AC, Zhang ZY. J Biol Chem. 2002;277:11190–11200. doi: 10.1074/jbc.M109636200. [DOI] [PubMed] [Google Scholar]
- 54.Bigeleisen J, Wolfsberg M. Adv Chem Phys. 1958;1:15–76. [Google Scholar]
- 55.O’Leary MH, Marlier JF. J Am Chem Soc. 1979;101:3300–3306. [Google Scholar]
- 56.Caldwell SR, Raushel FM, Weiss PM, Cleland WW. Biochemistry. 1991;30:7444–7450. doi: 10.1021/bi00244a011. [DOI] [PubMed] [Google Scholar]
- 57.Knight WB, Weiss PM, Cleland WW. J Am Chem Soc. 1986;108:2759–2761. [Google Scholar]
- 58.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 59.Vaguine AA, Richelle J, Wodak SJ. Acta Crystallogr. 1999;D55:191–205. doi: 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]
- 60.Goldberg J, Huang H, Kwon Y, Greengard P, Nairn AC, Kuriyan J. Nature. 1995;376:745–753. doi: 10.1038/376745a0. [DOI] [PubMed] [Google Scholar]
- 61.Kissinger CR, et al. Nature. 1995;178:641–644. doi: 10.1038/378641a0. [DOI] [PubMed] [Google Scholar]
- 62.Kaden TA, Holmquist B, Vallee BL. Biochem Biophys Res Commun. 1972;46:1654–1659. doi: 10.1016/0006-291x(72)90799-1. [DOI] [PubMed] [Google Scholar]
- 63.Lever ABP. Inorganic electronic spectroscopy. 2. Elsevier; Amsterdam; Oxford; New York: 1984. [Google Scholar]
- 64.Prescott JM, Wagner FW, Holmquist B, Vallee BL. Biochemistry. 1985;24:5350–5356. doi: 10.1021/bi00341a012. [DOI] [PubMed] [Google Scholar]
- 65.Larrabee JA, Leung CH, Moore RL, Thamrong-Nawasawat T, Wessler BSH. J Am Chem Soc. 2004;126:12316–12324. doi: 10.1021/ja0485006. [DOI] [PubMed] [Google Scholar]
- 66.Fitting the pH dependence of kcat for the hydrolysis of EtpNPP to Equation 4 resulted in seven parameters, the three acid dissociation constants Kes1 – Kes3 listed in Table 2 and the four turnover numbers for the different protonation states of the enzyme51 (i.e. kcat1 < 0.05 s−1, kcat2 = 2.1 ± 0.2 s−1, kcat3 = 3.6 ± 0.2 s−1 and kcat4 < 0.1 s−1).
- 67.Cleland WW. In: Enzyme mechanism from isotope effects. Kohen A, Limback H, editors. 2006. p. 917. [Google Scholar]
- 68.Lowther WT, Matthews BW. Biochim Biophys Acta. 2000;1477:157–167. doi: 10.1016/s0167-4838(99)00271-x. [DOI] [PubMed] [Google Scholar]
- 69.Griffith JP, Kim JL, Kim EE, Sintchak MD, Thomson JA, Fitzgibbon MJ, Fleming MA, Caron PR, Hsiao K, Navia MA. Cell. 1995;82:507–522. doi: 10.1016/0092-8674(95)90439-5. [DOI] [PubMed] [Google Scholar]
- 70.Voegtli WC, White DJ, Reiter NJ, Rusnak F, Rosenzweig AC. Biochemistry. 2000;39:15365–15374. doi: 10.1021/bi0021030. [DOI] [PubMed] [Google Scholar]
- 71.Mullen GP, Serpersu EH, Ferrin LJ, Loeb LA, Mildvan AS. J Biol Chem. 1990;265:14327–14334. [PubMed] [Google Scholar]
- 72.McLoughlin SY, Ollis DL. Chem Biol. 2004;11:735–737. doi: 10.1016/j.chembiol.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 73.Merkx M, Pinkse MWH, Averill BA. Biochemistry. 1999;38:9914–9925. doi: 10.1021/bi9904454. [DOI] [PubMed] [Google Scholar]
- 74.Schenk G, Peralta RA, Batista SC, Bortoluzzi AJ, Szpoganicz B, Dick AK, Herrald P, Hanson GR, Szilagyi RK, Riley MJ, Gahan LR, Neves A. J Biol Inorg Chem. 2008;13:139–155. doi: 10.1007/s00775-007-0305-z. [DOI] [PubMed] [Google Scholar]
- 75.Hengge AC. Acc Chem Res. 2002;35:105–112. doi: 10.1021/ar000143q. [DOI] [PubMed] [Google Scholar]
- 76.Zalatan JG, Catrina R, Mitchell R, Gryska PK, O’Brien PJ, Herschlag D, Hengge AC. J Am Chem Soc. 2007;129:9789–9798. doi: 10.1021/ja072196+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hoff RH, Mertz P, Rusnak F, Hengge AC. J Am Chem Soc. 1999;121:6382–6390. [Google Scholar]
- 78.Czyryca PG, Hengge AC. Biochim Biophys Acta. 2001;1547:245–253. doi: 10.1016/s0167-4838(01)00191-1. [DOI] [PubMed] [Google Scholar]
- 79.Hengge AC, Cleland WW. J Am Chem Soc. 1991;113:5835–5841. [Google Scholar]
- 80.Jackson CJ, Hadler KS, Carr PD, Oakley AJ, Yip S, Schenk G, Ollis DL. Acta Cryst. 2008;F64:681–685. doi: 10.1107/S1744309108017600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Truong NT, Naseri JI, Vogel A, Rompel A, Krebs B. Arch Biochem Biophys. 2005;440:38–45. doi: 10.1016/j.abb.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 82.Funhoff EG, Wang Y, Andersson G, Averill BA. FEBS J. 2005;272:2968–2977. doi: 10.1111/j.1742-4658.2005.04686.x. [DOI] [PubMed] [Google Scholar]
- 83.The data were also fit to an equation derived for a model with two protonation equilibria (Fig. S6). The magnitudes of pKes1 and pKes3 are not greatly altered, but the fit is poor resulting in the appearance of a hollow at low pH and a hump at higher pH (Fig. S6). While sticky substrates may lead to the occurrence of a hollow or, more rarely, a hump52 the occurrence of both argue against the fit in Fig. S6. Furthermore, the presence of a viscogen (up to 30% sucrose) does not affect kcat/KM of EtpNPP (Fig. S6), an observation that indicates that this substrate is not sticky.
- 84.Baes CF, Mesmer RE. The hydrolysis of cations. Wiley; New York: 1976. [Google Scholar]
- 85.Badarau A, Page MI. Biochemistry. 2006;45:10654–10666. doi: 10.1021/bi060934l. [DOI] [PubMed] [Google Scholar]
- 86.Bicknell R, Knott-Hunziker K, Waley SG. Biochem J. 1983;213:61–66. doi: 10.1042/bj2130061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marti-Arbona R, Fresquet V, Thoden JB, Davis ML, Holden HM, Raushel FM. Biochemistry. 2005;44:7115–7124. doi: 10.1021/bi050008r. [DOI] [PubMed] [Google Scholar]
- 88.Barrios AM, Lippard SJ. J Am Chem Soc. 1999;121:11751–11757. [Google Scholar]
- 89.Evdokimov AG, et al. Proteins: Struct, Funct, Bioinf. 2007;66:538–546. [Google Scholar]
- 90.Jackson CJ, Foo JL, Kim HK, Carr PD, Liu JW, Salem G, Ollis DL. J Mol Biol. 2008;375:1189–1196. doi: 10.1016/j.jmb.2007.10.061. [DOI] [PubMed] [Google Scholar]
- 91.Schenk G, Neidig ML, Zhou J, Holman TR, Solomon EI. Biochemistry. 2003;42:7294–7302. doi: 10.1021/bi027380g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.