

While gene conversion and classical nonhomologous end-joining are the most physiologically predominant forms of DNA repair mechanisms, emerging lines of evidence suggest the usage of several noncanonical homology-directed repair (HDR) pathways in both prokaryotes and eukaryotes in different contexts. Here, Verma and Greenberg review how these alternative HDR pathways are executed.
Keywords: ALT, DNA repair, homologous recombination, telomere, break-induced replication
Abstract
DNA repair is essential to maintain genomic integrity and initiate genetic diversity. While gene conversion and classical nonhomologous end-joining are the most physiologically predominant forms of DNA repair mechanisms, emerging lines of evidence suggest the usage of several noncanonical homology-directed repair (HDR) pathways in both prokaryotes and eukaryotes in different contexts. Here we review how these alternative HDR pathways are executed, specifically focusing on the determinants that dictate competition between them and their relevance to cancers that display complex genomic rearrangements or maintain their telomeres by homology-directed DNA synthesis.
Double-strand break (DSB) repair is essential to all forms of life. The canonical and predominant pathways of DSB repair include gene conversion (GC) and classical nonhomologous end-joining (c-NHEJ). These pathways are essential for the repair of DSBs that are generated during meiosis, DNA replication, and antigen receptor diversification (Dudley et al. 2005; Keeney and Neale 2006; Lieber 2010). Mutations in genes critical for these pathways result in a host of pathologies that speak to the importance of faithful DNA repair (Sung and Klein 2006; Heyer et al. 2010; Chapman et al. 2012; Jiang and Greenberg 2015; Prakash et al. 2015). While somewhat less appreciated, homology-directed DNA repair (HDR) encompasses many genetically distinct mechanisms beyond the canonical view of GC by Rad51 recombinase. Starting from 3′ ssDNA overhangs, numerous alternative forms of error-prone HDR pathways can be invoked. Emerging evidence indicates their central involvement in the genomic instability that is present in cancers and other congenital disorders (Hastings et al. 2009b; Rodgers and McVey 2015; Carvalho and Lupski 2016).
This review describes the basis for several different forms of noncanonical HDR and highlights the discovery and conservation of these DNA repair mechanisms across prokaryotic and eukaryotic organisms. Where possible, we attempt to delineate determinants of competition between these distinct DNA repair mechanisms. We begin with the two-ended DSB repair pathways (Fig. 1) and compare the biochemical mechanisms and the molecular machinery used by these and the GC pathway (Table 1). Finally, we focus on various forms of mutagenic one-ended DSB repair pathways and review recent studies on alternative forms of telomere maintenance and genomic rearrangements that arise from varied themes of one-ended break repair mechanisms.
Figure 1.
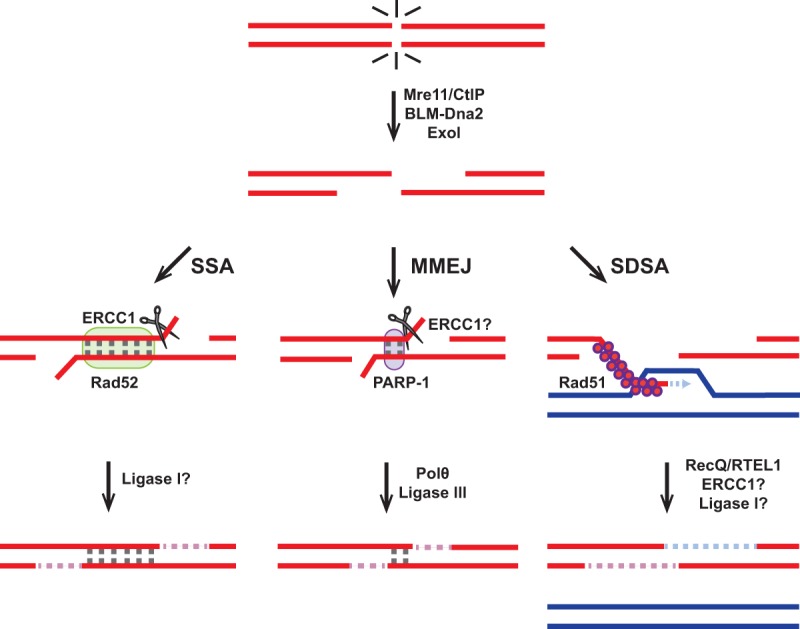
Model for the noncanonical HDR pathways at two-ended DSBs. Single-stranded annealing (SSA), microhomology-mediated end-joining (MMEJ), and synthesis-dependent strand annealing (SDSA) use the same end resection machinery as the GC pathway. SSA or MMEJ operates in the absence of 3′ single-stranded tail invasion into a homologous template. If the sequences revealed after resection are direct repeats, SSA is favored over MMEJ. Repair by SDSA occurs when the 3′ invading strand, after undergoing limited DNA synthesis, is displaced from the D loop by a helicase and anneals back to a complementary strand. Mammalian proteins required for each pathway are shown; those proteins for which direct evidence is currently lacking are followed by a question mark. For simplicity, nuclease ERCC1 is shown on only one strand.
Table 1.
Proteins involved in homology-directed repair

Single-stranded annealing (SSA)
The SSA pathway is initiated when a DSB occurs between homologous direct repeats. Extensive bidirectional resection reveals complementary ssDNA sequences that can then anneal. This is followed by nuclease-mediated removal of the unpaired 3′ tail, resulting in the deletion of the intervening region and one of the repeats. The gaps generated are filled in and ligated (Fig. 1). Because deletions are inherently associated with this form of DNA repair, SSA is, by definition, mutagenic.
Studies examining the mechanisms by which palindromic sequences stimulate genomic deletions in Escherichia coli suggested the existence of a bacterial SSA pathway. Cleavage of the cruciform structures formed between palindromic sequences by the nuclease component of the structure-specific nuclease SbcCD was proposed to be followed by resection and annealing to form a deletion product. This pathway was found to be RecA-independent (bacterial Rad51 homolog) (Bzymek and Lovett 2001). RecO, a functional homolog of the yeast Rad52 protein, may mediate the annealing of the complementary sequences (Kantake et al. 2002). A functional genomics study in radio-resistant Deinococcus radiodurans also provided evidence for a RecA-independent SSA pathway in generating deletions at the repeat regions and hence contributing to genomic instability (Ithurbide et al. 2015). Thus, SSA may be a prevalent repair mechanism for DSBs within repeat regions.
A modified version of SSA has also been proposed in D. radiodurans to account for its remarkable ability to accurately assemble a 3.28-Mb genome shattered in hundreds of fragments after 5-kGy γ radiation (Zahradka et al. 2006). A two-staged DNA repair process was suggested to accomplish this reassembly. The first stage involves a unique form of polymerase A (Pol A)-dependent assembly called extended synthesis-dependent strand annealing (ESDSA), which differs from classical SSA in that it involves a D-loop-dependent strand extension step that continues until a complementary strand is found. The subsequent annealing of the two complementary strands is similar to the SSA pathway. The second step involves RecA-dependent crossover of the linear fragments to form circular chromosomes (Zahradka et al. 2006). The SSA pathway has also been reported in mycobacteria, where the nuclease RecBCD, which has classically been implicated in RecA-mediated homologous recombination (HR), was shown to be exclusively required for SSA. In contrast, RecA-mediated HR in mycobacteria is RecBCD-independent, and the mycobacterial genome encodes another heterodimeric, helicase nuclease termed AdnAB for RecA-dependent homology synthesis (Gupta et al. 2011). In addition, a recent study in mycobacteria proposed that the regulation of the RecFOR system, which can either perform strand annealing and promote SSA or load RecA and support GC, might be a determinant for the pathway opted for the repair of DSBs (Gupta et al. 2015).
Indications for an SSA-like process in yeast came from studies on the repair of HO endonuclease-induced DSBs in rDNA repeats of rad52 mutant cells (Ozenberger and Roeder 1991). Most of the repair products were associated with the deletion of one or more repeat units. This observation was subsequently explained using an SSA mechanism that had already been proposed in a mammalian system (Lin et al. 1984). Ozenberger and Roeder (1991) showed that the DSB repair product of a plasmid with two copies of the same gene in direct orientation was associated with the annealing of the homologous sequences with concomitant deletion of one of the repeats and the intervening DNA. Unlike GC, the kinetics of SSA product formation were inversely related to the distance between the repeats (Fishman-Lobell and Haber 1992). A two-step end resection mechanism was proposed for SSA that involved (1) initial resection by the Mre11–Rad50–Xrs2 (MRX) complex–Sae2 followed by (2) extensive bidirectional resection by 5′–3′ exonuclease Dna2 and Sgs1, a Bloom (BLM) helicase homolog, and/or 5′–3′ exonuclease Exo1 (Mimitou and Symington 2008; Zhu et al. 2008; Garcia et al. 2011). This 3′ end resection machinery is used by other HDR pathways as well. While Rad52 is required for the annealing of RPA-covered ssDNA, SSA can operate independently of Rad52, albeit at a significantly lower efficiency (Ozenberger and Roeder 1991; Fishman-Lobell and Haber 1992). Rad52-mediated SSA is independent of several other members of the Rad52 epistasis group, including Rad51, Rad54, Rad55, and Rad57 (Ivanov et al. 1996), establishing it as a process distinct from GC. Rad59 is also required for SSA, particularly in the context of annealing shorter regions of homology (Sugawara et al. 2000; Agmon et al. 2009). After annealing of the complementary strands, the removal of the 3′ nonhomologous tails is executed by the Rad1–Rad10 endonuclease complex, which is facilitated by a heterodimer of mismatch repair proteins, MSH2/MSH3 (Fishman-Lobell and Haber 1992; Ivanov and Haber 1995; Saparbaev et al. 1996; Sugawara et al. 1997; Symington 2002). Based on recent in vitro biochemical studies, it was proposed that the structure-specific DNA-binding protein Saw1 recognizes the 3′ nonhomologous flap and recruits Rad1. In addition, Saw1 enhances the 3′ tail cleavage activity of Rad1–Rad10 (Li et al. 2013). It was demonstrated that, when two homologous segments are in direct orientation, SSA predominates over GC (Fishman-Lobell et al. 1992). However, in a rad51 deletion strain, there is a marked increase in SSA with a concomitant decrease in GC, exemplifying the competing nature of the two pathways (Ivanov et al. 1996; Agmon et al. 2009). Indeed, Rad51 has been shown to prevent Rad52-mediated annealing of complementary ssDNA in defined in vitro systems (Wu et al. 2008). SSA has been demonstrated to mediate nonallelic HR (NAHR) between rereplicated repeat sequences, resulting in copy number variations (CNVs) in the yeast genome (Finn and Li 2013).
Unlike most other DNA repair pathways that were discovered in bacteria and yeast, the SSA mode of recombination was first identified in mammalian cells. It was observed that the efficiency of both intermolecular and intramolecular recombination of plasmids with sequence repeats was dependent on the degree of homology and could be stimulated 10-fold to 100-fold by appropriately cutting the DNA relative to the recombining sequences. This led to a model in which these cut ends could be substrates for exonuclease activity, which would help expose the recombining sequences for annealing (Lin et al. 1984). A subsequent study in Xenopus oocyte extracts provided evidence for this annealing model by identifying reaction intermediates with full-length 3′ ending strands and shorter 5′ ending strands with overlaps between the 3′ complementary region (Sweigert and Carroll 1990). Interestingly, it was demonstrated that SSA could mediate translocations in human cells harboring DSBs in the repetitive Alu elements (Elliott et al. 2005), which can explain the deletions observed in the Alu elements in germline mutations of several tumor suppressor genes (Kolomietz et al. 2002). Because SSA relies on extensive base-pairing between homologous sequences, its contribution to translocations is limited even between the repetitive elements, which is most likely due to sequence variability in these repeats (Weinstock et al. 2006). Analysis of the genetic requirements for different DNA repair pathways revealed that while GC, microhomology-mediated end-joining (MMEJ), and SSA repair pathways share the same end resection machinery, Rad52 and ERCC1 (a homolog of Rad10) are more important for SSA. Additionally, as reported in yeast, Rad51 disruption causes a decrease in GC with a parallel increase in SSA and has no effect on MMEJ (Adair et al. 2000; Stark et al. 2004; Bennardo et al. 2008). Several questions arise from these studies as to how complementary sequences, particularly in the form of direct repeats, are shunted to the various forms of HDR. Notably, how does resection coordinate and hand off these repair substrates to the gene products that dictate whether GC or error-prone SSA or MMEJ is the repair pathway of choice? A point to note is that much of the molecular machinery used and the biochemical steps involved are similar among these different HDR pathways (Table 1). Therefore, it is important to identify the rate-limiting step in each pathway that would allow one to predominate over the other.
Alternative end-joining (alt-EJ)/MMEJ
While alt-EJ is a more generalized term for end-joining that occurs independently of the c-NHEJ machinery, MMEJ is a form of alt-EJ that has been characterized more extensively. MMEJ involves annealing of “microhomologous” sequences revealed after bidirectional end resection and removal of the overhanging 3′ single-stranded tail followed by error-prone filling of the gaps and subsequent ligation (Fig. 1). This pathway of repair is mutagenic and has been associated with chromosomal translocation events. While mechanistically similar to SSA in concept, MMEJ requires less homology and different gene products for its execution, firmly indicating that it is a process distinct from SSA.
Evidence for the presence of alt-EJ in bacteria emerged from studies in E. coli, which do not possess the multifunctional ligase-D and Ku-like proteins essential for bacterial c-NHEJ (Bowater and Doherty 2006; Chayot et al. 2010). Studies on the repair of linearized plasmids with noncomplementary ends revealed that E. coli could join these DNA ends, albeit in an error-prone manner. This end-joining was independent of the substrate ends but was found to be dependent on extensive bidirectional resection, the use of microhomology of mostly <3 base pairs (bp), and limited nontemplated DNA synthesis. Additionally, alt-EJ was shown to repair I-SceI endonuclease-induced chromosomal DSBs in a recombination-independent manner and in the presence of an intact HR machinery. Although the physiological importance of this pathway is unclear, it suggested that alt-EJ might contribute to the mending of DSBs that cannot be repaired by GC (Chayot et al. 2010). The error-prone nature of this repair can contribute to genome evolution for the adaptability of bacteria to changing environmental conditions. While it is unknown whether such an alternative repair pathway exists in other bacterial species, a Ku- and LigD-independent end-joining pathway was reported in mycobacteria, which, unlike E. coli alt-EJ, is specific to sticky 3′ overhangs, does not involve end-processing, and is nonmutagenic in nature (Aniukwu et al. 2008).
Early evidence for MMEJ came from studies in Saccharomyces cerevisiae, where the ligation product of a linearized plasmid transformed into a Ku-deficient strain showed sustained deletions with microhomologous sequences ranging from 3 to 16 bp at the ligated junctions (Boulton and Jackson 1996). Subsequent studies in S. cerevisiae identified the molecular machinery that drives MMEJ: (1) initiation of end resection by the MRX–Sae2; (2) if the microhomologous sequences are >2 kb away from the DSB ends, extensive bidirectional resection is performed by Sgs1–Dna2 and/or Exo1; (3) annealing of microhomologous sequences and removal of 3′ single-stranded flaps by Rad1–Rad10; (4) and filling of gaps by polymerases Pol λ (Pol 4) and Pol δ in conjunction with its nonessential subunit, Pol 32 (Ma et al. 2003; Decottignies 2007; Garcia et al. 2011; Symington and Gautier 2011; Villarreal et al. 2012; Cannavo and Cejka 2014; Deng et al. 2014; Meyer et al. 2015). Rad51 filament formation is considered to be a critical feature in dictating GC versus MMEJ events. Binding of RPA to ssDNA, generated by end resection, prevents spontaneous annealing and promotes Rad51 filament formation, leading to GC events (Deng et al. 2014). Indeed, the frequency of MMEJ is enhanced in yeast after removal of Rad51, suggesting a competition between the two repair pathways (Villarreal et al. 2012; Deng et al. 2014). Unlike SSA, Rad52 is not required for MMEJ. Specifically, Rad52 is dispensable for microhomologous sequences <14 bp (Villarreal et al. 2012). Although the basic steps in MMEJ are similar between mammals and yeast, the molecular machinery and the functional relevance are more elaborate in higher eukaryotes (discussed below). In contrast, MMEJ in yeast is assumed to be a backup mechanism to repair DSBs in the event that HR and NHEJ fail to do so (Sfeir and Symington 2015).
MMEJ in mammals was reported by studies in p53-null mice lacking the components of the NHEJ machinery. These mice developed pro-B-cell lymphomas resulting from chromosomal translocations between the IgH and the c-myc loci. The repair junctions were characterized by insertions, deletions, and microhomology (Difilippantonio et al. 2002; Zhu et al. 2002). Later studies performed in NHEJ-deficient mice showed that antigen receptor-associated translocations could still be observed in B cells at the regions of microhomology, suggesting a role of MMEJ in mediating these translocations (Yan et al. 2007). While MMEJ is the predominant pathway in mice for chromosomal translocations, recent studies demonstrated that translocations in humans are primarily mediated by c-NHEJ (Simsek and Jasin 2010; Ghezraoui et al. 2014). MMEJ has also been shown to mediate telomere fusion in cells lacking both the shelterin and the Ku70/80 complexes (Sfeir and de Lange 2012). Several independent studies have demonstrated that MMEJ can also operate in the presence of intact NHEJ (Corneo et al. 2007; Truong et al. 2013), suggesting that Ku deficiency is not a requisite feature for the usage of MMEJ. Similar to yeast, mammalian MMEJ and GC share the same end resection machinery, where Mre11 in complex with CtIP performs the initial resection. In case the microhomologous sequences are far apart, further resection is performed by BLM–Dna2 and Exo1 (Dinkelmann et al. 2009; Rass et al. 2009; Xie et al. 2009; Lee-Theilen et al. 2010; Zhang and Jasin 2011; Truong et al. 2013). The key players that drive MMEJ in mammalian cells include the highly mutagenic DNA polymerase Pol θ, ligase 3, and poly(ADP-ribose) polymerase 1 (PARP1) (Audebert et al. 2004; Wang et al. 2005; McVey and Lee 2008; Chan et al. 2010; Yu and McVey 2010; Simsek et al. 2011). Initial evidence for the role of Pol θ in this pathway came from studies in Drosophila, and this polymerase has since then been shown to be essential for MMEJ in all metazoan species examined to date (Chan et al. 2010; Yu and McVey 2010; Sfeir and Symington 2015). Pol θ has the unique ability to prime DNA synthesis from nonoptimal base-pairing and add nucleotides to the 3′ end of ssDNA (Hogg et al. 2012; Yousefzadeh et al. 2014; Kent et al. 2015). This promiscuous polymerase activity in the C-terminal domain of Pol θ arises from its ability to maintain a tight hold on the 3′-terminal phosphate of the primer via strong electrostatic interactions (Zahn et al. 2015). A recent crystal structure of the Pol θ helicase domain revealed its tetrameric organization and suggested its role in annealing the DNA strands for subsequent processing by the polymerase domain (Newman et al. 2015).
The competitive nature between GC and MMEJ was recently brought to light by several studies that demonstrated elevated Rad51-dependent GC in Pol θ-deficient cells. One study implicated Pol θ and Rad51 interactions as a mechanism to prevent Rad51–ssDNA nucleofilament assembly, thus inhibiting GC and promoting MMEJ (Ceccaldi et al. 2015). The accompanying study demonstrated a critical role for Pol θ polymerase activity in mediating this balance (Mateos-Gomez et al. 2015). In contrast to MMEJ in yeast, where ≥6 nucloetides (nt) of homology is required and no insertions are observed at the junction, in higher eukaryotes, Pol θ uses shorter (2- to 6-bp) microhomologies for annealing and introduces insertions at the break sites, making this pathway highly error-prone and, in contrast to SSA, readily available to most genomic locations (McVey and Lee 2008; Kent et al. 2015). HR-deficient ovarian cancer exhibits higher expression levels of Pol θ, and a synthetic lethal relationship has been demonstrated between HR and PARP1/Pol θ-mediated MMEJ (Ceccaldi et al. 2015, 2016; Mateos-Gomez et al. 2015). It will be interesting to examine whether MMEJ is the key target underlying PARP inhibitor efficacy in BRCA mutant cancer cells. Such issues are at the heart of developing Pol θ inhibitors as an additional approach to treat tumors that harbor HR deficiencies.
SDSA
The SDSA pathway involves bidirectional resection of DSBs, enabling either one or both 3′ single-stranded ends to invade a homologous template and form a D loop similar to GC. However, unlike GC, the invading strand is displaced during the D-loop migration (Ferguson and Holloman 1996). During one-ended invasion, the displaced strand now anneals to the other complementary end of the DSB. In the case of two-ended invasions, both the displaced strands now anneal via the newly synthesized complementary regions (Fig. 1; Nassif et al. 1994; Krogh and Symington 2004). SDSA primarily appears to be a pathway that preserves genomic integrity by suppressing crossovers and preventing the loss of heterozygosity. However, models for SDSA that result in crossover outcomes have also been proposed (Pâques et al. 1998; Pâques and Haber 1999; Allers and Lichten 2009).
Although the SDSA model has been used to explain the nature of recombinant products formed during the intron homing process in bacteriophage T4 (Bryk et al. 1995), the repair of DSBs via SDSA in bacteria still needs to be documented (Ayora et al. 2011). ESDSA (discussed earlier) is one rare example of a SDSA-related process in bacteria known to date (Zahradka et al. 2006). It is worthwhile to note that bacteria possess homologs of helicases that have been demonstrated to play an important role in promoting SDSA by disrupting the D loop. The Srs2-like bacterial protein UvrD can disrupt the RecA nucleoprotein filament (Veaute et al. 2005). An in vitro biochemical study reconstituting the early steps of the bacterial RecFOR repair pathway demonstrated that the bacterial RecQ protein, like its eukaryotic homologs, could disrupt the formation of joint DNA molecules (Handa et al. 2009). Additionally, proteins with an anti-recombination activity like MutS2, which can digest branch DNA structures, including Holliday junctions (HJs), have also been documented in bacteria (Fukui et al. 2008).
The idea of SDSA was first proposed in a theoretical study, where it was hypothesized that homology-mediated DSB repair can happen via heteroduplex unwinding after limited DNA synthesis without the formation of double HJs (dHJs) (Resnick 1976). A related pathway was used to describe a mating type conversion in yeast, where the exchange of DNA between the expressed and unexpressed alleles at the mating type locus was not associated with crossover (Strathern et al. 1982). Subsequently, an SDSA model was used to explain the presence of a heteroduplex region on a single chromatid during meiosis (Porter et al. 1993; Gilbertson and Stahl 1996). Most research on the molecular machinery driving SDSA has focused on the mechanisms that would lead to displacement of the invading strand from the D loop and hence favor SDSA. Initial evidence for the involvement of a helicase in promoting SDSA came from yeast genetic studies that demonstrated that deletion of either the sgs1 or srs2 gene increased crossover frequencies. It was suggested that Sgs1 and its associated topoisomerase, Top3, dissociate the dHJs, and Srs2 regulates Rad51 nucleofilament formation and facilitates invading strand displacement (Ira et al. 2003). In vitro biochemical studies demonstrated that Srs2 helicase activity can disrupt Rad51 presynaptic filaments (Krejci et al. 2003; Veaute et al. 2003) and unwind synthetic structures that mimic D loops (Dupaigne et al. 2008). It was also shown that the helicase activity of Srs2 was stimulated by the presence of Rad51 filaments on dsDNA, suggesting that Srs2 can unwind the duplex part of a D loop while removing Rad51 from the invading strand. This can eventually result in annealing of the invading strand to the ssDNA on the other break end (Dupaigne et al. 2008). Yeast Rad51 paralogs Rad55–Rad57 can counteract the anti-recombination activity of the Srs2 helicase and promote Rad51 filament formation (Liu et al. 2011a). Recent biochemical studies on Sgs1 helicase demonstrated that while this RecQ helicase can disrupt protein-free D loops, it could not disrupt D loops reconstituted with yeast RPA, Rad51, and Rad54 proteins. On the other hand, Top3 could dissolve yeast Rad51–Rad54-mediated D loops in an Sgs1-independent manner, and this activity was found to be stimulated by its binding partner, Rmi1. This study explains the basis for the stronger hyperrecombination phenotype of top3 mutants compared with sgs1 mutants (Fasching et al. 2015). Additionally, yeast helicase Mph1, an ortholog of human Fanconi anemia (FA) protein FANCM, has been shown to dissociate Rad51-generated D loops in vitro and promote the noncrossover SDSA pathway in vivo independently of Srs2 and Sgs1 (Prakash et al. 2009). The molecular machinery used in the later steps of SDSA that includes DNA strand annealing and removal of the extra 3′ ssDNA is the same as that used by SSA. These findings further emphasize that while these repair pathways are markedly different in the nature of the repair products formed, they tend to use the same repair proteins in mechanistically similar steps (Nassif et al. 1994; Lao et al. 2008; Mazon et al. 2012; Symington et al. 2014). A sequencing-based study analyzing the gap repair products suggested a model in which the length of the invading strand decides whether SDSA or GC is the repair pathway of choice. Interestingly, most noncrossovers generated during mitosis are produced by SDSA rather than by the canonical GC pathway (Mitchel et al. 2010). Like other repair pathways, SDSA can outcompete the kinetically slow process of break-induced replication (BIR) (discussed below) (Malkova et al. 2005).
The term SDSA was coined in a DSB repair study in Drosophila examining P-element transposition in mitotic cells. Sequences from various loci in the donor template were found in the repair product. This observation was explained by an SDSA model, according to which both the broken ends perform homology search, undergo extension, and dissociate prior to the formation of dHJs. The complementary single strands eventually anneal, completing the synthesis (Nassif et al. 1994). Subsequent studies in several higher eukaryotes have revealed the existence of the SDSA pathway (Belmaaza and Chartrand 1994; Adelman and Boulton 2010). Like in yeast, members of the RecQ helicase family have been documented to have a role in promoting SDSA in higher eukaryotes as well (Adams et al. 2003; Wu and Hickson 2003). Human BLM, along with TopIII α topoisomerase, promotes noncrossover formation by dissolution of dHJ intermediates and by preventing the exchange of flanking sequences (Wu and Hickson 2003). Recently, another member of the RecQ helicase family, RecQ5, a potential Srs2 ortholog in mammals, was shown to promote SDSA by disrupting Rad51–ssDNA filaments and counteracting the inhibitory effect of Rad51 on the strand-annealing activity of Rad52 (Paliwal et al. 2014). It will be interesting to examine whether FANCM can also perform a function similar to its yeast ortholog, Mph1, in dissociating Rad51-coated D loops and promoting SDSA. Indeed, FANCM has been biochemically shown to dissociate large recombination intermediates (α structures) containing a HJ (Gari et al. 2008). In higher eukaryotes, Rtel1 (regulator of telomere length 1), a replication fork-associated 5′-to-3′ helicase, has been shown to disrupt D loops in vitro and promote SDSA in vivo (Adelman and Boulton 2010; Youds et al. 2010). It will be important to examine how the activities of these helicases and their access to DSB repair intermediates are regulated and how this influences the HDR pathway outcome.
BIR
BIR, previously known as break–copy recombination or recombination-dependent replication, is a HDR pathway that comes into play when there is homology at only one end of the DSB. It involves invasion of 3′ single-stranded overhangs onto a homologous sequence followed by copying of the invaded sequences to the distal end, which can extend for up to several hundred kilobases in yeast (Fig. 2; Morrow et al. 1997).
Figure 2.
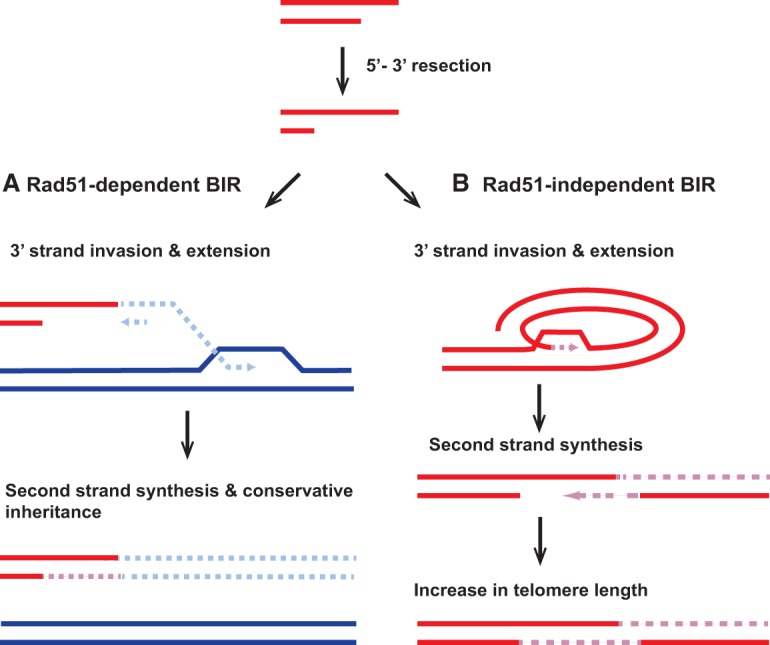
Model for HDR at one-ended DSBs by Rad51-dependent and -independent BIR. (A) A one-end break repair that involves intermolecular recombination (e.g., a broken replication fork) occurs primarily by Rad51-dependent BIR. The Rad51 nucleofilament invades a homologous template, and DNA synthesis can continue to the end of the donor template. As demonstrated in yeast, asynchronous synthesis of the leading and lagging strands has been shown. (B) Rad51-independent BIR at a one-ended break can occur when long-range strand invasion is not required. For instance, in the figure above, formation of a t-circle at the end of an ALT (alternative lengthening of telomeres) telomere can initiate BIR-mediated homology synthesis. The 3′ end is extended, which can then serve as a template for synthesis of the second strand.
In E. coli, a BIR-related pathway functions during the recovery of broken replication forks, which are formed particularly during SOS-induced replication called inducible stable DNA replication (iSDR) (Asai et al. 1993; Marians 2000). PriA, a SF2 family helicase, mediates this BIR-related process by recognizing the D-loop structures formed by RecA (Lee and Kornberg 1991; Nurse et al. 1991; Xu and Marians 2003). PriA recruits PriB, which stabilizes the PriA–DNA interactions and is followed by the recruitment of ssDNA-binding protein DnaT, loader/helicase DnaC/DnaB, and primase DnaG to the D loop. DNA Pol III finally joins this complex to form an active replisome. This replication restart is independent of DnaA, which is required for replication originating at OriC (Marians 2000; Gabbai and Marians 2010). Since there is only a single origin of replication in bacteria, this OriC-independent pathway is essential in rectifying any replication fork failure. Homologs for PriA are present across nearly all bacterial genomes with the exception of some endosymbionts and mollicutes (Rocha et al. 2005). This widespread conservation perhaps highlights the importance of Pri-A-directed replisome formation at the broken replication forks in bacterial physiology.
Early evidence for BIR in yeast came from meiotic and mitotic recombination studies where the repair products were associated with very long GC tracts that extended to the end of the chromosome (Esposito 1978; Judd and Petes 1988; Voelkel-Meiman and Roeder 1990). A direct demonstration of BIR in yeast was made in studies showing that if DSBs were generated on a chromosome in a haploid strain, then the centromere-proximal end near the break invaded a homologous sequence on the other end of the chromosome and copied it to the end, resulting in the formation of a nonreciprocal translocation product (Bosco and Haber 1998). BIR in yeast has been shown to be important for the recovery of collapsed replication forks and has an essential role in recombination-dependent telomere maintenance in cells lacking telomerase (McEachern and Haber 2006). How replication fork assembly occurs for BIR in the context of a failed replication fork or during telomere maintenance is not fully understood. No PriA homolog in eukaryotes has been identified to date. Like other recombination pathways in yeast, Rad52 is essential for BIR. However, while BIR in yeast mostly proceeds in a Rad51-dependent manner, it can also operate independently of Rad51, albeit with lower efficiency (Fig. 2; Malkova et al. 2005).
Rad51-dependent BIR
More than 95% of BIR events in S. cerevisiae are reported to be Rad51-dependent and do not require either Rad50 or Rad59. Rad51-dependent BIR has been suggested to be essential for recombination-dependent restoration of a collapsed replication fork (Davis and Symington 2004). When homology occurs at both ends of DSBs, GC outcompetes BIR. Similarly, a DSB flanked by a direct repeat is preferentially repaired by SSA rather than by BIR. This suppression of BIR has been attributed to the delayed initiation of new strand synthesis, making BIR a kinetically slow process. However, once BIR is initiated, its rate of synthesis becomes comparable with normal replication (Malkova et al. 2005; Jain et al. 2009). Formation of Rad51 filaments during BIR relies on 5′–3′ resection of DSBs, which is initiated by MRX–Sae2 and is continued later by Exo1 or Sgs1–Dna2. However, it is important to note that genetic deletion of either Exo1, Sgs1, or Sae2 increases BIR efficiency. The basis for this somewhat counterintuitive observation is currently unclear (Lydeard et al. 2010; Marrero and Symington 2010; Anand et al. 2013). It is important to note that, in these studies, the BIR assays used substrates with limited homology. Since the 3′ single-stranded tail generated after resection are unstable, it is possible that some sequences at the 3′ end were lost owing to the slow kinetics of BIR, leaving no homologous substrate and resulting in decreased efficiency (Marrero and Symington 2010). Following resection, the ssDNA initially associates with RPA. Rad52 facilitates RPA displacement by Rad51 to promote nucleofilament formation on the 3′ overhang (Sugiyama and Kowalczykowski 2002; Krogh and Symington 2004). Various components of the S-phase machinery have been shown to be required for BIR (Lydeard et al. 2010). Around the same time, Pif1, a nonreplicative helicase, was shown to be essential for long-range BIR in yeast (Chung et al. 2010). Subsequent studies suggested that Pif1 rather than the replicative helicases Mcm2–7 are the major drivers of BIR (Wilson et al. 2013). Mechanistic studies outside the context of replication have demonstrated that BIR proceeds via a migrating bubble-like replication fork that moves down the length of a chromosome, resulting in the conservative inheritance of genetic material (Smith et al. 2007; Donnianni and Symington 2013; Saini et al. 2013; Wilson et al. 2013). This replication fork was driven by Pif1 helicase, and there was asynchronous leading and lagging strand synthesis, resulting in accumulation of ssDNAs, which are prone to mutations (Saini et al. 2013). Pol α primase and Pol δ have been shown to be essential for the initiation of DNA synthesis in Rad51-dependent BIR. In contrast, Pol ε is not required for initiation but is assumed to be required for completion of DNA synthesis over long stretches of DNA (Lydeard et al. 2007). This switch in the DNA polymerase has been suggested to convert BIR to a highly processive process. Strong genetic and biochemical studies demonstrated that, during BIR, Pif1 is required for the proper recruitment of Pol δ to the D loops generated by Rad51 (Wilson et al. 2013). The Pol 32 subunit was found to be essential for Rad51-dependent BIR (Lydeard et al. 2007). How Pol 32 promotes this recombination-mediated replication is yet to be determined, given that Pol 32 is not required for Pol δ processivity or collision release (Langston and O'Donnell 2008). Interestingly, a recent study demonstrated that BIR-driven mutagenic synthesis at broken replication forks can be restricted by either a converging replication fork or Mus81, an endonuclease that cleaves the D loop. This study suggests an elegant strategy by which cells maintain their genomic integrity during error-prone DNA synthesis at breaks harboring one-sided homology (Mayle et al. 2015). This begs the question of how often BIR ensues at genomic locations that lie in between replication origins. It also brings up a potentially new concept of checkpoints that slow down the initial migrating D-loop bubble for convergence with an opposing replication fork.
Rad51-independent BIR
Early evidence for Rad51-independent BIR came from studies in a rad51Δ strain where a DSB with one-ended homology was repaired in such a way that a 100-kb region distal to the breakpoint was copied from the homologous chromosome, resulting in loss of heterozygosity (Malkova et al. 1996). Concomitantly, it was demonstrated that rad51Δ rad59Δ double-mutant strains were more defective in recombination than rad51Δ or rad59Δ single mutants, suggesting a Rad51-independent pathway for spontaneous and DSB-induced recombination as well (Bai and Symington 1996). Rad51-independent recombination requires much less homology (30 bp) for strand invasion compared with Rad51-dependent repair (Ira and Haber 2002). A Rad52 mutant lacking the C-terminal Rad51 interaction domain is essential for this form of BIR (Tsukamoto et al. 2003). Yeast genetic studies have established the requirement of the MRX complex, Rad59, Radh54/Tid1, and Rad52 for Rad51-independent BIR (Signon et al. 2001). The requirement of Rad50 and Rad59 suggests that this form of BIR requires extensive resection and a strand-annealing mechanism (Symington et al. 2014). The importance of Rad50 in BIR, however, was found to be dependent on the kind of assay performed. While the aforementioned studies used site-specific endonucleases to induce DSBs with one-ended homology and suggested the importance of Rad50 in Rad51-independent BIR, chromosome fragmentation-based assays showed no role for Rad50 in either Rad51-dependent or -independent BIR (Davis and Symington 2004). Based on earlier Rad51-independent intraplasmid recombination studies (Ira and Haber 2002), it was proposed that Rad50 could be important during BIR between short repeats, such as those found in type II survivors, which maintain their telomeres via amplification of TG1–3 telomeric repeats. Together, these studies propose that Rad51-independent BIR primarily operates during intramolecular recombination; however, intermolecular events mostly rely on Rad51 strand invasion and are independent of Rad50 and Rad59 (Ira and Haber 2002; Davis and Symington 2004).
The idea for the existence of a Rad51-dependent and -independent BIR is strongly supported by studies in yeast that maintain their telomeres via a telomerase-independent recombination mechanism (Lundblad and Blackburn 1993; Le et al. 1999). Two kinds of survivors are observed in yeast that lack telomerase and can bypass senescence (Lundblad and Blackburn 1993; Teng and Zakian 1999; Chen et al. 2001). Type I survivors are Rad51-dependent and maintain their telomeres by frequent recombination between the conserved subtelomeric Y′ repeat sequences, resulting in the formation of telomeres with amplified Y′ sequences. This pathway requires Rad54, Rad55, and Rad57 and might involve resection of the telomere into subtelomeric regions, such as X and Y′ elements, to generate a 3′ single strand, which can invade a homologous sequence on other telomeres to promote BIR. On the other hand, type II survivors are Rad51-independent and require Rad59, Rdh54, Sgs1, and the MRX complex. Here, the imperfect nucleotide TG1–3 telomere sequences have been proposed to elongate by the invasion of these TG1–3 sequences into another TG1–3 sequence on the same chromosome through the formation of a t-loop-like structure or by annealing to single-stranded regions on a telomere from a different chromosome (Walmsley et al. 1984; Griffith et al. 1999; Le et al. 1999; Teng and Zakian 1999; Chen et al. 2001; Huang et al. 2001; Johnson et al. 2001; Tsukamoto et al. 2001). In comparison with the telomerase-positive cells, both kinds of survivors contain copious amounts of extrachromosomal circular DNA with telomeric sequences, which could act as a template for recombination-based telomere maintenance (McEachern and Haber 2006). While type I survivors harbor circular Y′ elements (Horowitz and Haber 1985), type II survivors contain heterogeneously sized extrachromosomal circles of TG1–3 DNA, which are similar to what is observed in some telomerase-negative human cells (Teng et al. 2000; Cesare and Griffith 2004; Lin et al. 2005). Regardless of the clear differences in the genetic requirements and products generated, Pol 32 is required for telomere synthesis in both type I and type II survivors. In addition, the finding that Pol 32 and Pif1 are required for telomere synthesis in these survivors further supports that this telomerase-independent telomere maintenance uses a BIR-related mechanism that requires processive DNA synthesis and is most likely independent of the canonical replisome used during S phase (Lydeard et al. 2007; Dewar and Lydall 2010; Hu et al. 2013).
BIR in higher eukaryotes have been mentioned in two different contexts.
BIR-related mechanism in alternative lengthening of telomeres (ALT)
Similar to yeast survivors discussed above, 15% of human cancers bypass senescence by using telomerase-independent mechanisms collectively known as ALT (Shay and Bacchetti 1997; Cesare and Reddel 2010; Dilley and Greenberg 2015). Early evidence that human telomeres can be maintained by recombination came from the observation that telomeres of telomerase-negative cells can undergo a rapid change in their length (Murnane et al. 1994). It was subsequently demonstrated that telomerase-negative cell lines contained long, heterogeneous telomeres (Bryan et al. 1995), and a “tag” engineered into one telomere can get copied to other telomeres in an ALT cell, suggesting the role of HR in maintaining these telomeres (Dunham et al. 2000). However, the specific HR mechanism and the replication machinery used for ALT telomere maintenance are not clearly understood. Additionally, it appears that ALT telomeres can be maintained by more than one mechanism, and hence the ALT phenotype cannot be attributed to a given pathway (Muntoni and Reddel 2005). Many human ALT telomeres maintain themselves in a Rad51-independent manner similar to the type II survivors in yeast (Potts and Yu 2007). Several recent studies suggest that nuclease-induced breaks or sustained replication stress within ALT telomeres induce long-range telomere movements that culminate in intertelomere associations, suggesting the occurrence of a specialized homology-searching process. Telomere clustering was partially dependent on Rad51 and also required the Hop2–Mnd1 heterodimer, which is crucial for extensive homolog pairing during meiosis (Cho et al. 2014). Rad51-dependent telomere clustering was also reported in ALT cells undergoing replication stress after the depletion of annealing helicase SMARCAL1 (Cox et al. 2016). Additionally, induction of replication-induced damage by depletion of histone chaperon ASF1 resulted in the induction of ALT activity even in immortalized human cells with longer telomeres (O'Sullivan et al. 2014). Intertelomere recombination during ALT may also be assisted by other unique and alternative mechanisms. Recent studies showed that C-type (TCAGGG) variant repeats in ALT telomeres recruit NR2C/F orphan nuclear receptors, which lead to telomere clustering events that can promote telomere recombination (Conomos et al. 2012; Marzec et al. 2015). What is unclear is the extent to which the Rad51-dependent telomere homology search contributes to break-induced telomere synthesis. It is possible that both type I and type II mechanisms may be occurring in parallel during ALT, as it is commonly observed that a large percentage of ALT telomeres experience persistent DSB responses that recruit a myriad of DNA repair proteins with opposing functions, including BRCA1, Rad51, and 53BP1 (Cesare et al. 2009; Cho et al. 2014). It will also be interesting to investigate commonalities in function between replicative DNA synthesis during scheduled S-phase and break-induced synthesis mechanisms that are thought to proceed during ALT; for example, whether factors like the BLM helicase, which facilitates the movement of replication forks on G-rich telomeric sequences in telomerase-positive cells, can perform a similar function during ALT telomere synthesis (Drosopoulos et al. 2015).
Various kinds of donor templates have been suggested for homology-directed ALT telomere recombination, which can be either intertelomeric or intratelomeric (Cesare and Reddel 2010). Telomeric recombination can occur within the same chromosome either via formation of a t-loop (Fig. 2) or by using the telomere of a sister chromatid as a donor template (Muntoni et al. 2009). Another source of the donor template can be the extrachromosomal DNA found in ALT cells, which can be in the form of circular double-stranded telomeric repeats called t-circles (Cesare and Griffith 2004; Wang et al. 2004), partially single-stranded G- or C-rich circles of telomeric DNA (Henson et al. 2009; Nabetani and Ishikawa 2009), linear extrachromosomal telomeric repeats (ECTRs) (Tokutake et al. 1998), or high-molecular-weight “t-complex” DNA (Nabetani and Ishikawa 2009). Telomeric chromatin, along with DNA recombination and repair proteins, can also exist in ALT-associated promyelocytic bodies (ABPs). While ABPs are considered markers of ALT recombination, ALT telomeres can be maintained in the absence of ABPs (Henson and Reddel 2010). The formation of t-circles was reported to be dependent on Nbs1 and Xrcc3 (Compton et al. 2007). These proteins have also been implicated in the formation of functional ABPs and telomere maintenance in ALT cells (Natarajan and McEachern 2002; Jiang et al. 2005). Strikingly, prevention of t-circle formation in ALT cells after Nbs1 or Xrcc3 knockdown was not lethal. The nonessential nature of ABPs or t-circles suggests the presence of functionally redundant pathways in maintaining ALT telomeres. Partially single-stranded C-rich circles (C-circles) have been shown to be a quantifiable marker for ALT activity in cells (Henson et al. 2009). Based on studies in yeast, it has been suggested that these t-circles and C-circles can serve as a template for rolling circle replication of telomeres and hence allow telomere maintenance in the absence of telomerase (Cesare and Griffith 2004; Wang et al. 2004). However, a direct in vivo experiment supporting the rolling circle model for ALT telomere replication in humans is lacking.
BIR-mediated synthesis during replication stress
Studies in mammalian cells have demonstrated that a small proportion of HDR involves long-tract GC (LTGC), where the invading strand is extended much longer compared with the more frequently observed short-tract GC (Richardson et al. 1998; Willis et al. 2015). As discussed above, one-ended DSBs in yeast have been shown to result in a BIR pathway that leads to DNA synthesis extending up to several hundred kilobases (Morrow et al. 1997). Because LTGC in mammals also results in the formation of long recombination products, it has been suggested to be analogous to BIR in yeast (Willis et al. 2015). Interestingly, depletion of BRCA1 or the Rad51 paralog XRCC3 increased the tract length of GC products (Brenneman et al. 2002; Nagaraju et al. 2009; Chandramouly et al. 2013). The absolute frequency of such LTGC products after site-specific replication stalling was found to be enhanced after BRCA1 and BRCA2 knockdown in mammalian cells, suggesting that the loss of canonical HR pathways due to BRCA deficiency may enhance mutagenic recombination events at stalled replication forks (Willis et al. 2014). It will be interesting to determine the genetic requirements for such BRCA-independent LTGC and whether such mechanisms represent compensatory repair pathways used to navigate replication stress in BRCA mutant cancers.
A direct demonstration of a BIR-like recombination event in human cells was recently reported (Costantino et al. 2014). In a Cyclin E overexpression model of replication stress, Pol D3, the mammalian homolog of Pol 32, was shown to be essential for cell viability. It was observed that depletion of Pol D3 specifically prevented Cyclin E-overexpressing cells from entering S phase and resulted in the accumulation of terminated forks. A BIR reporter was used to demonstrate Pol D3-dependent BIR-like recombination events in human cells after induction of one-ended DSBs. Additionally, it was demonstrated that CNVs induced by Cyclin E overexpression was dependent on Pol D3. Based on these observations, it was proposed that human cells may use a BIR mechanism to repair and restart broken replication forks (Costantino et al. 2014). It will be interesting to examine whether LTGC products obtained at the stalled replication fork are dependent on Pol D3. A further question of interest is the relative usage of BIR in comparison with the repair of a locus by a converging replication fork, which has also been shown to limit BIR-mediated synthesis (Mayle et al. 2015).
HR-dependent fork restart (HoRReR)
HoRReR is a recombination-dependent replication pathway in Schizosaccharomyces pombe. HoRReR was reported in the context of HR-mediated resumption of DNA synthesis following fork arrest at a naturally occurring replication fork barrier called RTS1. In contrast to BIR, which occurs in non-S-phase cells and is initiated by a DSB followed by end resection, HoRReR is initiated in S phase and does not involve a DSB intermediate. There is unwinding of the nascent strand that can then undergo template exchange with homologous sequences, initiating DNA synthesis on a noncontinuous template (Lambert et al. 2010). Although the exact mechanism initiating this response is not understood, this pathway requires Rad22, Rph1, Rad50, and Srs2 helicase. Recent characterization of the RTS1 model for HoRReR demonstrated that it proceeds via a semiconservative mechanism and primarily uses Pol δ for both leading and lagging strand synthesis (Daigaku et al. 2015). These forks, which are restarted by HR in fission yeast, have a high propensity to execute a U-turn at small inverted repeats, contributing to the error-prone nature of this pathway. It was suggested that such a pathway might be responsible for gross chromosomal arrangements and gene amplifications in human cancers (Mizuno et al. 2012).
Fork stalling and template switching (FoSTeS)/microhomology-mediated BIR (MMBIR)
The MMBIR pathway is a DNA replication-based pathway associated with the repair of stalled or collapsed replication forks. According to this model, subsequent to the collapse of a replication fork, the lagging 3′ single-stranded tail disengages and switches to a nearby microhomologous template and primes DNA synthesis. Templated synthesis can then proceed in either direction with low processivity followed by eventual resumption at the original template (Fig. 3). The recurrent template switching at positions of microhomology results in complex genomic rearrangements. FoSTeS was initially proposed to explain template switching events at regions of microhomology after replication fork stalling (Lee et al. 2007). Eventually, MMBIR was proposed as a more general model encompassing FoSTeS to account for complex rearrangements in the genome (Lee et al. 2007; Hastings et al. 2009a,b; Carvalho and Lupski 2016).
Figure 3.
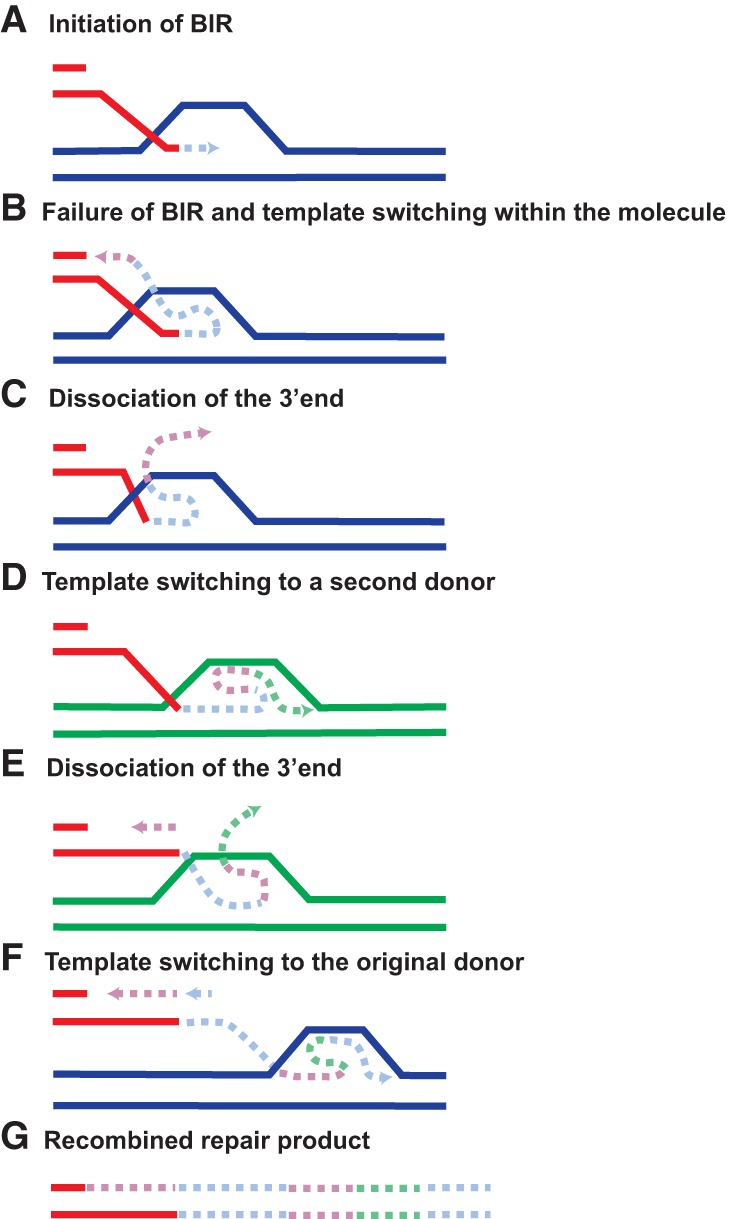
Proposed model for MMBIR. (A) Failure to repair collapsed replication forks by BIR has been shown to induce MMBIR. The 3′ end dissociates from the original donor template (in blue) and can anneal to microhomologous sequences present in the newly replicated ssDNA within the recipient molecule (in red) via a snapback mechanism (B,C) or at any other donor template (in green) (D,E). The 3′ strand extension proceeds with low processivity and involves multiple template switchings before annealing back to the original template (F), resulting in complex genomic rearrangements (G).
Studies focusing on stress-induced tandem gene amplifications in E. coli gave evidence for this interesting concept of a long-distance template switching model. The template switching was observed to occur between regions separated by 7–32 kb, and the breakpoint junctions had microhomology in the range of 4–15 bp. This pathway was found to be dependent on RecA, RecBC, the endonuclease domain of DNA Pol I, and the F-plasmid-encoded endonuclease TraI, which was proposed to initiate replication by generating a nick at the transfer origin of the plasmid (Slack et al. 2006). This study was performed on an F′ conjugative plasmid, and it is not clear whether a similar mechanism also operates on chromosomal DNA.
Evidence for an MMBIR-like process in yeast came from the genomic analysis of a temperature-sensitive S. cerevisae strain lacking an RFA1 allele that encodes for a RPA subunit. Large-scale deletions and nonreciprocal translocations were observed, and the breakpoints of these rearrangements were flanked by imperfect direct repeats of 2–20 bp. While rad10 and rad52 mutations eliminated this phenotype, a rad51 mutation increased the frequency of these events (Chen et al. 1998). Similarly, a subsequent study in a strain lacking the Sgs1 helicase showed frequent translocations and inversions associated with microhomology at sites of highly diverged genes. The group proposed that break-induced replication coupled with multiple cycles of template switching at regions of microhomology could explain these rearrangements (Schmidt et al. 2006). A related mechanism has also been suggested to account for segmental duplications in yeast. This microhomology-mediated duplication was found to be dependent on Pol 32, and the frequency of these events was enhanced by fork collapse and was negligibly affected by fork stalling (Payen et al. 2008).
Models of FoSTeS/MMBIR in higher eukaryotes, particularly humans, have been primarily invoked to account for complex genomic rearrangements and CNVs. Over 60%–69% of the human genome is repetitive in nature, making it susceptible to genomic rearrangement and CNVs (de Koning et al. 2011). Both canonical and noncanonical repair pathways, including NHEJ, NAHR, MMEJ, and FoSTeS/MMBIR, are involved in various genomic rearrangements. NAHR has been attributed to recurrent CNVs, which are common in regions of low-copy repeats. NAHR involves HR between DNA stretches that already exist as two or more copies. As a result, the sequences between these repeat stretches are either deleted or duplicated, resulting in copy number changes (Sasaki et al. 2010). NHEJ has been associated with various chromosomal translocations (Bunting and Nussenzweig 2013; Ghezraoui et al. 2014). NHEJ has also been proposed to be responsible for the recently discovered phenomena called chromothripsis, which is characterized by a massive number of chromosomal rearrangements in a single catastrophic event that are usually restricted to one chromosome. Chromothripsis was proposed to arise from chromosome shattering followed by restitching of DNA fragments in a rearranged fashion with loss of many intervening fragments (Berger et al. 2011). However, a recent study involving single-cell whole-genome sequencing of daughter cells derived from micronuclei (where shattering and rearrangement happen) revealed microhomology at the breakpoint junction of the rearranged genome, suggesting that both random joining of shattered chromosome fragments and MMBIR contribute to chromothripsis (Zhang et al. 2015).
FoSTeS/MMBIR has also been used to explain the process of chromoanasynthesis, which involves a combination of chromosomal rearrangements with both losses and gains in copy number (Liu et al. 2011b; Carvalho et al. 2011, 2013, 2015; Beck et al. 2015). This model was proposed to explain the nonrecurrent duplications of the PLP1 gene associated with the demyelinating disorder Pelizaeus-Merzbacher disease (PMD). Stretches of DNA with discontinuous duplications interspersed with deletions, inverted duplications, and triplications with 2- to 5-bp microhomology at the junctions were observed in patient samples (Lee et al. 2007). Subsequent studies on PMD and another neurological disease called MECP2 duplication syndrome, which is associated with rearrangements at the MECP2 gene locus, gave further evidence for the MMBIR pathway in complex genomic rearrangement. In patient samples, the PLP1 and MECP2 locus consisted of an inverted triplicated segment flanked by duplicated segments (DUP-TRP/INV-DUP). This kind of rearrangement in both genes was proposed to be facilitated by inverted repeats and was suggested to occur by a two-step process involving BIR followed by MMBIR (Carvalho et al. 2013; Beck et al. 2015). This hypothesis was recently proved in a pif1Δ yeast strain, where it was demonstrated that the collapse of a replication fork during BIR led to the initiation of MMBIR. This study also identified that translesion polymerases Rev1 and Pol ς drive MMBIR in yeast. In accordance with the earlier study by Fischer's group (Payen et al. 2008), Pol32, which is a subunit of both Pol ς and Pol δ, was also found to be required for MMBIR in yeast (Sakofsky et al. 2015). However, the DNA polymerases that drive MMBIR in mammals remain elusive.
An important question that arises is why cells switch to error-prone pathways such as FoSTeS/MMBIR. It has been suggested that hypoxia associated with the tumor microenvironment leads to repression of Rad51 and BRCA1 and reduces HR. It was suggested that, in the absence of these proteins required for GC, cells would switch to NHEJ. However, at sites where NHEJ is not possible, such as at single-ended DSBs, it was proposed that error-prone pathways like MMBIR might be initiated (Hastings et al. 2009a).
Template switching seems to be a recurrent theme to account for large-scale genomic rearrangements. Template switching has also been proposed in other DSB repair pathways, including BIR (Smith et al. 2007; Štafa et al. 2014) and GC (Hicks et al. 2010). What triggers these switches, what molecular machinery is involved, and how the processivity of different DNA polymerases contributes to these switching events remain to be examined.
Concluding remarks
With the continuous advancement of genome sequencing technology, the footprint of noncanonical HDR pathways is becoming increasingly large (Kidd et al. 2008; Liu et al. 2012; Carvalho and Lupski 2016). Therefore, it is not surprising that the concept of targeting alternative DSB repair pathways has been actively viewed as a promising therapeutic. One such example is the discovery of PARP inhibitors, which may target the homology-directed MMEJ pathway in HR-deficient cancers (Bryant et al. 2005; Farmer et al. 2005; Yousefzadeh et al. 2014; Ceccaldi et al. 2015; Mateos-Gomez et al. 2015). Similarly, preventing DNA recombination by trapping RPA–ssDNA via ATR inhibitors was found to selectively target ALT-positive cells (Flynn et al. 2015). Understanding the mechanisms that underlie the activation and maintenance of these pathways and thereby lead to cancer genome evolution is critical to understand how and in what genetic context they should be targeted. It is striking to note the evolutionary conservation of most of these noncanonical DNA repair pathways in prokaryotic and eukaryotic organisms, despite significant differences in their genome organization and chromatin structure. This highlights the importance of mutagenic repair mechanisms in the normal physiology of a cell and suggests that these repair pathways might confer adaptive advantages. While almost universally considered pathogenic, interestingly, a recent study reported the cure of WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis) syndrome in a patient via a chromothripsis-like pathway, resulting in the loss of 164 genes (McDermott et al. 2015) and suggesting a role in genome evolution that could be selected for in certain scenarios. Continuing to examine these pathways in experimentally tractable systems will provide new mechanistic and functional insights into these alternative DSB repair strategies.
Acknowledgments
This work was supported by National Institutes of Health grants GM101149 and CA17494 to R.A.G., who is also supported by funds from the Abramson Family Cancer Research Institute and the Basser Research Center for BRCA.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.280545.116.
References
- Adair GM, Rolig RL, Moore-Faver D, Zabelshansky M, Wilson JH, Nairn RS. 2000. Role of ERCC1 in removal of long non-homologous tails during targeted homologous recombination. EMBO J 19: 5552–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MD, McVey M, Sekelsky JJ. 2003. Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science 299: 265–267. [DOI] [PubMed] [Google Scholar]
- Adelman CA, Boulton SJ. 2010. Metabolism of postsynaptic recombination intermediates. FEBS Lett 584: 3709–3716. [DOI] [PubMed] [Google Scholar]
- Agmon N, Pur S, Liefshitz B, Kupiec M. 2009. Analysis of repair mechanism choice during homologous recombination. Nucleic Acids Res 37: 5081–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allers T, Lichten M. 2009. Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 106: 47–57. [DOI] [PubMed] [Google Scholar]
- Anand RP, Lovett ST, Haber JE. 2013. Break-induced DNA replication. Cold Spring Harb Perspect Biol 5: a010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aniukwu J, Glickman MS, Shuman S. 2008. The pathways and outcomes of mycobacterial NHEJ depend on the structure of the broken DNA ends. Genes Dev 22: 512–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai T, Sommer S, Bailone A, Kogoma T. 1993. Homologous recombination-dependent initiation of DNA replication from DNA damage-inducible origins in Escherichia coli. EMBO J 12: 3287–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audebert M, Salles B, Calsou P. 2004. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem 279: 55117–55126. [DOI] [PubMed] [Google Scholar]
- Ayora S, Carrasco B, Cardenas PP, Cesar CE, Canas C, Yadav T, Marchisone C, Alonso JC. 2011. Double-strand break repair in bacteria: a view from Bacillus subtilis. FEMS Microbiol Rev 35: 1055–1081. [DOI] [PubMed] [Google Scholar]
- Bai Y, Symington LS. 1996. A Rad52 homolog is required for RAD51-independent mitotic recombination in Saccharomyces cerevisiae. Genes Dev 10: 2025–2037. [DOI] [PubMed] [Google Scholar]
- Beck CR, Carvalho CMB, Banser L, Gambin T, Stubbolo D, Yuan B, Sperle K, McCahan SM, Henneke M, Seeman P, et al. 2015. Complex genomic rearrangements at the PLP1 locus include triplication and quadruplication. PLoS Genet 11: e1005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmaaza A, Chartrand P. 1994. One-sided invasion events in homologous recombination at double-strand breaks. Mutat Res 314: 199–208. [DOI] [PubMed] [Google Scholar]
- Bennardo N, Cheng A, Huang N, Stark JM. 2008. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet 4: e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, et al. 2011. The genomic complexity of primary human prostate cancer. Nature 470: 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco G, Haber JE. 1998. Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics 150: 1037–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton SJ, Jackson SP. 1996. Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J 15: 5093–5103. [PMC free article] [PubMed] [Google Scholar]
- Bowater R, Doherty AJ. 2006. Making ends meet: repairing breaks in bacterial DNA by non-homologous end-joining. PLoS Genet 2: e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneman MA, Wagener BM, Miller CA, Allen C, Nickoloff JA. 2002. XRCC3 controls the fidelity of homologous recombination: roles for XRCC3 in late stages of recombination. Mol Cell 10: 387–395. [DOI] [PubMed] [Google Scholar]
- Bryan TM, Englezou A, Gupta J, Bacchetti S, Reddel RR. 1995. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J 14: 4240–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434: 913–917. [DOI] [PubMed] [Google Scholar]
- Bryk M, Belisle M, Mueller JE, Belfort M. 1995. Selection of a remote cleavage site by I-tevI, the td intron-encoded endonuclease. J Mol Biol 247: 197–210. [DOI] [PubMed] [Google Scholar]
- Bunting SF, Nussenzweig A. 2013. End-joining, translocations and cancer. Nat Rev Cancer 13: 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bzymek M, Lovett ST. 2001. Evidence for two mechanisms of palindrome-stimulated deletion in Escherichia coli: single-strand annealing and replication slipped mispairing. Genetics 158: 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Cejka P. 2014. Sae2 promotes dsDNA endonuclease activity within Mre11–Rad50–Xrs2 to resect DNA breaks. Nature 514: 122–125. [DOI] [PubMed] [Google Scholar]
- Carvalho CMB, Lupski JR. 2016. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 17: 224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Zhang F, Lupski JR. 2011. Structural variation of the human genome: mechanisms, assays, and role in male infertility. Syst Biol Reprod Med 57: 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CMB, Pehlivan D, Ramocki MB, Fang P, Alleva B, Franco LM, Belmont JW, Hastings PJ, Lupski JR. 2013. Replicative mechanisms for CNV formation are error prone. Nat Genet 45: 1319–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CMB, Pfundt R, King DA, Lindsay SJ, Zuccherato LW, Macville MVE, Liu P, Johnson D, Stankiewicz P, Brown CW, et al. 2015. Absence of heterozygosity due to template switching during replicative rearrangements. Am J Hum Genet 96: 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MIR, O'Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, et al. 2015. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 518: 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Rondinelli B, D'Andrea AD. 2016. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol 26: 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare AJ, Griffith JD. 2004. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol Cell Biol 24: 9948–9957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare AJ, Reddel RR. 2010. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet 11: 319–330. [DOI] [PubMed] [Google Scholar]
- Cesare AJ, Kaul Z, Cohen SB, Napier CE, Pickett HA, Neumann AA, Reddel RR. 2009. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat Struct Mol Biol 16: 1244–1251. [DOI] [PubMed] [Google Scholar]
- Chan SH, Yu AM, McVey M. 2010. Dual roles for DNA polymerase theta in alternative end-joining repair of double-strand breaks in Drosophila. PLoS Genet 6: e1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramouly G, Kwok A, Huang B, Willis NA, Xie A, Scully R. 2013. BRCA1 and CtIP suppress long-tract gene conversion between sister chromatids. Nat Commun 4: 2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Taylor MRG, Boulton SJ. 2012. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 47: 497–510. [DOI] [PubMed] [Google Scholar]
- Chayot R, Montagne B, Mazel D, Ricchetti M. 2010. An end-joining repair mechanism in Escherichia coli. Proc Natl Acad Sci 107: 2141–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Umezu K, Kolodner RD. 1998. Chromosomal rearrangements occur in S. cerevisiae rfa1 mutator mutants due to mutagenic lesions processed by double-strand-break repair. Mol Cell 2: 9–22. [DOI] [PubMed] [Google Scholar]
- Chen Q, Ijpma A, Greider CW. 2001. Two survivor pathways that allow growth in the absence of telomerase are generated by distinct telomere recombination events. Mol Cell Biol 21: 1819–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho NW, Dilley RL, Lampson MA, Greenberg RA. 2014. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 159: 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W-H, Zhu Z, Papusha A, Malkova A, Ira G. 2010. Defective resection at DNA double-strand breaks leads to de novo telomere formation and enhances gene targeting. PLoS Genet 6: e1000948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton SA, Choi J-H, Cesare AJ, Özgür S, Griffith JD. 2007. Xrcc3 and Nbs1 are required for the production of extrachromosomal telomeric circles in human alternative lengthening of telomere cells. Cancer Res 67: 1513–1519. [DOI] [PubMed] [Google Scholar]
- Conomos D, Stutz MD, Hills M, Neumann AA, Bryan TM, Reddel RR, Pickett HA. 2012. Variant repeats are interspersed throughout the telomeres and recruit nuclear receptors in ALT cells. J Cell Biol 199: 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corneo B, Wendland RL, Deriano L, Cui X, Klein IA, Wong S-Y, Arnal S, Holub AJ, Weller GR, Pancake BA, et al. 2007. Rag mutations reveal robust alternative end joining. Nature 449: 483–486. [DOI] [PubMed] [Google Scholar]
- Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, Halazonetis TD. 2014. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343: 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox KE, Marechal A, Flynn RL. 2016. SMARCAL1 resolves replication stress at ALT telomeres. Cell Rep 14: 1032–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigaku Y, Keszthelyi A, Müller CA, Miyabe I, Brooks T, Retkute R, Hubank M, Nieduszynski CA, Carr AM. 2015. A global profile of replicative polymerase usage. Nat Struct Mol Biol 22: 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AP, Symington LS. 2004. RAD51-dependent break-induced replication in yeast. Mol Cell Biol 24: 2344–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decottignies A. 2007. Microhomology-mediated end joining in fission yeast is repressed by pku70 and relies on genes involved in homologous recombination. Genetics 176: 1403–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning APJ, Gu W, Castoe TA, Batzer MA, Pollock DD. 2011. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet 7: e1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng SK, Gibb B, de Almeida MJ, Greene EC, Symington LS. 2014. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat Struct Mol Biol 21: 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar JM, Lydall D. 2010. Pif1- and Exo1-dependent nucleases coordinate checkpoint activation following telomere uncapping. EMBO J 29: 4020–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio MJ, Petersen S, Chen HT, Johnson R, Jasin M, Kanaar R, Ried T, Nussenzweig A. 2002. Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J Exp Med 196: 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilley RL, Greenberg RA. 2015. ALTernative telomere maintenance and cancer. Trends Cancer 1: 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, Ferguson DO. 2009. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol 16: 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnianni RA, Symington LS. 2013. Break-induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci 110: 13475–13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosopoulos WC, Kosiyatrakul ST, Schildkraut CL. 2015. BLM helicase facilitates telomere replication during leading strand synthesis of telomeres. J Cell Biol 210: 191–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley DD, Chaudhuri J, Bassing CH, Alt FW. 2005. Mechanism and control of V(D)J recombination versus class switch recombination: similarities and differences. Adv Immunol 86: 43–112. [DOI] [PubMed] [Google Scholar]
- Dunham MA, Neumann AA, Fasching CL, Reddel RR. 2000. Telomere maintenance by recombination in human cells. Nat Genet 26: 447–450. [DOI] [PubMed] [Google Scholar]
- Dupaigne P, Le Breton C, Fabre F, Gangloff S, Le Cam E, Veaute X. 2008. The Srs2 helicase activity is stimulated by Rad51 filaments on dsDNA: implications for crossover incidence during mitotic recombination. Mol Cell 29: 243–254. [DOI] [PubMed] [Google Scholar]
- Elliott B, Richardson C, Jasin M. 2005. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol Cell 17: 885–894. [DOI] [PubMed] [Google Scholar]
- Esposito MS. 1978. Evidence that spontaneous mitotic recombination occurs at the two-strand stage. Proc Natl Acad Sci 75: 4436–4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921. [DOI] [PubMed] [Google Scholar]
- Fasching CL, Cejka P, Kowalczykowski SC, Heyer W-D. 2015. Top3-Rmi1 dissolve Rad51-mediated D loops by a topoisomerase-based mechanism. Mol Cell 57: 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson DO, Holloman WK. 1996. Recombinational repair of gaps in DNA is asymmetric in Ustilago maydis and can be explained by a migrating D-loop model. Proc Natl Acad Sci 93: 5419–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn KJ, Li JJ. 2013. Single-stranded annealing induced by re-initiation of replication origins provides a novel and efficient mechanism for generating copy number expansion via non-allelic homologous recombination. PLoS Genet 9: e1003192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman-Lobell J, Haber J. 1992. Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science 258: 480–484. [DOI] [PubMed] [Google Scholar]
- Fishman-Lobell J, Rudin N, Haber JE. 1992. Two alternative pathways of double-strand break repair that are kinetically separable and independently modulated. Mol Cell Biol 12: 1292–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RL, Cox KE, Jeitany M, Wakimoto H, Bryll AR, Ganem NJ, Bersani F, Pineda JR, Suva ML, Benes CH, et al. 2015. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347: 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui K, Nakagawa N, Kitamura Y, Nishida Y, Masui R, Kuramitsu S. 2008. Crystal structure of MutS2 endonuclease domain and the mechanism of homologous recombination suppression. J Biol Chem 283: 33417–33427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbai CB, Marians KJ. 2010. Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA Repair (Amst) 9: 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia V, Phelps SEL, Gray S, Neale MJ. 2011. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 479: 241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari K, Décaillet C, Stasiak AZ, Stasiak A, Constantinou A. 2008. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol Cell 29: 141–148. [DOI] [PubMed] [Google Scholar]
- Ghezraoui H, Piganeau M, Renouf B, Renaud J-B, Sallmyr A, Ruis B, Oh S, Tomkinson AE, Hendrickson EA, Giovannangeli C, et al. 2014. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol Cell 55: 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson LA, Stahl FW. 1996. A test of the double-strand break repair model for meiotic recombination in Saccharomyces Cerevisiae. Genetics 144: 27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T. 1999. Mammalian telomeres end in a large duplex loop. Cell 97: 503–514. [DOI] [PubMed] [Google Scholar]
- Gupta R, Barkan D, Redelman-Sidi G, Shuman S, Glickman MS. 2011. Mycobacteria exploit three genetically distinct DNA double-strand break repair pathways. Mol Microbiol 79: 316–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Shuman S, Glickman MS. 2015. RecF and RecR play critical roles in the homologous recombination and single-strand annealing pathways of mycobacteria. J Bacteriol 197: 3121–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa N, Morimatsu K, Lovett ST, Kowalczykowski SC. 2009. Reconstitution of initial steps of dsDNA break repair by the RecF pathway of E. coli. Genes Dev 23: 1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. 2009a. A microhomology-mediated break-induced replication model for the origin of human copy number variation. ed. I. Matic. PLoS Genet 5: e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Lupski JR, Rosenberg SM, Ira G. 2009b. Mechanisms of change in gene copy number. Nat Rev Genet 10: 551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson JD, Reddel RR. 2010. Assaying and investigating alternative lengthening of telomeres activity in human cells and cancers. FEBS Lett 584: 3800–3811. [DOI] [PubMed] [Google Scholar]
- Henson JD, Cao Y, Huschtscha LI, Chang AC, Au AYM, Pickett HA, Reddel RR. 2009. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat Biotech 27: 1181–1185. [DOI] [PubMed] [Google Scholar]
- Heyer W-D, Ehmsen KT, Liu J. 2010. Regulation of homologous recombination in eukaryotes. Annu Rev Genet 44: 113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks WM, Kim M, Haber JE. 2010. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 329: 82–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg M, Sauer-Eriksson AE, Johansson E. 2012. Promiscuous DNA synthesis by human DNA polymerase theta. Nucleic Acids Res 40: 2611–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz H, Haber JE. 1985. Identification of autonomously replicating circular subtelomeric Y′ elements in Saccharomyces cerevisiae. Mol Cell Biol 5: 2369–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Tang H-B, Liu N-N, Tong X-J, Dang W, Duan Y-M, Fu X-H, Zhang Y, Peng J, Meng F-L, et al. 2013. Telomerase-null survivor screening identifies novel telomere recombination regulators. PLoS Genet 9: e1003208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Pryde FE, Lester D, Maddison RL, Borts RH, Hickson ID, Louis EJ. 2001. SGS1 is required for telomere elongation in the absence of telomerase. Curr Biol 11: 125–129. [DOI] [PubMed] [Google Scholar]
- Ira G, Haber JE. 2002. Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol Cell Biol 22: 6384–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, Foiani M, Haber JE. 2003. Srs2 and Sgs1–Top3 suppress crossovers during double-strand break repair in yeast. Cell 115: 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ithurbide S, Bentchikou E, Coste G, Bost B, Servant P, Sommer S. 2015. Single strand annealing plays a major role in RecA-independent recombination between repeated sequences in the radioresistant Deinococcus radiodurans bacterium. PLoS Genet 11: e1005636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov EL, Haber JE. 1995. RAD1 and RAD10, but not other excision repair genes, are required for double-strand break-induced recombination in Saccharomyces cerevisiae. Mol Cell Biol 15: 2245–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov EL, Sugawara N, Fishman-Lobell J, Haber JE. 1996. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics 142: 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Sugawara N, Lydeard J, Vaze M, Tanguy Le Gac N, Haber JE. 2009. A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev 23: 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Greenberg R. 2015. Deciphering the BRCA1 tumor suppressor network. J Biol Chem 290: 17724–17732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W-Q, Zhong Z-H, Henson JD, Neumann AA, Chang AC-M, Reddel RR. 2005. Suppression of alternative lengthening of telomeres by Sp100-mediated sequestration of the MRE11/RAD50/NBS1 complex. Mol Cell Biol 25: 2708–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson FB, Marciniak RA, McVey M, Stewart SA, Hahn WC, Guarente L. 2001. The Saccharomyces cerevisiae WRN homolog Sgs1p participates in telomere maintenance in cells lacking telomerase. EMBO J 20: 905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judd SR, Petes TD. 1988. Physical lengths of meiotic and mitotic gene conversion tracts in Saccharomyces cerevisiae. Genetics 118: 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantake N, Madiraju MVVM, Sugiyama T, Kowalczykowski SC. 2002. Escherichia coli RecO protein anneals ssDNA complexed with its cognate ssDNA-binding protein: a common step in genetic recombination. Proc Natl Acad Sci 99: 15327–15332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S, Neale MJ. 2006. Initiation of meiotic recombination by formation of DNA double-strand breaks: mechanism and regulation. Biochem Soc Trans 34: 523–525. [DOI] [PubMed] [Google Scholar]
- Kent T, Chandramouly G, McDevitt SM, Ozdemir AY, Pomerantz RT. 2015. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat Struct Mol Biol 22: 230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, et al. 2008. Mapping and sequencing of structural variation from eight human genomes. Nature 453: 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolomietz E, Meyn MS, Pandita A, Squire JA. 2002. The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer 35: 97–112. [DOI] [PubMed] [Google Scholar]
- Krejci L, Van Komen S, Li Y, Villemain J, Reddy MS, Klein H, Ellenberger T, Sung P. 2003. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423: 305–309. [DOI] [PubMed] [Google Scholar]
- Krogh BO, Symington LS. 2004. Recombination proteins in yeast. Annu Rev Genet 38: 233–271. [DOI] [PubMed] [Google Scholar]
- Lambert S, Mizuno K, Blaisonneau J, Martineau S, Chanet R, Fréon K, Murray JM, Carr AM, Baldacci G. 2010. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol Cell 39: 346–359. [DOI] [PubMed] [Google Scholar]
- Langston LD, O'Donnell M. 2008. DNA polymerase δ is highly processive with proliferating cell nuclear antigen and undergoes collision release upon completing DNA. J Biol Chem 283: 29522–29531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao JP, Oh SD, Shinohara M, Shinohara A, Hunter N. 2008. Rad52 promotes postinvasion steps of meiotic double-strand-break repair. Mol Cell 29: 517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le S, Moore JK, Haber JE, Greider CW. 1999. RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 152: 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EH, Kornberg A. 1991. Replication deficiencies in priA mutants of Escherichia coli lacking the primosomal replication n′ protein. Proc Natl Acad Sci 88: 3029–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Carvalho CMB, Lupski JR. 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131: 1235–1247. [DOI] [PubMed] [Google Scholar]
- Lee-Theilen M, Matthews AJ, Kelly D, Zheng S, Chaudhuri J. 2010. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat Struct Mol Biol 18: 75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Dong J, Eichmiller R, Holland C, Minca E, Prakash R, Sung P, Yong Shim E, Surtees JA, Eun Lee S. 2013. Role of Saw1 in Rad1/Rad10 complex assembly at recombination intermediates in budding yeast. EMBO J 32: 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR. 2010. The mechanism of double-strand DNA break repair by the nonhomologous DNA end joining pathway. Annu Rev Biochem 79: 181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin FL, Sperle K, Sternberg N. 1984. Model for homologous recombination during transfer of DNA into mouse L cells: role for DNA ends in the recombination process. Mol Cell Biol 4: 1020–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C-Y, Chang H-H, Wu K-J, Tseng S-F, Lin C-C, Lin C-P, Teng S-C. 2005. Extrachromosomal telomeric circles contribute to Rad52-, Rad50-, and polymerase δ-mediated telomere–telomere recombination in Saccharomyces cerevisiae. Eukaryot Cell 4: 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Renault L, Veaute X, Fabre F, Stahlberg H, Heyer W-D. 2011a. Rad51 paralogues Rad55–Rad57 balance the antirecombinase Srs2 in Rad51 filament formation. Nature 479: 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Erez A, Sreenath Nagamani SC, Dhar SU, Kołodziejska KE, Dharmadhikari AV, Cooper ML, Wiszniewska J, Zhang F, Withers MA, et al. 2011b. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 146: 889–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Carvalho CMB, Hastings PJ, Lupski JR. 2012. Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev 22: 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad V, Blackburn EH. 1993. An alternative pathway for yeast telomere maintenance rescues est1− senescence. Cell 73: 347–360. [DOI] [PubMed] [Google Scholar]
- Lydeard JR, Jain S, Yamaguchi M, Haber JE. 2007. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448: 820–823. [DOI] [PubMed] [Google Scholar]
- Lydeard JR, Lipkin-Moore Z, Sheu Y-J, Stillman B, Burgers PM, Haber JE. 2010. Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev 24: 1133–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J-L, Kim EM, Haber JE, Lee SE. 2003. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol Cell Biol 23: 8820–8828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova A, Ivanov EL, Haber JE. 1996. Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc Natl Acad Sci 93: 7131–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova A, Naylor ML, Yamaguchi M, Ira G, Haber JE. 2005. RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51-mediated gene conversion. Mol Cell Biol 25: 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marians KJ. 2000. PriA-directed replication fork restart in Escherichia coli. Trends Biochem Sci 25: 185–189. [DOI] [PubMed] [Google Scholar]
- Marrero VA, Symington LS. 2010. Extensive DNA end processing by Exo1 and Sgs1 inhibits break-induced replication. PLoS Genet 6: e1001007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzec P, Armenise C, Perot G, Roumelioti F-M, Basyuk E, Gagos S, Chibon F, Dejardin J. 2015. Nuclear-receptor-mediated telomere insertion leads to genome instability in ALT cancers. Cell 160: 913–927. [DOI] [PubMed] [Google Scholar]
- Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, Sfeir A. 2015. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 518: 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayle R, Campbell IM, Beck CR, Yu Y, Wilson M, Shaw CA, Bjergbaek L, Lupski JR, Ira G. 2015. DNA REPAIR. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science 349: 742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazon G, Lam AF, Ho CK, Kupiec M, Symington LS. 2012. The Rad1–Rad10 nuclease promotes chromosome translocations between dispersed repeats. Nat Struct Mol Biol 19: 964–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott DH, Gao J-L, Liu Q, Siwicki M, Martens C, Jacobs P, Velez D, Yim E, Bryke CR, Hsu N, et al. 2015. Chromothriptic cure of WHIM syndrome. Cell 160: 686–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEachern MJ, Haber JE. 2006. Break-induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem 75: 111–135. [DOI] [PubMed] [Google Scholar]
- McVey M, Lee SE. 2008. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet 24: 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D, Fu BXH, Heyer W-D. 2015. DNA polymerases δ and λ cooperate in repairing double-strand breaks by microhomology-mediated end-joining in Saccharomyces cerevisiae. Proc Natl Acad Sci 112: E6907–E6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. 2008. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455: 770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchel K, Zhang H, Welz-Voegele C, Jinks-Robertson S. 2010. Molecular structures of crossover and noncrossover intermediates during gap repair in yeast: implications for recombination. Mol Cell 38: 211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno K, Miyabe I, Schalbetter SA, Carr AM, Murray JM. 2012. Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature 493: 246–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow DM, Connelly C, Hieter P. 1997. ‘Break copy’ duplication: a model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics 147: 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntoni A, Reddel RR. 2005. The first molecular details of ALT in human tumor cells. Hum Mol Genet 14: R191–R196. [DOI] [PubMed] [Google Scholar]
- Muntoni A, Neumann AA, Hills M, Reddel RR. 2009. Telomere elongation involves intra-molecular DNA replication in cells utilizing alternative lengthening of telomeres. Hum Mol Genet 18: 1017–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murnane JP, Sabatier L, Marder BA, Morgan WF. 1994. Telomere dynamics in an immortal human cell line. EMBO J 13: 4953–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabetani A, Ishikawa F. 2009. Unusual telomeric DNAs in human telomerase-negative immortalized cells. Mol Cell Biol 29: 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraju G, Hartlerode A, Kwok A, Chandramouly G, Scully R. 2009. XRCC2 and XRCC3 regulate the balance between short- and long-tract gene conversions between sister chromatids. Mol Cell Biol 29: 4283–4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassif N, Penney J, Pal S, Engels WR, Gloor GB. 1994. Efficient copying of nonhomologous sequences from ectopic sites via P-element-induced gap repair. Mol Cell Biol 14: 1613–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan S, McEachern MJ. 2002. Recombinational telomere elongation promoted by DNA circles. Mol Cell Biol 22: 4512–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JA, Cooper CDO, Aitkenhead H, Gileadi O. 2015. Structure of the helicase domain of DNA polymerase theta reveals a possible role in the microhomology-mediated end-joining pathway. Structure 23: 2319–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse P, Zavitz KH, Marians KJ. 1991. Inactivation of the Escherichia coli priA DNA replication protein induces the SOS response. J Bacteriol 173: 6686–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan RJ, Arnoult N, Lackner DH, Oganesian L, Haggblom C, Corpet A, Almouzni G, Karlseder J. 2014. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat Struct Mol Biol 21: 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozenberger BA, Roeder GS. 1991. A unique pathway of double-strand break repair operates in tandemly repeated genes. Mol Cell Biol 11: 1222–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliwal S, Kanagaraj R, Sturzenegger A, Burdova K, Janscak P. 2014. Human RECQ5 helicase promotes repair of DNA double-strand breaks by synthesis-dependent strand annealing. Nucleic Acids Res 42: 2380–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pâques F, Haber JE. 1999. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63: 349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pâques F, Leung W-Y, Haber JE. 1998. Expansions and contractions in a tandem repeat induced by double-strand break repair. Mol Cell Biol 18: 2045–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payen C, Koszul R, Dujon B, Fischer G. 2008. Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet 4: e1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter SE, White MA, Petes TD. 1993. Genetic evidence that the meiotic recombination hotspot at the His4 locus of Saccharomyces cerevisiae does not represent a site for a symmetrically processed double-strand break. Genetics 134: 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts PR, Yu H. 2007. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat Struct Mol Biol 14: 581–590. [DOI] [PubMed] [Google Scholar]
- Prakash R, Satory D, Dray E, Papusha A, Scheller J, Kramer W, Krejci L, Klein H, Haber JE, Sung P, et al. 2009. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev 23: 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, Zhang Y, Feng W, Jasin M. 2015. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol 7: a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. 2009. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol 16: 819–824. [DOI] [PubMed] [Google Scholar]
- Resnick MA. 1976. The repair of double-strand breaks in DNA: a model involving recombination. J Theor Biol 59: 97–106. [DOI] [PubMed] [Google Scholar]
- Richardson C, Moynahan ME, Jasin M. 1998. Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev 12: 3831–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EPC, Cornet E, Michel B. 2005. Comparative and evolutionary analysis of the bacterial homologous recombination systems. PLoS Genet 1: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers K, McVey M. 2015. Error-prone repair of DNA double-strand breaks. J Cell Physiol 231: 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, Ira G, Haber JE, Lobachev KS, Malkova A. 2013. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 502: 389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky CJ, Ayyar S, Deem AK, Chung W-H, Ira G, Malkova A. 2015. Translesion polymerases drive microhomology-mediated break-induced replication leading to complex chromosomal rearrangements. Mol Cell 60: 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saparbaev M, Prakash L, Prakash S. 1996. Requirement of mismatch repair genes MSH2 and MSH3 in the RAD1-RAD10 pathway of mitotic recombination in Saccharomyces cerevisiae. Genetics 142: 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Lange J, Keeney S. 2010. Genome destabilization by homologous recombination in the germ line. Nat Rev Mol Cell Biol 11: 182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt KH, Wu J, Kolodner RD. 2006. Control of translocations between highly diverged genes by Sgs1, the Saccharomyces cerevisiae homolog of the Bloom's syndrome protein. Mol Cell Biol 26: 5406–5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, de Lange T. 2012. Removal of shelterin reveals the telomere end-protection problem. Science 336: 593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, Symington LS. 2015. Microhomology-mediated end joining: a back-up survival mechanism or dedicated pathway? Trends Biochem Sci 40: 701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay JW, Bacchetti S. 1997. A survey of telomerase activity in human cancer. Eur J Cancer 33: 787–791. [DOI] [PubMed] [Google Scholar]
- Signon L, Malkova A, Naylor ML, Klein H, Haber JE. 2001. Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol Cell Biol 21: 2048–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek D, Jasin M. 2010. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4–ligase IV during chromosomal translocation formation. Nat Struct Mol Biol 17: 410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek D, Brunet E, Wong SY-W, Katyal S, Gao Y, McKinnon PJ, Lou J, Zhang L, Li J, Rebar EJ, et al. 2011. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet 7: e1002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack A, Thornton PC, Magner DB, Rosenberg SM, Hastings PJ. 2006. On the mechanism of gene amplification induced under stress in Escherichia coli. PLoS Genet 2: e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CE, Llorente B, Symington LS. 2007. Template switching during break-induced replication. Nature 447: 102–105. [DOI] [PubMed] [Google Scholar]
- Štafa A, Miklenić M, Žunar B, Lisnić B, Symington LS, Svetec I-K. 2014. Sgs1 and Exo1 suppress targeted chromosome duplication during ends-in and ends-out gene targeting. DNA Repair (Amst) 22: 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. 2004. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol 24: 9305–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathern JN, Klar AJ, Hicks JB, Abraham JA, Ivy JM, Nasmyth KA, McGill C. 1982. Homothallic switching of yeast mating type cassettes is initiated by a double-stranded cut in the MAT locus. Cell 31: 183–192. [DOI] [PubMed] [Google Scholar]
- Sugawara N, Pâques F, Colaiácovo M, Haber JE. 1997. Role of Saccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. Proc Natl Acad Sci 94: 9214–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Ira G, Haber JE. 2000. DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. Mol Cell Biol 20: 5300–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Kowalczykowski SC. 2002. Rad52 protein associates with replication protein A (RPA)–single-stranded DNA to accelerate Rad51-mediated displacement of RPA and presynaptic complex formation. J Biol Chem 277: 31663–31672. [DOI] [PubMed] [Google Scholar]
- Sung P, Klein H. 2006. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol 7: 739–750. [DOI] [PubMed] [Google Scholar]
- Sweigert SE, Carroll D. 1990. Repair and recombination of X-irradiated plasmids in Xenopus laevis oocytes. Mol Cell Biol 10: 5849–5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS. 2002. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev 66: 630–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Gautier J. 2011. Double-strand break end resection and repair pathway choice. Annu Rev Genet 45: 247–271. [DOI] [PubMed] [Google Scholar]
- Symington LS, Rothstein R, Lisby M. 2014. Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics 198: 795–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng SC, Zakian VA. 1999. Telomere–telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol Cell Biol 19: 8083–8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng SC, Chang J, McCowan B, Zakian VA. 2000. Telomerase-independent lengthening of yeast telomeres occurs by an abrupt Rad50p-dependent, Rif-inhibited recombinational process. Mol Cell 6: 947–952. [DOI] [PubMed] [Google Scholar]
- Tokutake Y, Matsumoto T, Watanabe T, Maeda S, Tahara H, Sakamoto S, Niida H, Sugimoto M, Ide T, Furuichi Y. 1998. Extra-chromosomal telomere repeat DNA in telomerase-negative immortalized cell lines. Biochem Biophys Res Commun 247: 765–772. [DOI] [PubMed] [Google Scholar]
- Truong LN, Li Y, Shi LZ, Hwang PY-H, He J, Wang H, Razavian N, Berns MW, Wu X. 2013. Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci 110: 7720–7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto Y, Taggart AK, Zakian VA. 2001. The role of the Mre11–Rad50–Xrs2 complex in telomerase-mediated lengthening of Saccharomyces cerevisiae telomeres. Curr Biol 11: 1328–1335. [DOI] [PubMed] [Google Scholar]
- Tsukamoto M, Yamashita K, Miyazaki T, Shinohara M, Shinohara A. 2003. The N-terminal DNA-binding domain of Rad52 promotes RAD51-independent recombination in Saccharomyces cerevisiae. Genetics 165: 1703–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F. 2003. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423: 309–312. [DOI] [PubMed] [Google Scholar]
- Veaute X, Delmas S, Selva M, Jeusset J, Le Cam E, Matic I, Fabre F, Petit M-A. 2005. UvrD helicase, unlike Rep helicase, dismantles RecA nucleoprotein filaments in Escherichia coli. EMBO J 24: 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal DD, Lee K, Deem A, Shim EY, Malkova A, Lee SE. 2012. Microhomology directs diverse DNA break repair pathways and chromosomal translocations. PLoS Genet 8: e1003026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelkel-Meiman K, Roeder GS. 1990. A chromosome containing HOT1 preferentially receives information during mitotic interchromosomal gene conversion. Genetics 124: 561–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walmsley RW, Chan CSM, Tye B-K, Petes TD. 1984. Unusual DNA sequences associated with the ends of yeast chromosomes. Nature 310: 157–160. [DOI] [PubMed] [Google Scholar]
- Wang RC, Smogorzewska A, de Lange T. 2004. Homologous recombination generates T-loop-sized deletions at human telomeres. Cell 119: 355–368. [DOI] [PubMed] [Google Scholar]
- Wang H, Rosidi B, Perrault R, Wang M, Zhang L, Windhofer F, Iliakis G. 2005. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res 65: 4020–4030. [DOI] [PubMed] [Google Scholar]
- Weinstock DM, Richardson CA, Elliott B, Jasin M. 2006. Modeling oncogenic translocations: distinct roles for double-strand break repair pathways in translocation formation in mammalian cells. DNA Repair (Amst) 5: 1065–1074. [DOI] [PubMed] [Google Scholar]
- Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, Scully R. 2014. BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510: 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, Rass E, Scully R. 2015. Deciphering the code of the cancer genome: mechanisms of chromosome rearrangement. Trends Cancer 1: 217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, Kwon Y, Xu Y, Chung W-H, Chi P, Niu H, Mayle R, Chen X, Malkova A, Sung P, et al. 2013. Pif1 helicase and Polδ promote recombination-coupled DNA synthesis via bubble migration. Nature 502: 393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Hickson ID. 2003. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426: 870–874. [DOI] [PubMed] [Google Scholar]
- Wu Y, Kantake N, Sugiyama T, Kowalczykowski SC. 2008. Rad51 protein controls Rad52-mediated DNA Annealing. J Biol Chem 283: 14883–14892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Kwok A, Scully R. 2009. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol 16: 814–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Marians KJ. 2003. PriA mediates DNA replication pathway choice at recombination intermediates. Mol Cell 11: 817–826. [DOI] [PubMed] [Google Scholar]
- Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, et al. 2007. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 449: 478–482. [DOI] [PubMed] [Google Scholar]
- Youds JL, Mets DG, McIlwraith MJ, Martin JS, Ward JD, ONeil NJ, Rose AM, West SC, Meyer BJ, Boulton SJ. 2010. RTEL-1 enforces meiotic crossover interference and homeostasis. Science 327: 1254–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefzadeh MJ, Wyatt DW, Takata K-I, Mu Y, Hensley SC, Tomida J, Bylund GO, Doublie S, Johansson E, Ramsden DA, et al. 2014. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genet 10: e1004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AM, McVey M. 2010. Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions. Nucleic Acids Res 38: 5706–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn KE, Averill AM, Aller P, Wood RD, Doublie S. 2015. Human DNA polymerase theta grasps the primer terminus to mediate DNA repair. Nat Struct Mol Biol 22: 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahradka K, Slade D, Bailone A, Sommer S, Averbeck D, Petranovic M, Lindner AB, Radman M. 2006. Reassembly of shattered chromosomes in Deinococcus radiodurans. Nature 443: 569–573. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Jasin M. 2011. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat Struct Mol Biol 18: 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C-Z, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, Pellman D. 2015. Chromothripsis from DNA damage in micronuclei. Nature 522: 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Mills KD, Ferguson DO, Lee C, Manis J, Fleming J, Gao Y, Morton CC, Alt FW. 2002. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell 109: 811–821. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Chung W-H, Shim EY, Lee SE, Ira G. 2008. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double strand break ends. Cell 134: 981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]