

Abstract
The objective of the present study was to determine whether guanine nucleotide-binding protein α stimulating (GNAS) gene expression correlates with pathognomonic signs by analyzing the mutations, methylation status and G-protein α subunit (Gsα) expression of GNAS in Ewing sarcoma (ES). Formalin-fixed paraffin-embedded tissue samples from 77 patients with primary ES were obtained in South Korea, Argentina and Brazil, and were studied via methylation chip assay and direct sequencing of the GNAS gene and immunohistochemical analysis of Gsα. The mutation and methylation statuses of the GNAS gene were examined. Immunohistochemical results were measured with respect to proportion and staining intensity. The results revealed that GNAS genes in ES tumor samples were less methylated compared with normal controls. No mutations were detected at exons 8 or 9 of the GNAS locus complex on chromosome 20q13.3, indicating that the pathogenesis of ES was not associated with GNAS mutation. Gsα expression correlated well with the methylation status of the GNAS gene. Notably, high Gsα expression was detected more frequently in samples from living patients than from decedents, although this was not statistically significant (P=0.055). In conclusion, GNAS mutation is not associated with the pathogenesis of ES tumors. This finding may be used to differentiate ES tumors from metastatic bone lesions with morphological similarity to ES tumors. Analysis of the methylation status of the GNAS gene and immunohistochemical Gsα expression suggests that hypermethylated GNAS (low Gsα expression) in ES may be associated with unfavorable progression with a non-significant trend.
Keywords: GNAS, Ewing sarcoma
Introduction
Ewing sarcoma (ES) is the second most common primary bone malignancy, and typically develops in children and adolescents, predominantly in white males (1). It is also referred to as Ewing sarcoma family tumor (ESFT), which includes extraskeletal ES and primitive neuroectodermal tumors. ESFT is a highly aggressive malignancy, with a rate of metastasis of 27% at the time of diagnosis (1). Chemotherapy with intercalated locoregional managements, such as surgery and radiation, is the generally recommended treatment (2). The advanced development of diagnostic tools and delicate understanding of transcriptional and translational factors associated with the pathogenetic Ewing sarcoma breakpoint region 1 (EWSR1)/Friend leukemia integration 1 transcription factor (FLI1) fusion protein (EWS-FLI1) gene have contributed to the improvement of targeted therapies for important oncoproteins. Therefore, the survival rate of patients suffering from ES tends to increase with better elucidation of the pathogenesis and the application thereof to the development of management strategies; the 5-year survival rate may increase from 15 to 39% for metastatic disease, and from 44 to 68% for localized disease (1). For this reason, it is important to determine and act based upon the pathognomonic signs of ES; however, the pathogenesis of ES remains to be elucidated.
There have been a number of suggestions attempting to explain the complicated pathogenesis of ES, which have included insulin-like growth factor-binding protein 3 downregulation, sonic hedgehog signaling and microsatellite-related signaling (3). Additionally, there is evidence to suggest that expression of the guanosine nucleotide-binding protein α stimulating (GNAS) gene, which encodes the G-protein α subunit (Gsα), is associated with the pathogenesis of ES: i) Insulin-like growth factor 1 receptor (IGF1R) expression is related to the early growth response 1 (EGR1) gene and its promoters, which consist of the EWS-FLI1 fusion protein and the cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB) (4–6); this cAMP/CREB signature is activated by Gsα and may result in EGR1 and IGF1R expression; ii) it has been shown that CREB-Smad6-Runx2 signaling promotes defective osteogenesis (7), and the EWS-FLI1 fusion protein also inhibits Runx2 (8); iii) our previous genome-wide methylation studies of ES revealed that the GNAS gene was hypomethylated (9), and proposed that a hypomethylated GNAS gene may be overexpressed in ES relative to normal mesenchymal cells of bone, such that ES tumors would have high expression of Gsα; and iv) activating mutations of the GNAS gene have been identified in pituitary tumors (10), ovarian granulosa cell tumors (11), renal cell carcinomas (12) and hepatocellular carcinomas (13), and these tumors exhibited morphological resemblances to neuroendocrine cells, similar to ES. On the basis of this evidence, we hypothesized that GNAS expression may be associated with the pathogenesis of ES.
The purposes of the present study were to analyze GNAS mutation and methylation statuses, and Gsα expression in ES, in order to determine whether GNAS expression is pathognomonically relevant. To the best of our knowledge, the current study is the first to examine the pathogenic role of the GNAS gene in ES.
Materials and methods
Clinical tumor samples
Formalin-fixed paraffin-embedded (FFPE) tissue samples from 77 patients with primary ES were obtained at the Kyung Hee University Hospital in Korea, Central Army Hospital in Argentina and SARAH Network of Rehabilitation Hospitals in Brazil between January 2000 and December 2005. Normal control samples were obtained from the remaining tissues following total knee replacement surgery due to degenerative osteoarthritis at the Kyung Hee University Hospital in Korea. At the time of tumor sampling, patients had no history of chemotherapy or radiation therapy and there was no evidence of metastatic disease. These tumor samples were diagnosed according to the World Health Organization criteria (14) which, in brief, consist of the following: Small round cell sarcomas, showing diffuse membranous CD99 immunostaining, cytoplasmic periodic acid-Schiff staining, and EWSR1 gene translocation demonstrated via fluorescence in situ hybridization (Zytolight SPEC ROS1 and RET Dual Color Break Apart Probes; ZytoVision, Bremerhaven, Germany). If an EWSR1 gene translocation is not identified but a tumor has a typical immunophenotype in differential diagnosis which is inconsistent with other small round cell tumors, such as lymphoma or rhabdomyosarcoma, such tumor samples are diagnosed as ES.
Patient demographics are presented in Table I. Full data, including follow-up periods and overall survival, were available for 45 patients. The study protocol was reviewed and approved by the Kyung Hee University Institutional Review Board (Seoul, South Korea).
Table I.
Demographics of Ewing sarcoma patients (n=77).
| Clinicopathological parameter | Value |
|---|---|
| Age at diagnosis, years | |
| Range | 1–57 |
| Median | 17 |
| Gender, n (%) | |
| Male | 45 (58.4) |
| Female | 32 (41.6) |
| Tumor site, n (%) | |
| Peripheral | 48 (62.3) |
| Central | 29 (37.7) |
| Follow-up, months | |
| Range | 6–96 |
| Median | 30.5 |
| Lung metastasis, n (%) | |
| Present | 6 (7.8) |
| Absent | 39 (50.6) |
| Not available | 32 (41.6) |
| Patient outcome, n (%) | |
| Survived | 25 (32.5) |
| Died | 20 (26.0) |
| Not available | 32 (41.6) |
Bisulfite conversion and methylation chip assay
Bisulfite conversions of all DNA samples were performed using an EZ-96 DNA methylation kit (Zymo Research, Orange, CA, USA) according to the manufacturer's instructions. For each bisulfite conversion, 500 ng of genomic DNA was used. Following bisulfite treatment, quantification of methylcytosine content was conducted using an Illumina GoldenGate Methylation Cancer Panel I microarray (Illumina, Inc., San Diego, CA, USA). The GoldenGate Panel was used to process 1,505 CpG sites from a panel of 807 cancer-related genes. Briefly, bisulfite-converted DNA was allowed to react with biotin and was then hybridized to assay oligos, after which allele-specific extensions and ligations were conducted at 45°C for 15 min. Ligated products were amplified via polymerase chain reaction (PCR) with the following parameters: 10 min at 37°C; 34 cycles of 35 sec at 95°C, 35 sec at 56°C and 2 min at 72°C; 10 min at 72°C; and 5 min at 4°C. Single-stranded PCR products were prepared by denaturation and hybridized to a Sentrix Array Matrix (GoldenGate Methylation Cancer Panel I). Array hybridization was conducted overnight in a temperature gradient program ranging from 45 to 60°C, and arrays were imaged using a BeadArray Reader scanner (Illumina, Inc.). Raw methylation ratios were calculated using the Methylation Module in Illumina's BeadStudio following background normalization, which was derived by averaging the signals of a built-in negative control. Each sample was examined in a duplicate manner in the chip assay.
Direct sequencing
Direct sequencing was performed to detect the mutational status of GNAS exons 8 and 9. Genomic DNA was extracted using the Magna Pure LC instrument (Roche Applied Science, Mannheim, Germany). PCR was performed using a thermal cycler (GeneAmp PCR system 9700; Thermo Fisher Scientific, Waltham, MA, USA). PCR ingredients containing 2.5 µl of 10X buffer [50 mM KCl, 10 mM Tris-HCl (pH 8.3), 15 mM MgCl2 and 0.001% gelatin], 2.0 µl of 2.5 mM dNTP, 1.0 µl of forward and reverse primers (10 pmol/µl; Bioneer Corporation, Daejeon, Korea), 0.5 µl of AmpliTaq® DNA Polymerase (5 U/µl; Thermo Fisher Scientific) and 1.0 µl of DNA (50 ng/µl) were mixed with deionized water, at a total volume of 25 µl. PCR conditions for amplifying GNAS exons 8 and 9 were as follows: Denaturation at 94°C for 2 min; 40 cycles of denaturation at 94°C for 30 sec, annealing at 62°C for 30 sec, and extension at 72°C for 1 min; and a final extension at 72°C for 10 min. PCR products were purified for sequencing analysis with the QIAquick PCR purification kit (Qiagen, Valencia, CA, USA). Cycle sequencing was performed with the BigDye® Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific) according to the manufacturer's protocols, and the reaction mixture was analyzed on an ABI Prism 3100 DNA Sequencer (Applied Biosystem; Thermo Fisher Scientific). Primer sequences for PCR and sequencing are listed in Table II.
Table II.
Primer sequences for GNAS exons 8 and 9.
| Primer sequence | |||
|---|---|---|---|
| GNAS exon | Forward | Reverse | Amplicon size (bp) |
| 8 | 5′-GTT TCG GTT GGC TTT GGT GA-3′ | 5′-TGG CTT ACT GGA AGT TGA CT-3′ | 129 |
| 9 | 5′-GAC ATT CAC CCC AGT CCC TCT-3′ | 5′-GAA GCA AAG CGT TCT TTA CGA-3′ | 155 |
Immunohistochemistry
Immunohistochemical stains for anti-G protein α S antibody were performed on FFPE human specimens. Immunohistochemistry procedures were performed on 5 µm tissue sections on a Leica Bond-Max automatic slide stainer (Leica Biosystems Melbourne Pty. Ltd., Melbourne, Australia) using the standard protocol. In brief, the 5 µm sections of FFPE tissues were deparaffinized using Bond Dewax Solution (Cat#. AR9222; Leica Biosystems Newcastle Ltd.), and an antigen retrieval procedure was performed using Proteinase K Solution (ready for use; Cat#. S3020; Dako Korea Co., Ltd., Seoul, Korea) for 7 min at room tempurature. The endogenous peroxidase was quenched by incubation with hydrogen peroxide for 15 min. Sections were incubated for 15 min at ambient temperate with a rabbit monoclonal anti-G protein α S antibody (Cat#. ab83735; Abcam, Cambridge, MA, USA) at a 1:600 dilution. Secondary antibodies goat anti-rabbit biotin-free polymeric horseradish peroxidase and rabbit anti-mouse linker antibody, contained in a Bond Polymer Refine Detection System (Cat#. DS9800; Leica Biosystems Newcastle Ltd., Newcastle, UK), were incubated for 8 min at room temperature. Bound primary antibodies were visualized using DAB with a Bond Polymer Refine Detection System (Cat#. DS9800; Leica Biosystems Newcastle Ltd.) and a Bond-Max automatic slide stainer (Leica Biosystems Melbourne Pty. Ltd.). The nuclei in these sections were counterstained with hematoxylin using a Bond Polymer detection System (Cat#. 9800; Leica Biosystems Newcastle, Ltd.). Pancreatic islet cells, obtained from the remaining tissues of patients that received pancreatectomies due to chronic pancreatitis, were used as a positive external control.
Pathological analysis of immunohistochemistry
The immunohistochemical results were measured and scored with respect to intensity and proportion in positive tumor cells, and were independently reviewed by three pathologists (Drs Byeong-Joo Noh, Ji-Youn Sung and Yong-Koo Park). The staining intensity was graded in a 4-tiered system as follows: No visible brown staining, 0; pale brown, 1+; non-homogeneous brown, 2+; homogeneous dark brown color, 3+. According to the cytoplasmic staining intensity and proportion of positive tumor cells, the final scores of Gsα were categorized as grade 0, 1, 2 or 3: Grade 0, absence of Gsα staining in 100% of tumor cells; grade 1, intensity 1+ in >70% of tumor cells or intensity 2+ in ≤30% of tumor cells; grade 2, intensity 1+ in >70% of tumor cells, intensity 2+ in >30% but ≤70% of tumor cells or intensity 3+ ≤30% of tumor cells; grade 3, intensity 2+ in >70% of tumor cells or intensity of 3+ in >30% of tumor cells (Fig. 1).
Figure 1.

Final immunohistochemical staining cores of G-protein α subunit (visible as brown staining) are classified into 4-tiered system: (A) Grade 0, (B) intensity 1+, (C) intensity 2+ and (D) intensity 3+. Original magnification, ×400.
Statistical analysis
Statistical analyses were conducted using SPSS version 12.0 (SPSS, Inc., Chicago, IL, USA). Pearson's χ2 and independent t-tests were conducted to determine correlations between tested values and clinicopathological parameters. Univariate survival analyses were performed to examine the prognostic significance of antibody expression and clinicopathological parameters, according to the Kaplan-Meier curve with a log-rank test. Statistical significance was considered to be indicated by P<0.05.
Results
Methylation analysis of the GNAS gene
The degree of methylation of the GNAS gene was assessed using the Illumina GoldenGate Methylation Cancer Panel I microarray. The GoldenGate DNA methylation method measures DNA methylation levels as β-values ranging from 0 (no DNA methylation detected) to 1 (complete DNA methylation). The results indicated that the GNAS gene in ES tumor samples was less methylated than in normal controls; the mean β-value was 0.48 in ES tumor samples vs. 0.83 in normal control samples, indicating that the GNAS gene was overexpressed in ES relative to normal tissue.
Mutation analysis of the GNAS gene
No mutations were detected in exons 8 or 9 of the GNAS locus complex on chromosome 20q13.3 in DNA extracted from any of the FFPE tumor samples from the ES patients (Fig. 2), demonstrating that the pathogenesis of ES was not associated with GNAS mutation.
Figure 2.

Direct sequencing of GNAS exon 8 and 9 detected no mutations, demonstrating that the pathogenesis of Ewing sarcoma was not connected to GNAS mutation. GNAS, guanine nucleotide-binding protein α stimulating.
Immunohistochemical analysis of Gsα expression
The correlation of Gsα expression with clinicopathological parameters was analyzed using a binary system approach, grouping low expression (grades 0–1) vs. high expression (grades 2–3). Clinicopathological cases involving missing values or without available clinical information were abbreviated for statistical analyses. Of the 52 ES tumor samples, 34 samples (65.4%) showed high Gsα expression, compared with 18 sample (34.6%) with low Gsα expression.
Gsα expression correlated well with the methylation status of the GNAS gene; β-values were 0.681±0.304 in tumor samples with low expression of Gsα, compared with 0.245±0.229 in samples with high Gsα expression (P=0.001) (Table III). High Gsα expression in tumor samples was found in 14/17 samples (82.4%) with a hypomethylated GNAS gene vs. 1/5 samples (20%) with a hypermethylated GNAS gene (P=0.009; Table III).
Table III.
Association of clinicopathological parameters with Gsα expression.
| Gsα expression | ||||
|---|---|---|---|---|
| Clinicopathological parametera | Low | High | Total | P-value |
| Age, mean ± SD | 21.17±11.03 | 18.62±10.60 | 19.16 ± 10.50 | 0.420b |
| Gender, n (%) | 0.727c | |||
| Male | 12 (36.4) | 21 (63.6) | 33 (100.0) | |
| Female | 6 (31.6) | 13 (68.4) | 19 (100.0) | |
| Site involved, n (%) | 0.451c | |||
| Peripheral | 13 (38.2) | 21 (61.8) | 34 (100.0) | |
| Central | 5 (27.8) | 13 (72.2) | 18 (100.0) | |
| β-value, mean ± SD | 0.681±0.304 | 0.245±0.229 | 0.478 ± 0.345 | 0.001b |
| Degree of methylation, n (%) | 0.009c | |||
| Hypomethylation | 3 (17.6) | 14 (82.4) | 17 (100.0) | |
| Hypermethylation | 4 (80.0) | 1 (20.0) | 5 (100.0) | |
| Outcome, n (%) | 0.055c | |||
| Survived | 2 (13.3) | 13 (86.7) | 15 (100.0) | |
| Died | 6 (46.2) | 7 (53.8) | 13 (100.0) | |
Clinicopathological cases involving missing values or without available clinicopathological values were removed for statistical analyses
Student's t-test
χ2 test. Gsα, G-protein α subunit; SD, standard deviation.
Notably, high Gsα expression was detected more frequently in samples from living patients than decedents, although this was not statistically significant; high Gsα was found in 12/15 samples (86.7%) from living patients, vs. 7/13 samples (53.8%) from decedents (P=0.055; Table III). Furthermore, Gsα levels were not found to significantly correlate with the survival rate (P=0.220; Fig. 3).
Figure 3.
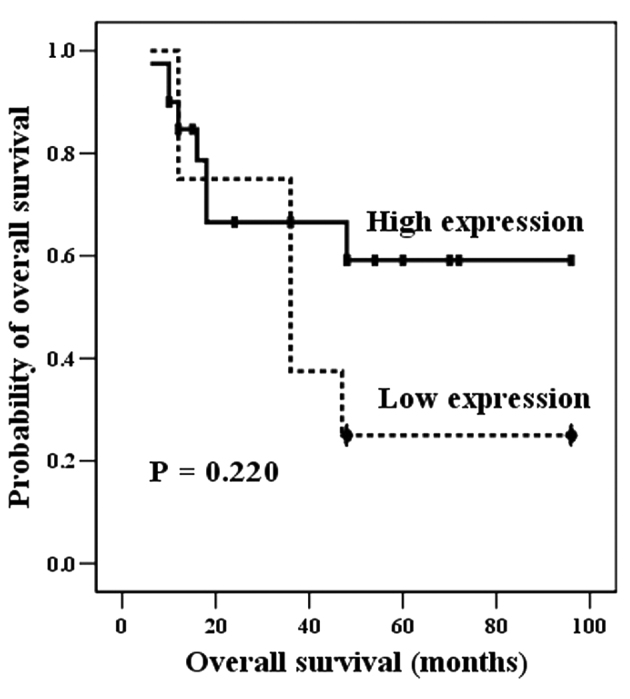
Univariate analysis (Kaplan-Meier curve) demonstrated that high Gsα expression was inclined to indicate more favorable clinical behavior, compared with low Gsα expression, although this was not statistically significant (P=0.220). Gsα, G-protein α subunit.
Discussion
The GNAS gene on chromosome 20q13.3 encodes the α subunit of the heterotrimeric G protein complex (Gsα) (15). Activating or inactivating mutations and epigenetic changes at the GNAS locus have been described in a variety of human diseases (15).
Activating mutations of the GNAS gene induce protein alterations as follows: A mutation at exon 8 of the GNAS gene is responsible for substitution of arginine at codon 201 with cysteine or histidine, termed R201C or R201H, respectively; and, more rarely, a mutation at exon 9 of GNAS results in substitution of the glutamine at codon 227 with leucine, arginine, lysine or histidine, termed Q227L, Q227R, Q227K or Q227H, respectively (16). These protein alterations may inhibit GTPase activity, maintaining an active form of Gsα. Activating mutations of the GNAS locus have been detected in McCune-Albright syndrome (17), pituitary adenoma (10), ovarian granulosa cell tumor (11), renal cell carcinoma (12), hepatocellular carcinoma (13), pancreatic intestinal-type intraductal papillary mucinous neoplasm (18,19) and myelodysplastic syndrome (20). As with mesenchymal bone tumors, fibrous dysplasia (2,21) and parosteal osteosarcoma (22) have also been linked to activating mutations of the GNAS gene. It has been documented that GNAS status has diagnostic utility for differentiating fibro-osseous lesions in morphologically challenging diagnoses: Fibrous dysplasia vs. ossifying fibroma, adamantinoma or osteofibrous dysplasia (16,22,23). GNAS mutation was not identified in any ES tumor sample in the current study, indicating that GNAS mutation is not associated with the pathogenesis of ES. For this reason, GNAS mutation analysis may be used to exclude metastatic bone lesions with GNAS mutations, including renal cell carcinoma or hepatocellular carcinoma, which exhibit morphological resemblances to ES tumor cells (24,25).
Inactivating mutations of the GNAS locus from the maternal or paternal germ-line are attributed to amino acid substitutions, nonsense mutations, inversions, splicing site mutations, insertions or deletions. Progressive osseous heterotopia has been verified to result from inactivating GNAS mutations of predominantly paternal origin (21,26). It has also been documented that epigenetic alterations of the GNAS gene are associated with deletion of the syntaxin 16 gene and hypomethylation of the A/B domain at the GNAS locus complex (27). Epigenetic changes in the GNAS gene have been demonstrated to be important in disease progression in pseudohypoparathyroidism type Ib (28). Until now, there has been no research demonstrating that activating or inactivating mutations or epigenetic changes of GNAS have prognostic and not diagnostic value. The T393C pleomorphism of the GNAS locus has been identified to have prognostic value in various malignant tumors, including clear cell renal cell carcinoma (29), bladder cancer (30), colorectal cancer (31), epithelial ovarian cancer (32), melanoma (33), glioblastoma (34) and non-small cell lung cancer (35).
In the current study, the epigenetic methylation status of the GNAS gene in ES tumor samples was significantly associated with Gsα expression: The less methylated GNAS gene in ES tumor samples overexpressed Gsα (Table III). However, ES patients with hypomethylated GNAS genes (high expression of Gsα) had increased survival probability relative to those with hypermethylated GNAS genes (low expression of Gsα), with a non-significant positive trend (P=0.055, Table III; P=0.220, Fig. 3). On the basis of these results, we speculate that the epigenetic transformation of the GNAS gene to hypermethylation status (low Gsα expression) in ES tumors may have a an association with more aggressive behavior of ES tumors, but is not significantly correlated with the overall survival.
In summary, GNAS mutation is not associated with the pathogenesis of ES tumors. This finding may be used to distinguish metastatic bone lesions with GNAS mutations that have morphological similarities to ES tumors. Analysis of the methylation status of the GNAS gene and immunohistochemical Gsα expression suggests that hypermethylated GNAS gene (low Gsα expression) in ES may be associated with unfavorable progression with a non-significant trend. Further studies with a larger sample of patients are required to verify these results.
References
- 1.Esiashvili N, Goodman M, Marcus RB., Jr Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance epidemiology and end results data. J Pediatr Hematol Oncol. 2008;30:425–430. doi: 10.1097/MPH.0b013e31816e22f3. [DOI] [PubMed] [Google Scholar]
- 2.Lee SE, Lee EH, Park H, Sung JY, Lee HW, Kang SY, Seo S, Kim BH, Lee H, Seo AN, et al. The diagnostic utility of the GNAS mutation in patients with fibrous dysplasia: Meta-analysis of 168 sporadic cases. Hum Pathol. 2012;43:1234–1242. doi: 10.1016/j.humpath.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 3.Lessnick SL, Ladanyi M. Molecular pathogenesis of Ewing sarcoma: New therapeutic and transcriptional targets. Annu Rev Pathol. 2012;7:145–159. doi: 10.1146/annurev-pathol-011110-130237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma Y, Cheng Q, Ren Z, Xu L, Zhao Y, Sun J, Hu S, Xiao W. Induction of IGF-1R expression by EGR-1 facilitates the growth of prostate cancer cells. Cancer Lett. 2012;317:150–156. doi: 10.1016/j.canlet.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 5.Wu X, Cheng J, Li P, Yang M, Qiu S, Liu P, Du J. Mechano-sensitive transcriptional factor Egr-1 regulates insulin-like growth factor-1 receptor expression and contributes to neointima formation in vein grafts. Arterioscler Thromb Vasc Biol. 2010;30:471–476. doi: 10.1161/ATVBAHA.109.184259. [DOI] [PubMed] [Google Scholar]
- 6.Watson DK, Robinson L, Hodge DR, Kola I, Papas TS, Seth A. FLI1 and EWS-FLI1 function as ternary complex factors and ELK1 and SAP1a function as ternary and quaternary complex factors on the Egr1 promoter serum response elements. Oncogene. 1997;14:213–221. doi: 10.1038/sj.onc.1200839. [DOI] [PubMed] [Google Scholar]
- 7.Fan QM, Yue B, Bian ZY, Xu WT, Tu B, Dai KR, Li G, Tang TT. The CREB-Smad6-Runx2 axis contributes to the impaired osteogenesis potential of bone marrow stromal cells in fibrous dysplasia of bone. J Pathol. 2012;228:45–55. doi: 10.1002/path.4033. [DOI] [PubMed] [Google Scholar]
- 8.Li X, McGee-Lawrence ME, Decker M, Westendorf JJ. The Ewing's sarcoma fusion protein, EWS-FLI, binds Runx2 and blocks osteoblast differentiation. J Cell Biochem. 2010;111:933–943. doi: 10.1002/jcb.22782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park HR, Jung WW, Kim HS, Park YK. Microarray-based DNA methylation study of Ewing's sarcoma of the bone. Oncol Lett. 2014;8:1613–1617. doi: 10.3892/ol.2014.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gadelha MR, Trivellin G, Hernández, Ramírez LC, Korbonits M. Genetics of pituitary adenomas. Front Horm Res. 2013;41:111–140. doi: 10.1159/000345673. [DOI] [PubMed] [Google Scholar]
- 11.Kalfa N, Ecochard A, Patte C, Duvillard P, Audran F, Pienkowski C, Thibaud E, Brauner R, Lecointre C, Plantaz D, et al. Activating mutations of the stimulatory g protein in juvenile ovarian granulosa cell tumors: A new prognostic factor? J Clin Endocrinol Metab. 2006;91:1842–1847. doi: 10.1210/jc.2005-2710. [DOI] [PubMed] [Google Scholar]
- 12.Kalfa N, Lumbroso S, Boulle N, Guiter J, Soustelle L, Costa P, Chapuis H, Baldet P, Sultan C. Activating mutations of Gsalpha in kidney cancer. J Urol. 2006;176:891–895. doi: 10.1016/j.juro.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 13.Nault JC, Fabre M, Couchy G, Pilati C, Jeannot E, Van Tran Nhieu J, Saint-Paul MC, De Muret A, Redon MJ, Buffet C, et al. GNAS-activating mutations define a rare subgroup of inflammatory liver tumors characterized by STAT3 activation. J Hepatol. 2012;56:184–191. doi: 10.1016/j.jhep.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 14.de alava E, Lessnick SL, Sorensen PH. Ewing sarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors. WHO classification of Tumours of Soft Tissue and Bone. 4th. IARC Press; Lyon: 2012. pp. 305–309. [Google Scholar]
- 15.Weinstein LS, Liu J, Sakamoto A, Xie T, Chen M. Minireview: GNAS: Normal and abnormal functions. Endocrinology. 2004;145:5459–5464. doi: 10.1210/en.2004-0865. [DOI] [PubMed] [Google Scholar]
- 16.Tabareau-Delalande F, Collin C, Gomez-Brouchet A, Decouvelaere AV, Bouvier C, Larousserie F, Marie B, Delfour C, Aubert S, Rosset P, et al. Diagnostic value of investigating GNAS mutations in fibro-osseous lesions: A retrospective study of 91 cases of fibrous dysplasia and 40 other fibro-osseous lesions. Mod Pathol. 2013;26:911–921. doi: 10.1038/modpathol.2012.223. [DOI] [PubMed] [Google Scholar]
- 17.Salpea P, Stratakis CA. Carney complex and McCune Albright syndrome: An overview of clinical manifestations and human molecular genetics. Mol Cell Endocrinol. 2014;386:85–91. doi: 10.1016/j.mce.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matthaei H, Wu J, Dal Molin M, Shi C, Perner S, Kristiansen G, Lingohr P, Kalff JC, Wolfgang CL, Kinzler KW, et al. GNAS sequencing identifies IPMN-specific mutations in a subgroup of diminutive pancreatic cysts referred to as ‘incipient IPMNs’. Am J Surg Pathol. 2014;38:360–363. doi: 10.1097/PAS.0000000000000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dal Molin M, Matthaei H, Wu J, Blackford A, Debeljak M, Rezaee N, Wolfgang CL, Butturini G, Salvia R, Bassi C, et al. Clinicopathological correlates of activating GNAS mutations in intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann Surg Oncol. 2013;20:3802–3808. doi: 10.1245/s10434-013-3096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, Ebert BL. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–2506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Regard JB, Malhotra D, Gvozdenovic-Jeremic J, Josey M, Chen M, Weinstein LS, Lu J, Shore EM, Kaplan FS, Yang Y. Activation of hedgehog signaling by loss of GNAS causes heterotopic ossification. Nat Med. 2013;19:1505–1512. doi: 10.1038/nm.3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carter JM, Inwards CY, Jin L, Evers B, Wenger DE, Oliveira AM, Fritchie KJ. Activating GNAS mutations in parosteal osteosarcoma. Am J Surg Pathol. 2014;38:402–409. doi: 10.1097/PAS.0000000000000144. [DOI] [PubMed] [Google Scholar]
- 23.Shi RR, Li XF, Zhang R, Chen Y, Li TJ. GNAS mutational analysis in differentiating fibrous dysplasia and ossifying fibroma of the jaw. Mod Pathol. 2013;26:1023–1031. doi: 10.1038/modpathol.2013.31. [DOI] [PubMed] [Google Scholar]
- 24.Kalfa N, Lumbroso S, Boulle N, Guiter J, Soustelle L, Costa P, Chapuis H, Baldet P, Sultan C. Activating mutations of Gsalpha in kidney cancer. J Urol. 2006;176:891–895. doi: 10.1016/j.juro.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 25.Nault JC, Fabre M, Couchy G, Pilati C, Jeannot E, Van Nhieu Tran J, Saint-Paul MC, De Muret A, Redon MJ, Buffet C, et al. GNAS-activating mutations define a rare subgroup of inflammatory liver tumors characterized by STAT3 activation. J Hepatol. 2012;56:184–191. doi: 10.1016/j.jhep.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 26.Pignolo RJ, Xu M, Russell E, Richardson A, Kaplan J, Billings PC, Kaplan FS, Shore EM. Heterozygous inactivation of Gnas in adipose-derived mesenchymal progenitor cells enhances osteoblast differentiation and promotes heterotopic ossification. J Bone Miner Res. 2011;26:2647–2655. doi: 10.1002/jbmr.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turan S, Ignatius J, Moilanen JS, Kuismin O, Stewart H, Mann NP, Linglart A, Bastepe M, Jüppner H. De novo STX16 deletions: An infrequent cause of pseudohypoparathyroidism type Ib that should be excluded in sporadic cases. J Clin Endocrinol Metab. 2012;97:E2314–E2319. doi: 10.1210/jc.2012-2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuno A, Usui T, Yambe Y, Higashi K, Ugi S, Shinoda J, Mashio Y, Shimatsu A. Genetic and epigenetic states of the GNAS complex in pseudohypoparathyroidism type Ib using methylation-specific multiplex ligation-dependent probe amplification assay. Eur J Endocrinol. 2013;168:169–175. doi: 10.1530/EJE-12-0548. [DOI] [PubMed] [Google Scholar]
- 29.Frey UH, Lümmen G, Jäger T, Jöckel KH, Schmid KW, Rübben H, Müller N, Siffert W, Eisenhardt A. The GNAS1 T393C polymorphism predicts survival in patients with clear cell renal cell carcinoma. Clin Cancer Res. 2006;12:759–763. doi: 10.1158/1078-0432.CCR-05-1722. [DOI] [PubMed] [Google Scholar]
- 30.Frey UH, Eisenhardt A, Lümmen G, Rübben H, Jöckel KH, Schmid KW, Siffert W. The T393C polymorphism of the G alpha s gene (GNAS1) is a novel prognostic marker in bladder cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:871–877. doi: 10.1158/1055-9965.EPI-04-0720. [DOI] [PubMed] [Google Scholar]
- 31.Frey UH, Alakus H, Wohlschlaeger J, Schmitz KJ, Winde G, van Calker HG, Jöckel KH, Siffert W, Schmid KW. GNAS1 T393C polymorphism and survival in patients with sporadic colorectal cancer. Clin Cancer Res. 2005;11:5071–5077. doi: 10.1158/1078-0432.CCR-05-0472. [DOI] [PubMed] [Google Scholar]
- 32.Tominaga E, Tsuda H, Arao T, Nishimura S, Takano M, Kataoka F, Nomura H, Hirasawa A, Aoki D, Nishio K. Amplification of GNAS may be an independent, qualitative and reproducible biomarker to predict progression-free survival in epithelial ovarian cancer. Gynecol Oncol. 2010;118:160–166. doi: 10.1016/j.ygyno.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 33.Frey UH, Fritz A, Rotterdam S, Schmid KW, Potthoff A, Altmeyer P, Siffert W, Brockmeyer NH. GNAS1 T393C polymorphism and disease progression in patients with malignant melanoma. Eur J Med Res. 2010;15:422–427. doi: 10.1186/2047-783X-15-10-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.El Hindy N, Lambertz N, Bachmann HS, Frey UH, Adamzik M, Zhu Y, Sure U, Siffert W, Sandalcioglu IE. Role of the GNAS1 T393C polymorphism in patients with glioblastoma multiforme. J Clin Neurosci. 2011;18:1495–1499. doi: 10.1016/j.jocn.2011.02.044. [DOI] [PubMed] [Google Scholar]
- 35.Xie FJ, Zhao P, Kou JY, Hong W, Fu L, Hu L, Hong D, Su D, Gao Y, Zhang YP. The T393C polymorphism of GNAS1 as a predictor for chemotherapy sensitivity and survival in advanced non-small-cell lung cancer patients treated with gemcitabine plus platinum. Cancer Chemother Pharmacol. 2012;69:1443–1448. doi: 10.1007/s00280-012-1849-3. [DOI] [PubMed] [Google Scholar]