

Abstract
The oxygen atom transfer (OAT) reactivity of two valence tautomers of a MnV(O) porphyrinoid complex was compared. The OAT kinetics of MnV(O)(TBP8Cz) (TBP8Cz = octakis(p-tert-butylphenyl)corrolazinato3−) reacting with a series of triarylphosphine (PAr3) substrates were monitored by stopped-flow UV-vis spectroscopy, and revealed second-order rate constants ranging from 16(1) to 1.43(6) × 104 M−1 s−1. Characterization of the OAT transition state analogs MnIII(OPPh3)(TBP8Cz) and MnIII(OP(o-tolyl)3)(TBP8Cz) was carried out by single-crystal X-ray diffraction (XRD). A valence tautomer of the closed-shell MnV(O)(TBP8Cz) can be stabilized by the addition of Lewis and Brønsted acids, resulting in the open-shell MnIV(O)(TBP8Cz•+):LA (LA = ZnII, B(C6F5)3, H+) complexes. These MnIV(O)(π-radical-cation) derivatives exhibit dramatically inhibited rates of OAT with the PAr3 substrates (k = 8.5(2) × 10−3 − 8.7 M−1 s−1), contrasting the previously observed rate increase of H-atom transfer (HAT) for MnIV(O)(TBP8Cz•+):LA with phenols. A Hammett analysis showed that the OAT reactivity for MnIV(O)(TBP8Cz•+):LA is influenced by the Lewis acid strength. Spectral redox titration of MnIV(O)(TBP8Cz•+):ZnII gives Ered = 0.69 V vs SCE, which is nearly +700 mV above its valence tautomer MnV(O)(TBP8Cz) (Ered = −0.05 V). These data suggest that the two-electron electrophilicity of the Mn(O) valence tautomers dominate OAT reactivity and do not follow the trend in one-electron redox potentials, which appear to dominate HAT reactivity. This study provides new fundamental insights regarding the relative OAT and HAT reactivity of valence tautomers such as MV(O)(porph) versus MIV(O)(porph•+) (M = Mn or Fe) found in heme enzymes.
TOC image
INTRODUCTION
Much attention has been given to the mechanistic aspects of biological oxidation reactions involving high-valent metal-oxo porphyrin species due to their invaluable roles in synthetic organic chemistry and heme enzyme mechanisms.1–9 Due to the non-innocent nature of porphyrinoid ligands in these biomimetic complexes, they often undergo an electronic redistribution between metal and ligand, which has been characterized as valence tautomerism. Valence tautomers are of fundamental interest because of their distinct optical, electronic, and magnetic properties.10,11 Heme-containing enzymes such as peroxidases, catalases, and cytochrome P450, take advantage of the facile valence tautomerism inherent to iron porphyrins in order to access formally high oxidation state species. In the case of P450, a wide range of spectroscopic methods was employed to conclusively show that the reactive Compound I intermediate is in the FeIV(O)(porph•+)] form, as opposed to the FeV(O)(porph) valence tautomer.12–15
Valence tautomers in metalloporphyrins involve electron-transfer between the bound metal and the aromatic π system of the ligand, and can be induced both by chemical and non-chemical (temperature changes, irradiation and pressure) means. Chemically-driven valence tautomerization has been observed in several synthetic porphyrin models. Early work showed that coordination of methoxide, a strong π-donor ligand, causes FeIII(TMP•+)(ClO4)2 (TMP = 5,10,15,20-tetramesitylporphyrinato2−) to convert to its valence tautomer FeIV(TMP)(OMe)2.16 A similar finding with iron corroles was observed for FeIV(TPFC)(Cl) (TPFC = 5,10,15-tris(pentafluorophenyl)corrolato3−), in which the replacement of the axial Cl− ligand with the weaker donor ClO4− forms FeIII(TPFC•+)(ClO4)17. Recently, Nakamura18 has shown that there is an equilibrium between FeIII(p-X-TPP•+)(N3)2 (TPP = 5,10,15,20-tetraphenylporphyrinato2−) and FeIV(p-X-TPP)(N3)2, and the position of the equilibrium is dependent on the nature of the para-X substituent of the meso-phenyl groups. Axial ligand-dependent valence tautomerism has also been observed in Mn porphyrins, where the addition of strong axial donors such as CH3O− to MnIII(TPP•+)(Cl) induces the formation of MnIV(TPP)(OCH3)2.19
Although the former complexes provide clear evidence for the propensity of porphyrin ligands to allow for valence tautomerism, there are few examples of metal-oxo porphyrinoid complexes for which both valence tautomers are known. Fujii and coworkers showed that protonation of the oxo ligand of FeIV(O)(TPFP•+) (TPFP = 5,10,15,20-tetrakis(pentafluorophenyl)porphyrinato2−) results in the formation of the electronic isomer FeIII(TPFP++)(L)2 by intramolecular electron transfer from the porphyrin π-radical cation to the Fe center. Subsequent addition of Cl− forms the FeIII meso-chloro-isoporphyrin, an excellent chlorinating agent.20 Acid-dependent valence tautomerization was observed in Mn(salen) complexes, where MnIII(H2O)(salen•+) was reacted with base to sequentially form MnIV(OH)(salen) and MnIV(O)(salen). Among the three species, MnIV(O)(salen) was observed to be the most reactive in H-atom transfer (HAT) and O-atom transfer (OAT) reactions.21 Besides these few studies, there is little known about the comparative reactivity of isoelectronic high-valent metal-oxo complexes that constitute valence tautomers.
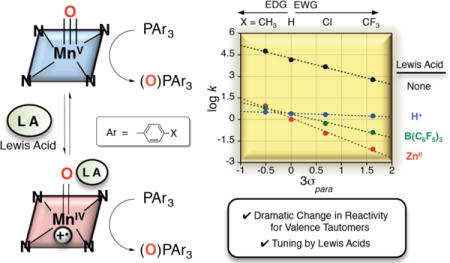
We have shown previously that reaction of MnV(O)(TBP8Cz) with Lewis acids (LA) (LA = ZnII, B(C6F5)3) leads to stabilization of the valence tautomer in dilute solution, in which an electron from the Cz ligand transfers to the metal and gives a metastable MnIV(O) π-radical-cation complex. The data suggested that the Lewis acids were most likely bound to the terminal oxo group, although direct structural information has not been obtained.22,23 The new species, MnIV(O)(TBP8Cz•+):ZnII, exhibited enhanced reactivity toward the one-electron oxidation of ferrocene, and the abstraction of hydrogen atoms from phenol O-H substrates. However, the reactivity of MnIV(O)(TBP8Cz•+):LA in two-electron, O-atom transfer reactions has not been investigated.
Model systems have been used to investigate the effect of Lewis acids on manganese-oxo complexes, including their influence on HAT, dioxygen activation,24 redox potential,25 and OAT reactivity. The addition of Lewis acids (e.g. ZnII, ScIII) to a nonheme MnV(O)(TAML) complex, in which a remote pyridyl-based LA binding site was incorporated, led to an increase in OAT reaction rates with PPh3 as substrate.26 Significant rate enhancements were generated by addition of LAs to MnO4− in the oxidation of alcohol27 and alkane28 substrates. Yin and coworkers29 have also reported on stoichiometric and catalytic rate enhancements for OAT produced by addition of Lewis acids to manganese cross-bridged cyclam complexes. Fukuzumi, Nam, and coworkers30 found that there is a 2,200-fold rate increase in OAT for [MnIV(O)(N4Py)]2+ to sulfide substrates upon binding of ScIII ions. These previous results point to the ability of the Lewis acid to increase the electrophilicity of the metal-oxo complex. In all of these former cases, however, valence tautomerism was not observed and the oxidation state of the Mn ion remained unchanged upon addition of Lewis acids.
Herein we provide a comparative study on the reactivity of two isoelectronic valence tautomers of a high-valent Mn-oxo porphyrinoid complex. The MnV(O)(TBP8Cz) and MnIV(O)(TBP8Cz•+):LA (LA = ZnII, B(C6F5)3, H+) valence tautomers were examined for their O-atom transfer reactivity with a wide range of triarylphosphine derivatives. Both valence tautomers react with PAr3 to give the respective two-electron reduced MnIII complexes. The MnV(O) species shows relatively rapid reaction kinetics, but remarkably, MnIV(O)(π-radical-cation):LA shows dramatically slower reaction rates. Kinetic analyses, including Hammett plots and substrate steric effects, provide insights into the origins of the difference in rate constants for the different valence tautomers. The reduction potential of MnIV(O)(TBP8Cz•+):ZnII was also determined by redox titration, and gives additional insight into the observed OAT and HAT reactivities. This comparison of O-atom transfer reactivity for Mn(O) valence tautomers can be considered as analogous to a comparison of heme Cpd I-type (FeIV(O)(porph•+) versus FeV(O)(porph)) valence tautomers.
RESULTS AND DISCUSSION
Reactivity of MnV(O)(TBP8Cz) in O-atom Transfer to Phosphines
Previously we showed that the two-electron oxidation of PPh3 by MnV(O)(TBP8Cz) is rapid and high-yielding, resulting in OPPh3 (83%) and MnIII(TBP8Cz).31 Evidence for a direct O-atom transfer mechanism between the terminal oxo ligand and PPh3 came from the isotopically labeled MnV(18O) complex, which reacted to give 18OPPh3. It was found that the stable MnV(O) complex could be oxidized by strong one-electron oxidants to give a novel [MnV(O)(TBP8Cz)]+ species in which the Cz ring was oxidized to a π-radical-cation. The O-atom transfer reactivity of the latter complex was compared to the starting MnV(O) complex, and exhibited a 125-fold rate enhancement for OAT with dimethyl sulfide to give the sulfoxide product. An Eyring analysis showed that unfavorable entropic factors attenuated the rate enhancement, but the highly favorable enthalpic term still gave a significant increase in the O-atom transfer reactivity for the π-radical-cation complex. This study was the first example of a direct comparison of the reactivity of a MnV(O) versus MnV(O)(π-radical-cation), in which two high-valent Mn-oxo complexes with the same structure differed by only one unit of charge.32 In other work, MnV(O)(TBP8Cz) was modified through attachment of anionic axial donors (X = CN−, F−) which dramatically enhanced OAT reactivity for thioether substrates.33 However, the kinetics for the oxidation of phosphine substrates by MnV(O) corrolazine has not been reported. Herein stopped-flow UV-vis spectroscopy was used to measure the rate constants of O-atom transfer between MnV(O)(TBP8Cz) and a wide range of triarylphosphine derivatives. This series of substrates allowed for systematic variation of both steric and electronic properties.
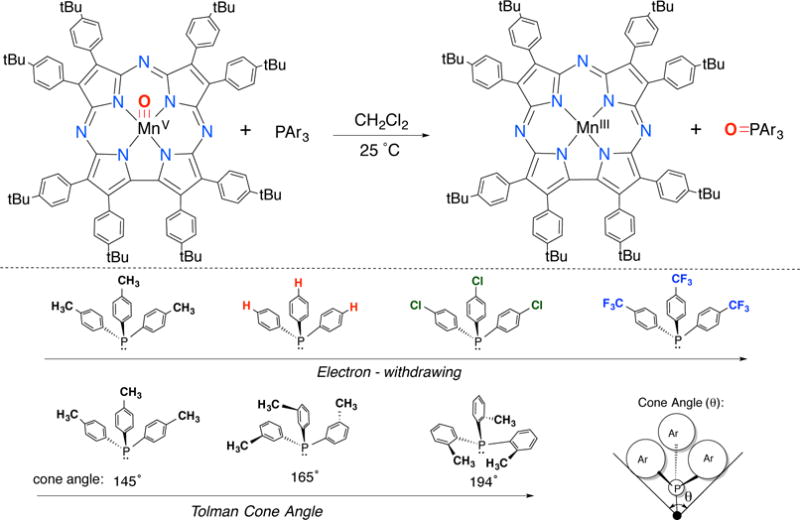
The reaction of MnV(O)(TBP8Cz) with the para-substituted triarylphosphines shown in Scheme 1 was monitored by UV-vis spectroscopy. The decrease in absorbance at 635 nm corrresponding to the decay of MnV(O)(TBP8Cz), was accompanied by an isosbestic growth at 685 nm due to the formation of MnIII(OPPh3)(TBP8Cz) (Figure 1a). Plots of absorbance versus time for both species fit a first-order kinetics model (inset, Figure 1a), and yielded pseudo-first-order rate constants (kobs). The kobs values for PPh3 and the other triarylphosphines shown in Scheme 1 increased linearly with increasing concentration of triarylphosphine. Second-order rate constants were obtained from the linear plots shown in Figures 1b–c for the various phosphine derivatives, and can be compared in Table 1.
Scheme 1.
Oxygen Atom Transfer Reaction Between MnVO(TBP8Cz) and a Series of Phosphine Derivatives
Figure 1.
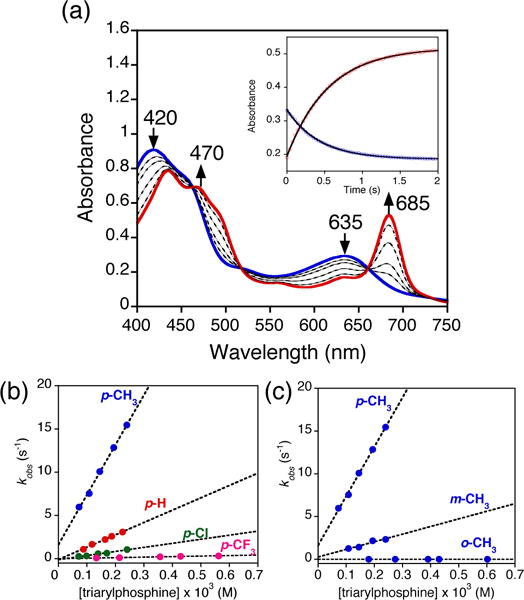
a) Time-resolved UV-vis spectral changes observed in the reaction of MnV(O)(TBP8Cz) (13 μM) with PPh3 (0.16 mM) in CH2Cl2 at 25 °C. Inset: changes in absorbance vs time for the growth of MnIII(TBP8Cz) (685 nm) (red circles) and decay of MnV(O)(TBP8Cz) (635 nm) (blue circles) with the best fit lines (black). Plots of pseudo-first order rate constants (kobs) vs [triarylphosphine] for b) para-substituted triarylphosphines and c) para, meta and ortho-tolyl substituted phosphines.
Table 1.
Hammett Constants, Cone Angles, and Second Order Rate Constants for the Oxidation of Triarylphosphines by MnV(O)(TBP8Cz)
For the para-substituted derivatives, there is a significant dependence on the electron-donating properties of the para-X substituent. This dependence is seen in the Hammett plot shown in Figure 2, where log(kX/kH) versus 3σp (where σp is the Hammett substituent constant34) gave a straight line with ρ = −0.91(5), confirming that the MnV(O) complex oxidizes PAr3 by an electrophilic mechanism.
Figure 2.
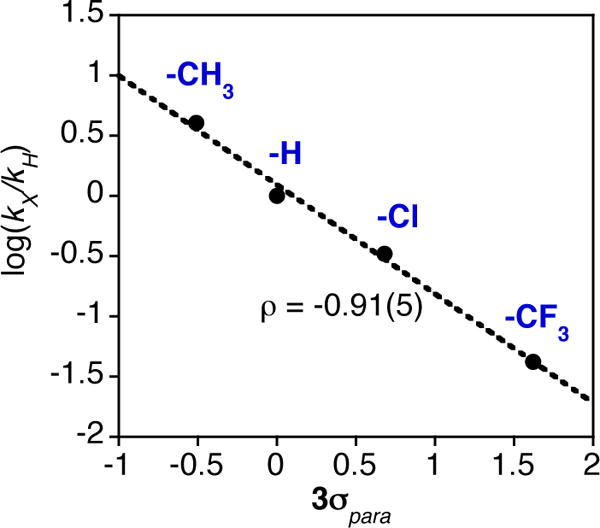
Hammett plot for the OAT reactions between MnV(O)(TBP8Cz) and PAr3 in CH2Cl2 at 25°C.
The influence of the steric properties of the triarylphosphines on the OAT reactivity was examined by employing the ortho, meta, and para-substituted tri(tolyl)phosphines. These phosphine derivatives vary significantly in their Tolman cone angles, which is a measure of the steric encumbrance around the phosphine center (Scheme 1). A significant decrease in rate of ~3700-fold was observed upon an increase in cone angle from 145° to 194° for tri(p-tolyl)phosphine versus tri(o-tolyl)phosphine. This strong steric effect supports a concerted OAT mechanism, in which a nucleophilic phosphorus substrate must attack the electrophilic oxo group while avoiding steric clash with the Cz ring and its peripheral substituents. It also helps to rule out other mechanisms that include outer-sphere electron-transfer as part of the rate-determining step. The least sterically hindered pathway likely involves the P atom approaching in an approximately collinear fashion with the Mn–O bond. This pathway could then be expected to give a MnIII(OPAr3) product with an Mn–O–P angle of ~180°.
Structural Characterization of MnIII(OPAr3)(TBP8Cz) Complexes as Transition State Analogs
Further insights were gained from the synthesis and structural characterization of MnIII(OPPh3)(TBP8Cz) (1) and MnIII(OP(o-tolyl)3)(TBP8Cz) (2), which were prepared by the addition of excess OPAr3 to MnIII(TBP8Cz) in CH2Cl2 followed by layering with CH3CN to yield crystals suitable for X-ray structure determination. The displacement ellipsoid plots for molecules 1 and 2 are shown in Figure 3, and selected bond distances and angles are summarized in Table 2.
Figure 3.
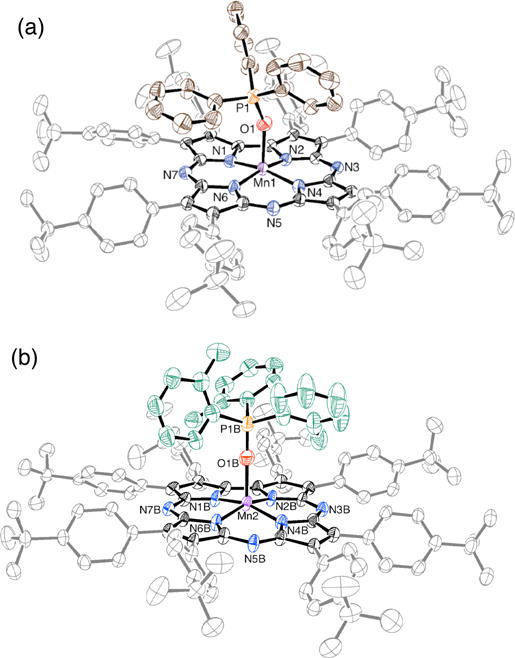
Displacement ellipsoid plot (50% probability level) of a) MnIII(OPPh3)(TBP8Cz) (1) and b) MnIII(OP(o-tolyl)3)(TBP8Cz) (2b) at 110(2) K. The disorder, H atoms and solvent molecules are omitted for clarity.
Table 2.
Selected Bond Distances (Å) and Angles (°) for 1 and 2a–b
| 1 | 2a | 2b | |
|---|---|---|---|
| Mn1 – N1 | 1.872(2) | 1.871(2) | 1.878(3) |
| Mn1– N2 | 1.876(2) | 1.873(3) | 1.885(3) |
| Mn1 – N4 | 1.884(2) | 1.897(3) | 1.895(3) |
| Mn1 – N6 | 1.879(2) | 1.884(3) | 1.879(4) |
| Mn1 – (Npyrrole)plane | 0.274 | 0.358 | 0.368 |
| Mn1 – (23-atom)core | 0.201 | 0.399 | 0.396 |
| Cβ – Cβ (av) | 1.394 | 1.391 | 1.395 |
| Cα – Cβ (av) | 1.449 | 1.445 | 1.446 |
| Cα – Cα (C4–C5) | 1.439(3) | 1.455(5) | 1.463(5) |
| Cα – Npyrrole (av) | 1.370 | 1.368 | 1.370 |
| Cα – Nmeso (av) | 1.341 | 1.344 | 1.336 |
| Mn1 – O1 | 2.0754(19) | 2.084(2) | 2.107(4) |
| P1 – O1 | 1.4900(19) | 1.498(3) | 1.496(5) |
| N1 – Mn1 – N2 | 80.51(9) | 80.56(11) | 80.45(11) |
| N2 – Mn1 – N4 | 90.27(8) | 88.82(11) | 88.61(11) |
| N4 – Mn1 – N6 | 94.07(9) | 93.86(11) | 93.67(11) |
| N6 – Mn1 – N1 | 90.63(9) | 88.76(11) | 88.85(11) |
| N1 – Mn1 – O1 | 97.63(9) | 102.12(11) | 100.1(3) |
| N2 – Mn1 – O1 | 96.81(8) | 105.34(11) | 101.6(2) |
| N4 – Mn1 – O1 | 99.09(8) | 99.83(11) | 102.4(3) |
| N6 – Mn1 – O1 | 98.53(9) | 96.29(11) | 100.0(2) |
| Mn1 – O1 – P1 | 155.57(13) | 158.15(17) | 174.5(5) |
Complexes 1 and 2 are both 5-coordinate MnIII complexes with one OPAr3 molecule bound in the axial position. Complex 2, which contains the OP(o-tolyl)3 axial ligand, crystallized with two crystallographically independent molecules (2a–b) in the asymmetric unit. The MnIII-N distances for 1 and 2 are comparable with those found in other MnIII corrolazines,36,37 and the Mn–O distances (2.075(2) – 2.107(4) Å) are slightly elongated compared to the FeIII analog FeIII(OPPh3)(TBP8Cz) (Fe–O = 2.001(2) Å).38 The Mn ion in 1 is displaced by ca. 0.20 Å from the plane described by the 23-atom Cz core, while this displacement is ca. 0.40 Å in both 2a and 2b. The latter displacement is consistent with the o-tolyl derivative having a larger steric demand, forcing the Mn ion further out of plane to allow for good binding of the phosphine oxide axial donor. The out-of-plane distance for Fe in the FeIII(OPPh3) analog is 0.26 Å, close to that seen for 1. The Mn–O–P angle in 1 is 155.57(13)°, nearly identical to that seen for the FeIII derivative (154.4(1)°). However, this angle deviates significantly for the major component of 2b, which shows Mn–O–P = 174.5(5)°. This nearly linear angle may be consistent with our hypothesis that the more sterically encumbered phosphine substrate would favor a collinear attack on the MnV(O) group. The other independent molecule 2a exhibits a slightly larger Mn–O–P angle (158.15(17)°) compared to 1, but much less than ~180° seen for 2b. Taken together, the structures for 1 and 2 are consistent with the strong influence of the steric properties of the phosphine substrates on the rate of O-atom transfer.
Valence Tautomerism of MnV(O)(TBP8Cz) to MnIV(O)(TBP8Cz•+) by addition of Lewis and Brønsted acids
We previously reported the stabilization of the MnIV(O)(TBP8Cz•+) valence tautomer by addition of the Lewis acids ZnII or B(C6F5)3.22,23 Reaction of Zn(OTf)2 or B(C6F5)3 with the MnV(O) complex led to the isosbestic conversion of MnV(O)(TBP8Cz) (λmax = 420, 635) to MnIV(O)(TBP8Cz•+) with a weakened and broadened Soret band, and a low-intensity band in the near-IR region (λmax = 419, 789), which are characteristic of porphyrin,39,40 corrole,41,42 and corrolazine32 π-radical cations. The Lewis acid adducts were not stable to isolation as solids, but the binding of the Lewis acids was reversible in solution, and spectral titrations yielded association constants of Ka (ZnII) = 4.0 × 106 M−1 and Ka (B(C6F5)3) = 2.0 × 107 M−1. These species exhibited paramagnetic 1H NMR spectra, and Evans method yielded μeff(ZnII) = 4.11 μB and μeff(B(C6F5)3) = 4.19 μB, both falling in between the predicted values for S = 1 (2.83 μB) and S = 2 (4.90 μB). A high spin MnIV (S = 3/2) π-radical cation (S = ½) could couple in either a ferromagnetic (Stotal = 2) or antiferromagnetic (Stotal = 1) manner. These complexes were also EPR silent, consistent with an integer-spin (Stotal = 1 or 2) assignment. Although the instability of these species precluded characterization by X-ray crystallography, the B(C6F5)3 adduct was characterized by ESI-MS (MnIV(O)(TBP8Cz•+):(B(C6F5)3), 1939.7619 m/z, (M+)). Without XRD characterization, we are unable to conclusively assign the binding site for Lewis acids on the Mn(O) complex. However, other metal-oxo-Lewis acid adducts,30 including an isoelectronic ReV(O)–B(C6F5)3 complex,43 bind Lewis acids at the terminal oxo ligand. In addition, the most likely way to stabilize the MnIV(O)(TBP8Cz+•) electronic configuration is by weakening the metal-oxo π-bonding, which can occur only if LA/H+ binds to the terminal oxo group. Our data, together with the literature precedent, suggests that binding of Lewis acids (or H+) occurs at the oxo position.
In this study, we sought to compare the OAT reactivity of the open-shell Lewis acid adducts with the closed-shell MnV(O) complex. We also wanted to examine the influence of Brønsted acids, and thus the strong H+ donor HBF4 was examined. Reaction of MnV(O)(TBP8Cz) and 1 equiv of HBF4 in CH2Cl2 led to the isosbestic conversion of the green MnV(O)(TBP8Cz) to brown MnIV(O)(TBP8Cz•+):H+ (Figure S12). A similar conversion was observed when H+[B(C6F5)4]− was used.44 The reversibility of this reaction was tested by addition of 2,6-lutidine, which led to 75% recovery of the starting MnV(O) complex (Figure S13). The 1H NMR spectrum following addition of HBF4 was paramagnetic, and Evans method gave μeff = 3.96 μB (Figure S14). Thus the spectroscopic data are similar to the data seen for the Lewis acid adducts, providing strong evidence that the Brønsted acid HBF4 also stabilizes the open-shell valence tautomer MnIV(O)(TBP8Cz•+):H+.
O-atom Transfer Reactivity of MnIV(O)(TBP8Cz•+):ZnII. Product Analysis
The valence tautomer MnIV(O)(TBP8Cz•+):ZnII was generated as described,22 by addition of Zn(OTf)2 to MnV(O)(TBP8Cz) (13 μM) in a 1:1 ratio in CH2Cl2/CH3CN (100:1 v/v). The previously measured association constant for ZnII (Ka = 4 × 106 M−1) indicates that MnIV(O)(TBP8Cz•+):ZnII should be quantitatively formed under these conditions. Addition of PPh3 initiated the OAT reaction, and caused the spectrum for the MnIV(O) π-radical-cation to undergo isosbestic conversion to a new spectrum with λmax = 419, 725 nm. (Figure 4a). The final spectrum matches that seen for an independently generated sample from the addition of Zn(OTf)2 to MnIII(TBP8Cz) in CH2Cl2/CH3CN (100:1 v/v) (Figure 4b). The MnIII oxidation state for the λmax = 443, 725 nm species was supported by the absence of an EPR signal (13 K, 9.44 GHz) for this product (Figure S5). A reasonable binding site for ZnII on the MnIII complex is one of the Lewis basic meso-N atoms of the Cz ligand. Both porphyrazine and phthalocyanine compounds are known to coordinate Lewis and Brønsted acids at the meso-N positions.45–48 In recent work, we provided structural evidence by XRD that the meso-N positions of MnIII(H2O)(TBP8Cz) can be protonated.44
Figure 4.
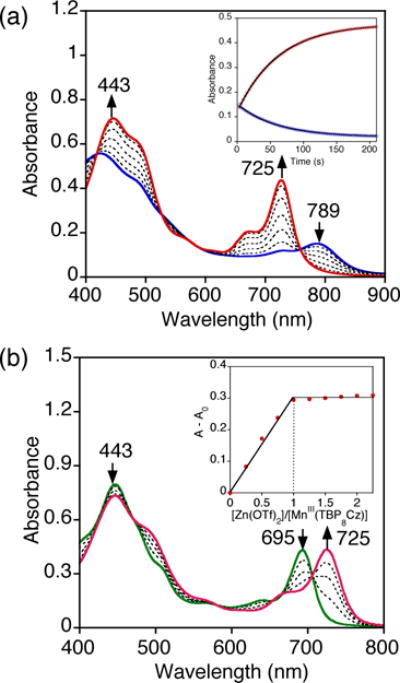
a) Time-resolved UV-vis spectral changes for the reaction of MnIV(O)(TBP8Cz•+):ZnII (13 μM) and PPh3 (0.02 M) in 100:1 v/v CH2Cl2/CH3CN at 25 °C. Inset: changes in absorbance vs time for the growth of MnIII(TBP8Cz):ZnII (725 nm) (red circles) and decay of MnIV(O)(TBP8Cz•+):ZnII (789 nm) (blue circles) with the best fit lines (black). b) UV-vis spectral changes for the decay of MnIII(TBP8Cz) (green solid line) upon titration with Zn(OTf)2 (0 – 2.25 equiv), forming MnIII(TBP8Cz):ZnII (red solid line). Inset: plot of A–Ao at 725 nm vs total equiv of Zn(OTf)2, showing maximal formation at 1 equiv of ZnII.
Direct O-atom transfer from MnIV(O)(TBP8Cz•+):ZnII to PPh3 was confirmed by 31P{1H} NMR and GC-MS, which revealed the phosphine oxide as the only product in 73% yield (Figure S6). The use of 18O-labeled starting material MnV(18O)(TBP8Cz) (70% 18O) followed by treatment with Zn(OTf)2 and PPh3 led to substantial labeling of the OPPh3 product (88% 18O incorporation from GC-MS) (Figure S7). A summary of these observations is shown in Scheme 2.
Scheme 2.
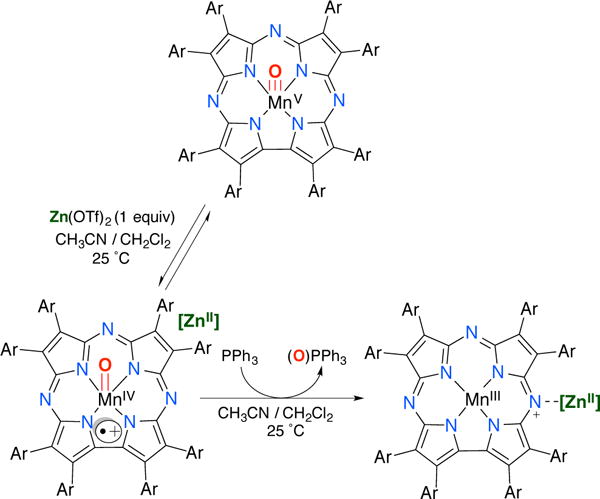
Kinetics of Oxygen Atom Transfer for MnIV(O)(TBP8Cz•+):(LA) and Phosphine Substrates
As seen in Figure 4a, the spectral changes versus time for the reaction between MnIV(O)(TBP8Cz•+):ZnII and PPh3 fit a single exponential model, yielding pseudo-first-order rate constants, kobs. The concentration of phosphine substrate was varied and led to a second order plot (Figure S8), which showed a linear dependence of kobs on [PPh3] and yielded a second-order rate constant k = 0.99(1) M−1 s−1 (Table 3). This rate constant can be compared to that for the MnV(O) complex in the absence of ZnII (Table 1), which shows that MnIV(O)(TBP8Cz•+):ZnII reacts at a rate 14,000-fold slower than the parent MnV(O) complex. The series of para-substituted phosphine derivatives (Scheme 1) was reacted with the MnIV(O) π-radical-cation complex, and each derivative gave good overall second-order kinetics. The second-order rate constants obtained for each of the PAr3 derivatives (Table 3) are dramatically slower than those seen for the MnV(O) complex, on the order of a 104-fold reduction in rate.
Table 3.
Second Order Rate Constants for the Oxidation of Triarylphosphines with MnIV(O)(TBP8Cz•+):(LA) (LA = ZnII, B(C6F5)3 or HBF4)
| P(X-Ph)3 | k (M−1 s−1)
|
||
|---|---|---|---|
| Zn(OTf)2a | B(C6F5)3b | HBF4b | |
| o-CH3 | 1.9(2) | 0.6(1) | 2.2(1) |
| m-CH3 | 1.8(1) | 1.2(1) | 0.9(1) |
| p-CH3 | 8.7(4) | 6.5(4) | 3.3(2) |
| p-H | 0.99(1) | 1.65(3) | 2.5(3) |
| p-Cl | 0.109(4) | 0.525(5) | 2.2(2) |
| p-CF3 | 0.0085(2) | 0.126(9) | 1.12(3) |
in 100:1 v/v CH2Cl2/CH3CN at 25 °C.
in CH2Cl2 at 25 °C
To gain further insights regarding the influence of the Lewis acid, the ZnII ion was replaced with B(C6F5)3, as well as the Brønsted acid HBF4. Reaction of MnIV(O)(TBP8Cz•+):LA (LA = B(C6F5)3, H+) with the para-substituted phosphine derivatives led to good second-order kinetics and production of the two-electron reduced MnIII complexes. The log k values for the MnIV(O)(TBP8Cz•+):LA complex were plotted versus Hammett σ parameters as shown in Figure 5. Linear trends are revealed for all three complexes, with a negative ρ value found for ZnII (−1.4) that is larger than the parent complex (Figure 2). The negative ρ value (−0.82) for B(C6F5)3 is also consistent with an electrophilic mechanism, but is smaller in magnitude than the ρ value seen for ZnII. In contrast, the small value of ρ = −0.13 for H+ shows that, for this complex, there is little correlation of reaction rate with the electron-rich nature of the phosphine substrates.
Figure 5.
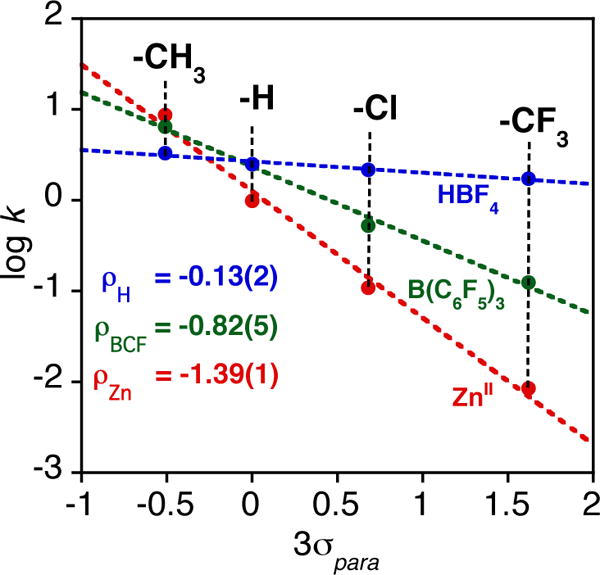
Plots of log k (second order rate constants) versus Hammett σ values for para-substituted triarylphosphines for the reactions with MnIV(O)(TBP8Cz•+):(LA) (LA = ZnII (red), B(C6F5)3 (green), HBF4 (blue)).
The trends in the Hammett data in Figure 5 can be rationalized by taking into consideration the strengths of the Lewis acids. Gutmann-Beckett acceptor numbers, which are a measure of the relative Lewis acidity of both Lewis and Brønsted acids, were determined for HBF4, B(C6F5)3, and Zn(OTf)2 by measuring their influence on the 31P chemical shift of OPEt3 with 31P{1H} NMR spectroscopy.49–54 Acceptor numbers of 122, 82, and 68 were obtained for HBF4, B(C6F5)3, and Zn(OTf)2, respectively, and the magnitudes of the ρ values for the Hammett plots in Figure 5 decrease as the acceptor number increases for the Lewis acids. The influence of the strength of the Lewis acid is most evident for the p-CF3 derivative, where the rate constants vary over two orders of magnitude from ZnII (lowest) to H+ (highest). These trends clearly follow the reactivity/selectivity principle, where the least reactive MnIV(O) π-radical-cation complex, which is generated from the weakest Lewis acid (ZnII), is the most selective toward O-atom transfer for the phosphine derivatives.
The influence of the steric bulk of the phosphine derivatives on reaction rates for OAT was also examined. The methyl-substituted tri(tolyl)phosphines in Scheme 1 were employed, and rate constants are given in Table 3. These data show that the MnIV(O)(π-radical-cation) formed with the Lewis acid B(C6F5)3 is most sensitive to the steric demand of the phosphine substrate, with an ~10-fold rate decrease for ortho- versus para-substituted tri(tolyl)phosphine. For the less sterically bulky Lewis acids ZnII and H+, the steric nature of the substrate has little influence on the rate of OAT. These data are consistent with the Lewis acid being coordinated to the terminal oxo group, where the relatively large B(C6F5)3 can be expected to have the largest steric clash with the incoming phosphine nucleophile.
Redox titration
Assessment of the redox potential of MnIV(O)(TBP8Cz•+):ZnII could be expected to shed light on its reactivity in the oxidation of organic substrates. Addition of the one-electron reductant acetylferrocene (Eox = 0.62 V vs SCE, where Eox is the redox potential of the species being oxidized)55 and monitoring by spectral redox titration resulted in ring reduction and isosbestic conversion of MnIV(O)(TBP8Cz•+):ZnII to MnIV(O)(TBP8Cz):ZnII, as seen previously with ferrocene.22 The titration curve (Figure S18) was fit to a 1:1 electron-transfer equilibrium model, yielding a KET value of 18.3. The Nernst equation (eq 1) was employed to obtain a redox potential of Ered = +0.69 V vs SCE for MnIV(O)(TBP8Cz•+):ZnII. In comparison, Ered (Ered is the redox potential of the species being reduced) for the valence tautomer MnV(O)(TBP8Cz) is −0.05 V vs SCE.36
| (1) |
The increased redox potential for MnIV(O)(TBP8Cz•+):ZnII suggests that this complex could show enhanced rates for one-electron oxidations such as H-atom abstraction, and indeed this complex, as well as a borane analog, exhibits faster rates of HAT with phenol O-H substrates.22,23 However, the redox potential for PPh3 is 2.08 V vs Cp2Co+/0 (converted to 2.97 V vs SCE),56 and therefore one-electron oxidation of PPh3 by the MnIV(O) complex remains strongly endergonic, helping to rule out either a pure ET or ET-OT type mechanism.30,57–58
A summary of HAT and OAT reactivity for the two valence tautomers is shown in Scheme 3. A major question remains; why are the rates of OAT to PAr3substrates dramatically slowed for the MnIV(O)(TBP8Cz•+):LA complexes? The increase in HAT noted for the MnIV(O) π-radical-cation tautomer may be attributed to the large increase in redox potential compared to the MnV(O) form, which should result in a larger driving force for HAT.59–64
Scheme 3.
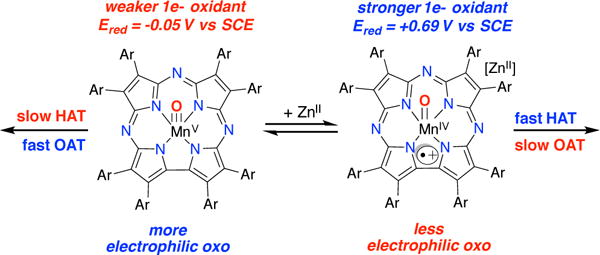
In contrast, the two-electron OAT process involving the phosphine substrates is dramatically slower for the MnIV(O) π-radical-cation species. If a concerted OAT process is invoked, then the two-electron electrophilicity of the metal-oxo unit should be a critical factor that influences the OAT rates. We suggest that the MnIV(O) π-radical-cation species is less electrophilic at the oxo ligand than the MnV(O) species, despite the addition of Lewis acid, because of the lower oxidation state at the metal. We hypothesize that this lowering of the electrophilicity is responsible for the slower OAT reaction rates.
It should be noted that for some metal-oxo complexes, a correlation has been observed between the redox potentials of the complex and OAT rates.30,57,58,65,66 In addition, DFT calculations on two-electron atom-transfer reactions have suggested that spin state may also be an important factor. For example, DFT calculations performed on OAT of MnV(O) corroles implicate a lower-barrier triplet state in the OAT reaction with thioanisole.67 Other calculations suggested that singlet versus triplet state energies could be responsible for the relative rates of related nitrogen-atom-transfer reactions involving Cu-(N-tosyl)-ScIII and Cu-(N-mesityl)-ScIII complexes.68 It is clear that more work is needed to determine the factors that control biomimetic, metal-mediated two-electron atom-transfer reactions.
SUMMARY AND CONCLUSIONS
The comparative O-atom transfer chemistry for MnV(O) and MnIV(O)(π-radical-cation) porphyrinoid complexes has been determined. The interconversion of these species is mediated by the addition of either Lewis or Brønsted acids. The two-electron OAT reactivity of the MnIV(O)(π-radical-cation):LA complexes with PAr3 derivatives is dramatically inhibited in comparison to the MnV(O) valence tautomer. This inhibition can be related to the difference in the electronic structures for MnIV(O)(π-radical-cation) vs MnV(O) valence tautomers, in which the former can be anticipated to have a less electrophilic terminal oxo group. The relative rate constants for one-electron processes such as HAT are found to be influenced in the opposite manner, with a significant increase in rate seen for the open-shell MnIV(O)(π-radical-cation) tautomer. This trend can be attributed to the large, measured increase in reduction potential for MnIV(O)(π-radical-cation), which increases the driving force for the HAT reaction.
In biological systems, it has been shown that a manganese-substituted cytochrome P450cam carries out efficient OAT to alkenes to give epoxide through a likely MnV(O)(porphyrin) intermediate, in line with our conclusions that MnV(O) porphyrinoid species are good electrophiles for OAT. However, the Mn analog of P450 was not capable of mediating hydroxylation, in contrast to the native FeIV(O)(porphyrin•+) intermediate, which can perform both oxygen transfer and hydroxylation reactions.69 Thus in the case of Fe, the valence tautomer containing the radical-cation appears inherently more reactive than the MnIV(O)(π-radical-cation) observed in this work.
Valence tautomers are prevalent in nature and are of key importance in heme enzymes, but there is still little known about the relative reactivities of heme-derived valence tautomers for high-valent metal-oxo species. This work provides fundamental information regarding the reactivity of two biomimetic valence tautomers of a high-valent Mn-oxo porphyrinoid complex. Determining the differences in reactivity of valence tautomeric species may help in our understanding of the preference for one valence tautomer over another in heme enzymes.
EXPERIMENTAL SECTION
Materials
All reactions were performed under an Ar atmosphere using dry solvents and standard Schlenk techniques. The complexes MnVO(TBP8Cz) and MnIII (TBP8Cz) (TBP8Cz = octakis(p-tert-butylphenyl)corrolazinato3−) were synthesized and purified according to previously published methods.20 Dichloromethane and acetonitrile were purified via a Pure-Solv solvent purification system from Innovative Technologies, Inc. H218O (97% 18O) and deuterated solvents for NMR measurements were obtained from Cambridge Isotopes, Inc. Tri(o-tolyl)phosphine oxide (OP(o-tolyl)3) was synthesized according to Granoth et al70 and was recrystallized from ethanol. All other reagents, except for tri(m-tolyl)phosphine (Alfa-Aesar, 98+%), were purchased from Sigma-Aldrich at the highest level of purity and used as received.
Instrumentation
Kinetics and other UV-vis measurements were performed on a Hewlett-Packard Agilent 8453 diode-array spectrophotometer with a 3.5 mL air-free quartz cuvette (path length = 1 cm) fitted with a septum. For reactions with total reaction time of <10 seconds, stopped-flow experiments were carried out using HiTech SHU-61SX2 (TgK scientific Ltd.) with a xenon light source and Kinetic Studio software. Gas chromatography mass spectrometry (GC-MS) was performed on an Agilent 6850 gas chromatograph fitted with a DB-5 5% phenylmethyl siloxane capillary column and equipped with an electron-impact (EI) mass spectrometer. Product yields were calculated from a dodecane internal standard. LDI-MS was conducted on a Bruker Autoflex III TOF/TOF instrument equipped with a nitrogen laser at 335 nm using an MTP 384 ground steel target plate. Electrospray ionization mass spectra (ESI-MS) were collected on a Thermo Finnigan LCQ Duo ion-trap mass spectrometer fitted with an electrospray ionization source in positive ion mode. Samples were infused into the instrument at a rate of 25 μL/min using a syringe pump via a silica capillary line. The spray voltage was set at 5 kV, and the capillary temperature was held at 250 °C. 1H NMR (400.13 MHz) and 31P{1H} NMR (161.9 MHz) spectra were recorded on a Bruker Avance 400 MHz NMR spectrometer at room temperature. Elemental analyses were performed at Atlantic Microlab, Inc., Norcross, GA. Electron paramagnetic resonance (EPR) spectra were recorded with a Bruker EMX spectrometer equipped with a Bruker ER 041 X G microwave bridge and a continuous-flow liquid helium cryostat (ESR900) coupled to an Oxford Instruments TC503 temperature controller.
Stopped-flow UV-vis Kinetics Studies
In a typical reaction, MnV(O)(TBP8Cz) (13 μM, CH2Cl2) was reacted with triarylphosphine (0.15 – 0.75 mM) [tris(para-X-phenyl)phosphine (X = CH3, H, Cl, CF3), tri(meta-tolyl)phosphine, and tri(ortho-tolyl)phosphine)]. The spectral changes showed isosbestic conversion of MnV(O)(TBP8Cz) (λmax = 420, 635 nm) to MnIII(TBP8Cz) (λmax = 435, 470, 685 nm). The pseudo-first-order rate constants, kobs, for these reactions were obtained by non-linear least-squares fitting of the plots of absorbance at 635 nm (Abst) versus time (t) according to the equation Abst = Absf + (Abs0 − Absf) exp(−kobst) where Abs0 and Absf are initial and final absorbance, respectively. Second order rate constants (k) were obtained from the slope of the best-fit line from a plot of kobs vs substrate concentration.
Formation of MnIV(O)(TBP8Cz•+):H+
To a solution of MnV(O)(TBP8Cz) (20 μM, 2 mL), successive amounts of HBF4•Et2O (0.2 equiv aliquots dissolved in 5 μL CH2Cl2) was added. A color change from green to brown was observed. Monitoring the reaction after each addition showed isosbestic conversion of MnV(O)(TBP8Cz) (λmax = 420, 635 nm) to MnIV(O)(TBP8Cz•+):H+ (λmax = 419, 785 nm) (Figure S12). Full formation of the product was observed after addition of 1 equiv HBF4. Reaction of this complex with 2,6-lutidine (1 equiv, 25 μL in CH2Cl2) gives back ~75% of the starting MnV(O)(TBP8Cz) complex (Figure S13).
Magnetic Susceptibility by Evans Method
To a solution of MnV(O)(TBP8Cz) (2.0 mM in 500 μL 0.5 % TMS in CD2Cl2), HBF4•Et2O (1 equiv in 20 μL CD2Cl2) was added. Complete formation of MnIV(O)(TBP8Cz•+):H+ was observed by UV-vis spectroscopy. The reaction mixture was transferred to an NMR tube, together with a second coaxial insert tube containing the solvent blank. 1H NMR spectra were recorded at 297.2 K, and the chemical shift of the TMS peak in the presence of the paramagnetic MnIV(O)(TBP8Cz•+):H+ complex was compared to that of the TMS peak in the inner tube containing only the TMS standard (Figure S14). The effective spin-only magnetic moment was calculated by a simplified Evans method analysis71 according to μeff = 0.0618(sqrt(ΔνT/2fM)), where Δν is the difference in frequency (Hz) between the two reference (TMS) signals, T is the temperature (K), f is the oscillator frequency (MHz) of the superconducting spectrometer, and M is the molar concentration of the paramagnetic metal complex. The number of unpaired electrons was calculated using the equation μ2 = n(n+2). A control experiment was performed using MnV(O)(TBP8Cz), where no shift in the TMS peak was observed (Figure S15).
UV-vis Kinetics Studies with MnIV(O)(TBP8Cz•+):LA
The valence tautomer MnIV(O)(TBP8Cz•+):LA was generated in situ by addition of Lewis acids (LA) [Zn(OTf)2 in CH3CN, B(C6F5)3 or HBF4•Et2O in CH2Cl2] to an amount of MnV(O)(TBP8Cz) (13 μM, CH2Cl2). Upon complete formation of the valence tautomer, varying amounts of triarylphosphine (1.5 – 45 mM) were added to start the reaction. The spectral change showed isosbestic conversion of MnIV(O)(TBP8Cz•+):LA (λmax = 419, 789 nm) to MnIII(TBP8Cz):LA (λmax = 443, 725 nm). The same kinetic analysis was used as employed for the stopped-flow UV-vis studies, following the growth in absorbance at 725 nm which corresponds to MnIII(TBP8Cz):LA. Pseudo-first-order kobs values were obtained and exhibited a linear correlation with substrate concentration for all triarylphosphine substrates.
OAT Product Analysis
In a custom-made 250 mL round bottom flask fitted with a 3 mL quartz cuvette under an Ar atmosphere, a solution of MnV(O)(TBP8Cz) (100 μM in 15 mL of CH2Cl2) was combined with a solution of Zn(OTf)2 (1 equiv in CH3CN) to form MnIV(O)(TBP8Cz•+):ZnII. An amount of PPh3 (1 equiv) was then added and the reaction was monitored by UV-vis spectroscopy, which showed complete conversion to MnIII(TBP8Cz):ZnII. The solution was concentrated to dryness, re-dissolved in toluene and injected directly onto the GC-MS for analysis. Yields were calculated from a calibration curve with dodecane as an internal standard. The obtained yield from this method (71%) is an average of three runs. Unreacted PPh3 was also measured by GC (0.33 equiv). The yield of OPPh3 was also independently measured by 31P{1H} NMR as follows: A solution of MnV(O)(TBP8Cz) (2 mM) was combined with Zn(OTf)2 (1 equiv) in CH2Cl2/CH3CN 10:1 v/v to form MnIV(O)(TBP8Cz•+):ZnII. An amount of PPh3 (10 equiv) was then added, and the reaction was monitored by UV-vis spectroscopy, which showed complete conversion to MnIII(TBP8Cz):ZnII. To release the OPPh3 bound to the paramagnetic MnIII species, a strong axial ligand donor, Bu4N+F− (tetrabutylammonium fluoride) (20 equiv) was added. A color change to a dark green solution typical of [MnIII(TBP8Cz)(F)]− (λmax = 428, 471, 680 nm) was noted.33 The solution was then concentrated under vacuum and re-dissolved in CD2Cl2:CD3CN (500 μL; 10:1 v/v) and immediately analyzed by 31P{1H} NMR (85% H3PO4 external standard). The delay time (D1) was set to 150 s to allow for complete relaxation of the 31P nucleus. Comparison of the integrations for the peak assigned to OPPh3 and the peak for PPh3 gave a yield of 74% for OPPh3, in good agreement with the GC method. Unreacted PPh3 (9.26 equiv) was also measured from the NMR spectra (Figure S6).
18O Labeling Studies
To a solution of MnV(O)(TBP8Cz) in dry CH2Cl2 (7 μmol, 6 mL), H218O (25 μL, 200 equiv, 97% 18O-enriched) was added via micro-syringe under an Ar atmosphere. Stirring was continued for 48 h and the solvent was removed under vacuum. The product was immediately analyzed by LDI-MS, which revealed 70% 18O incorporation in MnV(O)(TBP8Cz) (Figure S7a). The 18O-enriched MnV(O)complex was combined with Zn(OTf)2 (2 equiv) in CH2Cl2/CH3CN (10:1 v/v, 2 mL) to form MnIV(18O)(TBP8Cz•+):ZnII. The solution was again analyzed by LDI-MS which revealed that 40% of the 18O label was retained (Figure S7b). An amount of PPh3 (10 equiv) was added, and the reaction was monitored by UV-vis, which showed complete conversion to MnIII(TBP8Cz):ZnII. The OPPh3 product was extracted with CH3OH, dried, and re-dissolved in CH3OH. An aliquot (1 μL) was injected directly into the GC-MS, which showed 88% 18O incorporation (Figure S7c).
Lewis Acidity Measurements
(Gutmann-Beckett method).30,31 In an NMR tube, a 3:1 mixture of Lewis acid (HBF4•Et2O, B(C6F5)3, Zn(OTf)2) and triethylphosphine oxide was prepared in CD2Cl2 (or CD3CN for Zn(OTf)2). 31P{1H} NMR spectra were collected with an 85% H3PO4 external standard. The Acceptor Number (A.N.) was calculated using the relationship: A.N. = 2.21 × (δsample − 41.0), where δsample is the chemical shift for the OPEt3 – Lewis Acid adduct.
Measurement of Equilibrium Constant of Electron Transfer (KET)
Under strictly dry and anaerobic conditions, a solution of MnVO(TBP8Cz) in CH2Cl2 (15μM, 2.0 mL) was mixed with Zn(OTf)2 (20 equiv, 20 μL in CH3CN) to generate MnI− VO(TBP8Cz•+):ZnII. Successive aliquots of acetylferrocene (AcFc) (0–2.25 equiv, in CH2Cl2) were added and the reaction was monitored by UV-vis until no further change was observed. Monitoring the titration at 725 nm resulted in the plot in Figure S18, which was fitted to eq S3 to obtain a value for KET. The values of KET, [MnIV(O)(TBP8Cz•+)]0, and Δε = [ε(MnIV(O)(TBP8Cz)):ZnII – ε(MnIV(O)(TBP8Cz•+)):ZnII] were allowed to vary during the fitting procedure either simultaneously or in sequence, and the final values for the best fit were KET = 18.3; [MnIV(O)Cz•+]0 = 16.4 μM; Δε = 25651; r = 0.999. The refined value for [MnIV(O)Cz•+]0 is close to the starting concentration (15 μM), and the refined Δε value was physically reasonable.
Synthesis of MnIII(OPPh3)(TBP8Cz) (1)
To a CH2Cl2 solution of MnIII(TBP8Cz) (7 μmol, 1 mL), OPPh3 (10 equiv) was added. This solution was layered with CH3CN and set aside to undergo slow evaporation, giving X-ray quality single crystals (black blocks) in good yield (8 mg, 81%). UV-vis (CH2Cl2): λmax = 435, 470, 685 nm, ESI-MS (m/z): isotopic cluster centered at 1690.1 (M+H). (Figure S4) Anal. Calcd for C114H119MnN7OP: C, 81.06; H, 7.10; N, 5.80. Found: C, 81.00; H, 7.07; N, 5.80.
Synthesis of MnIII(OP(o-tolyl)3)(TBP8Cz) (2)
The complex was prepared similarly to 1. X-ray quality single crystals (black blocks) were obtained from slow evaporation of a CH2Cl2/CH3CN solution in good yield (6 mg, 62%). UV-vis (CH2Cl2): λmax = 435, 470, 685 nm, ESI-MS (m/z): isotopic cluster centered at 1732.0 (M+H). (Figure S4) Anal. Calcd for C117H125MnN7OP: C, 81.17; H, 7.28; N, 5.66. Found: C, 81.04; H, 7.21; N, 5.58.
Single Crystal X-ray Crystallography
All reflection intensities were measured at 110(2) K using a SuperNova diffractometer (equipped with Atlas detector) with Cu Kα radiation (λ = 1.54178 Å) under the program CrysAlisPro (Version 1.171.36.32 Agilent Technologies, 2013). The same program was used to refine the cell dimensions and for data reduction. The structure was solved with the program SHELXS-201372 and was refined on F2 with SHELXL-2013.72 Analytical numeric absorption corrections based on a multi-faceted crystal model were applied using CrysAlisPro. The temperature of the data collection was controlled using the system Cryojet (manufactured by Oxford Instruments). The H atoms were placed at calculated positions (unless otherwise specified) using the instructions AFIX 23, AFIX 43 or AFIX 137 with isotropic displacement parameters having values 1.2 or 1.5 times Ueq of the attached C atoms.
The structure for MnIII(OPPh3)(TBP8Cz) (1) is partly disordered. Six of the eight tert-butylphenyl (TBP) groups are found to be disordered over two orientations (only the t-butyl part is disordered). The occupancy factors of the six major components of the disorder refine to 0.53(2), 0.69(4), 0.768(7), 0.52(2), 0.695(6) and 0.57(3). The crystal lattice contains some amount of solvent molecules (CH3CN and CH2Cl2). The occupancy factors of the lattice CH3CN solvent molecules were refined using free variables, and there are ca. 3.76 CH3CN molecules per Mn complex. Some electron density in the asymmetric unit (i.e., a disordered solvent CH2Cl2 molecule (with at least 3 different orientations) with partial occupancy) has been taken out in the final refinement (SQUEEZE73 details are provided in the CIF file).
Data for MnIII(OPPh3)(TBP8Cz) (1) follow: Fw = 1843.47, irregular shaped crystal, 0.43 × 0.18 × 0.09 mm3, monoclinic, P21/c (no. 14), a = 17.17627(19), b = 33.6006(3), c = 19.6140(2) Å, β = 101.1080(11)°, V = 11107.8(2) Å3, Z = 4, Dx = 1.102 g cm−3, μ = 1.500 mm−1, abs. corr. range: 0.640−0.882. 82335 Reflections were measured up to a resolution of (sin θ/λ)max = 0.62 Å−1. 21779 Reflections were unique (Rint = 0.0392), of which 18304 were observed [I > 2σ(I)]. 1527 Parameters were refined using 855 restraints. R1/wR2 [I > 2σ(I)]: 0.0658/0.1749. R1/wR2 [all refl.]: 0.0768/0.1841. S = 1.043. Residual electron density found between −0.57 and 1.06 e Å−3.
The asymmetric unit for MnIII(OP(o-tolyl)3)(TBP8Cz) (2) contains two crystallographically independent Mn complexes 2a and 2b and some amount of lattice solvent molecules (CH2Cl2, CH3CN). The structure is significantly disordered. Six of the eight tert-butyl groups of 2a are disordered over two orientations, and the six occupancy factors of the major components of the disorder refine to 0.517(9), 0.779(8), 0.680(8), 0.60(2), 0.697(10), 0.816(8). The OP(o-tolyl)3 coordinated to 2b is disordered over two orientations, and the occupancy factor of the major component of the disorder refines to 0.694(4). Two ordered lattice CH3CN and one disordered (over two orientations) CH2Cl2 solvent molecules were found in the asymmetric unit. The occupancy factors were refined freely and their final values are 0.815(10), 0.956(9), 0.496(5) and 0.285(5). Other solvent molecules (most likely CH3CN and CH2Cl2) were found to be very disordered, and their contribution has been taken out in the final refinement using the SQUEEZE procedure73 (SQUEEZE details are provided in the CIF file).
Data for MnIII(OP(o-tolyl)3)(TBP8Cz) (2) follow: Fw = 1800.72, thick dark brown-black lath, 0.39 × 0.17 × 0.14 mm3, triclinic, P-1 (no. 2), a = 22.6324(3), b = 23.4125(3), c = 23.4458(4) Å, α = 118.4068(15), β = 98.4848(13), γ = 93.6179(11)°, V = 10680.4(3) Å3, Z = 4, Dx = 1.120 g cm−3, μ = 1.716 mm−1, Tmin–Tmax: 0.653–0.865. 139649 Reflections were measured up to a resolution of (sin θ/λ)max = 0.62 Å−1. 41851 Reflections were unique (Rint = 0.0292), of which 34261 were observed [I > 2σ(I)]. 2837 Parameters were refined using 1684 restraints. R1/wR2 [I > 2σ(I)]: 0.0861/0.2439. R1/wR2 [all refl.]: 0.0996/0.2572. S = 1.005. Residual electron density found between −0.84 and 1.97 e Å−3.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge research support of this work by the NIH (Grant GM101153 to D.P.G.). We also thank Prof. Kenneth D. Karlin (JHU) for instrumentation (stopped-flow UV-vis) use.
Footnotes
Supporting Information
UV-vis kinetics studies, X-ray crystal structure of 2a (Figure S3), crystallographic information files (CIF for 1 and 2), EPR, 31P{1H} and 1H NMR, and MS data, and redox titration studies. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Sono M, Roach MP, Coulter ED, Dawson JH. Chem Rev. 1996;96:2841. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 2.Meunier B, de Visser SP, Shaik S. Chem Rev. 2004;104:3947. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 3.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 4.Gunter MJ, Turner P. Coord Chem Rev. 1991;108:115. [Google Scholar]
- 5.Watanabe Y, Fujii H. In: Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidations. Meunier B, editor. Springer; Berlin; New York: 2000. p. 62. [Google Scholar]
- 6.McLain JL, Lee J, Groves JT. In: Biomimetic Oxidations Catalyzed by Transition Metal Complexes. Meunier B, editor. Imperial College Press; London: 2000. p. 91. [Google Scholar]
- 7.Liu H-Y, Mahmood MHR, Qiu S-X, Chang CK. Coord Chem Rev. 2013;257:1306. [Google Scholar]
- 8.Gross Z. J Biol Inorg Chem. 2001;6:733. doi: 10.1007/s007750100273. [DOI] [PubMed] [Google Scholar]
- 9.McGown AJ, Badiei YM, Leeladee P, Prokop KA, DeBeer S, Goldberg DP. In: The Handbook of Porphyrin Science. Kadish KM, Smith KM, Guilard R, editors. Vol. 14. World Scientific; New Jersey: 2011. p. 525. [Google Scholar]
- 10.Weiss R, Bulach V, Gold A, Terner J, Trautwein A. J Biol Inorg Chem. 2001;6:831. doi: 10.1007/s007750100277. [DOI] [PubMed] [Google Scholar]
- 11.Evangelio E, Ruiz-Molina D. C R Chim. 2008;11:1137. [Google Scholar]
- 12.Jung C. Biochim Biophys Acta. 2011;1814:46. doi: 10.1016/j.bbapap.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 13.Poulos TL. Chem Rev. 2014;114:3919. doi: 10.1021/cr400415k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rittle J, Green MT. Science. 2010;330:933. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 15.Rittle J, Younker JM, Green MT. Inorg Chem. 2010;49:3610. doi: 10.1021/ic902062d. [DOI] [PubMed] [Google Scholar]
- 16.Groves JT, Quinn R, McMurry TJ, Lang G, Boso B. J Chem Soc, Chem Commun. 1984:1455. [Google Scholar]
- 17.Pan Z, Harischandra DN, Newcomb M. J Inorg Biochem. 2009;103:174. doi: 10.1016/j.jinorgbio.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ikezaki A, Takahashi M, Nakamura M. Chem Commun. 2013;49:3098. doi: 10.1039/c3cc40319j. [DOI] [PubMed] [Google Scholar]
- 19.Spreer LO, Maliyackel AC, Holbrook S, Otvos JW, Calvin M. J Am Chem Soc. 1986;108:1949. [Google Scholar]
- 20.Cong Z, Kurahashi T, Fujii H. J Am Chem Soc. 2012;134:4469. doi: 10.1021/ja209985v. [DOI] [PubMed] [Google Scholar]
- 21.Kurahashi T, Kikuchi A, Tosha T, Shiro Y, Kitagawa T, Fujii H. Inorg Chem. 2008;47:1674. doi: 10.1021/ic702061y. [DOI] [PubMed] [Google Scholar]
- 22.Leeladee P, Baglia RA, Prokop KA, Latifi R, de Visser SP, Goldberg DP. J Am Chem Soc. 2012;134:10397. doi: 10.1021/ja304609n. [DOI] [PubMed] [Google Scholar]
- 23.Baglia RA, Dürr M, Ivanović-Burmazović I, Goldberg DP. Inorg Chem. 2014;53:5893. doi: 10.1021/ic500901y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park YJ, Ziller JW, Borovik AS. J Am Chem Soc. 2011;133:9258. doi: 10.1021/ja203458d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsui EY, Tran R, Yano J, Agapie T. Nat Chem. 2013;5:293. doi: 10.1038/nchem.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller CG, Gordon-Wylie SW, Horwitz CP, Strazisar SA, Peraino DK, Clark GR, Weintraub ST, Collins TJ. J Am Chem Soc. 1998;120:11540. [Google Scholar]
- 27.Du H, Lo P-K, Hu Z, Liang H, Lau K-C, Wang Y-N, Lam WWY, Lau T-C. Chem Commun. 2011;47:7143. doi: 10.1039/c1cc12024g. [DOI] [PubMed] [Google Scholar]
- 28.Lam WWY, Yiu S-M, Lee JMN, Yau SKY, Kwong H-K, Lau T-C, Liu D, Lin Z. J Am Chem Soc. 2006;128:2851. doi: 10.1021/ja0552951. [DOI] [PubMed] [Google Scholar]
- 29.Dong L, Wang Y, Lv Y, Chen Z, Mei F, Xiong H, Yin G. Inorg Chem. 2013;52:5418. doi: 10.1021/ic400361s. [DOI] [PubMed] [Google Scholar]
- 30.Chen J, Lee Y-M, Davis KM, Wu X, Seo MS, Cho K-B, Yoon H, Park YJ, Fukuzumi S, Pushkar YN, Nam W. J Am Chem Soc. 2013;135:6388. doi: 10.1021/ja312113p. [DOI] [PubMed] [Google Scholar]
- 31.Mandimutsira BS, Ramdhanie B, Todd RC, Wang H, Zareba AA, Czernuszewicz RS, Goldberg DP. J Am Chem Soc. 2002;124:15170. doi: 10.1021/ja028651d. [DOI] [PubMed] [Google Scholar]
- 32.Prokop KA, Neu HM, de Visser SP, Goldberg DP. J Am Chem Soc. 2011;133:15874. doi: 10.1021/ja2066237. [DOI] [PubMed] [Google Scholar]
- 33.Neu HM, Yang T, Baglia RA, Yosca TH, Green MT, Quesne MG, de Visser SP, Goldberg DP. J Am Chem Soc. 2014;136:13845. doi: 10.1021/ja507177h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165. [Google Scholar]
- 35.Tolman CA. Chem Rev. 1977;77:313. [Google Scholar]
- 36.Lansky DE, Mandimutsira B, Ramdhanie B, Clausén M, Penner-Hahn J, Zvyagin SA, Telser J, Krzystek J, Zhan R, Ou Z, Kadish KM, Zakharov L, Rheingold AL, Goldberg DP. Inorg Chem. 2005;44:4485. doi: 10.1021/ic0503636. [DOI] [PubMed] [Google Scholar]
- 37.Lansky DE, Narducci Sarjeant AA, Goldberg DP. Angew Chem Int Ed. 2006;45:8214. doi: 10.1002/anie.200603139. [DOI] [PubMed] [Google Scholar]
- 38.Leeladee P, Jameson GNL, Siegler MA, Kumar D, de Visser SP, Goldberg DP. Inorg Chem. 2013;52:4668. doi: 10.1021/ic400280x. [DOI] [PubMed] [Google Scholar]
- 39.Barzilay CM, Sibilia SA, Spiro TG, Gross Z. Chem – Eur J. 1995;1:222. [Google Scholar]
- 40.Ehlinger N, Scheidt WR. Inorg Chem. 1999;38:1316. doi: 10.1021/ic981041n. [DOI] [PubMed] [Google Scholar]
- 41.Simkhovich L, Mahammed A, Goldberg I, Gross Z. Chem – Eur J. 2001;7:1041. doi: 10.1002/1521-3765(20010302)7:5<1041::aid-chem1041>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 42.Meier-Callahan AE, Di Bilio AJ, Simkhovich L, Mahammed A, Goldberg I, Gray HB, Gross Z. Inorg Chem. 2001;40:6788. doi: 10.1021/ic010723z. [DOI] [PubMed] [Google Scholar]
- 43.Smeltz JL, Lilly CP, Boyle PD, Ison EA. J Am Chem Soc. 2013;135:9433. doi: 10.1021/ja401390v. [DOI] [PubMed] [Google Scholar]
- 44.Neu HM, Jung J, Baglia RA, Siegler MA, Ohkubo K, Fukuzumi S, Goldberg DP. J Am Chem Soc. 2015;137:4614. doi: 10.1021/jacs.5b00816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klyueva ME, Stuzhin PA, Berezin BD. Russ J Coord Chem. 2003;29:189. [Google Scholar]
- 46.Goslinski T, Tykarska E, Kryjewski M, Osmalek T, Sobiak S, Gdaniec M, Dutkiewicz Z, Mielcarek J. Anal Sci. 2011;27:511. doi: 10.2116/analsci.27.511. [DOI] [PubMed] [Google Scholar]
- 47.Freyer W, Minh LQ. J Porphyrins Phthalocyanines. 1997;01:287. [Google Scholar]
- 48.Graczyk A, Białkowskia E. Tetrahedron. 1978;34:3505. [Google Scholar]
- 49.Mayer U, Gutmann V, Gerger W. Monatsh Chem. 1975;106:1235. [Google Scholar]
- 50.Beckett MA, Strickland GC, Holland JR, Sukumar Varma K. Polymer. 1996;37:4629. [Google Scholar]
- 51.Bentivegna B, Mariani CI, Smith JR, Ma S, Rheingold AL, Brunker TJ. Organometallics. 2014;33:2820. [Google Scholar]
- 52.Mohr J, Durmaz M, Irran E, Oestreich M. Organometallics. 2014;33:1108. [Google Scholar]
- 53.Sivaev IB, Bregadze VI. Coord Chem Rev. 2014;270–271:75. [Google Scholar]
- 54.Welch GC, Cabrera L, Chase PA, Hollink E, Masuda JD, Wei P, Stephan DW. Dalton Trans. 2007:3407. doi: 10.1039/b704417h. [DOI] [PubMed] [Google Scholar]
- 55.Yoon H, Morimoto Y, Lee Y-M, Nam W, Fukuzumi S. Chem Commun. 2012;48:11187. doi: 10.1039/c2cc36291k. [DOI] [PubMed] [Google Scholar]
- 56.Bullock JP, Bond AM, Boeré RT, Gietz TM, Roemmele TL, Seagrave SD, Masuda JD, Parvez M. J Am Chem Soc. 2013;135:11205. doi: 10.1021/ja403555d. [DOI] [PubMed] [Google Scholar]
- 57.Park J, Morimoto Y, Lee Y-M, Nam W, Fukuzumi S. J Am Chem Soc. 2011;133:5236. doi: 10.1021/ja200901n. [DOI] [PubMed] [Google Scholar]
- 58.Kumar A, Goldberg I, Botoshansky M, Buchman Y, Gross Z. J Am Chem Soc. 2010;132:15233. doi: 10.1021/ja1050296. [DOI] [PubMed] [Google Scholar]
- 59.Mayer JM. Acc Chem Res. 1998;31:441. [Google Scholar]
- 60.Bordwell FG, Cheng J, Ji GZ, Satish AV, Zhang X. J Am Chem Soc. 1991;113:9790. [Google Scholar]
- 61.Mayer JM. Annu Rev Phys Chem. 2004;55:363. doi: 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]
- 62.Warren JJ, Tronic TA, Mayer JM. Chem Rev. 2010;110:6961. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cukier RI, Nocera DG. Annu Rev Phys Chem. 1998;49:337. doi: 10.1146/annurev.physchem.49.1.337. [DOI] [PubMed] [Google Scholar]
- 64.Gunay A, Theopold KH. Chem Rev. 2010;110:1060. doi: 10.1021/cr900269x. [DOI] [PubMed] [Google Scholar]
- 65.Wang D, Ray K, Collins MJ, Farquhar ER, Frisch JR, Gomez L, Jackson TA, Kerscher M, Waleska A, Comba P, Costas M, Que L., Jr Chem Sci. 2013;4:282. doi: 10.1039/C2SC21318D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Das S, Chakravorty A. Eur J Inorg Chem. 2006;2006:2285. [Google Scholar]
- 67.Zhu C, Liang J, Wang B, Zhu J, Cao Z. Phys Chem Chem Phys. 2012;14:12800. doi: 10.1039/c2cp41647f. [DOI] [PubMed] [Google Scholar]
- 68.Abram S-L, Monte-Perez I, Pfaff FF, Farquhar ER, Ray K. Chem Commun. 2014;50:9852. doi: 10.1039/c4cc03754e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gelb MH, Toscano WA, Sligar SG. Proc Natl Acad Sci USA. 1982;79:5758. doi: 10.1073/pnas.79.19.5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Segall Y, Granoth I. J Am Chem Soc. 1978;100:5130. [Google Scholar]
- 71.Evans DF, Jakubovic DA. J Chem Soc, Dalton Trans. 1988:2927. [Google Scholar]
- 72.Sheldrick GM. Acta Crystallogr, Sect A: Found Crystallogr. 2008;64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 73.Spek AL. J Appl Crystallogr. 2003;36:7. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.