

Abstract
miRNAs associate with Argonaute (AGO) proteins to silence the expression of mRNA targets by inhibiting translation and promoting deadenylation, decapping, and mRNA degradation. A current model for silencing suggests that AGOs mediate these effects through the sequential recruitment of GW182 proteins, the CCR4–NOT deadenylase complex and the translational repressor and decapping activator DDX6. An alternative model posits that AGOs repress translation by interfering with eIF4A function during 43S ribosomal scanning and that this mechanism is independent of GW182 and the CCR4–NOT complex in Drosophila melanogaster. Here, we show that miRNAs, AGOs, GW182, the CCR4–NOT complex, and DDX6/Me31B repress and degrade polyadenylated mRNA targets that are translated via scanning‐independent mechanisms in both human and Dm cells. This and additional observations indicate a common mechanism used by these proteins and miRNAs to mediate silencing. This mechanism does not require eIF4A function during ribosomal scanning.
Keywords: argonaute, DDX6, deadenylation, eIF4A, GW182
Subject Categories: Protein Biosynthesis & Quality Control, RNA Biology
Introduction
miRNAs are a class of small noncoding RNAs that assemble with Argonaute proteins into miRNA‐induced silencing complexes (miRISCs) to mediate post‐transcriptional repression of complementary mRNA targets (Ameres & Zamore, 2013). In animals, miRNA‐mediated silencing is effected by a combination of translational repression and mRNA destabilization, with the latter accounting for most of the steady‐state repression in mammalian cell cultures (Hendrickson et al, 2009; Guo et al, 2010; Eichhorn et al, 2014).
miRNA target degradation is catalyzed by the enzymes of the 5′‐to‐3′ mRNA decay pathway (Rehwinkel et al, 2005; Behm‐Ansmant et al, 2006; Eulalio et al, 2007, 2009; Chen et al, 2009; Piao et al, 2010). In this pathway, mRNAs are first deadenylated by the PAN2–PAN3 and the CCR4–NOT complexes, then decapped by the decapping enzyme DCP2, and finally degraded from the 5′‐end by the XRN1 exonuclease (Jonas & Izaurralde, 2015). The GW182/TNRC6 proteins play a central role in this process by interacting with AGOs and recruiting the PAN2–PAN3 and the CCR4–NOT deadenylase complexes to miRNA targets, thereby accelerating their degradation (Fabian & Sonenberg, 2012; Jonas & Izaurralde, 2015).
In addition to accelerating mRNA degradation, miRNAs also trigger translational repression, but the precise repressive mechanism remains poorly understood, and several models have been proposed (Fabian & Sonenberg, 2012). One possible model is that the recruitment of the CCR4–NOT complex is sufficient to mediate silencing (Jonas & Izaurralde, 2015). This model is based on the following observations: First, the interaction of GW182 proteins with the CCR4–NOT complex is not only required for degradation but also required for translational repression of miRNA reporters (Braun et al, 2011; Chekulaeva et al, 2011; Fabian et al, 2011; Huntzinger et al, 2013; Zekri et al, 2013; Chen et al, 2014; Mathys et al, 2014). Second, like miRISC, the CCR4–NOT complex represses translation in the absence of deadenylation (Cooke et al, 2010; Braun et al, 2011; Chekulaeva et al, 2011; Bawankar et al, 2013; Zekri et al, 2013). Translational repression by the CCR4–NOT complex can, at least in part, be explained by a direct interaction between the NOT1 subunit and the DEAD‐box ATPase DDX6 (also known as RCK). Human DDX6 and its Drosophila melanogaster (Dm) ortholog Me31B repress translation, activate decapping, and play a role in silencing (Chu & Rana, 2006; Eulalio et al, 2007; Nishihara et al, 2013; Chen et al, 2014; Mathys et al, 2014; Rouya et al, 2014).
An alternative model of silencing involves the translation initiation factor eIF4A. eIF4A proteins are DEAD‐box RNA helicases that unwind secondary structures within mRNA 5′‐UTRs, allowing the 43S preinitiation complex (PIC) to scan the 5′‐UTR toward the start codon (Jackson et al, 2010). Several studies have indicated that miRISCs inhibit 43S scanning by interfering with eIF4A function (Meijer et al, 2013; Ricci et al, 2013; Fukao et al, 2014; Fukaya et al, 2014). How this interference is achieved remains unclear. One study reported that NOT1 interacts with eIF4A2 but not with its paralog eIF4A1 in human cells (Meijer et al, 2013). It was suggested that this interaction locks eIF4A2 onto the mRNA 5′‐UTR and represses translation by blocking 43S scanning (Meijer et al, 2013). However, the interaction between NOT1 and eIF4A2 was not confirmed in subsequent studies (Chen et al, 2014; Mathys et al, 2014; Rouya et al, 2014) and the knockout of eIF4A2 in human cells does not suppress silencing (Galicia‐Vazquez et al, 2015).
Furthermore, in contrast to the model proposed by Meijer et al, two recent studies indicated that miRNAs repress translation by releasing rather than recruiting eIF4A2 and that both eIF4A1 and eIF4A2 are dislodged from silenced mRNAs (Fukao et al, 2014; Fukaya et al, 2014). Remarkably, Dm AGO1 promoted eIF4A displacement independently of GW182, and Dm GW182 caused eIF4A and eIF4E displacement independently of AGO1 (Fukaya et al, 2014). These and additional observations suggested that Dm AGO1 exhibits silencing activity independently of GW182 proteins in D. melanogaster (Fukaya & Tomari, 2012; Wu et al, 2013; Fukaya et al, 2014). Whether this is also the case for human AGOs has not been investigated.
In sum, the mechanism by which miRNAs repress translation is still not fully understood, and it is not known whether the recruitment of the CCR4–NOT complex fully explains silencing or whether parallel and potentially species‐specific repressive mechanisms exist. To clarify these open questions, we adopted a comparative approach and investigated the silencing mechanisms in human and Dm cells. We found that miRNAs, AGO, GW182, and NOT1 do not require 43S ribosomal scanning to repress translation but require DDX6 in human cells. We further show that the repressive activity of AGOs depends on the integrity of the W‐binding pockets, which in turn recruit GW182 proteins and the CCR4–NOT complex in both human and D. melanogaster cells. Collectively, our data indicate that CCR4–NOT is a major downstream effector complex in the miRNA pathway.
Results
The W‐binding pockets of AGOs mediate binding to the GW182/TNRC6 proteins
Structural studies of human (Hs) AGO2 revealed the presence of tandem tryptophan (W)‐binding pockets (pockets P1 and P2) on the surface of the AGO2 PIWI domain opposite to the miRNA‐binding site (Appendix Fig S1A; Schirle & MacRae, 2012). These pockets represent potential binding sites for the GW182/TNRC6 proteins and are conserved in Hs AGO1, AGO3, and AGO4 as well as in Dm AGO1 (Appendix Fig S1B; Schirle & MacRae, 2012). To determine the contribution of the W‐binding pockets to the interaction of AGOs with GW182 proteins, we designed mutations to disrupt the pockets in Hs AGO2 and Dm AGO1 (mutants P1 and P2, Appendix Fig S1A and B, Appendix Table S1).
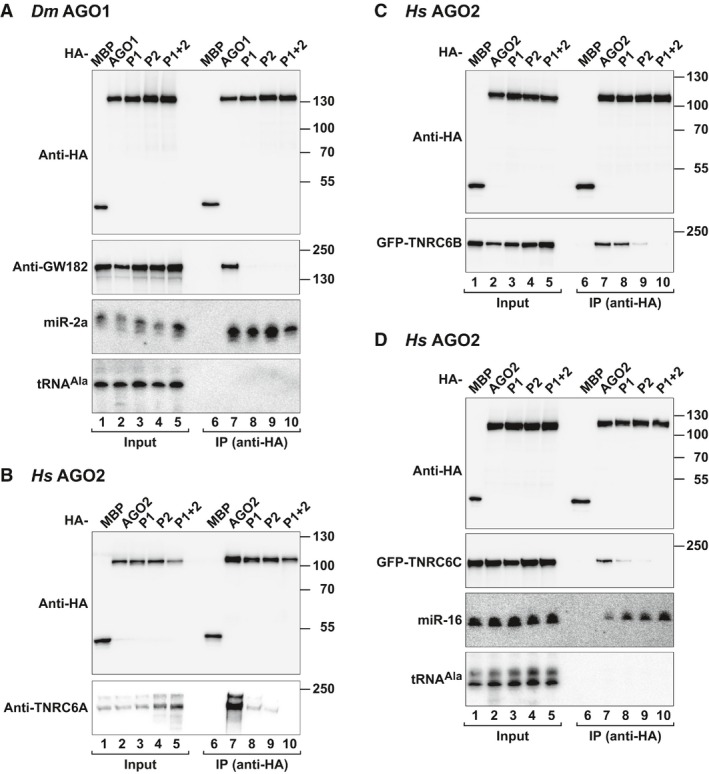
Mutations in either of the two Dm AGO1 pockets abolished the interaction with endogenous Dm GW182 in coimmunoprecipitation assays in Dm Schneider cells (S2 cells; Fig 1A, lanes 8 and 9 versus 7). The mutant proteins were expressed at levels comparable to wild type and associated with endogenous miR‐2a, indicating that the mutations do not disrupt the fold of the PIWI domain (Fig 1A). Similarly, combined mutations in the two pockets of Hs AGO2 abolished its interaction with endogenous TNRC6A and GFP‐tagged TNRC6B and TNRC6C in HEK293T cells (Fig 1B–D, lanes 10 versus 7). This AGO2 mutant no longer accumulated in P‐bodies as expected because P‐body localization requires the interaction with the TNRC6 proteins (Fig EV1A; Lazzaretti et al, 2009). Mutations in only one of the two pockets disrupted the interaction of Hs AGO2 with TNRC6C and strongly reduced TNRC6A binding (Fig 1B and D). The binding of TNRC6B was impaired by mutations in P2 but not in P1 (Fig 1C). These results reveal differences in the binding properties of the three TNRC6 proteins. Importantly, the mutations in the Hs AGO2 W‐binding pockets did not interfere with miR‐16 loading (Fig 1D). We concluded that the integrity of the W‐binding pockets is required for Hs AGO2 and Dm AGO1 to interact with GW182/TNRC6 proteins.
Figure 1. The W‐binding pockets are required for AGOs to bind GW182/TNRC6 proteins.
-
ALysates from S2 cells expressing HA‐tagged versions of MBP or AGO1 (wild type or pocket mutants) were immunoprecipitated using an anti‐HA antibody. Inputs (1% for the HA‐tagged proteins and 2.5% for GW182) and immunoprecipitates (20 and 35%, respectively) were analyzed by Western blotting using an anti‐HA antibody. Endogenous GW182 was detected using anti‐GW182 antibodies. The association between HA‐AGO1 and endogenous miR‐2a was analyzed by Northern blotting. tRNAA la served as a loading control.
-
BInteraction of HA‐tagged AGO2 (wild type or pocket mutants) with endogenous TNRC6A in HEK293T cells. HA‐tagged MBP served as a negative control.
-
C, DInteraction of HA‐tagged AGO2 (wild type or pocket mutants) with GFP‐tagged TNRC6B and TNRC6C in HEK293T cells. HA‐tagged MBP served as a negative control. Inputs (1.5% for the HA‐tagged proteins and 2% for the GFP‐tagged proteins) and immunoprecipitates (20 and 35%, respectively) were analyzed by Western blotting using the corresponding antibodies. The presence of endogenous miR‐16 in the immunoprecipitates was determined by Northern blotting (D). tRNAA la served as a loading control. AGO mutants are described in Appendix Fig S1 and Appendix Table S1.
Source data are available online for this figure.
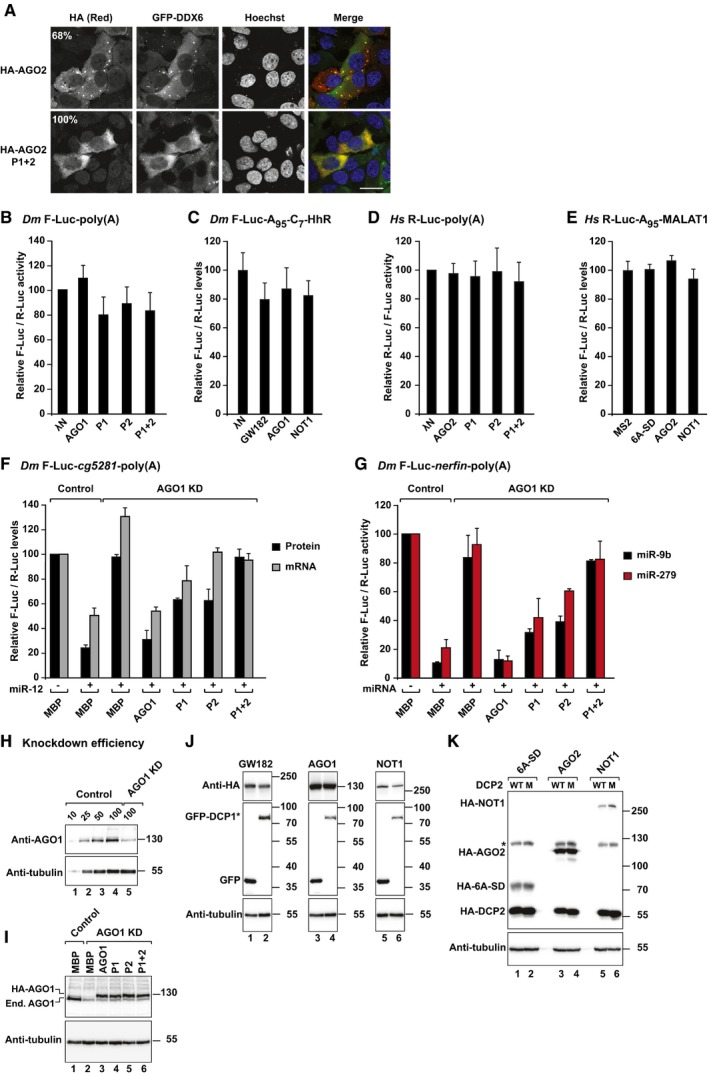
Figure EV1. The silencing activity of AGOs depends on the integrity of the W‐binding pockets.
-
ALocalization of HA‐AGO2 (wild type or P1+2 mutant) in HeLa cells expressing GFP‐DDX6. The merged images show the GFP signal in green and HA signal in red. Percentages indicate the fraction of cells exhibiting a staining identical to that shown in the representative panel (> 100 cells were counted per experiment). Scale bar, 10 μm.
-
B–ETethering assay using the indicated luciferase reporters lacking tethering sites (5BoxB or 6xMS2 sites) in Dm and human cells expressing λN‐HA‐ or MS2‐tagged proteins as indicated. The assays with the corresponding reporters containing 5BoxB or 6xMS2 sites are shown in Figs 2A–D and 3B–G. Samples were analyzed as described in Fig 2.
-
FComplementation assay. Control S2 cells (treated with β‐Gal siRNA) or cells depleted of endogenous AGO1 were transfected with a mixture of three plasmids: one expressing the F‐Luc‐cg5281 reporter; another expressing miR‐12 primary transcript or the corresponding empty vector; and a third expressing Renilla luciferase (R‐Luc). Plasmids encoding HA‐AGO1 (wild type or the indicated pocket mutants) or HA‐MBP (as negative control) were included in the transfection mixtures as indicated. For each condition, firefly luciferase activities and mRNA levels were normalized to those of the Renilla luciferase transfection control and set at 100% in the absence of the miR‐12. Normalized firefly luciferase activities and mRNA levels are shown.
-
GA complementation assay was carried out using the F‐Luc‐nerfin‐1 reporter and miR‐9b or miR‐279. F‐Luc activity was analyzed as described in (F). Plasmids expressing miR‐9b or miR‐279 primary transcript or the corresponding empty vector were included in the transfection mixtures as indicated.
-
HWestern blot showing AGO1 knockdown efficiency. Dilutions of control cell lysates were loaded in lanes 1–4 to estimate the efficacy of the depletion. Tubulin served as a loading control.
-
IWestern blots showing the efficiency of the AGO1 knockdown and the expression levels of endogenous and recombinant AGO1. α‐tubulin served as a loading control.
-
J, KWestern blots showing the expression of the tethered proteins in the experiments shown in Fig 2M and N, respectively. Tubulin served as a loading control. The asterisk in (K) indicates a crossreactivity of the anti‐HA antibody with an endogenous human protein.
The silencing activity of AGOs requires the integrity of the W‐binding pockets
The availability of AGO mutants that do not interact with GW182 proteins enabled us to reinvestigate the question of whether AGOs exhibit silencing activity independently of GW182 using an approach orthogonal to knockdowns, which cannot be unambiguously interpreted (Fukaya & Tomari, 2012). We examined the silencing activity of the AGO mutants using previously described λN‐ or MS2‐based tethering assays (Pillai et al, 2004; Rehwinkel et al, 2005). Tethered Dm AGO1 and Hs AGO2 promoted degradation of polyadenylated mRNA reporters, as shown previously (Fig 2A–D; Pillai et al, 2004; Rehwinkel et al, 2005; Eulalio et al, 2008; Chen et al, 2009; Piao et al, 2010). Mutations in P2 were sufficient to abolish the activity of the Dm and Hs AGOs in tethering assays (Fig 2A–D). The activity of Dm AGO1 was also abolished by mutations in P1 (Fig 2A and B).
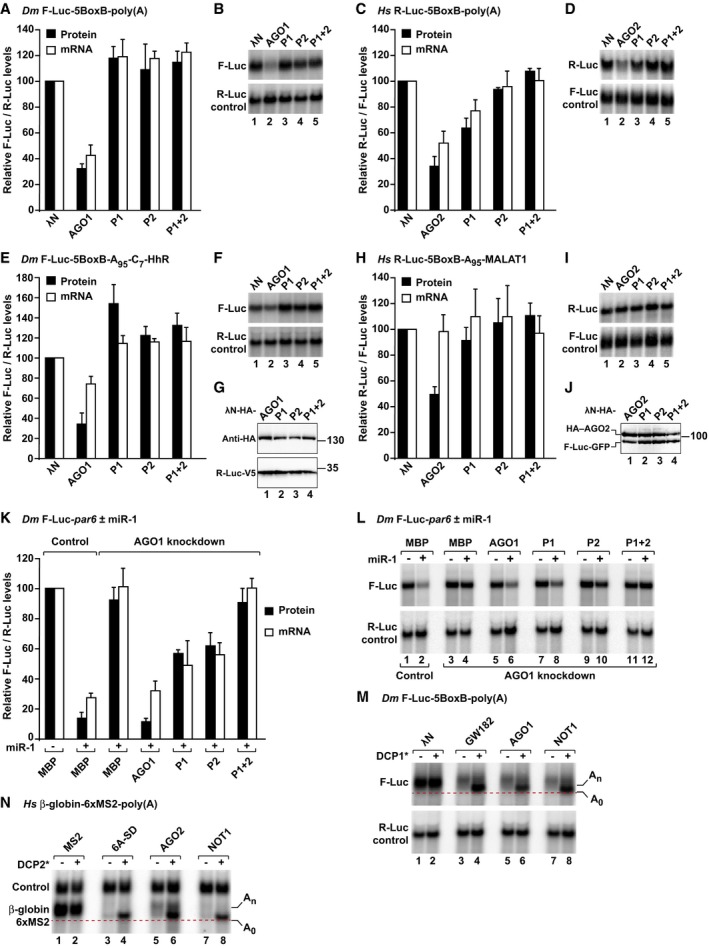
Figure 2. The silencing activity of AGOs depends on the integrity of the W‐binding pockets.
-
ATethering assay using the F‐Luc‐5BoxB‐poly(A) reporter and λN‐HA‐AGO1 (wild type or pocket mutants) in S2 cells. A plasmid expressing R‐Luc served as a transfection control. (A) F‐Luc activity and mRNA levels were normalized to those of the R‐Luc transfection control and set to 100% in cells expressing the λN‐HA peptide.
-
BNorthern blot of representative RNA samples corresponding to the experiment shown in (A).
-
C, DTethering assay using the R‐Luc‐5BoxB‐poly(A) reporter and λN‐HA‐AGO2 (wild type or mutants) in human HEK293T cells. A plasmid expressing F‐Luc served as a transfection control. R‐Luc activity and mRNA levels were normalized to those of the F‐Luc transfection control and analyzed as described in panels (A, B).
-
E, FTethering assay using the F‐Luc‐5BoxB‐A95‐C7‐HhR reporter and λN‐HA‐AGO1 (wild type or mutants) in S2 cells. Samples were analyzed as described in panels (A, B).
-
GWestern blot analysis showing the equivalent expression of the λN‐HA‐AGO1 proteins used in the tethering assays shown in panels (A, B, E, F).
-
H, ITethering assay using the R‐Luc‐5BoxB‐A95‐MALAT1 reporter and λN‐HA‐AGO2 (wild type or mutants) in HEK293T cells.
-
JWestern blot analysis showing the equivalent expression of the λN‐HA‐AGO2 proteins used in the tethering assays shown in (C, D, H, I).
-
K, LComplementation assays using F‐Luc‐par6 reporter in S2 cells depleted of endogenous AGO1. Plasmids encoding HA‐AGO1 (wild type or mutants) or HA–MBP (as negative control) and miR‐1 primary transcript (or empty vector) were included in the transfection mixtures as indicated. For each condition, firefly luciferase activities and mRNA levels were normalized to those of the Renilla luciferase transfection control and set to 100% in the absence of miR‐1 (100% values are only shown for control cells). (K) Normalized firefly luciferase activities and mRNA levels. (L) Northern blot of representative RNA samples.
-
MA tethering assay using the F‐Luc‐5BoxB‐poly(A) reporter was performed in S2 cells. The transfection mixtures included plasmids expressing GFP or GFP‐DCP1* as indicated. The panel shows a Northern blot of representative RNA samples. The positions of the polyadenylated (An) and deadenylated (A0, dashed red line) mRNA reporter are indicated on the right.
-
NA tethering assay using the β‐globin‐6xMS2 reporter and MS2‐tagged proteins was performed in HEK293T cells. The transfection mixtures contained plasmids expressing wild‐type DCP2 (−) or the catalytic DCP2* mutant (+). The panel shows a Northern blot of representative RNA samples analyzed as described in (M).
Because the reporters were degraded, it was possible that the recruitment of deadenylases by GW182/TNRC6 masked any additional repressive activity that AGOs might have independently of these proteins. Therefore, we next tested the activity of the AGO mutants using mRNA reporters that are refractory to deadenylation and subsequent decay. In Dm cells, we used an mRNA reporter with a 3′‐end generated by a self‐cleaving hammerhead ribozyme (HhR). Immediately upstream of the ribozyme cleavage site, the reporter contains an internal poly(A) stretch of 95 residues followed by a poly(C) stretch of 7 residues, which blocks deadenylation (F‐Luc‐5BoxB‐A95‐C7‐HhR; Zekri et al, 2013). Tethered Dm AGO1 repressed this reporter predominantly at the translational level (Fig 2E and F). The mutations in either P1 or P2 abolished Dm AGO1 repressive activity (Fig 2E and F). The protein mutants were expressed at levels comparable to wild type (Fig 2G).
In human HEK293T cells, we used an R‐Luc‐5BoxB reporter containing an internal poly(A) stretch of 95 residues followed by the 3′‐end of the MALAT1 noncoding RNA, which is processed by RNase P, and thus, it is not polyadenylated (Wilusz et al, 2012). Hs AGO2 repressed the expression of this reporter (Fig 2H–J). The repression was relieved by mutations in either of the two W‐binding pockets (Fig 2H–J). The Dm or Hs AGO proteins did not repress the corresponding reporters that lacked the BoxB or MS2‐binding sites (Fig EV1B–E). Taken together, our results indicate that the activity of tethered AGO proteins depends on the integrity of the W‐binding pockets.
The W‐binding pockets are required for Dm AGO1 to silence miRNA targets
Because tethering assays bypass some steps in silencing, we next tested the activity of the AGO mutants in complementation assays in Dm cells, wherein miRNAs predominantly associated with AGO1. For these assays, we used previously characterized firefly luciferase reporters containing 3′‐UTRs of natural miRNA targets (e.g. par‐6 and cg5281, silenced by miR‐1 and miR‐12, respectively; and nerfin‐1 silenced by miR‐9b and miR‐279; Eulalio et al, 2007). The depletion of endogenous Dm AGO1 suppressed the silencing of all reporters, as expected, and both F‐Luc expression and mRNA levels were restored (Figs 2K and L and EV1F and G). Western blot analysis indicated that AGO1 levels were reduced below 25% of their control levels in depleted cells (Fig EV1H and I).
In depleted cells, a siRNA‐resistant version of Dm AGO1 fully restored silencing of all reporters (Figs 2K and L and EV1F and G). By contrast, the double P1+P2 mutant did not restore silencing. The single‐pocket mutants partially rescued silencing to different extents depending on the reporters (Figs 2K and L and EV1F and G), suggesting that although these mutants do not bind to GW182 in coimmunoprecipitation assays, they may still interact transiently in vivo. The AGO1 proteins were expressed at similar levels (Fig EV1I). These levels were comparable to the levels of endogenous AGO1 in control cells (Fig EV1I, lane 3 versus 1). We concluded that the integrity of the two W‐binding pockets is also required for Dm AGO1 to silence miRNA targets in complementation assays.
AGOs, GW182/TNRC6s, and CCR4–NOT share a common mechanism to degrade mRNAs
It is well established that GW182/TNRC6 proteins and AGOs induce mRNA degradation through the 5′‐to‐3′ decay pathway (Rehwinkel et al, 2005; Behm‐Ansmant et al, 2006; Eulalio et al, 2007; Chen et al, 2009; Piao et al, 2010). In this pathway, deadenylation precedes decapping. Consequently, mRNAs degraded through this pathway accumulate in a deadenylated form in cells in which decapping is inhibited (Eulalio et al, 2007).
To investigate whether GW182/TNRC6, AGOs, and NOT1 all elicit first deadenylation and then decapping, we sought to inhibit decapping. To this end, we overexpressed a DCP1 mutant that inhibits decapping in a dominant negative manner (DCP1* mutant; Chang et al, 2014; Appendix Table S1). Tethered Dm GW182, AGO1, and NOT1 degraded the F‐Luc‐5BoxB‐poly(A) reporter (Fig 2M, lanes 3, 5, and 7 versus 1). Overexpression of the DCP1 mutant prevented this degradation in S2 cells (Fig 2M, lanes 4, 6, and 8 versus 2), without affecting the expression of the tethered proteins (Fig EV1J). The reporter accumulated as a fast migrating form, which corresponds to the deadenylated decay intermediate (Eulalio et al, 2007). Thus, as observed previously for GW182 and AGOs, tethered NOT1 caused deadenylation‐dependent decapping.
Likewise, in HEK293T cells, MS2‐tagged TNRC6A silencing domain (SD), AGO2, and NOT1 degraded a β‐globin reporter containing 6 binding sites for the MS2 protein in the 3′‐UTR (Fig 2N, lanes 3, 5, and 7 versus 1). This degradation was prevented in cells expressing a DCP2 catalytically inactive mutant (DCP2*), and the reporter accumulated in the deadenylated form (Fig 2N, lanes 4, 6, and 8). The expression of the tethered proteins was not affected (Fig EV1K). Our results indicate that miRNAs, AGOs, GW182/TNRC6 proteins, and the CCR4–NOT complex share a common mechanism to degrade target mRNAs.
AGOs, GW182/TNRC6s, and CCR4–NOT silence mRNAs translated via a scanning‐independent mechanism
Several studies reported that miRNAs silence gene expression by targeting eIF4A and interfering with ribosome scanning (Meijer et al, 2013; Ricci et al, 2013; Fukao et al, 2014; Fukaya et al, 2014). However, there were considerable disagreements between these studies. Ricci et al found that translation driven by the EMCV IRES is relatively refractory to miRNA silencing, whereas Meijer et al found that it is efficiently silenced by miRNAs. Furthermore, the results of experiments in which miRNA‐mediated translational repression is measured in the presence of drugs that target eIF4A and inhibit scanning (e.g. silvestrol or hippuristanol) are difficult to interpret because the transfection control is also inhibited. Therefore, to test whether AGO, GW182/TNRC6, and CCR4–NOT all require ribosome scanning to repress translation, we opted for an orthogonal approach and generated reporters translated via a scanning‐independent mechanism.
The reporter for expression in Dm cells was derived from the F‐Luc‐5BoxB‐A95‐C7‐HhR reporter but contained a 5′‐UTR of only eight nucleotides (8nt‐F‐Luc‐5BoxB‐A95‐C7‐HhR (Fig 3A, Appendix Tables S2 and S3)). The transcription start site used in S2 cells was confirmed by 5′ RACE (Fig EV2A). Notably, the translation efficiency of this reporter (F‐Luc activity normalized to mRNA levels) was reduced only 1.4‐fold relative to the parental reporter containing a 109‐nt 5′‐UTR (Fig EV2B–E). Given that F‐Luc proteins lacking the N‐terminal 3–10 amino acids are inactive (i.e. when translation starts at the next in‐frame methionine, Met31; Fig EV2F–H; Sung & Kang, 1998), and considering that the translation efficiencies of the reporters with a short and a long 5′‐UTR are comparable (Fig EV2B–E), it is reasonable to assume that a significant fraction of ribosomes initiate translation at the first AUG (or minimally at codon 3) on the mRNA with a 8‐nt 5′‐UTR. Consequently, translation of the 8nt‐F‐Luc‐5BoxB‐A95C7‐HhR occurs without scanning. Accordingly, when the F‐Luc ORF was replaced by HA–GST, the HA‐tagged protein could be detected by Western blotting using anti‐HA antibodies further validating the conclusion that a substantial fraction of ribosomes start translation at the first AUG (Fig EV2I).
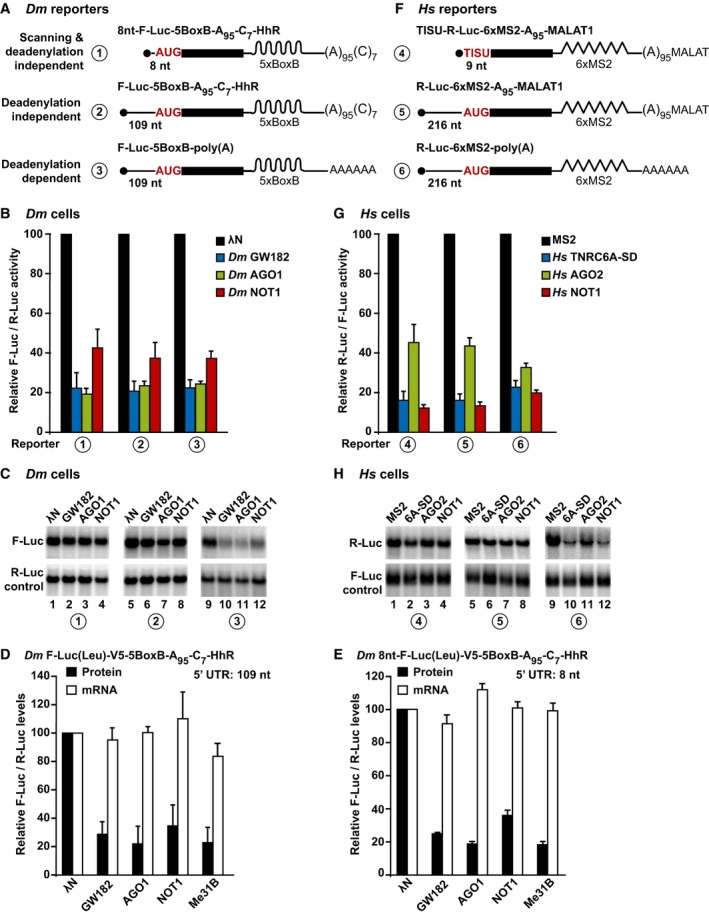
Figure 3. AGO, GW182/TNRC6, and NOT1 silence mRNA reporters translated via a scanning‐independent mechanism.
-
ASchematic representation of the Dm reporters used in panels (B, C).
-
B, CTethering assay using the F‐Luc‐5BoxB reporters shown in panel (A) and λN‐HA‐tagged GW182, AGO1, and NOT1 in S2 cells. F‐Luc activity and mRNA levels were normalized to those of the R‐Luc transfection control and analyzed as described in Fig 2A and B. Quantification of the corresponding Northern blots is shown in Fig EV2J.
-
D, ETethering assay with the indicated reporters containing a F‐Luc(Leu)‐V5 fusion in which all in‐frame methionines were replaced with leucine. The corresponding Western and Northern blots are shown in Fig EV2K–N.
-
FSchematic representation of the reporters used in panels (G, H).
-
G, HTethering assay using the R‐Luc‐6xMS2 reporters shown in panel (F) and HA‐MS2‐tagged TNRC6A‐SD, AGO2, and NOT1 in HEK293T cells. R‐Luc activity and mRNA levels were normalized to those of the F‐Luc transfection control and analyzed as described in Fig 2C and D. The corresponding quantification of mRNA levels is shown in Fig EV3F.
Figure EV2. Translation efficiencies of the reporters used in Fig 3A and B.
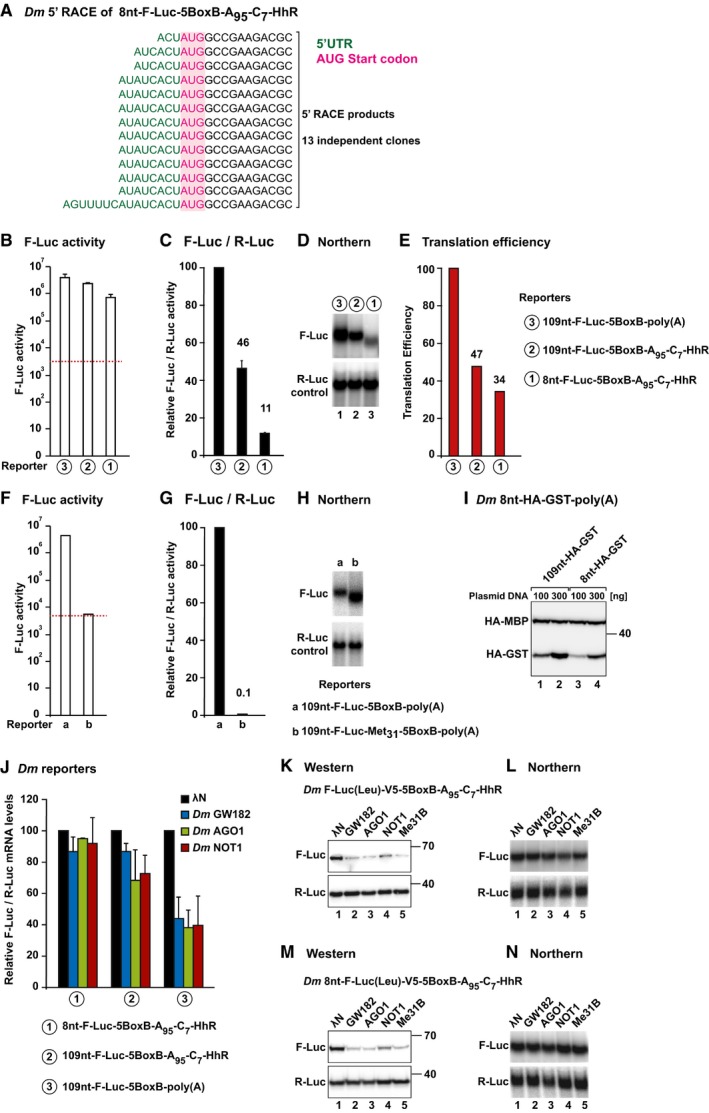
-
A5′ RACE sequences obtained in S2 cells expressing the 8nt‐F‐Luc‐5BoxB‐A95C7‐Hhr reporter after cloning the RACE products.
-
B–ES2 cells were transfected with the F‐Luc‐5BoxB reporters shown in Fig 3A. (B) Absolute values of luciferase activity (not normalized). Background levels are indicated by a dashed red line. (C) Luciferase activity normalized to that of the R‐Luc transfection control and set to 100 for the polyadenylated reporter containing a 109‐nt 5′‐UTR. (D) Northern blot of representative RNA samples. (E) The translation efficiency for each reporter was calculated by dividing the normalized values for F‐Luc activity shown in (C) by the normalized mRNA levels, shown in (D). Translation efficiencies were set to 100% for the polyadenylated reporter containing a 109‐nt 5′‐UTR. Note that although the translation efficiency of the reporters lacking a poly(A) tail is reduced, the absolute F‐Luc values are three orders of magnitude greater than background levels (B).
-
F–HA 109‐nt‐F‐Luc‐5BoxB‐poly(A) reporter containing a N‐terminally truncated F‐Luc ORF starting at Met31 is expressed, but no F‐Luc activity is detected above background (red dashed line). These results indicate that F‐Luc activity will not be detectable if ribosomes bypass the first AUG and start translation at the first in‐frame Met31.
-
IWestern blot analysis showing the expression of HA‐GST reporters containing 5′‐UTRs of 109 nt (lanes 1 and 2) and 8 nt (lanes 3 and 4). Transfection mixtures contained either 100 or 300 ng of reporter plasmids. HA‐MBP served as a transfection control.
-
JNormalized luciferase mRNA levels corresponding to the experiment shown in Fig 3B.
-
K–NRepresentative Western and Northern blots corresponding to the experiments shown in Fig 3D and E.
Remarkably, the reporter containing a 8‐nt 5′‐UTR was repressed by tethered AGO1, GW182, and NOT1 (Figs 3B and C and EV2J). The repression was comparable to that observed for the parental reporter containing a 109‐nt long 5′‐UTR as well as for the reporter also containing a poly(A) tail, which was consequently degraded (Figs 3B and C and EV2J).
To further confirm that the reporter was translated via a scanning‐independent mechanism and to prevent leaky initiation, we replaced all in‐frame methionines (except the initiating Met) in the ORF of the F‐Luc‐5BoxB‐A95C7‐HhR reporter with leucine and inserted a V5 tag C‐terminally to detect the protein product (F‐Luc(Leu)‐V5) because the substitutions abrogated F‐Luc activity. Dm AGO1, GW182, and NOT1 (as well as Me31B, see below) repressed the expression of the F‐Luc(Leu)‐V5 protein independently of whether it was translated from a mRNA reporter containing a 109‐nt or 8‐nt 5′‐UTR. mRNA levels were not affected (Figs 3D and E and EV2K–N).
To analyze the requirement for 43S scanning in human cells, we replaced the 5′‐UTR of the R‐Luc‐6xMS2‐A95‐MALAT1 reporter with a TISU motif (translation initiator of short 5′‐UTR, Fig 3F, Appendix Tables S2 and S3), which directs efficient cap‐dependent translation initiation of very short 5′‐UTRs (9‐nt) via a scanning‐ and eIF4A‐independent mechanism (Elfakess et al, 2011; Sinvani et al, 2015). The 5′‐UTR of the TISU reporter was confirmed by 5′ RACE (Fig EV3A and Appendix Fig S2).
Figure EV3. AGO, GW182/TNRC6, and NOT1 silence mRNA reporters translated via a scanning‐independent mechanism.
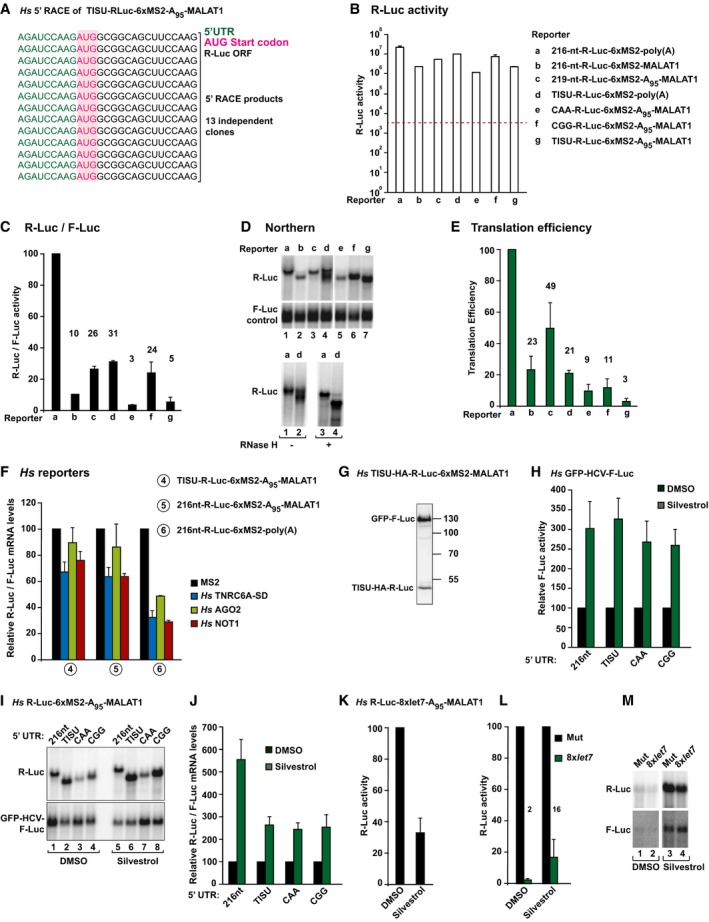
-
A5′ RACE sequences obtained in HEK293 cells expressing the TISU‐R‐Luc‐6xMS2‐A95‐MALAT reporter after cloning the RACE products.
-
B–EThe translation efficiency of the MS2 reporters shown in Figs 3F and 4 was analyzed as described in Fig EV2B–E. (B) Absolute values of Renilla luciferase activity (not normalized). Background levels are indicated by a dashed red line. (C) R‐Luc activity normalized to that of the F‐Luc transfection control and set to 100% for the polyadenylated reporter containing a 216‐nt 5′‐UTR. (D) Upper panel: Northern blot of representative RNA samples. Lower panel: RNase H assay for reporters a and d. (E) The translation efficiency for each reporter was calculated by dividing the normalized values for F‐Luc activity shown in (C) by the normalized mRNA levels, shown in (D). Translation efficiencies were set to 100% for the polyadenylated reporter containing a 109‐nt 5′‐UTR. Note that although the translation efficiency of the reporters lacking a poly(A) tail is reduced, the absolute F‐Luc values are three orders of magnitude greater than background levels (B). Note that although the translation efficiency of the TISU reporters is very low, the absolute R‐Luc values are 2–3 orders of magnitude greater than background levels (B). In all panels, bars represent mean values, which are indicated above the bars.
-
FNormalized luciferase mRNA levels corresponding to the experiment shown in Fig 3G.
-
GWestern blot analysis showing the expression of TISU‐HA‐R‐Luc‐6xMS2‐MALAT1. F‐Luc‐GFP served as a transfection control.
-
HLuciferase activity for a bicistronic GFP‐IRESHCV‐F‐Luc reporter, which served as a transfection control in the experiment shown in Fig 4A. Note that when cap‐dependent translation is inhibited by silvestrol treatment, IRES‐mediated translation increases, as reported before (Cope et al, 2014), which precludes the use of IRES reporters as normalization controls. Therefore, R‐Luc and F‐Luc activities measured in silvestrol‐treated cells were normalized to the respective values obtained in control cells treated with DMSO.
-
I, JNorthern blot analysis of representative RNA samples corresponding to the experiment shown in Fig 4A. R‐Luc mRNA levels were normalized to those of the F‐Luc transfection control and set to 100% in control cells treated with DMSO. Error bars represent standard deviations from four independent experiments.
-
K–MHeLa cells were transfected with plasmids expressing R‐Luc‐let‐7‐A95‐MALAT1 reporter or the corresponding reporter carrying mutations in the let‐7‐binding sites. HeLa cells were treated with silvestrol (300 nM) or DMSO for 16 h. Panel (K) shows the inhibitory effect of silvestrol on the translation of R‐Luc reporter. Renilla luciferase activities were measured and set to 100 in cells transfected with the reporter carrying mutations in the let‐7‐binding sites (L). A representative Northern blot is shown in panel (M). Note the effect of silvestrol on mRNA levels. Although the panels are separated, the source data for Fig EV3M clearly shows that the samples were analyzed on the same Northern blot.
The translation efficiency of the TISU reporter was 16‐fold lower relative to the parental reporter containing a 216‐nt long 5′‐UTR and the MALAT1 3′‐end (Fig EV3B–E, compare reporters c and g). Nevertheless, TNRC6A‐SD, AGO2, and NOT1 repressed the expression of these two reporters and that of a corresponding polyadenylated reporter to comparable extents. The polyadenylated reporter was degraded, whereas the reporters containing the MALAT1 3′‐end were resistant to degradation (Figs 3G and H and EV3F). Notably, insertion of an HA‐tag immediately downstream of the first AUG in the TISU reporter enabled detection of R‐Luc by Western blotting using anti‐HA antibodies (Fig EV3G), confirming that TISU directs translation initiation at the first AUG and therefore in the absence of scanning.
We further confirmed that translation of the TISU reporter was independent of scanning, and thus of eIF4A activity, using silvestrol, a compound that stimulates eIF4A1/2 RNA‐binding activity, reducing the pool of the protein available for translation (Bordeleau et al, 2008; Cencic et al, 2009). Silvestrol inhibited cap‐dependent translation of Renilla luciferase, but the translation driven by the TISU sequence was only marginally inhibited (Fig 4A) as expected (Elfakess et al, 2011). In contrast, silvestrol stimulated HCV IRES‐driven translation of an F‐Luc reporter, which was included as a transfection control (Fig EV3H). Notably, the inhibitory effect of silvestrol in translation was observed despite a two‐ to fivefold increase of reporter mRNA levels in HEK293 cells (Fig EV3I and J).
Figure 4. eIF4A2 binding to mRNAs is not influenced by silencing.
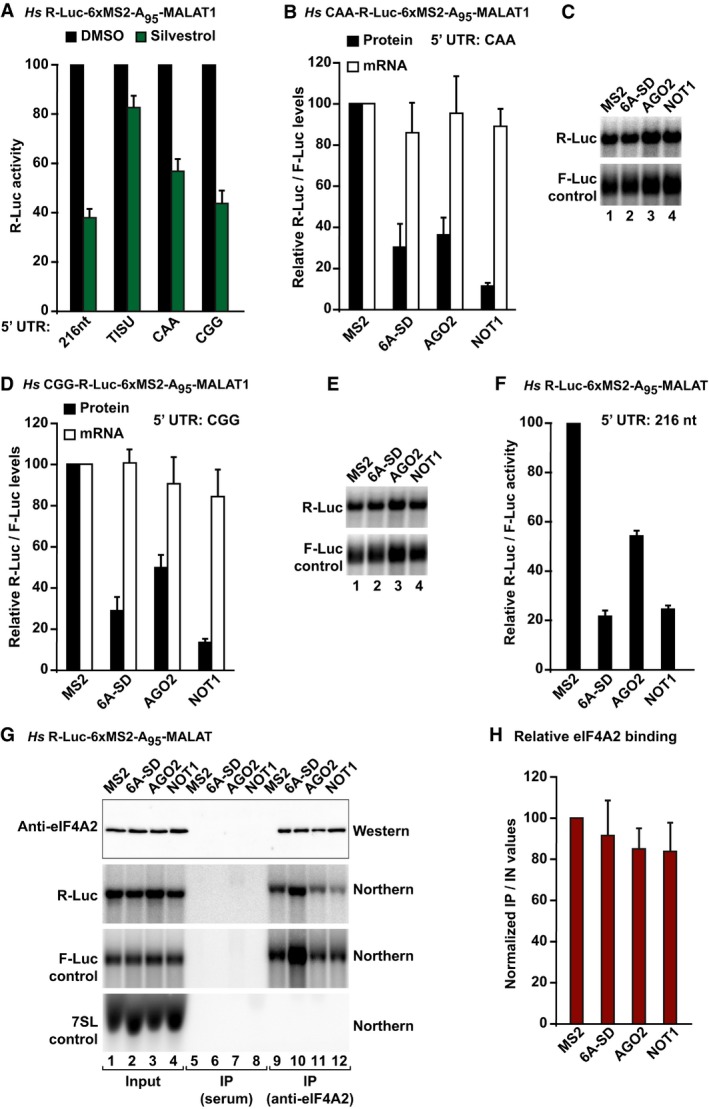
-
AHEK293T cells were transfected with the indicated R‐Luc‐6xMS2‐A95‐MALAT1 reporters containing the indicated 5′‐UTRs. An F‐Luc reporter whose translation was dependent on the hepatitis C virus (HCV) IRES was included as a control. One day after transfection, cells were treated with DMSO or silvestrol. R‐Luc activity in silvestrol‐treated cells was normalized to that measured in control cells. The corresponding values for the HCV IRES‐F‐Luc reporter are shown in Fig EV3H. The panel shows mean values ± standard deviations from three independent experiments.
-
B–ETethering assays using the indicated R‐Luc‐MS2‐A95‐MALAT1 reporters and MS2‐HA‐tagged TNRC6A‐SD, AGO2, and NOT1 in HEK293T cells. R‐Luc activity and mRNA levels were normalized to those of the F‐Luc transfection control and set to 100% in cells expressing MS2–HA. Panels (B, D) show mean values ± standard deviations from three independent experiments. Panels (C, E) show Northern blots of representative RNA samples.
-
F–HLysates from HEK293T cells expressing R‐Luc‐MS2‐A95‐MALAT1 reporter and MS2‐HA‐tagged proteins were immunoprecipitated using anti‐eIF4A2 antibody. The RNAs coimmunoprecipitating with eIF4A2 were analyzed by Northern blot. 7SL RNA served as a loading control. R‐Luc mRNA levels were normalized to those of the F‐Luc control. The normalized values in the IP were divided by those in the input and set to 100 for cells expressing MS2 peptide. (F) Normalized R‐Luc activities. The panel shows mean values ± standard deviations from five independent experiments. (G) Representative Western and Northern blots of input and IP fractions. For the Western blot, 2% of the input and 6% of the IP fraction were analyzed. For the Northern blots, 2% of the input and 40% of the IP were analyzed. (H) Efficacy of the immunoprecipitation. Error bars represent standard errors from five independent experiments.
Source data are available online for this figure.
Silencing does not correlate with 5′‐UTR secondary structure
To further investigate the dependence on eIF4A for silencing, we generated reporters containing 5′‐UTRs with different degrees of secondary structure, which were expected to exhibit different eIF4A requirements (Appendix Tables S2 and S3). In particular, we generated a reporter containing a 5′‐UTR consisting of 18 CAA repeats, which is unlikely to adopt secondary structures, and its translation is thought to be independent of eIF4A (Meijer et al, 2013). This 5′‐UTR was also shown to confer resistance to silencing (Meijer et al, 2013). Unexpectedly, translation of this reporter was partially inhibited by silvestrol (Figs 4A and EV3H). TNRC6A‐SD, AGO2, and NOT1 repressed the CAA reporter without causing mRNA degradation due to the presence of the MALAT1 3′‐end (Fig 4B and C).
We also tested a 5′‐UTR that contains four 12‐nt guanine quartet (CGG)4 motifs in tandem and is predicted to form G‐quadruplex structures and thus requires eIF4A for translation (Cencic et al, 2009; Wolfe et al, 2014; Appendix Tables S2 and S3). Accordingly, the translation of this reporter was sensitive to silvestrol treatment (Figs 4A and EV3H). TNRC6A‐SD, AGO2, and NOT1 repressed the CGG reporter to an extent comparable to that of the CAA reporter (Fig 4D and E versus Fig 4B and C), despite a eightfold difference in translation efficiency (Fig EV3B–E).
In sum, AGO, GW182/TNRC6s, and NOT1 silence mRNA targets independently of whether their translation requires ribosome scanning and independently of the 5′‐UTR secondary structure.
eIF4A2 binding to mRNAs does not correlate with silencing efficiency
eIF4A2 was reported to be either recruited to or dislodged from silenced miRNA targets (Meijer et al, 2013; Fukao et al, 2014; Fukaya et al, 2014). To resolve this discrepancy, we investigated eIF4A2 association with the R‐Luc‐6xMS2‐A95‐MALAT1 reporter containing a 5′‐UTR of 216 nt. The translation of this reporter is sensitive to silvestrol treatment (Fig 4A). The reporter was silenced to different extents by tethered AGO2, TNRC6A‐SD, or NOT1 (Fig 4F). Nevertheless, eIF4A2 associated with the reporter to comparable levels (Fig 4G and H). Moreover, the association was comparable to that observed for the unrepressed reporter in cells expressing the MS2 protein alone (Fig 4G and H and Appendix Fig S3). The possibility that the observed eIF4A2 binding is unspecific is unlikely, as eIF4A2 did not associate with 7SL RNA, and the reporters were not precipitated by an unrelated rabbit serum (Fig 4G). Thus, eIF4A2 in association with mRNAs is neither enhanced nor reduced by silencing.
Ribosome scanning is not required for mRNA degradation caused by tethered AGOs, GW182/TNRC6s, and NOT1
Because it has been reported that ribosome scanning is a prerequisite for miRNAs to degrade their targets (Meijer et al, 2013), we next analyzed whether GW182/TNRC6, AGO, and NOT1 degraded polyadenylated mRNA reporters that are translated via a scanning‐independent mechanism. In Dm cells, a polyadenylated reporter containing an 8‐nt 5′‐UTR was degraded by tethered GW182, AGO1, and NOT1 (Fig 5A and B). Similarly, the TISU‐R‐Luc‐6xMS2‐poly(A) reporter was also degraded by AGO2, TNRC6A‐SD, and NOT1 in HEK293T cells (Fig 5C and D). Thus, in the absence of scanning, deadenylation and subsequent mRNA decay still occur.
Figure 5. Ribosome scanning is not required for degradation of silenced mRNAs.
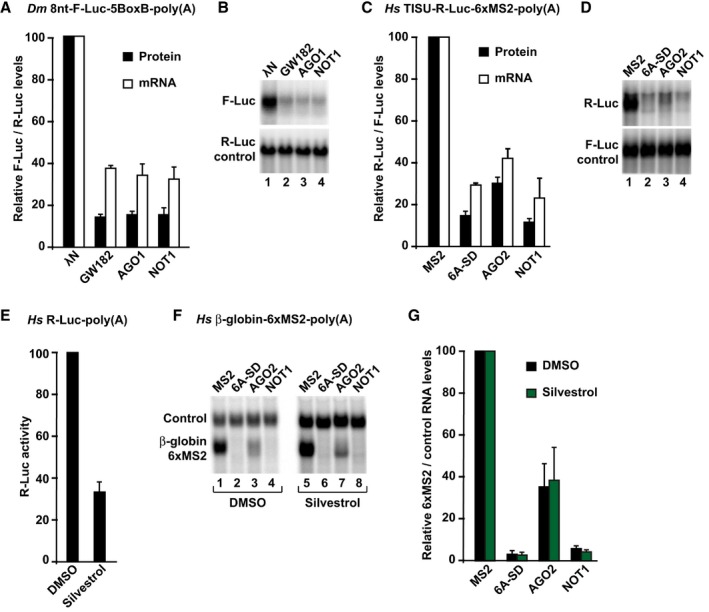
-
A, BTethering assay using the 8nt‐F‐Luc‐5BoxB‐poly(A) reporter in S2 cells. Firefly luciferase activities and mRNA levels were normalized to those of the Renilla luciferase and analyzed as described in Fig 2A and B.
-
C, DTethering assay using the TISU‐R‐Luc‐6xMS2‐poly(A) reporter in human HEK293T cells. Samples were analyzed as described in Fig 2C and D.
-
E–GTethering assay using the β‐globin‐6xMS2‐poly(A) reporter in human HEK293T cells treated with silvestrol or DMSO. Panel (E) shows the inhibitory effect of silvestrol on the translation of a cotransfected R‐Luc reporter. In panels (F, G), samples were analyzed as described in Fig 2C and D.
We also investigated whether a β‐globin reporter containing a 114‐nt 5′‐UTR could be degraded in the presence of silvestrol. This reporter was selected because it is very efficiently degraded by tethered TNRC6A‐SD, AGO2, and NOT1 in human cells. This degradation was not prevented by silvestrol (Fig 5E–G), although translation of a cotransfected R‐Luc reporter was inhibited 2.5‐fold (Fig 5E) and the mRNA reporters were stabilized by the silvestrol treatment in the absence of the tethered proteins (Fig 5F, lane 5 versus 1). Taken together, our results indicate that mRNA degradation induced by tethered AGO, GW182, and NOT1 does not require prior ribosomal scanning.
miRNAs silence reporters translated via a scanning‐independent mechanism
Next, we investigated whether miRNAs (as opposed to tethered silencing factors) also repress reporters translated via a scanning‐independent mechanism. To this end, we generated F‐Luc‐nerfin‐1 and F‐Luc‐par6 reporters containing an 8‐nt 5′‐UTR. These reporters were efficiently silenced by the corresponding miRNAs as the parental reporters, which contained 109‐nt long 5′‐UTRs (Fig 6A–D). Furthermore, the nerfin‐1 reporter was repressed predominantly at the translational level whereas the par‐6 reporter was predominantly degraded independently of the length of the 5′‐UTR (Fig 6A–D).
Figure 6. Ribosome scanning is not required for miRNA target degradation.
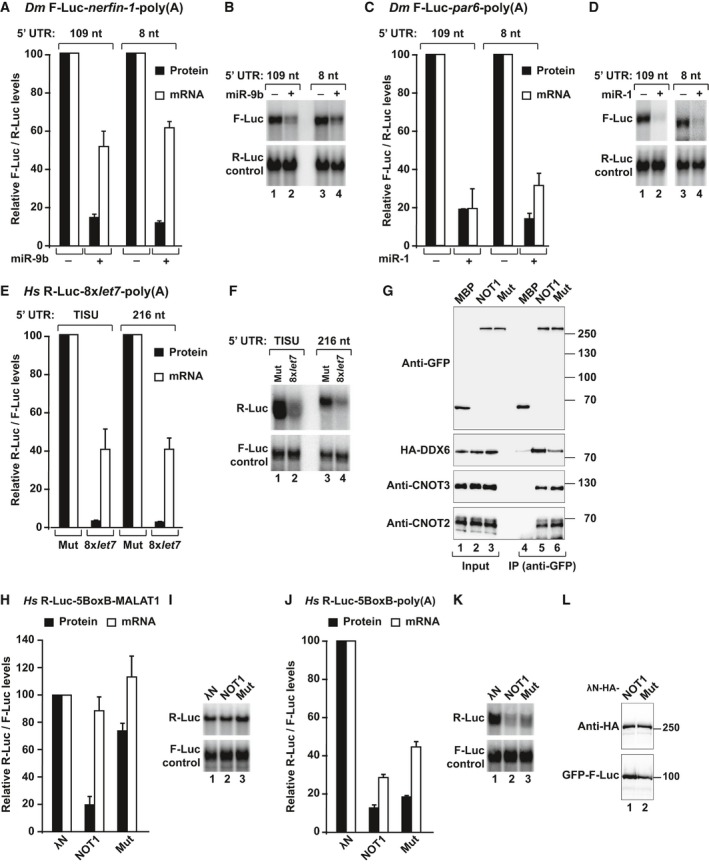
-
A–DS2 cells were transfected with polyadenylated F‐Luc‐nerfin‐1 or F‐Luc‐par6 reporters containing an 8‐nt or 109‐nt 5′‐UTR and the corresponding miRNAs. A plasmid expressing Renilla luciferase (R‐Luc) served as transfection control. Panels (A, C) show normalized firefly luciferase activities and mRNA levels. Northern blot analyses of representative RNA samples are shown in (B, D).
-
E, FHeLa cells were transfected with plasmids expressing the indicated R‐Luc‐let‐7 reporters or the corresponding reporters carrying mutations in the let‐7‐binding sites and a plasmid expressing F‐Luc as a transfection control. Renilla luciferase activities were normalized to those of the F‐Luc and set to 100% in cells transfected with the reporter lacking the let‐7‐binding sites and analyzed as described in (A–D).
-
GInteraction of GFP–NOT1 (wild type or a mutant that does not bind DDX6, Mut) with HA‐DDX6 and endogenous CNOT2 and CNOT3 in HEK293T cells.
-
H–KTethering assay using the R‐Luc‐5BoxB reporters containing the MALAT1 3′‐UTR (H, I) or a poly(A) tail (J, K) and λN‐HA‐NOT1 (wild type or Mut). R‐Luc activity and mRNA levels were normalized to those of the F‐Luc transfection control and analyzed as described in Fig 2C and D.
-
LExpression levels of wild type and mutant NOT1.
For expression in human cells, we generated a polyadenylated R‐Luc reporter containing the TISU motif or a 216‐nt 5′‐UTR and eight let‐7 miRNA‐binding sites in the 3′‐UTR. Both of these reporters were efficiently silenced and partially degraded by endogenous let‐7 independently of the length of the 5′‐UTR in HeLa cells (Fig 6E and F). We conclude that miRNAs also silence mRNA reporters translated via scanning‐independent mechanisms in both human and Dm cells.
Finally, we also tested the silencing of a let‐7 reporter containing the MALAT1 3′ end (R‐Luc‐8xlet‐7‐A95‐MALAT reporter) in the presence of silvestrol in HeLa cells. The silvestrol treatment partially suppressed silencing, as observed for hippuristanol treatment in previous studies. However, we observed a more than 10‐fold increase in mRNA levels both for the R‐Luc reporter and the F‐Luc control in HeLa cells treated with silvestrol (Fig EV3K–M). These results indicate that experiments in which miRNA‐mediated translational repression is measured in the presence of drugs that inhibit general translation should be interpreted with caution due to indirect effects on mRNA stability and the lack of appropriate transfection controls that are not affected by the treatment.
The CCR4–NOT complex requires DDX6 to repress translation
We and others have shown that DDX6 acts downstream of the CCR4–NOT complex to mediate translational repression and stimulate decapping in human cells (Chen et al, 2014; Mathys et al, 2014; Rouya et al, 2014). To further confirm that DDX6 function lies downstream of CCR4–NOT, we used tethering assays to analyze the repressive activity of a NOT1 mutant that it is impaired in binding to DDX6 but binds to CNOT2 and CNOT3 (Fig 6G, NOT1 Mut; Appendix Table S1). The NOT1 mutant was impaired in repressing the translation of the R‐Luc‐5BoxB‐MALAT1 mRNA, which is not degraded (Fig 6H and I). These results indicate that full‐length NOT1 requires interaction with DDX6 to effectively repress translation of a reporter that is resistant to deadenylation. However, the NOT1 mutant was active when tethered to the corresponding polyadenylated reporter, and the reporter was degraded (Fig 6J and K). The NOT1 proteins were expressed at comparable levels (Fig 6L). Thus, the deadenylase subunits degrade mRNA poly(A) tails even when the interaction with DDX6 is impaired.
Additionally, DDX6 depletion suppressed NOT1‐mediated silencing of the R‐Luc‐5BoxB‐MALAT1 reporter in HeLa cells (Fig EV4A and B), supporting the conclusion that NOT1 requires DDX6 to repress translation. In contrast to the results obtained in HeLa cells, depletion of Me31B in S2 cells did not completely suppress the silencing activity of AGO1, GW182, and NOT1 tethered to a reporter that is not degraded (Fig EV4C and D). These results suggest the existence of alternative silencing mechanisms in these cells. Alternatively, the residual amounts of Me31B in depleted cells (< 10% of control) might still be sufficient for silencing (Fig EV4D).
Figure EV4. DDX6 represses translation initiation.
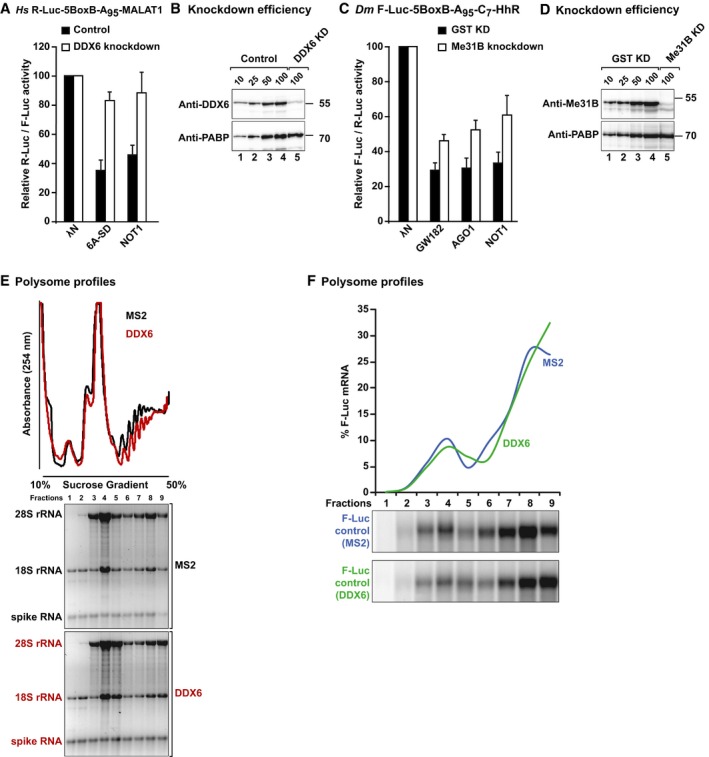
- Tethering assay using the R‐Luc‐5BoxB‐A95‐MALAT1 reporter and λN‐HA‐tagged TNRC6A‐SD and NOT1 in control HeLa cells or cells depleted of DDX6. Samples were analyzed as described in Fig 2. Error bars represent standard deviations from at least three independent experiments.
- Western blot showing DDX6 knockdown efficiency. Dilutions of control cell lysates were loaded in lanes 1–4 to estimate the efficacy of the depletion. PABP served as a loading control.
- Tethering assay using the F‐Luc‐5BoxB‐A95‐C7‐HhR reporter and λN‐HA‐tagged GW182, AGO1, and NOT1 in control S2 cells or cells depleted of Me31B. Samples were analyzed as described in Fig 2. Error bars represent standard deviations from at least three independent experiments.
- Western blot showing Me31B knockdown efficiency. Dilutions of control cell lysates were loaded in lanes 1–4 to estimate the efficacy of the depletion. PABP served as a loading control.
- Polysome profiles corresponding to Fig 7G. The absorbance at 254 nm of each fraction was quantitated and normalized to the total intensity across all fractions. The presence of 18S and 28S rRNAs in each fraction was analyzed on denaturing agarose gels. The spike RNA added to the fractions prior RNA isolation is indicated.
- The amount of F‐Luc control reporter corresponding to Fig 7G was quantified in each fraction of the gradient and normalized to the total amount across all fractions.
Source data are available online for this figure.
DDX6 represses translation initiation independently of 43S scanning
If DDX6/Me31B acts downstream of AGOs, GW182/TNRC6, and CCR4–NOT, it is expected that they also repress reporters translated via a scanning‐independent mechanism. Consistent with this expectation, tethered DDX6 repressed the expression of all reporters containing the MALAT1 3′‐end, including the TISU reporter, in HEK293T cells (Fig 7A–C). Thus, DDX6 represses translation in the absence of 43S scanning and deadenylation. DDX6 activity was only slightly impaired by the mutations that prevent binding to NOT1 (DDX6 Mut1), consistent with the notion that DDX6 acts downstream of the CCR4–NOT complex (Fig 7A–D). Similarly, Dm Me31B repressed the F‐Luc‐5BoxB‐A95‐C7‐HhR reporter independently of the length of the 5′‐UTR (Fig 7E), indicating that the ability to repress translation in the absence of scanning is conserved among DDX6 orthologs.
Figure 7. DDX6 represses translation independently of ribosome scanning.
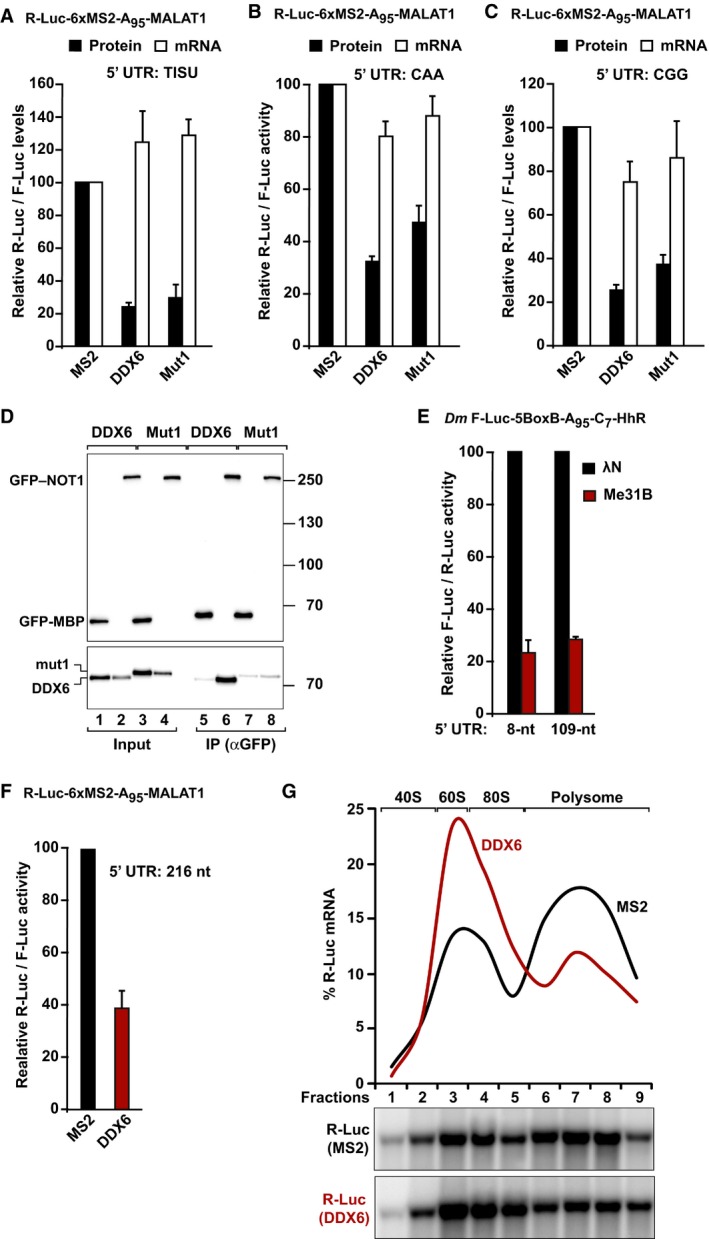
-
A–CTethering assay using the R‐Luc‐6xMS2‐A95‐MALAT1 reporters containing the indicated 5′‐UTRs and MS2‐HA‐tagged DDX6 or a DDX6 mutant (Mut1) that does not bind to NOT1 in HEK293T cells. R‐Luc activity and mRNA levels were normalized to those of the F‐Luc control and analyzed as described in Fig 2C and D.
-
DImmunoprecipitation assay showing the interaction of DDX6 or DDX6 Mut1 with GFP‐NOT1 in HEK293T cells. Inputs (1% for the HA‐tagged proteins and 3% for the GFP‐tagged proteins) and immunoprecipitates (20 and 15%, respectively) were analyzed by Western blotting using anti‐HA and anti‐GFP antibodies. Endogenous CNOT2 and CNOT3 (input 0.5% and IP 10%) were detected using specific antibodies (Appendix Table S4).
-
ETethering assay using the F‐Luc‐5BoxB‐A95‐C7‐Hhr reporters containing an 8‐nt or a 109‐nt 5′‐UTR and λN‐HA‐Me31B in S2 cells. F‐Luc activity and mRNA levels were analyzed as described in Fig 2A and B.
-
F, GTethering assay using the R‐Luc‐6xMS2‐A95‐MALAT1 reporter and MS2‐DDX6 in HEK293T. In panel (F), luciferase activity was analyzed as described in Fig 2C. The association of the R‐Luc reporter and the F‐Luc control with polysomes was analyzed by sedimentation through a sucrose gradient. Panel (G) shows the amount of the R‐Luc mRNA in each fraction normalized to the total amount across all fractions in cells expressing MS2 (black curve) or MS2‐DDX6 (red curve). Lower panels show representative Northern blots. The data corresponding to the F‐Luc control and the corresponding ribosomal RNA profiles are shown in Fig EV4E and F.
In yeast, Dhh1 (the DDX6 ortholog) was shown to repress translation at initiation (Coller & Parker, 2005) or at elongation (Sweet et al, 2012). To help to resolve this discrepancy, we analyzed the association of the R‐Luc‐6xMS2‐A95‐MALAT1 reporter with polysomes upon repression by DDX6. Sucrose gradient analyses indicated that tethered DDX6 changed the distribution of the reporter in the polysome profile toward lighter polysomes/nonpolysomal fractions without causing mRNA degradation, indicating that DDX6 represses translation initiation in HEK293T cells (Figs 7G and EV4E and F).
DDX6 requires interaction with NOT1 and the integrity of the FDF‐binding surface to silence miRNA reporters
To obtain further insight on how DDX6 represses translation, we compared the ability of DDX6 mutants to rescue silencing in HeLa cells depleted of DDX6. In particular, we used a DDX6 mutant (Mut1, Appendix Table S1) that does not interact with NOT1 but interacts with 4E‐T and decapping factors (namely PatL1, EDC3, and LSm14; Fig 8A–E). We also tested a mutant with substitutions on the FDF‐binding surface of DDX6 (Mut2, Appendix Table S1). These mutations prevent the interaction with 4E‐T and decapping factors, which all bind to the FDF‐binding surface of DDX6 in a mutually exclusive manner (Fig 8A–D; Tritschler et al, 2009; Ozgur et al, 2015). However, DDX6 Mut2 still binds to NOT1 (Fig 8E), indicating that the mutations do not alter the protein fold. We observed that only wild‐type DDX6 rescued silencing of the R‐Luc‐8xlet7‐poly(A), whereas DDX6 Mut1 or Mut2 were inactive (Fig 8F). Similarly, a DDX6 DEAD‐box mutant (E236Q) did not rescue silencing, as reported before (Mathys et al, 2014). DDX6 was depleted to < 25% of wild type (Fig 8G). Together, these results indicate that DDX6 requires interaction with NOT1 and additional factors that bind to the FDF‐binding surface to mediate silencing of miRNA targets.
Figure 8. DDX6 requires interaction with NOT1 and the integrity of the FDF‐binding surface to mediate silencing.
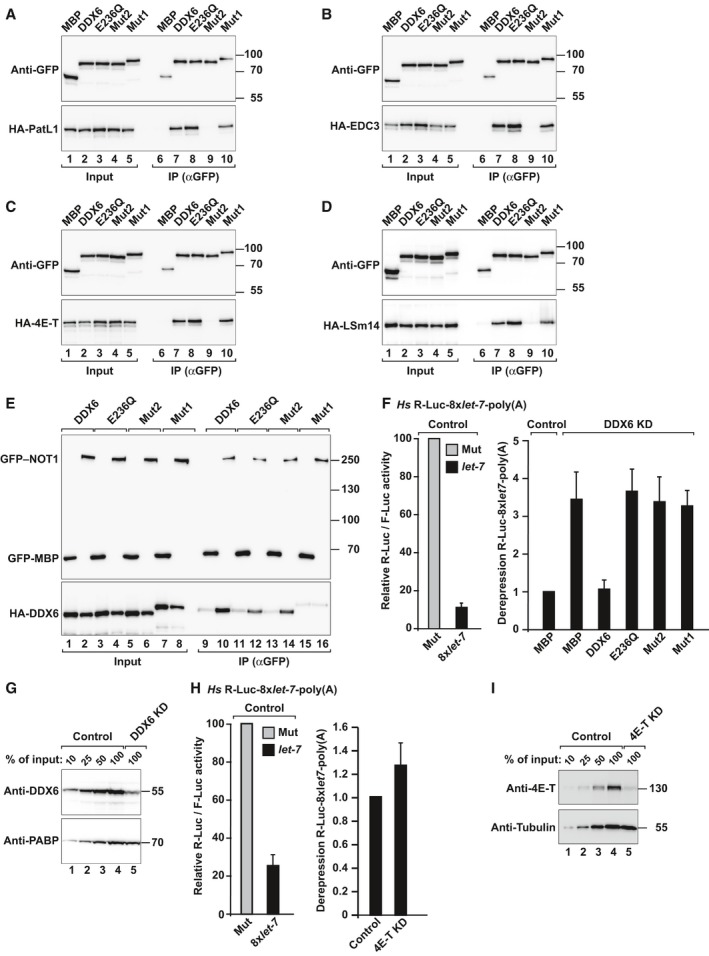
-
A–DImmunoprecipitation assay showing the interaction of GFP‐tagged DDX6 (wild type or the indicated mutants) with HA‐tagged decapping factors and 4E‐T in HEK293T cells. Inputs (1% for the HA‐tagged proteins and 3.5% for the GFP‐tagged proteins) and immunoprecipitates (20 and 7%, respectively) were analyzed by Western blotting using anti‐HA and anti‐GFP antibodies. GFP‐MBP served as a negative control.
-
EImmunoprecipitation assay showing the interaction of GFP‐tagged NOT1 with HA‐tagged DDX6 (wild type or the indicated mutants) in HEK293T cells. Samples were analyzed as described in (A–D).
-
FControl HeLa cells (transfected with a scrambled shRNA) or cells depleted of DDX6 were transfected with a mixture of three plasmids: the R‐Luc‐8xlet‐7‐poly(A) or the corresponding reporter carrying mutations in let‐7‐binding sites (R‐Luc‐Mut, Mut), a plasmid expressing F‐Luc as a transfection control, and a plasmid expressing shRNA‐resistant versions of GFP–DDX6 (wild type or the indicated mutants) or MBP. For each condition, Renilla luciferase activity was measured, normalized to that of the F‐Luc transfection control and set at 100% in cells expressing R‐Luc‐Mut. The left panel shows the normalized Renilla luciferase activities in control cells (i.e. cells treated with scrambled shRNA and expressing MBP). The right panel shows relative fold derepression for each condition for the R‐Luc‐8xlet‐7‐poly(A) after normalization. Mean values ± standard deviations from 4 independent experiments are shown.
-
GWestern blot showing the efficiency of the DDX6 knockdown. Dilutions of control cell lysates were loaded in lanes 1–4 to estimate the efficacy of the depletion. PABP served as a loading control.
-
HControl HeLa cells (transfected with a scrambled shRNA) or cells depleted of 4E‐T were transfected with a mixture of three plasmids as described in panel (F). The left panel shows normalized Renilla luciferase activities in control cells (i.e. cells treated with scrambled shRNA). The right panel shows the relative fold derepression for the R‐Luc‐8xlet‐7‐poly(A) (after normalization) in 4E‐T‐depleted cells relative to control cells. Mean values ± standard deviations from three independent experiments are shown.
-
IWestern blot showing the efficiency of the 4E‐T knockdown. Dilutions of control cell lysates were loaded in lanes 1–4 to estimate the efficacy of the depletion. Tubulin served as a loading control.
Source data are available online for this figure.
Previous studies indicated that depletion of decapping factors prevents miRNA target degradation but does not suppress miRNA‐mediated translational repression because the reporters accumulated in a deadenylated form, which is not translated efficiently (Eulalio et al, 2007). Therefore, we tested whether 4E‐T could mediate the repression. However, depletion of 4E‐T did not suppress silencing of this reporter (Fig 8H). 4E‐T was depleted to < 25% of wild type (Fig 8I). Thus, it is possible that other, not‐yet‐identified, factors interact with the FDF‐binding surface of DDX6 to mediate silencing or that the known binding partners act redundantly to promote silencing.
Discussion
AGOs require the W‐binding pockets to mediate silencing
Previous studies reported that Dm AGO1 can repress miRNA targets independently of GW182/TNRC6 proteins and of the CCR4–NOT complex (Fukaya & Tomari, 2012; Wu et al, 2013; Fukaya et al, 2014). Here, we show that amino acid substitutions in the two W‐binding pockets present on the surface of the PIWI domain of Dm AGO1 and Hs AGO2 (Schirle & MacRae, 2012) abolish the binding of AGOs to GW182/TNRC6 proteins without disrupting miRNA loading. These mutations also abolish the silencing activity of AGOs in both tethering and complementation assays. The most parsimonious explanation for our results is that AGOs require interaction with GW182/TNRC6 proteins to mediate silencing. Although we cannot rule out the possibility that AGOs interact with another, yet‐unidentified, protein partner using a mode of interaction similar to that used with GW182/TNRC6, we consider this possibility unlikely because AGOs, GW182/TNRC6, NOT1, and miRNAs degrade polyadenylated mRNA targets via the 5′‐to‐3′ decay pathway. Furthermore, all three proteins as well as miRNAs repress the translation of mRNA targets that are refractory to deadenylation. Finally, we show that all three proteins and miRNAs repress and/or degrade mRNA targets that do not require 43S ribosomal scanning for translation. These results point to a common mechanism used by these proteins and miRNAs to mediate silencing.
Ribosomal scanning is not a prerequisite for silencing
A previous study reported that mRNAs with highly structured 5′‐UTRs, which depend on eIF4A for translation, are susceptible to silencing whereas mRNAs with unstructured 5′‐UTRs are refractory to silencing in human cells (Meijer et al, 2013). By contrast, Ricci et al (2013) failed to detect a direct correlation between silencing and 5′‐UTR secondary structure in rabbit reticulocyte lysates. Furthermore, it has been reported that 5′‐UTR features such as length and secondary structure do not reliably correlate with the extent of miRNA‐mediated repression (Agarwal et al, 2015). We found no evidence for the requirement of structured 5′‐UTRs for silencing in either human or Dm cells. Our conclusions are based on the use of reporters that have very short 5′‐UTRs (in Dm cells) or contain the TISU motif (human cells). These reporters direct translation independently of scanning. We could also not confirm that a reporter containing 18 CAA repeats in the 5′‐UTR is resistant to silencing, as reported by Meijer et al (2013). Furthermore, the CAA reporter was sensitive to silvestrol treatment, indicating that it partially depends on eIF4A activity for translation as reported by Pestova and Kolupaeva (2002).
In summary, all reporters tested in our study were partially degraded when they contained a poly(A) tail or were repressed predominantly at the translational level when they contained 3′ termini that conferred resistance to deadenylation. Similar results were obtained for tethered human AGO2, TNRC6, NOT1, DDX6 and their Dm orthologs as well as for miRNAs in the absence of tethering, indicating that 43S scanning is not a prerequisite for translational repression, deadenylation, and decay of miRNA targets.
Silencing does not require eIF4A function during scanning
Previous studies made use of eIF4A inhibitors (hippuristanol, silvestrol, or pateamine A) to demonstrate a role for eIF4A in silencing. However, this approach has yielded conflicting results, as these drugs were reported either to have no effect (Petersen et al, 2006) or to partially inhibit miRNA‐mediated silencing (Meijer et al, 2013; Fukao et al, 2014). These discrepancies most likely result from the difficulty of finding appropriate normalization controls that are not affected by the treatment or by indirect effects on mRNA stability that have been overlooked. By contrast, our conclusion that the scanning function of eIF4A is not required for silencing is based on the results obtained with the reporters containing a very short 5′‐UTR (in Dm cells) or the TISU motif (human cells), which are not subjected to the normalization problem linked to the use of translational inhibitors. Using these reporters, we demonstrated that 43S scanning and thus eIF4A activity in this process are not required for silencing.
We further show that AGO2, GW182/TNRC6, and NOT1 degrade polyadenylated reporters in the presence of silvestrol, although the treatment causes an increase in reporter mRNA levels in the absence of the tethered proteins. These results indicate that silencing complexes assemble and degrade the mRNA target when translation is inhibited by silvestrol. In accordance with these results, miRNAs, AGO, GW182/TNRC6, and NOT1 degrade polyadenylated reporters that do not require scanning for translation.
Finally, we show that miRNAs can repress translation by a mechanism that does not involve either eIF4A2 dissociation or recruitment. Together with the observation that the knockout of eIF4A2 in human cells does not suppress silencing (Galicia‐Vazquez et al, 2015), our results indicate that eIF4A2 is unlikely to act as a repressor in the miRNA pathway.
Our results do not rule out an involvement of eIF4A1 in silencing (Fukao et al, 2014; Fukaya et al, 2014) but reveal the existence of a mechanism that acts independently of eIF4A1 (i.e. when eIF4A1 is inhibited) or downstream of eIF4A1 dissociation.
The role of DDX6 in silencing
Our data together with previous studies indicate that human DDX6 is required for translational repression mediated by the CCR4–NOT complex in human cells (Chen et al, 2014; Mathys et al, 2014; Rouya et al, 2014). The contribution of DDX6 to this repression becomes apparent in particular for reporters that lack a poly(A) tail and hence are not degraded. In the presence of a poly(A) tail, the dominant effect of the CCR4–NOT complex is deadenylation and decay in human and Dm cells. mRNA destabilization is observed even when the NOT1–DDX6 interaction is impaired, suggesting that the recruitment of decapping factors and XRN1 still occurs. Therefore, it is possible that in addition to DDX6, other factors are involved in coupling deadenylation with decapping. For example, the decapping activator PatL1 interacts with the CCR4–NOT complex (Jonas & Izaurralde, 2013) and can also facilitate the recruitment of decapping factors to mRNAs undergoing deadenylation. Alternatively, the residual amount of DDX6 in depleted cells may be sufficient for deadenylation by the CCR4–NOT complex but not for translational repression.
A question that remains unanswered is how DDX6 represses translation. Our polysome profiles together with previous studies indicate that DDX6 orthologs inhibit translation predominantly at initiation (Coller & Parker, 2005; Minshall et al, 2009). Furthermore, we found that DDX6‐mediated repression does not require ribosomal scanning, suggesting that DDX6 may interfere with eIF4E or eIF4G function. In this context, it has been shown that DDX6 interacts with the eIF4E‐transporter protein (4E‐T; Nishimura et al, 2015; Ozgur et al, 2015), an eIF4E‐binding protein that competes with eIF4G for binding to eIF4E and represses translation initiation. However, depletion of 4E‐T only partially alleviates silencing (this study; Kamenska et al, 2014; Nishimura et al, 2015), which implies that DDX6 may employ additional mechanisms to repress translation that are thus far unknown. Accordingly, we observed that the integrity of the FDF‐binding surface of DDX6 is required for silencing. Because this surface interacts with decapping factors in addition to 4E‐T, it is possible that these factors play a role in translational repression in addition to mRNA degradation. It is also possible that other, not‐yet‐identified, DDX6 partners also use the FDF‐binding surface to interact with DDX6.
Through its interaction with the CCR4–NOT complex, DDX6 is likely to be involved in the repression of many mRNAs in addition to miRNA targets. Indeed, the CCR4–NOT complex is recruited to specific mRNAs by numerous RNA‐associated proteins, including Bicaudal‐C, Smaug, CUP, Nanos, Pumilio, and tristetraprolin (TTP; Jonas & Izaurralde, 2015). Thus, elucidating the precise mechanism by which DDX6 represses translation is expected to provide valuable insight into a widespread post‐transcriptional repressive mechanism operating in eukaryotic cells.
Materials and Methods
DNA constructs
Plasmids for the expression of λN‐HA‐, GFP‐, and MS2‐tagged proteins in Dm S2 and human cells, as well as miRNA reporters and luciferase reporters for tethering assays, have been previously described (Rehwinkel et al, 2005; Eulalio et al, 2007, 2008; Braun et al, 2011; Zekri et al, 2013; Chen et al, 2014). We generated Hs AGO2 and Dm AGO1 mutants via site‐directed mutagenesis. Protein mutants used in this study are listed in Appendix Table S1. The reporters lacking the 5′‐UTR or containing the TISU motif, the (CAA)18 repeats, and the 12‐nucleotide guanine quartet (CGG)4 were generated by site‐directed mutagenesis using the corresponding parental plasmids as template. The sequences of the corresponding 5′‐UTRs are listed in Appendix Table S3. Additional constructs used in this study are described in the Appendix Table S2.
Cell culture and transfections
S2 cells were transfected in 6‐well plates using Effectene transfection reagent (Qiagen). Human cells were transfected using Lipofectamine 2000 or TurboFect reagents (Life Technologies). In all experiments, firefly and Renilla luciferase activities were measured using the Dual‐Luciferase Reporter Assay System (Promega), 3 days after transfection of the S2 cells and 48 h after transfection of the human cells. Total RNA was isolated from the S2 and human cells using TriFast (Peqlab Biotechnologies) and analyzed as described previously (Behm‐Ansmant et al, 2006). All Western blots were developed using the ECL Western blotting detection system (GE Healthcare). Antibodies used in this study are listed in Appendix Table S4. Silvestrol (MedChem Express, HY‐13251) was resuspended in DMSO at 1 mg/ml concentration and added to the cells at a final concentration of 0.25 μM. The treatment was for 16 h.
Tethering, complementation, and coimmunoprecipitation assays
The interaction of AGO (wild type or mutants) with endogenous miRNAs and GW182/TNRC6 was tested as described previously (Rehwinkel et al, 2005; Eulalio et al, 2008). Tethering assays in S2 and HEK293T cells were performed as described before (Braun et al, 2011). Detailed protocols can be found in the Appendix Supplementary Methods. AGO1 complementation assays in S2 cells were performed as described previously (Huntzinger et al, 2013).
Polysome profiling in HEK293T cells
HEK293T cells (9 × 106/15‐cm dish) were transfected with Lipofectamine 2000. The transfection mixtures contained 20 μg of the R‐Luc‐6xMS2‐(A)95‐MALAT1 reporter, 4 μg of the pEGFP‐N3‐F‐Luc transfection control, and 6 μg of the plasmids expressing MS2–HA or MS2‐HA‐DDX6. Cells were treated with cycloheximide 48 h after transfection at a final concentration of 50 μg/ml for 30 min. Cell lysis and sucrose gradients were performed as described in the Appendix Supplementary Methods.
RNA immunoprecipitation in HEK293T cells
To study the association of eIF4A2 with mRNA reporters in HEK293T cells, cells (4 × 106/10‐cm dish) were transfected with Lipofectamine 2000. The transfection mixtures contained 5 μg of the pEGFP‐N3‐F‐Luc transfection control reporter and any one of the following reporters: pCIneo‐R‐Luc‐6xMS2‐A95‐MALAT1 (10 μg), pCIneo‐TISU‐R‐Luc‐6xMS2‐MALAT1 (8 μg), pCIneo‐CAA‐R‐Luc‐6xMS2‐A95‐MALAT1 (15 μg), or pCIneo‐CGG‐R‐Luc‐6xMS2‐A95‐MALAT1 (8 μg). A detailed description of the precipitation procedure is found in the Appendix Supplementary Methods.
Author contributions
DKO developed the reporters used in this study and coordinated the experimental work. DKO, DB, EH, MF, and SH performed the experiments. DKO and DB analyzed and interpreted the data. EI designed and supervised the project. EI wrote the manuscript with the contribution from DKO and DB.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
The β‐globin MS2 reporters used for the tethering assays in human cells and the psiCHECK2‐let‐7 8x reporter were a kind gift from Dr. Jens Lykke‐Andersen and Dr. Yukihide Tomari, respectively. We thank Dr. Andreas Boland for designing the Argonaute mutants. This work was supported by the Max Planck Society and by grants from the Deutsche Forschungsgemeinschaft (DFG, FOR855, and the Gottfried Wilhelm Leibniz Program awarded to E.I.).
The EMBO Journal (2016) 35: 1186–1203
See also: T Nishimura & MR Fabian (June 2016)
References
- Agarwal V, Bell GW, Nam JW, Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. eLife 4: e05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameres SL, Zamore PD (2013) Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol 14: 475–488 [DOI] [PubMed] [Google Scholar]
- Bawankar P, Loh B, Wohlbold L, Schmidt S, Izaurralde E (2013) NOT10 and C2orf29/NOT11 form a conserved module of the CCR4‐NOT complex that docks onto the NOT1N‐terminal domain. RNA Biol 10: 228–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behm‐Ansmant I, Rehwinkel J, Doerks T, Stark A, Bork P, Izaurralde E (2006) mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev 20: 1885–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordeleau ME, Robert F, Gerard B, Lindqvist L, Chen SMH, Wendel HG, Brem B, Greger H, Lowe SW, Porco JA Jr, Pelletier J (2008) Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J Clin Invest 118: 2651–2660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun JE, Huntzinger E, Fauser M, Izaurralde E (2011) GW182 proteins directly recruit cytoplasmic deadenylase complexes to miRNA targets. Mol Cell 44: 120–133 [DOI] [PubMed] [Google Scholar]
- Cencic R, Carrier M, Galicia‐Vazquez G, Bordeleau ME, Sukarieh R, Bourdeau A, Brem B, Teodoro JG, Greger H, Tremblay ML, Porco JA Jr, Pelletier J (2009) Antitumor activity and mechanism of action of the cyclopenta[b]benzofuran, silvestrol. PLoS ONE 4: e5223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CT, Bercovich N, Loh B, Jonas S, Izaurralde E (2014) The activation of the decapping enzyme DCP2 by DCP1 occurs on the EDC4 scaffold and involves a conserved loop in DCP1. Nucleic Acids Res 42: 5217–5233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chekulaeva M, Mathys H, Zipprich JT, Attig J, Colic M, Parker R, Filipowicz W (2011) miRNA repression involves GW182‐mediated recruitment of CCR4–NOT through conserved W‐containing motifs. Nat Struct Mol Biol 18: 1218–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CYA, Zheng D, Xia Z, Shyu AB (2009) Ago‐TNRC6 triggers microRNA‐mediated decay by promoting two deadenylation steps. Nat Struct Mol Biol 16: 1160–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Boland A, Kuzuoğlu‐Öztürk D, Bawankar P, Loh B, Chang CT, Weichenrieder O, Izaurralde E (2014) A DDX6‐CNOT1 complex and W‐binding pockets in CNOT9 reveal direct links between miRNA target recognition and silencing. Mol Cell 54: 737–750 [DOI] [PubMed] [Google Scholar]
- Chu CY, Rana TM (2006) Translation repression in human cells by microRNA‐induced gene silencing requires RCK/p54. PLoS Biol 4: e210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller J, Parker R (2005) General translational repression by activators of mRNA decapping. Cell 122: 875–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke A, Prigge A, Wickens M (2010) Translational repression by deadenylases. J Biol Chem 285: 28506–28513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope CL, Gilley R, Balmanno K, Sale MJ, Howarth KD, Hampson M, Smith PD, Guichard SM, Cook SJ (2014) Adaptation to mTOR kinase inhibitors by amplification of eIF4E to maintain cap‐dependent translation. J Cell Sci 127: 788–800 [DOI] [PubMed] [Google Scholar]
- Eichhorn SW, Guo H, McGeary SE, Rodriguez‐Mias RA, Shin C, Baek D, Hsu SH, Ghoshal K, Villén J, Bartel DP (2014) mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol Cell 56: 104–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfakess R, Sinvani H, Haimov O, Svitkin Y, Sonenberg N, Dikstein R (2011) Unique translation initiation of mRNAs‐containing TISU element. Nucleic Acids Res 39: 7598–7609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A, Rehwinkel J, Stricker M, Huntzinger E, Yang SF, Doerks T, Dorner S, Bork P, Boutros M, Izaurralde E (2007) Target‐specific requirements for enhancers of decapping in miRNA‐mediated gene silencing. Genes Dev 21: 2558–2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A, Huntzinger E, Izaurralde E (2008) GW182 interaction with Argonaute is essential for miRNA‐mediated translational repression and mRNA decay. Nat Struct Mol Biol 15: 346–353 [DOI] [PubMed] [Google Scholar]
- Eulalio A, Huntzinger E, Nishihara T, Rehwinkel J, Fauser M, Izaurralde E (2009) Deadenylation is a widespread effect of miRNA regulation. RNA 15: 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian MR, Cieplak MK, Frank F, Morita M, Green J, Srikumar T, Nagar B, Yamamoto T, Raught B, Duchaine TF, Sonenberg N (2011) miRNA‐mediated deadenylation is orchestrated by GW182 through two conserved motifs that interact with CCR4–NOT. Nat Struct Mol Biol 18: 1211–1217 [DOI] [PubMed] [Google Scholar]
- Fabian MR, Sonenberg N (2012) The mechanics of miRNA‐mediated gene silencing: a look under the hood of miRISC. Nat Struct Mol Biol 19: 586–593 [DOI] [PubMed] [Google Scholar]
- Fukao A, Mishima Y, Takizawa N, Oka S, Imataka H, Pelletier J, Sonenberg N, Thoma C, Fujiwara T (2014) MicroRNAs trigger dissociation of eIF4AI and eIF4AII from target mRNAs in humans. Mol Cell 56: 79–89 [DOI] [PubMed] [Google Scholar]
- Fukaya T, Tomari Y (2012) MicroRNAs mediate gene silencing via multiple different pathways in Drosophila . Mol Cell 48: 825–836 [DOI] [PubMed] [Google Scholar]
- Fukaya T, Iwakawa HO, Tomari Y (2014) MicroRNAs block assembly of eIF4F translation initiation complex in Drosophila . Mol Cell 56: 67–78 [DOI] [PubMed] [Google Scholar]
- Galicia‐Vazquez G, Chu J, Pelletier J (2015) eIF4AII is dispensable for miRNA‐mediated gene silencing. RNA 21: 1826–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson DG, Hogan DJ, McCullough HL, Myers JW, Herschlag D, Ferrell JE, Brown PO (2009) Concordant regulation of translation and mRNA abundance for hundreds of targets of a human microRNA. PLoS Biol 7: e1000238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E, Kuzuoğlu‐Öztürk D, Braun JE, Eulalio A, Wohlbold L, Izaurralde E (2013) The interactions of GW182 proteins with PABP and deadenylases are required for both translational repression and degradation of miRNA targets. Nucleic Acids Res 41: 978–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CUT, Pestova TV (2010) The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11: 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas S, Izaurralde E (2013) The role of disordered protein regions in the assembly of decapping complexes and RNP granules. Genes Dev 27: 2628–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas S, Izaurralde E (2015) Towards a molecular understanding of microRNA‐mediated gene silencing. Nat Rev Genet 16: 421–433 [DOI] [PubMed] [Google Scholar]
- Kamenska A, Lu WT, Kubacka D, Broomhead H, Minshall N, Bushell M, Standart N (2014) Human 4E‐T represses translation of bound mRNAs and enhances microRNA‐mediated silencing. Nucleic Acids Res 42: 3298–3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaretti D, Tournier I, Izaurralde E (2009) The C‐terminal domains of human TNRC6A, TNRC6B, and TNRC6C silence bound transcripts independently of Argonaute proteins. RNA 15: 1059–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Basquin J, Ozgur S, Czarnocki‐Cieciura M, Bonneau F, Aartse A, Dziembowski A, Nowotny M, Conti E, Filipowicz W (2014) Structural and biochemical insights to the role of the CCR4‐NOT complex and DDX6 ATPase in microRNA repression. Mol Cell 54: 751–765 [DOI] [PubMed] [Google Scholar]
- Meijer HA, Kong YW, Lu WT, Wilczynska A, Spriggs RV, Robinson SW, Godfrey JD, Willis AE, Bushell M (2013) Translational repression and eIF4A2 activity are critical for microRNA‐mediated gene regulation. Science 340: 82–85 [DOI] [PubMed] [Google Scholar]
- Minshall N, Kress M, Weil D, Standart N (2009) Role of p54 RNA helicase activity and its C‐terminal domain in translational repression, P‐body localization and assembly. Mol Biol Cell 20: 2464–2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara T, Zekri L, Braun JE, Izaurralde E (2013) miRISC recruits decapping factors to miRNA targets to enhance their degradation. Nucleic Acids Res 41: 8692–8705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Padamsi Z, Fakim H, Milette S, Dunham WH, Gingras AC, Fabian MR (2015) The eIF4E‐binding protein 4E‐T is a component of the mRNA decay machinery that bridges the 5′ and 3′ termini of target mRNAs. Cell Rep 11: 1425–1436 [DOI] [PubMed] [Google Scholar]
- Ozgur S, Basquin J, Kamenska A, Filipowicz W, Standart N, Conti E (2015) Structure of a human 4E‐T/DDX6/CNOT1 complex reveals the different interplay of DDX6‐binding proteins with the CCR4‐NOT complex. Cell Rep 13: 703–711 [DOI] [PubMed] [Google Scholar]
- Pestova TV, Kolupaeva VG (2002) The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev 16: 2906–2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CP, Bordeleau ME, Pelletier J, Sharp PA (2006) Short RNAs repress translation after initiation in mammalian cells. Mol Cell 21: 533–542 [DOI] [PubMed] [Google Scholar]
- Piao X, Zhang X, Wu L, Belasco JG (2010) CCR4‐NOT deadenylates mRNA associated with RNA‐induced silencing complexes in human cells. Mol Cell Biol 30: 1486–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai RS, Artus CG, Filipowicz W (2004) Tethering of human Ago proteins to mRNA mimics the miRNA‐mediated repression of protein synthesis. RNA 10: 1518–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J, Behm‐Ansmant I, Gatfield D, Izaurralde E (2005) A crucial role for GW182 and the DCP1:DCP2 decapping complex in miRNA‐mediated gene silencing. RNA 11: 1640–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci EP, Limousin T, Soto‐Rifo R, Rubilar PS, Decimo D, Ohlmann T (2013) miRNA repression of translation in vitro takes place during 43S ribosomal scanning. Nucleic Acids Res 41: 586–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouya C, Siddiqui N, Morita M, Duchaine TF, Fabian MR, Sonenberg N (2014) Human DDX6 effects miRNA‐mediated gene silencing via direct binding to CNOT1. RNA 20: 1398–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirle NT, MacRae IJ (2012) The crystal structure of human Argonaute2. Science 336: 1037–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinvani H, Haimov O, Svitkin Y, Sonenberg N, Tamarkin‐Ben‐Harush A, Viollet B, Dikstein R (2015) Translational tolerance of mitochondrial genes to metabolic energy stress involves TISU and eIF1‐eIF4GI cooperation in start codon selection. Cell Metab 21: 479–492 [DOI] [PubMed] [Google Scholar]
- Sung D, Kang H (1998) The N‐terminal amino acid sequences of the firefly luciferase are important for the stability of the enzyme. Photochem Photobiol 68: 749–753 [DOI] [PubMed] [Google Scholar]
- Sweet T, Kovalak C, Coller J (2012) The DEAD‐box protein Dhh1 promotes decapping by slowing ribosome movement. PLoS Biol 10: e1001342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritschler F, Braun JE, Eulalio A, Truffault V, Izaurralde E, Weichenrieder O (2009) Structural basis for the mutually exclusive anchoring of P body components EDC3 and tral to the DEAD box protein DDX6/Me31B. Mol Cell 33: 661–668 [DOI] [PubMed] [Google Scholar]
- Wilusz JE, JnBaptiste CK, Lu LY, Kuhn CD, Joshua‐Tor L, Sharp PA (2012) A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack polyA tails. Genes Dev 26: 2392–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe AL, Singh K, Zhong Y, Drewe P, Rajasekhar VK, Sanghvi VR, Mavrakis KJ, Jiang M, Roderick JE, Van der Meulen J, Schatz JH, Rodrigo CM, Zhao C, Rondou P, de Stanchina E, Teruya‐Feldstein J, Kelliher MA, Speleman F, Porco JA Jr, Pelletier J et al (2014) RNA G‐quadruplexes cause eIF4A‐dependent oncogene translation in cancer. Nature 513: 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu PH, Isaji M, Carthew RW (2013) Functionally diverse microRNA effector complexes are regulated by extracellular signaling. Mol Cell 52: 113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zekri L, Kuzuoğlu‐Öztürk D, Izaurralde E (2013) GW182 proteins cause PABP dissociation from silenced miRNA targets in the absence of deadenylation. EMBO J 32: 1052–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8