

Abstract
TRPV4 ion channels represent osmo-mechano-TRP channels with pleiotropic function and wide-spread expression. One of the critical functions of TRPV4 in this spectrum is its involvement in pain and inflammation. However, few small-molecule inhibitors of TRPV4 are available. Here we developed TRPV4-inhibitory molecules based on modifications of a known TRPV4-selective tool-compound, GSK205. We not only increased TRPV4-inhibitory potency, but surprisingly also generated two compounds that potently co-inhibit TRPA1, known to function as chemical sensor of noxious and irritant signaling. We demonstrate TRPV4 inhibition by these compounds in primary cells with known TRPV4 expression - articular chondrocytes and astrocytes. Importantly, our novel compounds attenuate pain behavior in a trigeminal irritant pain model that is known to rely on TRPV4 and TRPA1. Furthermore, our novel dual-channel blocker inhibited inflammation and pain-associated behavior in a model of acute pancreatitis – known to also rely on TRPV4 and TRPA1. Our results illustrate proof of a novel concept inherent in our prototype compounds of a drug that targets two functionally-related TRP channels, and thus can be used to combat isoforms of pain and inflammation in-vivo that involve more than one TRP channel. This approach could provide a novel paradigm for treating other relevant health conditions.
Transient receptor potential Vanilloid 4 (TRPV4) ion channels were initially discovered as osmotically-activated channels1,2. Discussing the channel’s possible role as mechanosensor, and its expression in sensory neurons in the trigeminal and dorsal root ganglion1,3,4, led to postulation and eventual experimental validation of a possible function in pain sensing and signaling1,3,4,5. This medically-relevant role was corroborated over time6,7,8,9,10,11,12,13,14,15, as was the mechano-sensory role of TRPV411,16,17,18,19,20. The pro-nociceptive prostanoid PGE2, activation of PAR-2 signaling, inflammation and nerve injury were found to augment TRPV4-mediated pain signaling in various systems5,6,9,12,21,22, including a novel model of temporo-mandibular joint (TMJ) pain14. In a shift of paradigm, TRPV4 was found to function as a relevant sensing molecule in epidermal keratinocytes for UVB overexposure15. UVB-exposed keratinocytes, depending on their TRPV4 expression and signaling, were functioning as organismal pain generators, supported by the finding that deletion of Trpv4 exclusively in these cells sufficed to greatly attenuate the organismal pain response. TRPV4 was also found to play a role in visceral pain, e.g. of the colon and pancreas7,8,18,23,24,25, the latter two conditions also co-involving TRPA18,24,26,27,28. The co-involvement of TRPV4 and TRPA1 was also noted in our TMJ model14, as well as in formalin-mediated irritant pain of the trigeminal territory, which serves as a generic model of cranio-facial pain13.
Importantly, blocking TRPV4 with selective inhibitors shows similar results as those obtained with genetic knockouts13,14,25,29,30,31,32,33,34, particular in models of TMJ pain or formalin-induced trigeminal formalin pain13,14. These findings suggest that TRPV4 could serve as a critical pain target, thus incentivizing the development of more potent and selective small-molecule inhibitors as new clinically-relevant therapeutic drugs. This direction has advantageous features because genetic approaches are currently limited to experimental conditions and TRPV4 inhibitors are not yet clinically available
The goal of this study was to develop TRPV4 inhibitors with increased potency over a previously used tool compound, GSK20532,33,34. Our results indicate that we have successfully developed compounds with significantly increased TRPV4-inhibitory potency as compared to the tool compound. Interestingly, our approach led to the development of two novel inhibitor molecules that simultaneously target TRPV4 and TRPA1, a potentially advantageous property that we successfully applied in two exemplary in-vivo preclinical models of pain, irritation and inflammation.
Results
Chemical synthesis of GSK205 derivatives and assessment of their TRPV4-inhibitory potency in cell-based assays
We modified compound GSK205 by generating 7 primary modifications, as shown in Fig. 1. One additional compound (16-19) that had the combined respective modifications of the two most potent compounds, as defined in primary screens, was also synthesized. We assessed TRPV4-inhibitory potency of these synthetic compounds in a Ca++ imaging assay in neuronal 2a (N2a) permanent tissue culture cells with directed expression of mammalian (rat) TRPV4. TRPV4 channels were stimulated with a selective activator compound, GSK1016790A (GSK101), used at 5 nM. For first round assessment, all TRPV4-inhibitory compounds were used at 5 μM (Fig. 2A). Compound 16-43C did not inhibit Ca++ influx, and its effect was similar to vehicle control. All other compounds inhibited TRPV4-mediated Ca++ influx, with compounds 16-8 and 16-18 emerging as the two most potent. Compound 16-19 which incorporated the modifications of both 16-8 and 16-18, was also effective in inhibiting TRPV4-mediated currents. However, we did not find a significant difference between compound 16-19 and 16-8, both of which virtually eliminated Ca++ influx.
Figure 1. Modifications of tool compound GSK205 for improved targeting of TRPV4.

The synthesized compounds differed in the highlighted part of the molecule, changed residue indicated with arrow. Compound 16-19 compound was synthesized to incorporate two modifications from two compounds, 16-8 and 16-18, found most potent in anti-TRPV4 screening assays (see Fig. 2).
Figure 2. Assessment of 16-… compounds in N2a cells with directed expression of TRPV4.

(A) Ca++ imaging screening of all compounds in N2A cells with directed expression of TRPV4 (rat). The cells were stimulated with TRPV4-selective activator compound, GSK101 (5 nM) in the presence of 5 μM of the respective inhibitor. The number on each bar corresponds to average peak ∆Ca++ concentrations in ≈100 cells. Inset: micrographs of pseudo-colored cells before and after activation with 5nM GSK101, in addition note the corresponding time course of the averaged Ca++ signal (fura-2 Ca++ imaging). Except for compound 16-43C, the difference to vehicle control reach the level of statistical significance p < 0.01 (one-way ANOVA). (B) Dose-response of the most potent, “winner” compounds in TRPV4-expressing N2a cells. The IC50 were; 0.45 ± 0.05 μM (16-8), 0.59 ± 0.12 μM (16-18), 0.81 ± 0.1 μM (16-19), 4.19 ± 0.71 μM (GSK205). Plot generated from averaged peak ∆Ca++ concentration of ≥75 cells per data-point.
We then conducted more detailed dose-response assessments for compounds 16-8, 16-18 and 16-19, which yielded an IC50 of 0.45, 0.81 and 0.59 μM, respectively, vs. an IC50 of 4.19 μM for parental compound GSK205. These findings represent an increased potency of the GSK205-derivative compounds by approximately 10-fold for 16-8, 8-fold for 16-19 and 5-fold for 16-18. Surprisingly, compound 16-19 was not significantly more potent than 16-8, whereas 16-8 was more potent than 16-18. Based on these results, we tested 16-8 and 16-19 vs GSK205 (as a control) in patch-clamp studies (Fig. 3). Our results indicate significantly increased potency of compounds 16-8 and 16-19 as compared to the parental molecule, GSK205 (all applied at 5 μM) in attenuating TRPV4-mediated currents.
Figure 3. TRPV4 channel inhibition by compounds 16-8 and 16-19 – patch-clamp e-phys.

(A) Current-voltage relationship of TRPV4-mediated currents after activation with 5 nM GSK101. Recordings were performed in TRPV4-GFP+ N2a cells. The representative traces represent an average of ≈12 sweeps. In all experiments, cells were pre-incubated with the respective compound (5 μM) for 5 minutes. (B) Average current densities at −100mV/+100 mV were significantly diminished by inhibitors (*P < 0.05; one-way ANOVA; n ≥ 5 cells/group).
We next decided to assess potency of the most potent compound 16-8 vs. parental compound GKS205 in two types of primary cells that are known to express TRPV4 and in which TRPV4 function has been demonstrated in a relevant biological context. We examined articular chondrocytes, which have prominent TRPV4 expression, where TRPV4 serves as the mechanotransducer of physiologic mechanical loads to regulate the cells’ anabolic response, and thus tissue homeostasis, in cartilage19. In addition, we studied brain astrocytes, where TRPV4 expression and relevant function has been previously demonstrated in regulating astrocyte cellular edema, in the coupling of neuronal activity to cerebral blood flow, and in mediating CNS traumatic injury35,36,37. Fulfilling our main objective, in both cell types we observed significantly increased potency of compound 16-8 as compared to the parental molecule, GSK205 (Fig. 4). Evaluation of the inhibitory potency of GSK205 derivatives in these primary cells, which express functional and biologically-relevant TRPV4 without directed over-expression of the channel (Fig. 4), directly corroborate the findings of more basic studies using heterologously TRPV4-overexpressing immortalized cell lines (Fig. 2), strongly supporting our conclusions on the increased potency of the newly derived compounds. Taken together, we identified compound 16-8 as a TRPV4-inhibitory compound with sub-micromolar potency in heterologous systems, with approximately a 10-fold increase in potency as compared to its parental molecule, GSK205. Moreover, 16-8 proved more effective in TRPV4-expressing primary skeletal and CNS-derived cells. However, the rational modification to compound 16-19, intended to further enhance potency, did not yield the intended effect.
Figure 4. Compound 16-8 inhibits TRPV4 in I˚ cells more potently than GSK205.

(A) I˚ articular chondrocytes (pig); dose-response comparison between the most potent compound, 16-8, and GSK205 in response to stimulation with 5 nM GSK101. Inset: Chondrocytes responding to activation with GSK101, fura-2 Ca++ imaging; right-hand image taken at 5 sec after GSK101 application. 16-8 was significantly more potent than GSK205 (mean ± SEM, n = 6 independent expts, n ≥ 25 cells/expt; *p < 0.05, t-test). Ordinate shows average peak ∆Ca++ concentrations. (B) I˚ astrocytes (rat); dose-response comparison between 16-8 and GSK205 in response to 5 nM GSK101. Inset: Astrocytes responding to activation with GSK101; right-hand image taken at 5 sec after GSK101 application (mean ± SEM, n = 5 independent expts, n ≥ 200 cells/expt; *p < 0.05, t-test). Ordinate shows average peak ∆Ca++ concentrations.
Before testing these compounds in relevant in-vivo animal models, we next tested their cellular toxicity as well as the specificity of these compounds against other selected TRP ion channels.
Novel TRPV4 inhibitors are selective, with benign toxicity profile, yet display potent inhibition of TRPA1
In heterologously transfected permanent N2a cells, we did not observe inhibitory potency of compounds 16-8 or 16-19 toward TRPV1, TRPV2 and TRPV3 (Fig. 5A). However, we made the unexpected discovery of sub-micromolar inhibitory potency vs TRPA1 for compounds 16-8 and 16-19, micromolar potency for GSK205, and, remarkably, no significant activity for compound 16-18 (Fig. 5B). We recorded IC50 of 0.41, 0.43, 5.56 μM and >25 μM for compounds 16-8, 16-19, GSK205 and 16-18, respectively.
Figure 5. Compounds 16-8 and 16-19 also potently inhibit TRPA1, not TRPV1-3.

(A) Specificity vs TRPV1-3. Both 16-8 and 16-19 (5 μM each) compounds did not inhibit TRPV1, −2 or −3 channels (all mouse isoforms), directed over-expression in N2a cells and subsequent Ca++ imaging. Mean±SEM is shown, ≥100 cells per condition. (B) Dose-dependent inhibition of TRPA1 (mouse, directed expression in N2a cells) by GSK 205, 16-8 and 16-19, activation with 100 μM mustard oil, resulting in IC50 of 5.56 ± 0.4 μM (GSK205), 0.41 ± 0.37 μM (16-19), 0.43 ± 0.3 μM (16-8). Plot generated from averaged peak ∆Ca++ concentration of ≥75 cells per data-point.
In terms of cellular toxicity, we found first evidence of toxicity using a sensitive cell viability assay over a time-course of 2 days, at 20 μM, and more pronounced effects at 40 μM (Fig. 6).
Figure 6. Cellular toxicity studies of compounds 16-8 and 16-19.

N2a cells were subjected to increasing concentrations of compounds 16-8 and 16-19, resulting cell viability was analyzed for the next 48 h. (A) Time course of cell viability in the presence of various concentrations of 16-8. Note clear reduction at 40 and 80 μM. (B) As in (A), for compound 16-19, with similar outcome. Representative result of 2 independent experiments.
Taken together, our results indicate that compounds 16-8 and 16-19 also inhibit TRPA1 at sub-micromolar potency, and their cellular toxicity vs their inhibitory potency against TRPV4/TRPA1 ranges at a factor of 50–100. Assessment of specificity of 16-8, 16-18 and 16-19 against a wider spectrum of receptors and ion channels will be the subject of dedicated future studies directed toward translation of these compounds to the clinic.
We next evaluated these compounds to an in-vivo model of irritant pain known to rely on both, TRPV4 and TRPA1.
TRPV4/TRPA1 dual-inhibitors are effective in containing trigeminal irritant pain
We have previously described TRPV4 as a cellular receptor for formalin13, and demonstrated its involvement in formalin irritant-evoked pain behavior, with a focus on trigeminally-mediated irritant pain behavior. In this recent study, we also demonstrate the co-contribution of TRPV4 together with TRPA1 in trigeminal formalin-evoked pain. We also showed the effective attenuation of trigeminal formalin-evoked pain behavior using GSK205 in a dose-dependent manner. Moreover, we found irritant pain behavior in response to selective activation of TRPV4 in the trigeminal territory, which was blocked by GSK205, and the absence of such an effect in Trpv4−/− pan-null mice.
With this pertinent background as a rationale, we applied TRPV4/TRPA1 dual-inhibitory compounds 16-8 and 16-19 systemically, using GSK205 as control, at 10 mg/kg dosage. We prioritized the dual-inhibitors over testing of compound 16-18 (TRPV4-only inhibitor) because (i) the trigeminal formalin pain model relies on both TRPV4 and TRPA1, (ii) we intended to develop TRPV4/TRPA1 dual-inhibitory molecules toward translational use in the first place. None of the compounds were effective at significantly diminishing pain behavior in the early phase after formalin whisker-pad injection, which represents an acute chemical tissue injury pain. In the delayed phase, which is understood as neurally-mediated pain indicative of early maladaptive neural plasticity, there was a significant attenuation of formalin-evoked pain behavior in response to compound 16-8 and 16-19, with compound 16-8 diminishing pain behavior at a remarkable >50%13, and compound 16-19 also showing a robust effect (Fig. 7A,B). Of note, at 10 mg/kg systemic application, there was no significant effect of GSK205, which was effective previously in a dose-dependent manner when applied by intradermal injection13. Thus, compounds 16-8 and 16-19, upon systemic application, effectively attenuate the late, neurally-mediated phase of trigeminal formalin pain, and these compounds are more potent in-vivo than their parental compound, GSK205.
Figure 7. 16-8 and 16-19 effectively attenuate formalin-evoked trigeminal irritant pain.

(A) Time-course of nocifensive behavior in WT mice following whisker-pad injection of 4% formalin. The mice were pre-injected (i.p., 10 mg/kg; 15 min before formalin) with GSK205, 16-8 or 16-19. Note effective reduction of nocifensive behavior in the late “neural” phase by compounds 16-8, 16-19, not by GSK205. (B) Cumulative response binned into 3 phases: acute phase (0–5 min), interphase (5–15 min), and late “neural” phase (15–45 min). Note significant reduction of nocifensive behavior in the late phase by 16-8, 16-19, not GSK205 (*P < 0.01 vs vehicle and GSK205, one-way ANOVA). (C) As in (A), but also including Trpv4−/− mice. Compounds were applied i.p. 15 min before formalin challenge, at 10 mg/kg except established TRPA1 blocker, A967079 (25 mg/kg). Previously-established attenuated nocifensive behavior in early and late phase in Trpv4−/− mice was recapped, which was reduced further by TRPA1 blocker, A967079. (D) As in (B), plus inclusion of Trpv4−/− mice. Robust effects of TRPA1-blocker, A967079, were mimicked equi-potently by 16-8 and 16-19 for early phase, and by 16-8 for late phase, partially by 16-19 for late phase. (A,C) show averaged behavioral metrics per time-point, bars in (B,D) represent mean ± SEM; for (D) *P < 0.05; #P < 0.005, one-way ANOVA; for all panels n = 5–8 mice/group.
In view of these in-vivo findings, taken together with the results from heterologous cellular expression systems that indicate an additional TRPA1-inhibitory effect of compounds 16-8 and 16-19, we decided to assess effectiveness of these compounds in a setting of genetically-encoded absence of Trpv4 (Trpv4−/− mouse), in order to better define their TRPA1-inhibitory potency in-vivo. We observed significant residual irritant-pain behavior in all phases of the formalin model in Trpv4−/− mice, consistent with our previous report13 (Fig. 7C,D). Immediate-phase pain behavior was virtually eliminated with compounds 16-8 and 16-19, both applied again at 10 mg/kg body-weight. Late-phase pain behavior was strikingly reduced when applying compound 16-8, and still significantly reduced vs vehicle-treated Trpv4−/− when applying compound 16-19, although not as potently as 16-8. A reference TRPA1-inhibitory compound, A967079, was used at 25 mg/kg body weight as a positive control to inhibit TRPA1, based on a previous report38. Reduction of pain behavior in Trpv4−/− mice was striking, more than 50% in the late neural phase. We noted equal potency of compound 16-8 at 10 mg/kg body weight vs. reference TRPA1-inhibitory compound A967079 at 25 mg/kg body weight, both reducing formalin-evoked trigeminal pain behavior to similarly robust degree (Fig. 7C,D). We conclude that compound 16-8 also functions as a potent TRPA1-inhibitor in an in-vivo irritant pain model specifically designed to assess the contribution of TRPA1 to trigeminal irritant pain, and at least as potent as an established reference TRPA1-antagonistic compound.
Potent TRPV4/TRPA1 dual-inhibitor, 16-8, is effective at controlling inflammation and pain in acute pancreatitis
These findings define compound 16-8 as a potent TRPV4/TRPA1 dual-inhibitor molecule, based on cell-based and live-animal results. We therefore decided to test it in a more specific preclinical pain model that relies on both, TRPV4 and TRPA1, in order to establish proof-of-principle that a dual-inhibitor can effectively treat pain and inflammation in pancreatitis. We used a pancreatitis model because it provides high translation potential due to unmet clinical need for new effective treatments in this condition39, as well as the known role of both TRPV4 and TRPA1 in pancreatitis pain and inflammation8,25,40.
Pancreatitis was induced with caerulein, a well-characterized model for acute pancreatitis41. Animals were treated with 10 mg/kg bw 16-8 by intraperitoneal injection, 30 min before induction of inflammation. We found significant attenuation of inflammatory parameters, namely edema, which was virtually eliminated in 16-8 treated animals. Furthermore, serum amylase, a marker of inflammatory injury of the pancreas, was significantly reduced by 16-8 treatment, as was myelo-peroxidase content of the pancreas, as a marker of inflammatory cell infiltration of the pancreas. The histopathological score for pancreas inflammation was also significantly reduced (Fig. 8A–E). Of note, pain behavior, similar to the effect of 16-8 on pancreas edema, was virtually eliminated upon treatment with compound 16-8. Thus, compound 16-8 was found to be highly effective in attenuating pain and inflammation of acute chemically-evoked pancreatitis.
Figure 8. Compound 16-8 attenuates acute pancreatitis and improves pain behavior.

(A) Caerulein-evoked acute pancreatitis causes pancreatic edema, which is eliminated by compound 16-8 (10 mg/kg, applied at 30 min before first exposure to caerulein). (B) Caerulein-evoked acute pancreatitis strongly elevates cellular toxicity marker amylase in serum. Amylase is reduced, but not significantly, in 16-8 treated animals. (C) caerulein-evoked acute pancreatitis causes elevated myelo-peroxidase (MPO) activity in serum, a marker for infiltration of inflammatory cells into the pancreas. MPO activity is significantly reduced in 16-8 treated mice. (D) caerulein-evoked acute pancreatitis can be readily demonstrated histologically, exemplified in the micrograph panels shown. Note increased pancreas inflammation in the middle-panel vs non-inflamed pancreas in vehicle-control challenged mice, and its attenuation by treatment with compound 16-8. (E) Bar diagram shows quantitation of inflammatory histologic parameters as shown in (D). Note significant increase of inflammation-index in caerulein acute pancreatitis mice, and its significant reduction upon treatment with compound 16-8. (F) Caerulein-evoked acute pancreatitis causes pain behavior, significantly reduced by compound 16-8. Note greatly reduced activity over the 6 h test period in caerulein-induced acute pancreatitis. This nocifensive behavior is greatly improved in response to systemic application of compound 16-8. Results are expressed as mean ± SEM; n = 6 mice/group; *P < 0.05 (one-way ANOVA).
Benign preliminary toxicity and pharmacokinetics of novel TPRV4/TRPA1 inhibitors
Promising properties of potent 16-… compounds prompted us to attempt to define their initial preliminary features in terms of in-vivo toxicity and pharmacokinetics, which will be followed by more extensive and in-depth investigations in future studies. For this, we chose compound 16-8 as the all-around most potent dual TRPV4/TRPA1 inhibitor, and also compound 16-19 with its potentially increased lipophilicity (Suppl Table 1). We measured compound concentration in several organs and plasma, and detected micromolar/submicromolar concentrations in liver and kidney, less than 100 nM concentrations in heart, brain, brainstem, trigeminal ganglion and skin. Of note, compounds were virtually undetectable in plasma (Suppl Fig. 1A). We detected higher concentrations of 16-19 in non-nervous tissue, especially liver and kidney, a pattern perhaps related to compound 16-19’s increased lipophilicity. Based on this finding, we next examined 6 and 24 h time-points and detected 10-20 fold higher concentrations of 16-19 at the 6 h time-point, compared to the 1 h time-point, indicative of compound sequestration into solid organs (Suppl Fig. 1B–D). Values at the 24 h time-point were lower than at the 6 h but higher than at the 1 h time-point, indicating ongoing protracted compound clearance. We next confirmed that low/non-detectable concentrations in plasma were not caused by compound denaturation/degradation in plasma, as indicated in Suppl Fig. 1E, which shows no loss of detectable compound after 4 h incubation in plasma at 37 °C, conducted using compound 16-19.
We next performed basic preliminary in-vivo toxicity studies for compounds 16-8 and 16-19, both at 10 mg/kg bw, which was the effective concentration in both in-vivo pain models. We did not detect first evidence of cardiac, hepatic and renal toxicity, when comparing compounds 16-8 and 16-19 with vehicle (Suppl Fig. 2). For cardiac assessment, we did not detect differences and changes in heart rate over 1 h, conducted by EKG at the 6 h time-point. Serum creatinin as marker of renal function and alanine-amino-transferase (ALT) as marker of hepato-cellular integrity were not significantly elevated in animals treated with compounds 16-8 or 16-19. Thus, initial evidence for potent 16-… compounds highlights their acceptable preliminary pharmacokinetics properties as well as lack of gross systemic toxicity. Future studies will be necessary for more detailed assessment of the pharmacokinetics and toxicity of these compounds.
Discussion
Here we describe novel compounds that simultaneously inhibit both TRPV4 and TRPA1 ion channels. Both targets were inhibited by the novel “dual-inhibitors” at sub-micromolar potency in heterologous cellular channel activation assays. Furthermore, these compounds showed potent activity against TRPV4 in primary cells with native TRPV4 expression, and were more potent than their related parent-compound. The most potent compound identified here, compound 16-8, also showed a favorable activity profile in two pain-inflammation models, one of them a general irritant-pain model in the trigeminal system, the trigeminal formalin model, the other a visceral pain and inflammation assay with specificity for the pancreas, the caerulein-induced acute pancreatitis model. Of note, both in-vivo pain models have been shown previously to rely on co-contribution of TRPV4 and TRPA1. In this regard, several other relevant medical conditions, discussed in more detail below, also rely on TRPV4/TRPA1 and represent important unmet clinical needs that need to be addressed by translational-medical approaches. Therefore a potent “dual-inhibitor” for a specific combination of TRP ion channels, such as TRPV4 and TRPA1, could be highly beneficial in these indications.
More specifically, primary chondrocytes represent a cellular model for joint disease with involvement of TRPV4 in cartilage maintenance as well as arthritis/osteoarthritis. Recent evidence also suggests a potential role for TRPA1 in mediating joint pain in osteoarthritis42. In addition, TRPV4-expressing astrocytes are involved in many neurologic and psychiatric diseases such as pain, epilepsy, multiple sclerosis and other CNS autoimmune conditions, stroke, traumatic brain/spinal cord injury, brain edema, CNS infections, and more35,36,37. TRPA1 expression in astrocytes has also been reported43. Therefore, novel dual TRPV4/TRPA1 inhibitors might be suitable compounds for treatment of a number of diseases, from a spectrum of disorders affecting the nervous system as well as degenerative or inflammatory musculoskeletal conditions. For both types of disorders, we view compartmentalized application of compounds, i.e. intra-thecal or intra-articular delivery, as feasible future routes of delivery, in order to affect target cells more directly without affecting - TRPV4 and TRPA1 systemically.
In terms of in-vivo pain models, the trigeminal formalin model is rather a general model, not a direct pre-clinical translational-medical model. However, formalin models have a very robust track record in the identification of efficacious new compounds against pain44. Our findings with compound 16-8 in the trigeminal formalin model can be interpreted along these lines, especially for trigeminal pain including headaches45,46. In the context of these most prevalent neurological diseases, involvement of TRPV4 and TRPA1 has been reported previously in preclinical models, underscoring the case for their involvement with some compelling preclinical insights for both channels47,48,49,50.
Pancreatitis in mice represents a more specific preclinical visceral inflammatory pain model, helpful to elucidate pathophysiology of this specific pain in order to better address a significant unmet medical need. In the current study pain and edema were strongly reduced by compound 16-8, the compound identified as showing the most advantageous features when tested in the trigeminal formalin pain model vs another high-potency dual-inhibitor, 16-19, and vs parent molecule, GSK205. This is in keeping with previous studies which demonstrated that both TRPA1 and TRPV4 contributed to pancreatic inflammation and pancreatic pain25. More precisely TRPA1 was shown to contribute to both pain and inflammation while TRPV4 contributed more selectively toward pain8. In our current study, there was significant attenuation of inflammatory parameters, but the reduction was not as robust as for pain and edema. This finding begets two important issues, namely (i) this could be a feature of the 2 targeted TRP channels, that they are more significant for pain than for inflammation (as concluded from experimental evidence in14), and (ii) edema of the pancreas, which is readily measurable by imaging techniques in patients, could possibly serve as a bio-marker for pancreatic pain. Beyond its role as biomarker, pancreas edema could sustain pancreas pain. At the pathophysiological level, edema will encompass edematous distension of the inflamed organ causing mechanical pain. This pain, amplified by inflammation of the painful organ, will in turn cause neurogenic inflammation, which will give rise to increased level of edema. This could evolve into a detrimental feed-forward mechanism. The specific effect of compound 16-8 on pancreatitis pain could be due to the compound interfering potently and efficiently with such a feed-forward mechanism as in (ii), by potently inhibiting both channels, as laid out in (i).
Beyond the two pain-inflammation conditions tested, TRPV4/TRPA1 co-involvement appears to play a role in several health-relevant conditions, such as colitis, itch, injury to airway and lungs via the inhalatory route and chronic cough28,34,51,52,53,54,55,56,57,58. In addition, interesting recent findings point toward a prominent role for TRPV4 in conditions as diverse as fibrotic disorders, UVB skin injury, and premature birth15,59,60. In these conditions, possibly via TRPA1-expressing innervating sensory neurons, a co-contribution by TRPA1 could be an important element. For such cases, 16-… compounds represent attractive candidates for effective treatment, to address the significant underlying unmet clinical need. Except pancreatitis, compound access to relevant target cells could also be readily accomplished by topical, non-systemic delivery via transdermal, transmucosal, inhalatory, intra-articular or intra-thecal formulations.
Our study presented here was strongly geared toward a translational medical agenda, meaning demonstration of effect of TRPV4/TRPA1 dual inhibitors, combined with a first-pass at pharmaco-tox assessment were our priority, rather than in-depth mechanistic studies. In addition, our goal was to demonstrate that modified 16-… compounds were more potent than the parent compound, GSK205. For future studies, in addition to continuation of a translational-medical agenda based on 16-… compounds, e.g. in-depth assessment of 16-… compounds at human isoforms of TRPV4 and TRPA1, effect of 16-… compounds in TRPV4/TRPA1-expressing primary human cells, our current results raise the following important questions/issues, namely a mandate to conduct mechanistic studies that address how TRPV4/TRPA1 dual inhibitors act on their respective target channels, and whether there is perhaps a shared mechanism between TRPV4 and TRPA1 of channel inhibition by potent 16-… compounds. In this context, it will be rewarding to zero in on a potential mechanism as to why compounds 16-8, 16-19 and to minor degree GSK205 are active against TRPA1 whereas compound 16-18 interestingly is not. Furthermore, the question why compound 16-19 fails to show increased potency over 16-8 will be an interesting one to address in future studies.
Materials and Methods
Compound synthesis
Compound synthesis is explicitly described in Supplementary Information (Suppl Fig. 3 and Suppl Tables 2–5).
Animals
8–12 week old mice were used throughout the experiments. Trpv4−/− mice3 have been outcrossed to WT (C57BL/6J) background and genotyped by PCR3,15. Animals were housed in climate-controlled rooms on a 12/12 h light/dark cycle with water and standardized rodent diet that was available ad libitum. All experiments were conducted in compliance and accordance with the guidelines of the NIH and the Institutional Animals’ Care and Use Committee (IACUC) of Duke University, and under a valid IACUC protocol of the Duke University IACUC. All animal methods described in this publication were approved by the Duke University IACUC.
Trigeminal formalin irritant behavior – mouse model
Implementation of this model was conducted as described previously13.
Video-taped nocifensive behavior was assessed by investigators blinded to genotype and treatment.
Acute pancreatitis mouse model
C57BL/6J male mice 8–10 weeks of age were subjected to acute pancreatitis by intraperitoneal injections of supramaximal doses of caerulein (50 μg/kg) every hour for a total of 6 h, as previously described in61. Control animals received 25% DMSO-saline solution by intraperitoneal injection every hour for 6 h. Compound 16-8 was dissolved in this vehicle (10 mg/kg) and injected i.p. 30 min prior to the first injection of caerulein. Animals were sacrificed 1 h after the last injection. Blood was collected and pancreatic tissue was promptly isolated, weighed for determination of pancreas wet weight/body weight ratio. Samples of tissues were fixed overnight in 10% neutral-buffered formalin, paraffin embedded and H&E-stained, or pancreatic tissue was quickly frozen and assessed for myeloperoxidase (MPO) activity. Serum amylase, MPO and histologic evaluation were conducted as described previously61.
Assessment of nocifensive behavior8: Mice were housed in individual cages and video-recorded during the entire experiment. Two mice at a time were observed. Linear movement was measured as one event when mice passed through the median plane of the cage. Analysis began immediately after the first caerulein/vehicle injection and continued until the end of the experiment. Results were expressed as the sum of the movement events spanning the 6 h time-period following the first injection.
Cell cultures
N2a cells were used for directed expression of TRP ion channels as described previously13. TRPV4-eGFP from rat was used, previously found to respond to stimulation with GSK101 and hypotonicity in similar manner as native, non-fused TRPV4. All other channels were native channels from mouse, eGFP was co-transfected. Stimulation of over-expressed TRPV4 was conducted with GSK101 (5 nM), TRPV1 with capsaicin (10 μM), TRPV2 with hypotonicity (270 mosmol/L), TRPV3 with camphor (100 μM) and TRPA1 with mustard oil (100 μM). eGFP control-transfected N2a cells did not respond to these stimuli. Ca++ imaging was performed as described previously13,14,34.
To visualize dose-response relationships, Hill plots were conducted using the Igor Pro software program, which derived the plots based on the following equation:
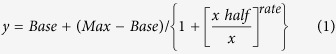 |
Primary porcine chondrocytes derived from femoral condyles of skeletally mature pigs were cultured and subjected to Ca++ imaging as described previously19,32,62,63.
Astrocyte cultures were conducted following established protocols64,65,66. Astrocytes were prepared from Sprague Dawley rat embryos (E18). Briefly, the isolated cortices were minced, and then incubated with trypsin and DNase. Dissociated cells were suspended in Dulbeccco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal calf serum and penicillin/streptomycin (100 U/ml and 100 μg/ml, respectively). Thereafter, cell suspensions were plated in 75 cm2 tissue culture flasks (10 × 106 cells/flask) which were pre-coated with poly-L-lysine (10 μg/ml). The cells were maintained in a 10%CO2 incubator at 37 °C. After 10–12 days, the media was removed and adherent cells were trypsinized (0.25%) and plated out onto coverslips for subsequent Ca++ imaging34,67. >95% of the cells were found to express astrocyte marker, glial fibrillary acidic protein (GFAP)68.
Cell viability in culture
N2a cells were cultured in 96 well plates for 24–48 h. Cell viability studies relied on metabolic capability monitored with the indicator dye resazurin. Its reduction to resorufin (indicated by color change dark blue to pink) was monitored over time. Changes in absorbance at λ = 570 nm were recorded using a microplate reader (Molecular Devices). Metabolically active and viable cells shared the ability to reduce resazurin to resorufin whereas dead cells did not. Eight replicate cultures per experimental point were studied.
Assessment of hepatic, renal and cardiac function in mice treated with 16-… compounds
Mice were treated i.p. with compounds 16-8 and 16-19 (10 mg/kg). Hepatic and renal integrity were analyzed by alanine amino-transferase- and creatinine assays (Sigma), both relying on measurement of absorbance at λ = 570 nm in 96-well micro-titerplates. 8 technical replicates per animal were performed.
For heart rate assessment in mice treated with 16-8 and 16-19, animals were fitted with two electrodes, one to the ear, via clip, one to the rib-cage, using firm adhesive. Heart rate was monitored and analyzed using axoscope and clampfit 9.2 software (Molecular Devices)
Liquid Chromatography – Tandem Mass Spectrometry (LC-MS/MS)
Mice were treated with 10 mg/kg i.p. of the respective inhibitor. Post-euthanasia harvested tissue was frozen in liquid nitrogen and stored at −80 °C for further analysis.
Frozen tissue samples were partially thawed and cut into 1 mm slices, 5–15 mg tissue, 2-fold excess water (mass/vol.), 6-fold excess acetonitrile (16-… compounds) or methanol (GSK205) containing appropriate amount of internal standard, and 2.5 mm zirconia/silica bead (Biospec Products Inc.) were added to 500- μL polypropylene (PP) conical tube, homogenized in a Fast-Prep apparatus (Thermo-Savant) at speed “4” for 20 sec at room temp, and centrifuged at 13,600 g for 5 min at room temp. Depending on the expected concentration range of the measured compound, the supernatant was diluted 1/4–1/20 (in Mobile phase A, see below) and placed in autosampler for LC-MS/MS analysis.
The LC-MS/MS assay for 16-… compounds and GSK205 was developed on an Agilent 1200 series LC system interfaced with Applied Biosystems API 5500QTrap, a hybrid triple quadruple-linear trap MS/MS spectrometer. Analyst (version 1.6.1) software was used for mass parameters tuning, data acquisition, and quantification. LC column: 3 × 4 mm RP C18 (Phenomenex, AJ0-4287) was operated at 35 °C. Mobile phase A: 0.1% formic acid, 2% acetonitrile, in LC/MS-grade water; mobile phase B: acetonitrile; flow rate: 1 mL/min, 1:1 MS/MS:waste split. Run time was 4 min. Diverter valve was used to send flow to MS/MS only between 1.2 and 2.5 min. The elution gradient was: 0–0.5 min, 1%B; 0.5–1.2 min, 1–95%B; 1.2–1.5 min, 95%B; 1.5–1.6 min, 95–1%B. Autosampler was operated at 4 °C; injection volume was kept at 10–50 μL. Electrospray ionization (ESI) source parameters were: positive ionization mode, curtain gas flow = 30, ionization potential = 5500 V, temperature = 500 °C, nebulizing gas 1 flow = 30, nebulizing gas 2 = 30, declustering potential = 20 V. 16-… compounds and GSK205 were individually infused as 100 nM solutions in 50%A/50%B at 10 μL/min flow rate and parameters optimized to provide maximal ion count for “parent” and collision-produced (“daughter”) MS/MS ions. Parent/daughter quantifier [qualifier] ions utilized: GSK205 (401.1/280[370]), 16-8 (400.1/279.1[91.1], 16-16 (387.1/280[105]), 16-18 (415.2/280[370], 16-19 (414.1/279.2[91.1]. Standard (analyte of interest)/internal standard pairs utilized: GSK205/16-16, 16-8/16-16, 16-18/GSK205, 16-19/16-8.
Calibration samples (n = 6) were prepared by adding pure standard of the measured compound to tissue homogenate (tissue + 2-fold excess water, mass/vol) in the appropriate range needed for the particular dosing regime. Organs studied were analyzed alongside the study samples. The following are typical ranges used (the lower value representing also the LLOQ at 80% accuracy limit, all other calibrator levels at 85% accuracy limit): 0.38–6 nM (plasma), 6–100 nM (skin), 6–48 nM (heart), 7.5–120 nM (brain), 19–300 or 1500–24000 nM (liver), 56–900 or 1500–12000 nM (kidney), 500–8000 nM (fat). Peak integration, calibration, and quantification was performed within Analyst software. The response of the peak area standard/int. std. to nominal concentration was linear with r = 0.999 or better.
Patch Clamp Recordings
Heterologously transfected N2a cells were subjected to patch clamp electrophysiological recordings. Briefly, 24 h after transfection cells were prewashed with extracellular fluid (ECF) which contained (in mM) 1 MgCl2, 10 Glucose, 10 HEPES, 145 NaCl and 2 CaCl2 (pH 7.4, 310mOsM). Cells were then incubated with or without TRPV4 inhibitors in ECF for 5 min before whole cell recording. Cover slips were transferred to a recording chamber mounted on the stage of a Leica inverted microscope that was equipped with fluorescent filters. Transfected cells were identified before patching by their green florescent color. Cells were patched with a 2.5–3.0 MΩ glass electrode pulled from borosilicate glass capillaries using pipette puller (Sutter instruments). The intracellular solution contained (mM) 140 CsCl, 10 HEPES, 1 EGTA, 0.3 Na-GTP, 2 Na2-ATP, and 2 MgCl2 (pH 7.4, 295 mOsm). Whole cell currents were recorded using pclamp 9.2 software and Axopatch 200B amplifier (Molecular Devices). The cells were first clamped at −65 mV before applying a 1 s voltage ramp from −110 mV to +120 mV. The voltage ramp was applied every 2 seconds for 15 to 20 sweeps. Capacitance was monitored throughout the experimental recordings. Reported data was within ±3 pF.
Statistical Analysis
Data are expressed as mean ± SEM. Two-tail t-tests or one-way ANOVA followed by Tukey post-hoc test were used for group comparisons. P < 0.05 indicated statistically significant differences
Additional Information
How to cite this article: Kanju, P. et al. Small molecule dual-inhibitors of TRPV4 and TRPA1 for attenuation of inflammation and pain. Sci. Rep. 6, 26894; doi: 10.1038/srep26894 (2016).
Supplementary Material
Acknowledgments
This work was supported in part by NIH/NCI Comprehensive Cancer Center Core Grant 2P30-CA014236-41 (IS), National Institutes of Health (grants DE018549 (WBL), AR48182 (FG&WBL), AR48182-S1 (FG&WBL, Minority Supplement PK), AR48852 (FG), AG15768 (FG), AR50245 (FG), AG46927 (FG), DK064213 (RAL), DK091946 (RAL), DK098796 (RAL), F33DE024668 (YC, mentor WBL), K12DE022793 (YC), K12CA100639 (RAM)), US Department of Defense (W81XWH-13-1-0299 (WBL)), US Department of Veterans Affairs (RAL), NSF grant #1445792 (FG), the Arthritis Foundation (FG), and a Harrington Discovery Institute (Cleveland OH) Scholar-Innovator Award (WBL). We would like to thank Dr. Holly Leddy (Duke University, Durham NC) for providing isolated porcine chondrocytes for this study.
Footnotes
Content as reported in this paper has been included in patent applications by the Duke University Office of Licensing and Ventures.
Author Contributions P.K. conducted experiments, analyzed data, wrote paper, conceptual input. Y.C. conducted experiments, analyzed data. W.L. conducted experiments, analyzed data. M.Y. conducted experiments, analyzed data, wrote paper. S.H.L. conducted experiments. J.R. conducted experiments, analyzed data. R.S. conducted experiments, analyzed data. P.F. conducted experiments, analyzed data. D.M.G. conducted experiments, analyzed data. S.A.S. wrote paper, conceptual input. I.S. conducted experiments, conceptual input. R.A.M. wrote paper, conceptual input, analyzed data. R.A.L. wrote paper, conceptual input, analyzed data. F.G. wrote paper, conceptual input, analyzed data. W.B.L. wrote paper, conceptual input, analyzed data.
References
- Liedtke W. et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103, 525–535 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strotmann R., Harteneck C., Nunnenmacher K., Schultz G. & Plant T. D. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol. 2, 695–702 (2000). [DOI] [PubMed] [Google Scholar]
- Liedtke W. & Friedman J. M. Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci. USA 100, 13698–13703 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W., Tobin D. M., Bargmann C. I. & Friedman J. M. Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorhabditis elegans. Proc Natl Acad Sci. USA 100, 14531–14536, doi: 10.1073/pnas.2235619100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessandri-Haber N., Joseph E., Dina O. A., Liedtke W. & Levine J. D. TRPV4 mediates pain-related behavior induced by mild hypertonic stimuli in the presence of inflammatory mediator. Pain 118, 70–79 (2005). [DOI] [PubMed] [Google Scholar]
- Alessandri-Haber N. et al. Transient receptor potential vanilloid 4 is essential in chemotherapy-induced neuropathic pain in the rat. J Neurosci 24, 4444–4452 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley S. M. et al. Selective role for TRPV4 ion channels in visceral sensory pathways. Gastroenterology 134, 2059–2069, doi: 10.1053/j.gastro.2008.01.074 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceppa E. et al. Transient receptor potential ion channels V4 and A1 contribute to pancreatitis pain in mice. American journal of physiology. Gastrointestinal and liver physiology 299, G556–571, doi: 10.1152/ajpgi.00433.2009 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant A. D. et al. Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. The Journal of physiology 578, 715–733 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine J. D. & Alessandri-Haber N. TRP channels: Targets for the relief of pain. Biochim Biophys Acta (2007). [DOI] [PubMed] [Google Scholar]
- McNulty A. L., Leddy H. A., Liedtke W. & Guilak F. TRPV4 as a therapeutic target for joint diseases. Naunyn Schmiedebergs Arch Pharmacol 388, 437–450, doi: 10.1007/s00210-014-1078-x (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segond von Banchet G. et al. Neuronal IL-17 receptor upregulates TRPV4 but not TRPV1 receptors in DRG neurons and mediates mechanical but not thermal hyperalgesia. Mol Cell Neurosci 52, 152–160, doi: 10.1016/j.mcn.2012.11.006 (2013). [DOI] [PubMed] [Google Scholar]
- Chen Y. et al. TRPV4 is necessary for trigeminal irritant pain and functions as a cellular formalin receptor. Pain 155, 2662–2672, doi: 10.1016/j.pain.2014.09.033 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. et al. Temporomandibular joint pain: a critical role for Trpv4 in the trigeminal ganglion. Pain 154, 1295–1304, doi: 10.1016/j.pain.2013.04.004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore C. et al. UVB radiation generates sunburn pain and affects skin by activating epidermal TRPV4 ion channels and triggering endothelin-1 signaling. Proc Natl Acad Sci. USA 110, E3225–3234, doi: 10.1073/pnas.1312933110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes J. et al. IP3 sensitizes TRPV4 channel to the mechano- and osmotransducing messenger 5′-6′-epoxyeicosatrienoic acid. J Cell Biol. 181, 143–155, doi: 10.1083/jcb.200712058 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil R. G. & Heller, S. The mechanosensitive nature of TRPV channels. Pflugers Arch 451, 193–203 (2005). [DOI] [PubMed] [Google Scholar]
- Sipe W. E. et al. Transient receptor potential vanilloid 4 mediates protease activated receptor 2-induced sensitization of colonic afferent nerves and visceral hyperalgesia. American journal of physiology. Gastrointestinal and liver physiology 294, G1288–1298 (2008). [DOI] [PubMed] [Google Scholar]
- O’Conor C. J., Leddy H. A., Benefield H. C., Liedtke W. B. & Guilak F. TRPV4-mediated mechanotransduction regulates the metabolic response of chondrocytes to dynamic loading. Proc Natl Acad Sci. USA 111, 1316–1321, doi: 10.1073/pnas.1319569111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews B. D. et al. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integrative biology: quantitative biosciences from nano to macro 2, 435–442, doi: 10.1039/c0ib00034e (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P. et al. Cathepsin S causes inflammatory pain via biased agonism of PAR2 and TRPV4. The Journal of biological chemistry 289, 27215–27234, doi: 10.1074/jbc.M114.599712 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning L. et al. Role of colchicine-induced microtubule depolymerization in hyperalgesia via TRPV4 in rats with chronic compression of the dorsal root ganglion. Neurological research 36, 70–78, doi: 10.1179/1743132813Y.0000000261 (2014). [DOI] [PubMed] [Google Scholar]
- Cenac N. et al. Transient receptor potential vanilloid-4 has a major role in visceral hypersensitivity symptoms. Gastroenterology 135, 937–946, 946 e931-932, doi: 10.1053/j.gastro.2008.05.024 (2008). [DOI] [PubMed] [Google Scholar]
- Mueller-Tribbensee S. M. et al. Differential Contribution of TRPA1, TRPV4 and TRPM8 to Colonic Nociception in Mice. PLoS One 10, e0128242, doi: 10.1371/journal.pone.0128242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. P. et al. Alcohol and high fat induced chronic pancreatitis: TRPV4 antagonist reduces hypersensitivity. Neuroscience 311, 166–179, doi: 10.1016/j.neuroscience.2015.10.028 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz E. S. et al. TRPV1 and TRPA1 antagonists prevent the transition of acute to chronic inflammation and pain in chronic pancreatitis. J Neurosci 33, 5603–5611, doi: 10.1523/JNEUROSCI.1806-12.2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaruzza F. et al. Transient receptor potential ankyrin 1 mediates chronic pancreatitis pain in mice. American journal of physiology. Gastrointestinal and liver physiology 304, G1002–1012, doi: 10.1152/ajpgi.00005.2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel M. A. et al. TRPA1 and substance P mediate colitis in mice. Gastroenterology 141, 1346–1358, doi: 10.1053/j.gastro.2011.07.002 (2011). [DOI] [PubMed] [Google Scholar]
- Everaerts W. et al. Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc Natl Acad Sci. USA 107, 19084–19089, doi: 10.1073/pnas.1005333107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feetham C. H., Nunn N. & Barrett-Jolley R. The depressor response to intracerebroventricular hypotonic saline is sensitive to TRPV4 antagonist RN1734. Frontiers in pharmacology 6, 83, doi: 10.3389/fphar.2015.00083 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent F. et al. Identification and characterization of novel TRPV4 modulators. Biochem Biophys Res Commun 389, 490–494, doi: 10.1016/j.bbrc.2009.09.007 (2009). [DOI] [PubMed] [Google Scholar]
- Phan M. N. et al. Functional characterization of TRPV4 as an osmotically sensitive ion channel in porcine articular chondrocytes. Arthritis Rheum 60, 3028–3037, doi: 10.1002/art.24799 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L. et al. TRPV4 is a regulator of adipose oxidative metabolism, inflammation, and energy homeostasis. Cell 151, 96–110, doi: 10.1016/j.cell.2012.08.034 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. et al. TRPV4-Mediated Calcium Influx into Human Bronchial Epithelia upon Exposure to Diesel Exhaust Particles. Environmental health perspectives 119, 784–793 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki K., Ikenaka K., Tamalu F., Tominaga M. & Ishizaki Y. A novel subtype of astrocytes expressing TRPV4 regulates neuronal excitability via release of gliotransmitters. The Journal of biological chemistry, doi: 10.1074/jbc.M114.557132 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn K. M., Hill-Eubanks D. C., Liedtke W. B. & Nelson M. T. TRPV4 channels stimulate Ca2+-induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc Natl Acad Sci. USA 110, 6157–6162, doi: 10.1073/pnas.1216514110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati V. et al. An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc Natl Acad Sci. USA 108, 2563–2568, doi: 10.1073/pnas.1012867108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. et al. Selective blockade of TRPA1 channel attenuates pathological pain without altering noxious cold sensation or body temperature regulation. Pain 152, 1165–1172, doi: 10.1016/j.pain.2011.01.049 (2011). [DOI] [PubMed] [Google Scholar]
- Forsmark C. E. & Liddle R. A. The challenging task of treating painful chronic pancreatitis. Gastroenterology 143, 533–535, doi: 10.1053/j.gastro.2012.07.029 (2012). [DOI] [PubMed] [Google Scholar]
- Kowal J. M., Yegutkin G. G. & Novak I. ATP release, generation and hydrolysis in exocrine pancreatic duct cells. Purinergic signalling 11, 533–550, doi: 10.1007/s11302-015-9472-5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddle R. A. Pancreatitis: the acid test. Gastroenterology 139, 1457–1460, doi: 10.1053/j.gastro.2010.09.021 (2010). [DOI] [PubMed] [Google Scholar]
- Moilanen L. J. et al. Monosodium iodoacetate-induced inflammation and joint pain are reduced in TRPA1 deficient mice–potential role of TRPA1 in osteoarthritis. Osteoarthritis and cartilage/OARS, Osteoarthritis Research Society 23, 2017–2026, doi: 10.1016/j.joca.2015.09.008 (2015). [DOI] [PubMed] [Google Scholar]
- Shigetomi E., Tong X., Kwan K. Y., Corey D. P. & Khakh B. S. TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nature neuroscience 15, 70–80, doi: 10.1038/nn.3000 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil J. S. Animal models of pain: progress and challenges. Nat Rev Neurosci 10, 283–294, doi: 10.1038/nrn2606 (2009). [DOI] [PubMed] [Google Scholar]
- Khan A. & Hargreaves K. M. Animal models of orofacial pain. Methods Mol Biol. 617, 93–104, doi: 10.1007/978-1-60327-323-7_8 (2010). [DOI] [PubMed] [Google Scholar]
- Trevisan G. et al. TRPA1 mediates trigeminal neuropathic pain in mice downstream of monocytes/macrophages and oxidative stress. Brain, doi: 10.1093/brain/aww038 (2016). [DOI] [PubMed] [Google Scholar]
- Dussor G. et al. Targeting TRP channels for novel migraine therapeutics. ACS chemical neuroscience 5, 1085–1096, doi: 10.1021/cn500083e (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassini R., Materazzi S., Benemei S. & Geppetti P. The TRPA1 channel in inflammatory and neuropathic pain and migraine. Rev Physiol Biochem Pharmacol 167, 1–43, doi: 10.1007/112_2014_18 (2014). [DOI] [PubMed] [Google Scholar]
- Edelmayer R. M. et al. Activation of TRPA1 on dural afferents: a potential mechanism of headache pain. Pain 153, 1949–1958, doi: 10.1016/j.pain.2012.06.012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassini R. et al. The ‘headache tree’ via umbellulone and TRPA1 activates the trigeminovascular system. Brain 135, 376–390, doi: 10.1093/brain/awr272 (2012). [DOI] [PubMed] [Google Scholar]
- Brierley S. M. et al. The ion channel TRPA1 is required for normal mechanosensation and is modulated by algesic stimuli. Gastroenterology 137, 2084–2095 e2083 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Aldebert E. et al. Transient receptor potential vanilloid 4 activated inflammatory signals by intestinal epithelial cells and colitis in mice. Gastroenterology 140, 275–285, doi: 10.1053/j.gastro.2010.09.045 (2011). [DOI] [PubMed] [Google Scholar]
- Wilson S. R. et al. The ion channel TRPA1 is required for chronic itch. J Neurosci 33, 9283–9294, doi: 10.1523/JNEUROSCI.5318-12.2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessac B. F. et al. TRPA1 is a major oxidant sensor in murine airway sensory neurons. J Clin Invest 118, 1899–1910, doi: 10.1172/JCI34192 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon S. A. & Liedtke W. How irritating: the role of TRPA1 in sensing cigarette smoke and aerogenic oxidants in the airways. J Clin Invest 118, 2383–2386, doi: 10.1172/JCI36111 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppetti P., Patacchini R., Nassini R. & Materazzi S. Cough: The Emerging Role of the TRPA1 Channel. Lung 188 Suppl 1, S63–68, doi: 10.1007/s00408-009-9201-3 (2010). [DOI] [PubMed] [Google Scholar]
- Bonvini S. J. et al. Transient receptor potential cation channel, subfamily V, member 4 and airway sensory afferent activation: Role of adenosine triphosphate. J Allergy Clin Immunol, doi: 10.1016/j.jaci.2015.10.044 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonvini S. J., Birrell M. A., Smith J. A. & Belvisi M. G. Targeting TRP channels for chronic cough: from bench to bedside. Naunyn Schmiedebergs Arch Pharmacol 388, 401–420, doi: 10.1007/s00210-014-1082-1 (2015). [DOI] [PubMed] [Google Scholar]
- Rahaman S. O. et al. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J Clin Invest, doi: 10.1172/JCI75331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying L. et al. The transient receptor potential vanilloid 4 channel modulates uterine tone during pregnancy. Science translational medicine 7, 319ra204, doi: 10.1126/scitranslmed.aad0376 (2015). [DOI] [PubMed] [Google Scholar]
- Romac J. M., McCall S. J., Humphrey J. E., Heo J. & Liddle R. A. Pharmacologic disruption of TRPV1-expressing primary sensory neurons but not genetic deletion of TRPV1 protects mice against pancreatitis. Pancreas 36, 394–401, doi: 10.1097/MPA.0b013e318160222a (2008). [DOI] [PubMed] [Google Scholar]
- Leddy H. A. et al. Follistatin in chondrocytes: the link between TRPV4 channelopathies and skeletal malformations. Faseb J 28, 2525–2537, doi: 10.1096/fj.13-245936 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. et al. Synergy between Piezo1 and Piezo2 channels confers high-strain mechanosensitivity to articular cartilage. Proceedings of the National Academy of Sciences 111, E5114–E5122, doi: 10.1073/pnas.1414298111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C. The role of astrocyte-secreted matricellular proteins in central nervous system development and function. Journal of cell communication and signaling 3, 167–176, doi: 10.1007/s12079-009-0078-y (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukdereli H. et al. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci. USA 108, E440–449, doi: 10.1073/pnas.1104977108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risher W. C. & Eroglu C. Thrombospondins as key regulators of synaptogenesis in the central nervous system. Matrix Biol. 31, 170–177, doi: 10.1016/j.matbio.2012.01.004 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo M., Berglund K., Augustine G. & Liedtke W. Novel repression of Kcc2 transcription by REST-RE-1 controls developmental switch in neuronal chloride. J Neurosci 29, 14652–14662 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W. et al. GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron 17, 607–615 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.