

Abstract
Calcification occurs in atherosclerotic vascular lesions and in the aortic valve. Calcific aortic valve disease (CAVD) is a slow, progressive disorder that ranges from mild valve thickening without obstruction of blood flow, termed aortic sclerosis, to severe calcification with impaired leaflet motion, termed aortic stenosis. In the past, this process was thought to be ‘degenerative’ because of time-dependent wear and tear of the leaflets, with passive calcium deposition. The presence of osteoblasts in atherosclerotic vascular lesions and in CAVD implies that calcification is an active, regulated process akin to atherosclerosis, with lipoprotein deposition and chronic inflammation. If calcification is active, via pro-osteogenic pathways, one might expect that development and progression of calcification could be inhibited. The overlap in the clinical factors associated with calcific valve disease and atherosclerosis provides further support for a shared disease mechanism. In our recent research we used an in vitro porcine valve interstitial cell model to study spontaneous calcification and potential promoters and inhibitors. Using this model, we found that denosumab, a human monoclonal antibody targeting the receptor activator of nuclear factor-κB ligand may, at a working concentration of 50 μg/mL, inhibit induced calcium deposition to basal levels.
Keywords: Aortic valve calcification disease, aortic valve interstitial cells, markers of calcification, gene regulation, aortic porcine model, denosumab, atorvastatin
Progressive thickening of the aortic valve leaflets and narrowing of the aortic annulus leads to increased mechanical stress on the left ventricle and reduces cardiac output, resulting in further complications.[1–3] The proportion of the population affected increases as the median age of a country or region rises. Approximately 2–4 % of people aged over 65 will develop calcific aortic stenosis, with 25 % of people in this age group presenting with signs of the disease, leading to a 50 % increased risk of cardiovascular related events. Furthermore, there is an associated risk of 80 % over 5 years of progression to heart failure, aortic valve replacement or death.[4]
Anatomy and Histology
The normal aortic valve maintains unidirectional blood flow from the left ventricle into the aorta. It is a supple membrane that opens and closes with each heartbeat more than 100,000 times a day. The healthy aortic valve comprises three leaflets and is located at the junction between the left ventricular outflow tract and the aortic root.
The internal collagen framework of the leaflets is arranged in three distinct layers, which – from the aortic to ventricular surface – are the fibrosa, spongiosa, and ventricularis (see Figure 1). This leaflet structure is covered on both the ventricular and aortic surfaces by endothelium in continuity with both the ventricular endocardium and the aortic endothelium. Each layer of the aortic valve has a distinct structure and function. The fibrosa, with its dense connective tissue, contains circumferentially oriented collagen fibres that provide most of the strength of the leaflets. The spongiosa is found at the bases of the leaflets. It contains a loose matrix of mucopolysaccharides, and provides a cushion to resist compressive forces and facilitate movements between the fibrosa and ventricularis during leaflet motion. The ventricularis layer contains radially oriented elastin and contributes to flexibility, allowing for changes in leaflet shape during opening and closing. Under normal conditions, all three layers are avascular with no cellular infiltrates and are innervated by adrenergic and cholinergic neural networks.[5–7] To remain pliable, the aortic valve must undergo continuous repair throughout life. Accumulation of fibrotic tissue and calcium in a valve leads to decreased pliability and narrowing of the valve orifice.[8,9]
Figure 1: Cellular Architecture of the Aortic Valve.
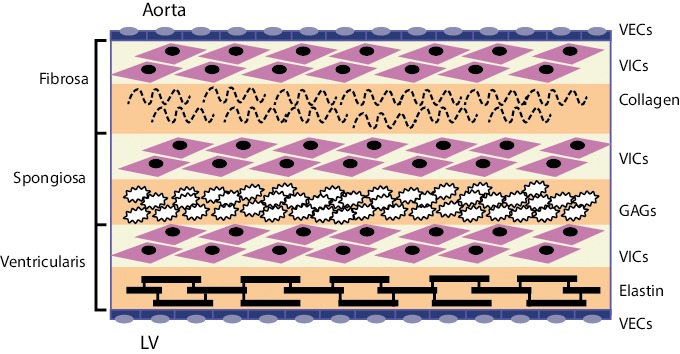
Valve endothelial cells (VECs) line the outer surface of the valve and function as a barrier to limit inflammatory cell infiltration and lipid accumulation. The three middle layers of the valve are the fibrosa, spongiosa, and ventricularis. These layers contain valve interstitial cells (VICs) as the predominant cell type. The fibrosa is nearest the aortic side of the valve, contains Type I and Type III fibrillar collagen, and serves a load-bearing function. The spongiosa contains glycosaminoglycans (GAGs) that lubricate the fibrosa and ventricularis layers as they shear and deform during the cardiac cycle. The ventricularis contains elastin fibres to decrease radial strain. Source: Rajamannan, 2011.[10]
Valve interstitial cells (VICs) are found in each of these layers, and have distinct sub-populations that regulate homeostasis within the valve leaflets.[10–12] In addition to the common tricuspid anatomy of the aortic valve, a congenital bicuspid valve is found in 0.5–1.4 % of the general population, giving rise to differential biomechanical forces – both on the valve and the aortic wall.[13–15]
Pathophysiology and Mechanism of Calcification
Over the past several decades, the aetiology of calcific aortic valve disease (CAVD) has changed considerably. The lower prevalence of rheumatic heart disease and increased longevity in industrialised countries has resulted in a pattern shift from rheumatic to degenerative calcification as the most common cause of CAVD and subsequent calcific aortic stenosis.[16–18] CAVD is the third most common heart disease in the western world,[19] following coronary heart disease and hypertension. Its prevalence in the elderly (≥65 years of age) ranges from 2–4 % when considering only severe aortic stenosis, increasing to 25 % when aortic sclerosis is included.[9] However, a relative minority of elderly individuals develop aortic valve calcification, suggesting pathological influences other than age play a role.
Calcific aortic stenosis is the second most prevalent cause for heart surgery and is responsible for approximately 15,000 deaths annually in North America.[18] Calcific aortic stenosis is a well-known disease entity and we are able to assess numerous haemodynamic parameters using cardiac catheterisation or ultrasonography as well as cardiac computed tomography and cardiac magnetic resonance imaging.[20] In CAVD, calcified nodules are initially observed at the base of the cusps and their presence gradually extends towards the orifice. All three cusps are usually usually affected, but one or more may be dominant. When blood flow through the stenotic aortic orifice becomes significantly restricted, haemodynamic impairment associated with serious symptoms of congestive heart failure and sudden cardiac death may occur. Severe symptomatic aortic stenosis is a Class I indication for surgical valve replacement according to the American Heart Association and American College of Cardiology guidelines for valvular heart disease.[21]
CAVD is currently considered as an actively regulated and progressive disease, characterised by a cascade of cellular changes that initially cause fibrotic thickening, followed by extensive calcification of the aortic valve leaflets. This in turn leads to significant aortic valve stenosis and eventual left ventricular outflow obstruction (see Figure 2),[10,22] for which surgical replacement remains the only viable treatment option. Currently there is no approved pharmacological treatment to stop the progression of CAVD.[23] Descriptive studies using human specimens have demonstrated the hallmark features of this disease, including early atherosclerosis, cell proliferation and osteoblast expression. [24–26]
Figure 2: Inflammatory Process of Calcific Aortic Stenosis.
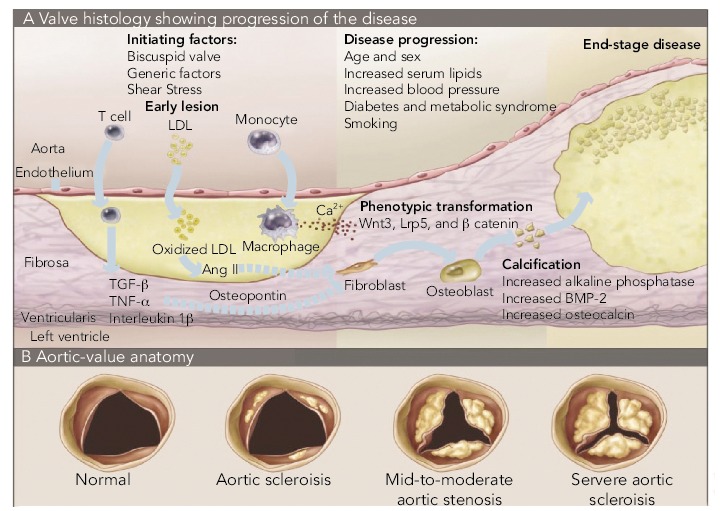
A: Progression of histological changes during the process of calcific aortic stenosis. B: Tricuspid aortic valve, showing increasing deposition of calcium and reduction of the aortic annulus. BMP = bone morphogenetic protein; LDL = low-density lipoprotein; TNF = tumour necrosis factor; TGF = transforming growth factor. Source: Otto, 2008.[22]
CAVD and Traditional Risk Factors for Atherosclerosis
Aortic valve stenosis was first described by Lazare Riviere in 1663.[27] In the early 1900s, eminent pathologists such as Monckeberg, described CAVD as a passive degenerative process associated with rheumatic fever or aging, during which serum calcium attaches to the valve surface and binds to the leaflet to form nodules.[28]
In more recent decades, several studies have implicated the traditional risk factors for cardiovascular atherosclerosis in the development of CAVD. Atherosclerosis is a complex and multifactorial process that produces a lesion composed of lipids,[29,30] macrophages,[31] proliferating smooth muscle cells[32] and apoptosis.[33] It is regulated by endothelial nitric oxide synthase,[34–38] and over time causes occlusion of the vessel diameter. Total cholesterol, increased low-density lipoprotein (LDL) cholesterol, increased lipoprotein(a), increased triglycerides, decreased high-density lipoprotein cholesterol, male sex, cigarette smoking, hypertension, and diabetes mellitus have been reported to increase the incidence of aortic stenosis, and are likely contribute to endothelial dysfunction and leaflet damage.[2,3,39–43] The presence of LDL and atherosclerosis in calcified valves in surgical pathological studies supports the hypothesis of a common cellular mechanism.[44,45] Furthermore, patients with familial hypercholesterolaemia develop aggressive peripheral vascular disease, coronary artery disease and aortic valve lesions, which calcify with age.[39,46–48]
Oxidised LDL (oxLDL) is implicated in vascular calcification associated with atherosclerosis.[49,50] Elevated blood levels of oxLDL correlate with aortic valve calcification and fibrosis,[51] and oxLDL accumulation in calcific, stenotic aortic valves is well described.[52–56] Metabolic bone diseases – including Paget’s disease, secondary hyperparathyroidism and renal disease – as well as increased serum creatinine and calcium are also linked to progression of valve calcification, but include only a relative minority of patients who have aortic stenosis.[57–59] Understanding of these clinical risk factors provides the foundation for cellular studies and the potential for targeted medical therapies for this disease, similar to vascular atherosclerosis. However, the overall evidence indicated by the presence of atherosclerotic risk factors may partly explain why some patients who have congenitally abnormal valves develop aortic stenosis and require valve replacement sooner than those without risk factors. If atherosclerotic risk factors are important in the development of valvular heart disease, then experimental models of atherosclerosis are important in the understanding of this process. Studies in mice and rabbits have confirmed that experimental hypercholesterolaemia causes both atherosclerosis and calcification in the aortic valves.[60–64] Two months of cholesterol diet treatment in an experimental rabbit model induced marked thickening and complex calcification in the aortic valve leaflets. The model was extended to test the pharmacological effect of atorvastatin and angiotensin receptor antagonists on the inhibition of atherosclerosis pathways and calcification.[65–69] Other pathways, such as Wnt signalling and increased calcium concentration via kallikrein-kinin signalling, are involved in CAVD. Wnt proteins interact with trans-membrane receptors, in particular LDL receptors, and inhibit the effect of the degradation of the intracellular protein β catenin. In turn, β catenins mediate osteoblastic transformation of VICs and bone production. in vitro, atorvastatin – an inhibitor of LDL-cholesterol in blood – can neutralise this signal pathway in mice models.[66,70–72]
The molecular and cellular processes that contribute to aortic valve stenosis are not fully characterised, but could provide insights into the development of new therapeutic approaches.
Heart valves comprise a heterogeneous population of valvular endothelial cells and VICs, which maintain valve homeostasis and structural leaflet integrity. VICs, the most abundant cell type in the heart valve, play a key role in CAVD progression. [73] Various VIC phenotypes have been identified in diseased human heart valves,[74] including quiescent fibroblast-like VICs, which upon pathological cues can differentiate into activated myofibroblast-like VICs; and osteoblast-like VICs, which are responsible for the active deposition of calcium in CAVD.[53,62,74] Additionally, several studies have demonstrated the ability of VICs to undergo osteogenic differentiation.[26,67,75]
CAVD and Shear Stress
Although atherosclerotic coronary artery disease and CAVD share common features, they do have differences in rheology. This difference may provide at least a partial explanation for the differences in pathophysiology and response to therapy.[76–80] CAVD is characterised by pulsatile shear stress on the ventricular side and low and reciprocating shear stress on the aortic side,[81] whereas the coronary artery is exposed to sustained laminar blood flow under normal circumstances.[82] As stenosis progresses, wall shear stress across the aortic valve dramatically increases.[76] Ahamed and colleagues have demonstrated that in vitro shear stress can activate latent transforming growth factor (TGF)-β1,[82] a critical pro-fibrotic growth factor that can induce fibrosis and calcification.[83] They also showed that active TGF-β1 could be eluted from thrombi formed in response to vascular injury in the carotid artery of mice where partial occlusion may have led to high local shear stress.[82] Subsequently, Albro et al. independently confirmed that shear stress can activate latent TGF-β1 in synovial fluid.[83] These data raise the possibility of an association between the activation of circulating latent TGF-β1 under high shear stress and the development of CAVD. Because platelets contribute ~45 % of the baseline circulating TGF-β1 level[84] and have 40–100 times more latent TGF-β1 than any other cells,[85] it is possible that shear stress has two separate effects – inducing release of latent TGF-β1 from platelets and activating the released latent TGF-β1. This mechanism may contribute to the progression of CAVD, because aortic valve narrowing increases shear stress resulting in greater release of platelet TGF-β1 and TGF-β1 activation. This in turn may lead to progressive valve narrowing and fibrosis, and thus even greater shear stress.
Calcifying valves initially have macrophage and T-cell infiltrates as a result of endothelial injury.[74] Bone morphogenetic protein (BMP)-2 and BMP-4 are then expressed by myofibroblasts and preosteoblasts adjacent to these lymphocytic infiltrates.[74] Furthermore, cardiac valves express markers of osteoblastic differentiation, including core-binding factor alpha 1 and osteocalcin.[26] These valves also calcify in a manner similar to osteogenesis, with lamellar bone evident in the majority of pathological specimens examined.[85] Congenitally bicuspid aortic valves uniformly show signs of calcification by the time individuals reach age 30,[86] which may, in part, be attributable to the particular mechanical stressors to which these valves are subjected.[87] Recently, the molecular mechanism underlying bicuspid aortic valve calcification was solved. Mutations in the transcriptional regulator NOTCH1 resulted in aortic valve anomalies and severe calcification, owing to impaired repression of the osteoblast stimulator runt-related transcription factor 2 (RUNX2).[88]
Recent evidence suggests that CAVD is the result of an active inflammatory process affecting the valve and leading to osteoblastic transformation with bone formation of VICs by activation of the receptor activator of nuclear factor-κB (RANK).[89]
Regulatory Pathways
There is increasing evidence that regulatory pathways that control heart valve development also are active with valve pathogenesis later in life. CAVD includes the activation of VICs in addition to increased expression of transcription factors that regulate the earliest events of valvulogenesis in the developing embryo.[90] In addition to valve developmental pathways, regulatory proteins that promote the development of cartilage and bone lineages also are active in diseased valves.[91] Thus, knowledge of the molecular regulatory pathways that control valve development will likely be informative in determining the molecular mechanisms of valve pathogenesis.
Aetiology
CAVD has multifactorial aetiology. Many factors are centered on an inflammatory process affecting the valve and leading to calcification,[74,85] including deposition of LDLs,[44,45] osteoblastic transformation with bone formation of valvular interstitial cells, connective tissue synthesis and tissue remodelling. On a microscopic level, the aortic leaflets contain disorganised collagen fibres, chronic inflammatory cells, extracellular bone matrix proteins, lipidic proteins and bone minerals.[5] Calcification of the valve occurs following trans-differentiation of the VICs through a myofibroblast stage and into osteoblast cells.[71,92]
Half of adults undergoing aortic valve replacement have a bicuspid aortic valve associated, and nearly all of them will need to have a new valve inserted.[93] Shear stress occurring with each cardiac systole is greater in a bicuspid valve than in a tri leaflet structure and these valves calcify earlier.[93]
Interestingly, the expression of RANK ligand (RANKL) by osteoblast cells will be actively involved in the activation and differentiation of osteoclast cells.[89] RANKL levels normally rise with age and can predict cardiovascular events in humans, while osteoprotegerin (a physiological inhibitor of RANK) deficit can lead to vascular calcification in animal models.[94,95] This study highlights an in vitro model to assess the mechanisms of aortic valve calcification.[95]
Molecular Mechanisms of Calcification
The processes of aortic valve stenosis and calcification share many similarities with atherosclerosis, and the pathologies of both conditions have similar risk factors and histopathology.[2] Activation of VICs and pathways of calcific aortic stenosis is the result of mechanical and shear stress, endothelial damage and deposition of LDLs, triggering inflammatory events and attracting inflammatory cells (monocytes, macrophages and T cells).
These cells produce cytokines, including TGF-β, which regulates cell proliferation and differentiation; tumour necrosis factor-α, whose primary function is the regulation of the immune cells; and interleukin 2, which is produced by activated T lymphocytes with growth factor activity.[1]
VICs activated by the inflammatory process are designated myofibroblasts.[5] These cells will develop angiogenic activity and produce matrix metalloproteinases, proteins that are involved in tissue remodelling and support VIC activation and transformation.[96,97] During this process activated VICs differentiate into osteoblasts.
In Vitro Studies
Initial studies in our laboratory have involved the establishment and validation of porcine VIC isolation, culture and calcification procedures and the effect of denosumab on in vitro calcification. During the characterisation of porcine VICs, the first objective was to determine the expression level of a common marker of myofibroblast phenotype, α-actin, to demonstrate that active VICs were present in the samples. The expression of RUNX2, a major regulator of osteoblast differentiation, was analysed to corroborate that CardiologyCardiology the effect of the complete transdifferentiation of VICs had taken place and that the osteoblast phenotype was present. Furthermore, changes in the expression of TGF-β (a promoter of osteogenesis), were detected and recorded. Additionally, RhoA, a regulator of nodule formation in myofibroblasts, was analysed, followed by examining changes in the expression of RANKL, a key regulator of bone metabolism. Finally, calponin, a protein with potential capability to inhibit bone formation, was measured to complete the genetic studies. TGF-β can increase calcium and collagen deposition.[98] It is known that TGF-β can also stimulate the expression of RANK on pre-osteoclastic cells, and in this way increase osteoclastic sensitivity to RANKL.[99] RANKL is expressed in the membrane of osteoblasts and monocytes. As yet there is still no evidence that TGF-β promotes calcification in porcine VICs by increasing RANK expression levels.
Our recent unpublished studies demonstrated the upregulation of key molecules during the spontaneous calcification of porcine VICs with an increase of calcium, collagen and alkaline phosphatase (ALP) activity. in vitro calcification was determined using standard staining and enzyme activity assays. Calcification in pig VICs was induced with sodium phosphate. The cells expressed markers for both vascular smooth muscle cells and osteoblasts, suggesting a transdifferentiation of the phenotype. Upregulation of α-actin, RUNX2, TGF-β and RhoA and downregulation of calponin were noted, with no changes seen in RANKL expression. Sodium phosphate increased nodular formation by day 7 and ALP activity of porcine VICs by day 14. The findings suggest that porcine VICs may be a good model to study the process of CAVD.[100]
Denosumab as a Potential Inhibitor of VIC Calcification In Vitro
Denosumab is a human IgG2 monoclonal antibody designed to target RANKL,[101] which is expressed on the membrane of the osteoblasts and osteoclasts. Denosumab is used in the treatment of osteoporosis. Additionally, owing to its mechanism that blocks the receptor RANKL, it neutralises the activation of RANK receptors on the membrane of pre-osteoclast cells.More research is needed to address the interaction between RANK receptor and denosumab in porcine VICs.
Our recent unpublished studies showed that 50 μg/mL denosumab inhibited induced calcium deposition to basal levels in porcine VIC culture.[100] Although associated with bone loss and shown to reduce vascular calcification, the effect of denosumab on calcification of human VICs is unknown. Recently, denosumab has been shown to reduce calcium deposition in the aorta, although the mechanisms by which it affects ectopic calcification are poorly understood.[102] Furthermore, osteoprotegerin (a signalling protein receptor and a member of the tumour necrosis factor receptor family) has been shown to stop ectopic calcification in vitro via a similar mechanism to denosumab, but there is still not enough evidence of any effect in reverting the process of calcification. Osteprotegerin’s mechanism of action is to block RANKL-RANK receptor interaction.[94,95] A fuller understanding of the mechanisms of action of denosumab may identify a novel therapeutic approach for clinical treatment, supplementing the current surgical approach. It should be noted that extrapolation of the results obtained in an in vitro porcine model to humans should be cautious, as species variations are likely to exist. Although it is not possible to include all mechanisms involved in CAVD in a single model, experimental models can contribute towards identifying the role several factors may play in the development of CAVD.
Acknowledgments
We wish to give thanks for all the support of Dr Neil Mackenzie, who unfortunately passed away after a fall while ice climbing in Canada (RIP).
References
- 1.Olson LJ, Edwards WD, Tajik AJ. Aortic valve stenosis: etiology, pathophysiology, evaluation, and management, Curr Probl Cardiol. 1987;12:455–508. doi: 10.1016/0146-2806(87)90024-7. [DOI] [PubMed] [Google Scholar]
- 2.Mohler ER, Sheridan MJ, Nichols R et al. Development and progression of aortic valve stenosis: atherosclerosis risk factors‚ Äì a causal relationship? A clinical morphologic study, Clin Cardiol. 1991;14:995–9. doi: 10.1002/clc.4960141210. [DOI] [PubMed] [Google Scholar]
- 3.Stewart BF, Siscovick D, Lind BK et al. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study, J Am Coll Cardiol. 1997;29:630–4. doi: 10.1016/s0735-1097(96)00563-3. [DOI] [PubMed] [Google Scholar]
- 4.Otto CM, Burwash IG, Legget ME et al. Prospective study of asymptomatic valvular aortic stenosis. Clinical, echocardiographic, and exercise predictors of outcome, Circulation. 1997;95:2262–70. doi: 10.1161/01.cir.95.9.2262. [DOI] [PubMed] [Google Scholar]
- 5.Otto CM, Kuusitoa J, Reichenbach DD. Characteristics of the early lesion of degenerative valvular aortic stenosis: Histological and Immunohistochemical studies, Circulation. 1994;90:844–53. doi: 10.1161/01.cir.90.2.844. [DOI] [PubMed] [Google Scholar]
- 6.Xu S, Liu AC, Gotlieb AI. Common pathogenic features of atherosclerosis and calcific aortic stenosis: role of transforming growth factor-[beta], Cardiovascular Pathology. 2010;19:236–47. doi: 10.1016/j.carpath.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 7.Lange FJ, Moorman AF, Anderson RH et al. Lineage and morphogenetic analysis of the cardiac valves, Circ Res. 2004;95:645–54. doi: 10.1161/01.RES.0000141429.13560.cb. [DOI] [PubMed] [Google Scholar]
- 8.Yetkin E, Waltenberger J. Molecular and cellular mechanisms of aortic stenosis, Int J Cardiol. 2009;135:4–13. doi: 10.1016/j.ijcard.2009.03.108. [DOI] [PubMed] [Google Scholar]
- 9.O’Brien KD. Pathogenesis of calcific aortic valve disease: a disease process comes of age (and a good deal more), Arterioscler Thromb Vasc Biol. 2006;26:1721–28. doi: 10.1161/01.ATV.0000227513.13697.ac. [DOI] [PubMed] [Google Scholar]
- 10.Rajamannan NM, Evans FJ, Aikawa E et al. Calcific aortic valve disease: not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease – 2011 update, Circulation. 2011;124:1783–91. doi: 10.1161/CIRCULATIONAHA.110.006767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dweck MR, Boon NA, Newby DE. Calcific aortic stenosis: a disease of the valve and the myocardium, J Am Coll Cardiol. 2012;60:1854–63. doi: 10.1016/j.jacc.2012.02.093. [DOI] [PubMed] [Google Scholar]
- 12.Leopold JA. Cellular mechanisms of aortic valve calcification, Circ Cardiovasc Interv. 2012;5:605–14. doi: 10.1161/CIRCINTERVENTIONS.112.971028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wauchope G. The clinical importance of variations in the number of cusps forming the aortic and pulmonary valves, QJM. 1928;21:383–99. [Google Scholar]
- 14.Larson EW, Edwards WD. Risk factors for aortic dissection: a necropsy study of 161 cases, Am J Cardiology. 1984;53:849–55. doi: 10.1016/0002-9149(84)90418-1. [DOI] [PubMed] [Google Scholar]
- 15.Pauperio HM, Azevedo AC, Ferreira CS. The aortic valve with two leaflets – a study in 2,000 autopsies, Cardiol Young. 1999;9:488–98. doi: 10.1017/s1047951100005400. [DOI] [PubMed] [Google Scholar]
- 16.Selzer A. Changing aspects of the natural history of valvular aortic stenosis, N Engl J Med. 1987;317:91–8. doi: 10.1056/NEJM198707093170206. [DOI] [PubMed] [Google Scholar]
- 17.Takkenberg JJ, Rajamannan NM, Rosenhek R et al. The need for a global perspective on heart valve disease epidemiology. The SHVD working group on epidemiology of heart valve disease founding statement, J Heart Valve Dis. 2008;17:135–9. [PubMed] [Google Scholar]
- 18.Virmani R, Burke A, Farb A, Atkinson JB. Cardiovascular Pathology. Second edition. Vol. 40. Philadelphia: WB Saunders Company; 2001. Pathology of Cardiac Valves. pp. 231–79. Vol. [Google Scholar]
- 19.Pibarot P, Dumesnil JG. New concepts in valvular hemodynamics: implications for diagnosis and treatment of aortic stenosis, Can J Cardiol. 2007;23(Suppl B):40B–47B. doi: 10.1016/s0828-282x(07)71009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chun EJ, Choi SI, Lim C et al. Aortic stenosis: evaluation with multidetector CT angiography and MR imaging, Korean J Radiol. 2008;9:439–48. doi: 10.3348/kjr.2008.9.5.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishimura RA, Otto CM, Bonow RO et al. 2014 AHA/ACC Guideline for the Management of Patients With Valvular Heart Disease, J Am Coll Cardiol. 2014;63:e71–9. doi: 10.1016/j.jacc.2014.02.536. [DOI] [PubMed] [Google Scholar]
- 22.Otto CM. Calcific aortic stenosis – time to look more closely at the valve, N Engl J Med. 2008;359:1395–8. doi: 10.1056/NEJMe0807001. [DOI] [PubMed] [Google Scholar]
- 23.Pawade TA, Newby DE, Dweck MR. Calcification in Aortic Stenosis: The Skeleton Key, J Am Coll Cardiol. 2015;66:561–77. doi: 10.1016/j.jacc.2015.05.066. [DOI] [PubMed] [Google Scholar]
- 24.O’Brien KD, Kuusisto J, Reichenbach DD et al. Osteopontin is expressed in human aortic valvular lesions [comment], Circulation. 1995;92:2163–68. doi: 10.1161/01.cir.92.8.2163. [DOI] [PubMed] [Google Scholar]
- 25.Mohler ER 3rd, Gannon F, Reynolds C et al. Bone formation and inflammation in cardiac valves, Circulation. 2001;102:1522–28. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 26.Rajamannan NM, Subramaniam M, Rickard D et al. Human aortic valve calcification is associated with an osteoblast phenotype, Circulation. 2003;107:2181–84. doi: 10.1161/01.CIR.0000070591.21548.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vaslef SN, Roberts WC. Early descriptions of aortic valve stenosis [editorial], Am Heart J. 1993;125:1465–74. doi: 10.1016/0002-8703(93)91036-e. [DOI] [PubMed] [Google Scholar]
- 28.Monckeberg JG. Der normale histologische Bau und die Sklerose der Aortenklappen, Virchows Arch Pathol Anat Physiol. 1904;176:472–514. [Google Scholar]
- 29.Desai MY, Rodriguez A, Wasserman BA et al. Association of cholesterol subfractions and carotid lipid core measured by MRI, Arterioscler Thromb Vasc Biol. 2005;25:e110–1. doi: 10.1161/01.ATV.0000166599.78182.6c. [DOI] [PubMed] [Google Scholar]
- 30.Subbaiah PV, Gesquiere LR, Wang K. Regulation of the selective uptake of cholesteryl esters from high density lipoproteins by sphingomyelin, J Lipid Res. 2005;46:2699–705. doi: 10.1194/jlr.M500263-JLR200. [DOI] [PubMed] [Google Scholar]
- 31.Kim WJ, Chereshnev I, Gazdoiu M et al. MCP-1 deficiency is associated with reduced intimal hyperplasia after arterial injury, Biochem Biophys Res Commun. 2003;310:936–42. doi: 10.1016/j.bbrc.2003.09.088. [DOI] [PubMed] [Google Scholar]
- 32.Tanner FC, Boehm M, Akyurek LM et al. Differential effects of the cyclin-dependent kinase inhibitors p27(Kip1), p21(Cip1), and p16(Ink4) on vascular smooth muscle cell proliferation, Circulation. 2000;101:2022–5. doi: 10.1161/01.cir.101.17.2022. [DOI] [PubMed] [Google Scholar]
- 33.Zhang R, Luo D, Miao R et al. Hsp90-Akt phosphorylates ASK1 and inhibits ASK1-mediated apoptosis, Oncogene, 2005;24:3954–63. doi: 10.1038/sj.onc.1208548. [DOI] [PubMed] [Google Scholar]
- 34.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase, J Biol Chem. 1998;273:24266–71. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 35.Venema RC, Sayegh HS, Kent JD, Harrison DG. Identification, characterization, and comparison of the calmodulin-binding domains of the endothelial and inducible nitric oxide synthases, J Biol Chem. 1996;271:6435–40. doi: 10.1074/jbc.271.11.6435. [DOI] [PubMed] [Google Scholar]
- 36.Rajamannan NM, Subramaniam M, Stock SR et al. Atorvastatin inhibits calcification and enhances nitric oxide synthase production in the hypercholesterolaemic aortic valve, Heart. 2005;91:806–10. doi: 10.1136/hrt.2003.029785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Charest A, Pepin A, Shetty R et al. Distribution of SPARC During Neovascularization of Degenerative Aortic Stenosis, Heart. 2006;92:1844–49. doi: 10.1136/hrt.2005.086595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ngo DT, Heresztyn T, Mishra K et al. Aortic stenosis is associated with elevated plasma levels of asymmetric dimethylarginine (ADMA), Nitric Oxide. 2007;16:197–201. doi: 10.1016/j.niox.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 39.Sprecher DL, Schaefer EJ, Kent KM et al. Cardiovascular features of homozygous familial hypercholesterolemia: analysis of 16 patients, Am J Cardiol. 1984;54:20–30. doi: 10.1016/0002-9149(84)90298-4. [DOI] [PubMed] [Google Scholar]
- 40.Deutscher S, Rockette HE, Krishnaswami V. Diabetes and hypercholesterolemia among patients with calcific stenosis, J Chron Dis. 1994;37:407–15. doi: 10.1016/0021-9681(84)90108-5. [DOI] [PubMed] [Google Scholar]
- 41.Gotoh T, Kuroda T, Yamasawa M et al. Correlation between lipoprotein(a) and aortic valve sclerosis assessed by echocardiography (the JMS Cardiac Echo and Cohort Study), Am J Cardiol. 1995;76:928–32. doi: 10.1016/s0002-9149(99)80263-x. [DOI] [PubMed] [Google Scholar]
- 42.Aronow WS, Schwartz KS, Koenigsberg M. Correlation of serum lipids, calcium, and phosphorus, diabetes mellitus and history of systemic hypertension with presence or absence of calcified or thickened aortic cusps or root in elderly patients, Am J Cardiol. 1987;59:998–9. doi: 10.1016/0002-9149(87)91144-1. [DOI] [PubMed] [Google Scholar]
- 43.Lindroos M, Kupari M, Valvanne J et al. Factors associated with calcific aortic valve degeneration in the elderly, Eur Heart J. 1994;15:865–70. doi: 10.1093/oxfordjournals.eurheartj.a060602. [DOI] [PubMed] [Google Scholar]
- 44.O’Brien KD, Reichenbach DD, Marcovina SM et al. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis, Arterioscler Thromb Vasc Biol. 1996;16:523–32. doi: 10.1161/01.atv.16.4.523. [DOI] [PubMed] [Google Scholar]
- 45.Olsson M, Thyberg J, Nilsson J. Presence of oxidized low density lipoprotein in nonrheumatic stenotic aortic valves, Arterioscler Thromb Vasc Biol. 1999;19:1218–22. doi: 10.1161/01.atv.19.5.1218. [DOI] [PubMed] [Google Scholar]
- 46.Goldstein JL, Brown MS. Familial hypercholesterolemia: identification of a defect in the regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated with overproduction of cholesterol, Proc Natl Acad Sci USA. 1973;70:2804–8. doi: 10.1073/pnas.70.10.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawaguchi A, Miyatake K, Yutani C et al. Characteristic cardiovascular manifestation in homozygous and heterozygous familial hypercholesterolemia, Am Heart J. 1999;137:410–8. doi: 10.1016/s0002-8703(99)70485-0. [DOI] [PubMed] [Google Scholar]
- 48.Rajamannan NM, Edwards WD, Spelsberg TC. Hypercholesterolemic aortic-valve disease, N Engl J Med. 2003;349:717–8. doi: 10.1056/NEJMc031360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Galle J, Hansen-Hagge T, Wanner C, Seibold S. Impact of oxidized low density lipoprotein on vascular cells, Atherosclerosis. 2006;185:219–26. doi: 10.1016/j.atherosclerosis.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 50.Doherty TM, Fitzpatrick LA, Inoue D et al. Molecular, endocrine, and genetic mechanisms of arterial calcification, Endocr Rev. 2004;25:629–72. doi: 10.1210/er.2003-0015. [DOI] [PubMed] [Google Scholar]
- 51.Côté C, Pibarot P, Després JP, Mohty D, Cartier A, Arsenault BJ, Couture C, Mathieu P. Association between circulating oxidised lowdensity lipoprotein and fibrocalcific remodelling of the aortic valve in aortic stenosis, Heart. 2008;94:1175–80. doi: 10.1136/hrt.2007.125740. [DOI] [PubMed] [Google Scholar]
- 52.Derbali H, Bossé Y, Côté N et al. Increased biglycan in aortic valve stenosis leads to the overexpression of phospholipid transfer protein via Toll-like receptor 2, Am J Pathol. 2010;176:2638–45. doi: 10.2353/ajpath.2010.090541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Otto CM, Kuusisto J, Reichenbach DD et al. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies, Circulation. 1994;90:844–53. doi: 10.1161/01.cir.90.2.844. [DOI] [PubMed] [Google Scholar]
- 54.Mehrabi MR, Sinzinger H, Ekmekcioglu C et al. Accumulation of oxidized LDL in human semilunar valves correlates with coronary atherosclerosis, Cardiovasc Res. 2000;45:874–82. doi: 10.1016/s0008-6363(99)00389-2. [DOI] [PubMed] [Google Scholar]
- 55.Mohty D, Pibarot P, Després JP et al. Association between plasma LDL particle size, valvular accumulation of oxidized LDL, and inflammation in patients with aortic stenosis, Arterioscler Thromb Vasc Biol. 2008;28:187–93. doi: 10.1161/ATVBAHA.107.154989. [DOI] [PubMed] [Google Scholar]
- 56.Côté N, Pibarot P, Pépin A et al. Oxidized low-density lipoprotein, angiotensin II and increased waist cirumference are associated with valve inflammation in prehypertensive patients with aortic stenosis, Int J Cardiol. 2010;145:444–9. doi: 10.1016/j.ijcard.2009.05.054. [DOI] [PubMed] [Google Scholar]
- 57.Strickberger SA, Schulman SP, Hutchins GM. Association of Paget’s disease of bone with calcific aortic valve disease, Am J Med. 1987;82:953–6. doi: 10.1016/0002-9343(87)90157-4. [DOI] [PubMed] [Google Scholar]
- 58.Maher ER, Pazianas M, Curtis JR. Calcific aortic stenosis: A complication of chronic uraemia. Nephron. 1987;47:119–22. doi: 10.1159/000184472. [DOI] [PubMed] [Google Scholar]
- 59.Palta S, Pai AM, Gill KS, Pai RG. New insights into the progression of aortic stenosis : implications for secondary prevention, Circulation. 2000;101:2497–502. doi: 10.1161/01.cir.101.21.2497. [DOI] [PubMed] [Google Scholar]
- 60.Rajamannan NM, Sangiorgi G, Springett M et al. Experimental hypercholesterolemia induces apoptosis in the aortic valve, J Heart Valve Dis. 2001;10:371–4. [PubMed] [Google Scholar]
- 61.Drolet MC, Arsenault M, Couet J. Experimental aortic valve stenosis in rabbits, J Am Coll Cardiol. 2003;41:1211–7. doi: 10.1016/s0735-1097(03)00090-1. [DOI] [PubMed] [Google Scholar]
- 62.Weiss RM, Ohashi M, Miller JD et al. Calcific aortic valve stenosis in old hypercholesterolemic mice, Circulation. 2006;114:2065–9. doi: 10.1161/CIRCULATIONAHA.106.634139. [DOI] [PubMed] [Google Scholar]
- 63.Aikawa E, Nahrendorf M, Sosnovik D et al. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease, Circulation. 2007;115:377–86. doi: 10.1161/CIRCULATIONAHA.106.654913. [DOI] [PubMed] [Google Scholar]
- 64.Drolet MC, Roussel E, Deshaies Y et al. A high fat/high carbohydrate diet induces aortic valve disease in C57BL/6J mice, J Am Coll Cardiol. 2006;47:850–5. doi: 10.1016/j.jacc.2005.09.049. [DOI] [PubMed] [Google Scholar]
- 65.Rajamannan NM, Subramaniam M, Springett M et al. Atorvastatin inhibits hypercholesterolemia-induced cellular proliferation and bone matrix production in the rabbit aortic valve, Circulation. 2002;105:2260–5. doi: 10.1161/01.cir.0000017435.87463.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rajamannan NM, Subramaniam M, Caira F et al. Atorvastatin inhibits hypercholesterolemia-induced calcification in the aortic valves via the Lrp5 receptor pathway, Circulation. 2005;112(Suppl 9):I229–34. doi: 10.1161/01.CIRCULATIONAHA.104.524306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Osman L, Yacoub MH, Latif N et al. Role of human valve interstitial cells in valve calcification and their response to atorvastatin, Circulation. 2006;114(Suppl 1):I547–52. doi: 10.1161/CIRCULATIONAHA.105.001115. [DOI] [PubMed] [Google Scholar]
- 68.Osman L, Chester AH, Amrani M et al. A novel role of extracellular nucleotides in valve calcification: a potential target for atorvastatin, Circulation. 2006;114(Suppl 1):I566–72. doi: 10.1161/CIRCULATIONAHA.105.001214. [DOI] [PubMed] [Google Scholar]
- 69.Arishiro K, Hoshiga M, Negoro N et al. Angiotensin receptor-1 blocker inhibits atherosclerotic changes and endothelial disruption of the aortic valve in hypercholesterolemic rabbits, J Am Coll Cardiol. 2007;49:1482–9. doi: 10.1016/j.jacc.2006.11.043. [DOI] [PubMed] [Google Scholar]
- 70.Caira FC, Stock SR, Gleason TG et al. Human degenerative valve disease is associated with upregulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation, J Am Coll Cardiol. 2006;47:1707–12. doi: 10.1016/j.jacc.2006.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akat K, Borggrefe M, Kaden JJ. Aortic valve calcification: basic science to clinical practice. Heart. 2009;95:616. doi: 10.1136/hrt.2007.134783. Äì23. [DOI] [PubMed] [Google Scholar]
- 72.Salas MJ, Santana O, Escolar E, Lamas GA. Medical therapy for calcific aortic stenosis, 2012;17:133–8. doi: 10.1177/1074248411416504. [DOI] [PubMed] [Google Scholar]
- 73.Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology, Am J Pathol. 2007;171:1407–18. doi: 10.2353/ajpath.2007.070251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Peacock JD, Levay AK, Gillaspie DB et al. Reduced sox9 function promotes heart valve calcification phenotypes in vivo, Circ Res. 2010;106:712–9. doi: 10.1161/CIRCRESAHA.109.213702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gould ST, Srigunapalan S, Simmons CA, Anseth KS. Hemodynamic and cellular response feedback in calcific aortic valve disease, Circ Res. 2013;113:186–97. doi: 10.1161/CIRCRESAHA.112.300154. [DOI] [PubMed] [Google Scholar]
- 76.Sun L, Rajamannan NM, Sucosky P. Design and validation of a novel bioreactor to subject aortic valve leaflets to side-specific shear stress, Ann Biomed Eng. 2011;39:2174–85. doi: 10.1007/s10439-011-0305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sucosky P, Balachandran K, Elhammali A et al. Altered shear stress stimulates upregulation of endothelial vcam-1 and icam-1 in a bmp-4- and tgf-beta1-dependent pathway, Arterioscler Thromb Vasc Biol. 2009;29:254–60. doi: 10.1161/ATVBAHA.108.176347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun L, Chandra S, Sucosky P. Ex vivo evidence for the contribution of hemodynamic shear stress abnormalities to the early pathogenesis of calcific bicuspid aortic valve disease, PLoS One. 2012;7:e48843. doi: 10.1371/journal.pone.0048843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sun L, Rajamannan NM, Sucosky P. Defining the role of fluid shear stress in the expression of early signaling markers for calcific aortic valve disease, PLoS One. 2013;8:e84433. doi: 10.1371/journal.pone.0084433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chandra S, Rajamannan NM, Sucosky P. Computational assessment of bicuspid aortic valve wallshear stress: Implications for calcific aortic valve disease, Biomech Model Mechanobiol. 2012;11:1085–96. doi: 10.1007/s10237-012-0375-x. [DOI] [PubMed] [Google Scholar]
- 81.Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives, Physiol Rev. 2011;91:327–87. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ahamed J, Burg N, Yoshinaga K et al. in vitro and in vivo evidence for shear-induced activation of latent transforming growth factor-beta1, Blood. 2008;112:3650–60. doi: 10.1182/blood-2008-04-151753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Albro MB, Cigan AD, Nims RJ et al. Shearing of synovial fluid activates latent tgf-beta, Osteoarthritis Cartilage. 2012;20:1374–82. doi: 10.1016/j.joca.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Assoian RK, Komoriya A, Meyers CA et al. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization, J Biol Chem. 1983;258:7155–60. [PubMed] [Google Scholar]
- 85.Mohler ER 3rd, Gannon F, Reynolds C, Zimmerman R et al. Bone formation and inflammation in cardiac valves, Circulation. 2001;103:1522–8. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 86.Yener N, Oktar GL, Erer D et al. Bicuspid aortic valve, Ann Thorac Cardiovasc Surg. 2002;8:264–7. [PubMed] [Google Scholar]
- 87.Robicsek F, Thubrikar MJ, Cook JW, Fowler B. The congenitally bicuspid aortic valve: how does it function? Why does it fail? Ann Thorac Surg. 2004;77:177–85. doi: 10.1016/s0003-4975(03)01249-9. [DOI] [PubMed] [Google Scholar]
- 88.Garg V, Muth AN, Ransom JF et al. Mutations in NOTCH1 cause aortic valve disease, Nature. 2005;437:270–4. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 89.Kaden JJ, Bickelhaupt S, Grobholz R. Receptor activator of nuclear factor Kappab ligand and osteoprotegerin regulate aortic valve calcification, J Mol Cell Cardiol. 2004;36:57–66. doi: 10.1016/j.yjmcc.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 90.Combs MD, Yutzey KE. Heart valve development: Regulatory networks in development and disease, Circ Res. 2009;105:408–21. doi: 10.1161/CIRCRESAHA.109.201566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wirrig EE, Hinton RB, Yutzey KE. Differential expression of cartilage and bone-related proteins in pediatric and adult diseased aortic valves, J Mol Cell Cardiol. 2011;50:561–9. doi: 10.1016/j.yjmcc.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cohn H. Cardiac surgery in the adult, Third edition. Copyright 2009, 2003, 1997. Mc Graw-Hill.Collin-Osdoby P. Regulation of vascular calcification by osteoclast regulatory factors RANKL and osteoprotegerin, Circulation Research. 2004;95:1046–57. doi: 10.1161/01.RES.0000149165.99974.12. [DOI] [PubMed] [Google Scholar]
- 93.Nalini M Rajamannan, Frank J Evans. Calcific aortic valve disease: not simply a degenerative process, Circulation. 2011;124:1783–91. doi: 10.1161/CIRCULATIONAHA.110.006767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Min H, Morony S, Sarosi I. Osteoprotegerin reverses osteoporosis by inhibiting endosteal osteoclasts and prevents vascular calcification by blocking a process resembling osteoclastogenesis, J Exp Med. 2000;192:463–74. doi: 10.1084/jem.192.4.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Price PA, June HH, Buckley JR. Osteoprotegerin inhibits artery calcification induced by warfarin and by vitamin D, Arterioscler Thromb Vasc Biol. 2001;21:1610–6. doi: 10.1161/hq1001.097102. [DOI] [PubMed] [Google Scholar]
- 96.Soiny Y, Satta J, Maata M. Expression of MMP2, MMP9, MT1-MMP, TIMP1 and TIMP2 m RNA in valvular lesions of the heart, J Pathol. 2001;194:225–31. doi: 10.1002/path.850. [DOI] [PubMed] [Google Scholar]
- 97.Kaden JJ, Demflie CE, Grobholz R. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis, Cardiovasc Pathol. 2005;14:80–7. doi: 10.1016/j.carpath.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 98.Roman-Blas JA, Stokes DG, Jimenez SA. Modulation of TGF-β signaling by proinflammatory cytokines in articular chondrocytes, Osteoarthritis and Cartilage. 2007;5:1367–77. doi: 10.1016/j.joca.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Khosla S. Mini-review: The OPG/RANKL/RANK System, Endocrinology. 2001;142:5050–5. doi: 10.1210/endo.142.12.8536. [DOI] [PubMed] [Google Scholar]
- 100.Lerman D. Msc Thesis, Inhibitors and promoters of calcific aortic stenosis. University of Edinburgh. Edinburgh: 2012. [Google Scholar]
- 101.Baron R, Ferrari, Graham R, Russell G. Denosumab and biphosphonates: Different mechanisms of action and effects, Bone. 2011;48:677–92. doi: 10.1016/j.bone.2010.11.020. [DOI] [PubMed] [Google Scholar]
- 102.Receptor Activator of NF-κB Ligand by Denosumab Attenuates Vascular Calcium Deposition in Mice, Am J Pathol. 2009;175:473–8. doi: 10.2353/ajpath.2009.080957. [DOI] [PMC free article] [PubMed] [Google Scholar]