

Abstract
Scavenger receptor BI (SR-BI) is the major receptor for high-density lipoprotein (HDL) cholesterol (HDL-C). In humans, high amounts of HDL-C in plasma are associated with a lower risk of coronary heart disease (CHD). Mice that have depleted Scarb1 (SR-BI knockout mice) have markedly elevated HDL-C levels but, paradoxically, increased atherosclerosis. The impact of SR-BI on HDL metabolism and CHD risk in humans remains unclear. Through targeted sequencing of coding regions of lipid-modifying genes in 328 individuals with extremely high plasma HDL-C levels, we identified a homozygote for a loss-of-function variant, in which leucine replaces proline 376 (P376L), in SCARB1, the gene encoding SR-BI. The P376L variant impairs posttranslational processing of SR-BI and abrogates selective HDL cholesterol uptake in transfected cells, in hepatocyte-like cells derived from induced pluripotent stem cells from the homozygous subject, and in mice. Large population-based studies revealed that subjects who are heterozygous carriers of the P376L variant have significantly increased levels of plasma HDL-C. P376L carriers have a profound HDL-related phenotype and an increased risk of CHD (odds ratio = 1.79, which is statistically significant).
The strong inverse association between amounts of high-density lipoprotein (HDL) cholesterol (HDL-C) and coronary heart disease (CHD) risk has generated interest in a potential causal relationship between HDL metabolism and CHD. However, clinical trials with drugs that raise HDL-C levels, niacin and cholesteryl ester transfer protein (CETP) inhibitors, have produced disappointing results (1). Furthermore, recent studies of human genetic variants that are associated with HDL-C levels have generally failed to show association with CHD (2, 3). Most notably, a loss-of-function variant in LIPG, a gene encoding an endothelial lipase that, in the heterozygous state, raises HDL-C by ~5 mg/dl, was found to have no association with CHD (4). Although these previous studies suggest that higher HDL-C levels may not be causally protective against CHD, we reasoned that additional human genetic analyses might provide mechanistic insight into the complex relationship between HDL and CHD.
The scavenger receptor class BI (SR-BI), encoded by the gene SCARB1, was discovered to be an HDL receptor two decades ago (5). SR-BI promotes the selective uptake of HDL cholesteryl esters (HDL-CEs) into cells, particularly hepatocytes and steroidogenic cells (5, 6). In mice, overexpression of SR-BI in the liver reduces levels of HDL-C (7–10), and genetic deletion of SR-BI results in higher HDL-C levels (11–13). Remarkably, these genetic manipulations in mice have effects on atherosclerosis opposite to those predicted by human epidemiological data: Overexpression reduces atherosclerosis despite the lower HDL-C levels (14–16), and gene deletion increases atherosclerosis despite the higher HDL-C levels (17–20). One potential explanation relates to the flux of cholesterol from macrophages through the reverse cholesterol transport (RCT) pathway; SR-BI overexpression increases macrophage RCT, and SR-BI knockout reduces macrophage RCT (21). The human relevance of these observations has been unclear.
Identification of SCARB1 P376L homozygote and association with extremely high HDL-C
We hypothesized that humans with extremely high levels of HDL-C may harbor loss-of-function variants in SCARB1 and undertook a targeted resequencing discovery experiment in 328 participants with very high HDL-C (>95th percentile, mean HDL-C of 106.8 mg/dl) and a control group of 398 subjects with low HDL-C (<25th percentile, mean HDL-C of 30.4 mg/dl). In this cohort, we sequenced the exons of ~990 genes located within 300 kb of each of the 95 loci with significant associations (P < 5 × 10−8) with plasma lipid levels identified by the Global Lipids Genetics Consortium as of 2010 (22). Among the high HDL-C subjects, we identified a homozygote for SCARB1 P376L (g.125284671 G>A, c.1127 C>T, p.P376L, rs74830677), a 67-year-old female with an HDL-C of 152 mg/dl, and confirmed this finding by Sanger sequencing. This subject harbored no mutations in other high HDL-C genes such as CETP and LIPG. In addition to this homozygote, four P376L heterozygotes were identified by targeted sequencing in the high HDL-C group; no heterozygotes were found in the low HDL-C group (P =0.008, Fisher’s exact test).
To identify additional P376L carriers, we genotyped an expanded cohort of very high versus low HDL-C subjects. Among 524 additional subjects with very high HDL-C (mean HDL-C 95.0 mg/dl), we identified 11 heterozygotes for P376L; whereas among 758 subjects with low HDL-C (mean HDL-C 33.5 mg/dl), we identified 3 heterozygotes. In total, our combined sequencing and genotyping for discovery of the P376L variant showed that this variant is significantly overrepresented in subjects with high HDL-C [minor allele frequency (MAF) = 0.010 in high HDL-C versus 0.0013 in low HDL-C controls, P = 0.000127, Fisher’s exact test, Table 1].
Table 1. Association of SCARB1 P376L with HDL-C in high versus low HDL-C cohorts.
Carriers of the P376L variant were ascertained from the Penn High HDL Study through two approaches, targeted sequencing of the SCARB1 gene in a total of 726 subjects (328 high HDL-C and 398 low HDL-C subjects) and genotyping on the exome array (Illumina) in an additional 1282 subjects (524 high HDL-C subjects and 758 low HDL-C subjects). The association of the P376L variant with the high HDL-C cohort from both approaches individually and combined together was tested using Fisher’s exact test. N, number of participants; NonC, noncarriers; Het, heterozygotes; Hom, homozygotes.
| Discovery cohort | High HDL-C (>95th percentile) (N) |
Low HDL-C (<25th percentile) (N) |
Association (P) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Total | NonC | Het | Hom | Total | NonC | Het | Hom | ||
| Targeted sequencing of SCARB1 | 328 | 323 | 4 | 1 | 398 | 398 | 0 | 0 | 0.008398 |
| Exome array genotyping | 524 | 513 | 11 | 0 | 758 | 755 | 3 | 0 | 0.005296 |
| Combined | 852 | 836 | 15 | 1 | 1156 | 1153 | 3 | 0 | 0.000127 |
Because this variant is present on the exome array, we expanded our analysis to the Global Lipid Genetics Consortium exome array data in >300,000 individuals. The P376L variant was very rare in this population (MAF of ~0.0003). It was significantly associated with higher HDL-C levels with a relatively large effect size (beta = 8.4 mg/dl; P =1.4 × 10−15). Notably, this variant was not associated with plasma levels of low-density lipoprotein cholesterol (LDL-C) or triglycerides (TGs) (table S1). Thus, we conclude that SCARB1 P376L is associated specifically with elevated HDL-C levels.
HDL-related phenotypes of SCARB1 P376L homozygote and heterozygotes
We next recruited the P376L homozygote, eight heterozygous carriers, and both high HDL-C and normal HDL-C noncarrier controls for deep phenotyping of HDL metabolism and related traits. All of the P376L study participants were of European ancestry, almost exclusively of Ashkenazi Jewish descent. Clinical characteristics and lipid profiles of the subjects are reported in Table 2. Fast protein liquid chromatography (FPLC) analysis of plasma lipoproteins confirmed the increase in large HDL particles in the homozygote (Fig. 1A). Cholesterol and apolipoprotein A-I (apoA-I) levels in HDL were significantly increased in the homozygote and heterozygotes compared with controls, but HDL apoA-II levels were not elevated (Table 2 and Fig. 1B). There were no differences between P376L carriers and controls in the absolute amount of HDL free cholesterol or the ratio of free-to-esterified cholesterol in their HDL (Fig. 1C). P376L heterozygotes had a 2.8-fold increase and the homozygote a 6.1-fold increase in large HDL-2b particles compared with noncarrier controls (Fig. 1D). There was more apoA-I (Fig. 1E and fig. S2) and apoC-III (Fig. 1F) in large HDL particles in the homozygote and heterozygous carriers. Cholesterol efflux capacity was similar in carriers and controls (Fig. 1G). In contrast to the infertility phenotype of Scarb1-deficient female mice (18), the P376L homozygote had two healthy children and reported no fertility impairment. We also did not observe the steroidogenic or platelet phenotypes reported in Scarb1-deficient mice (see supplementary materials).
Table 2. Characteristics of SCARB1 P376L carriers and controls recruited for deep phenotyping.
Demographic, plasma lipid, and apolipoprotein traits measured from one P376L homozygote, eight heterozygotes, and noncarrier controls from subjects identified from sequencing or genotyping of the Penn High HDL Study cohort for deep phenotyping. Lipid measurements from plasma were performed using an autoanalyzer. Where applicable, data are presented as means ± SD. Numbers correspond to groups for comparison. Group 1, normal HDL-C controls; group 2, high HDL-C controls; group 3, SCARB1 P376L heterozygotes.Tested: ANOVA or chi-square. Groups: Comparison between groups by number with Tukey’s multiple comparison.
| Measure | Group | P376L Hom | Significance | |||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | Tested | Groups | ||
| Number of subjects | 11 | 10 | 8 | 1 | – | |
| Age (years) | 61.6 (9.7) | 64.2 (12.5) | 67.5 (15.3) | 65 | n.s. | – |
| Sex (M/F) | 6/5 | 5/5 | 6/2 | 0/1 | n.s.** | – |
| BMI (kg/m2) | 26.4 (2) | 22.9 (1.3) | 25.6 (3.9) | 21 | * | 1/2 |
| TC (mg/dl) | 185.8 (22.3) | 215.8 (29.9) | 228 (33.2) | 280 | * | 1/3 |
| Glucose (mg/dl) | 93.5 (2.9) | 91.6 (7.0) | 98.8 (5.3) | 86 | n.s. | – |
| LDL-C (mg/dl) | 109.1 (17.3) | 97.4 (21.6) | 116.6 (27.1) | 109 | n.s. | – |
| HDL-C (PTA) (mg/dl) | 51 (11.4) | 110.1 (19.8) | 86.9 (19.9) | 152 | * | 1/2, 1/3, 2/3 |
| TG (mg/dl) | 121.2 (35) | 71.5 (32.3) | 99.5 (23.7) | 57 | * | 1/2 |
| Alcohol >1/day (n) | 4 | 4 | 2 | 0 | n.s.** | – |
| VLDL-C (mg/dl) | 26.9 (8.8) | 19 (6.2) | 23.1 (9.2) | 13 | n.s. | – |
| Lp(a) (mg/dl) | 22.3 (18.8) | 19 (22.7) | 15.9 (21.2) | 17 | n.s. | – |
| apoA-I (mg/dl) | 172.2 (33.3) | 241.7 (41.2) | 229.6 (36.1) | 327 | * | 1/2, 1/3 |
| apoA-II (mg/dl) | 40.5 (7) | 49.5 (11.5) | 46.6 (5.5) | 45 | n.s. | – |
| apoB (mg/dl) | 99.7 (13.4) | 82.8 (17.1) | 95.9 (18.2) | 92 | n.s. | – |
| apoC-II (mg/dl) | 4.32 (1.55) | 6.09 (2.69) | 4.49 (2.17) | 5.3 | n.s. | – |
| apoC-III (mg/dl) | 11.4 (4.3) | 15.5 (6.9) | 13.7 (2.7) | 16.1 | – | |
| apoE (mg/dl) | 4.52 (0.89) | 6.03 (1.86) | 4.94 (1.12) | 6.4 | * | 1/2 |
Significant at P < 0.05.
Significant at P < 0.05 by chi-square but not ANOVA. Dash indicates no significant comparison.
BMI, body mass index; PTA, phosphotungstate precipitation method; VLDL, very low density lipoprotein; Lp(a), lipoprotein a.
Fig. 1. HDL composition and functionality in a SCARB1 P376L homozygote, heterozygous carriers, and controls.
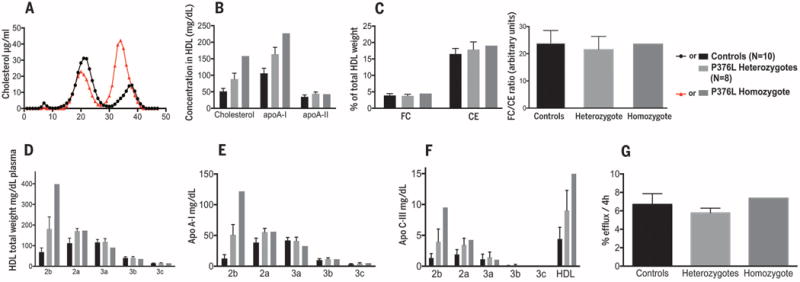
(A) FPLC fractionation of plasma lipoproteins from the P376L homozygote subject (red) and from a control with normal HDL-C. (B) Cholesterol, apoA-I, and apoA-II content in total HDL. (C) Free cholesterol (FC) and esterified cholesterol (CE) in total HDL (left) and the FC/CE ratio in total HDL (right). (D) HDL subclass concentrations after separation by density-gradient ultracentrifugation. (E) ApoA-I content in the same HDL subclasses. (F) ApoC-III content in the same HDL subclasses. (G) Cholesterol efflux capacity from macrophages of the THP-1 cell line. All data are reported as means ± SD.
SCARB1 P376L results in complete loss of function of SR-BI
Given the profound HDL phenotype of the P376L carriers, we sought to understand the impact of the variant on SR-BI function. We generated induced pluripotent stem cells (iPSCs) using peripheral blood mononuclear cells from the P376L homozygote and a noncarrier control. We next differentiated these cells into hepatocyte-like cells (HLCs) to study HDL metabolism in the setting of endogenous cellular SCARB1 expression. HLCs differentiated through this protocol recapitulate phenotypes of cultured primary hepatocytes such as albumin and VLDL (very low density lipoprotein) secretion (23–26). The cell lines from the control donor and the P376L homozygous subject demonstrated expression of hepatocyte-specific genes ALB (albumin) and AFP (alpha-fetoprotein) and exhibited comparable SCARB1 gene expression (fig. S3). Compared with control iPSC hepatocyte lines, those from the P376L homozygote demonstrated a profound reduction in selective cholesterol uptake from HDL in vitro (Fig. 2A). Similar results were observed in experiments with COS7 cells transfected with plasmids expressing wild-type (WT) or the P376L variant of SCARB1 (fig. S3, A and B), along with defective binding to HDL in vitro at 4°C (fig. S4, C and D).
Fig. 2. SCARB1 P376L is a null variant in vitro and in vivo.
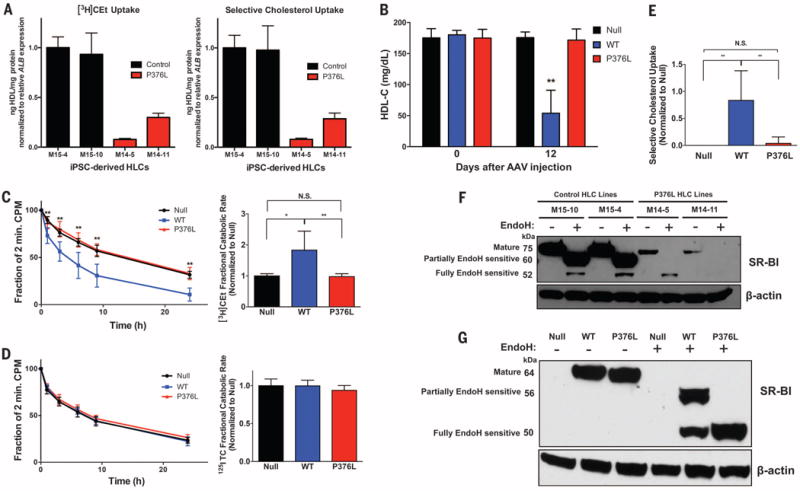
(A) [3H] Cholesterol ether (CEt) uptake (left) and selective cholesterol uptake from HDL (right) in iPSC-derived HLCs from the P376L homozygote versus a noncarrier control. Cells were incubated with [3H]CEt and 125I-labeled tyramine cellobiose (TC) dual-labeled human HDL. All values are normalized to relative ALB gene expression in each cell line. All data represent mean values for wells of respective cell lines ± SD. (B) Plasma HDL cholesterol levels before and 12 days after AAV administration to Scarb1 KO mice. (C) [3H]Cholesterol ether (CEt) clearance (left) and fractional catabolic rate (right) from plasma of Scarb1 KO mice injected with null or SR-BI AAVs after administration of [3H]CE/125I-labeled TC dual-labeled human HDL. (D) 125I-labeled TC clearance (left) and fractional catabolic rate (right) from plasma after administration of dual-labeled HDL. (E) Selective cholesterol uptake in mice expressing null, SR-BI WT, or P376L measured by relative differences in 3H- and 125I-labeled fractional catabolic rates. (F) Sensitivity to Endo-H in P376L homozygous versus noncarrier iPSC-derived HLCs. Cell lysates of each genotype were treated with Endo-H to remove complex N-linked glycans from mature forms of proteins and then immunoblotted for SR-BI. Molecular weights of different forms of SR-BI after Endo-H treatment are given on the left. (G) SR-BI Endo-H sensitivity from liver lysates from mice expressing null, SR-BI WT, or SR-BI P376L AAV. Lysates were treated with Endo-H, followed by immunoblotting for SR-BI. Molecular weights of different forms of SR-BI after Endo-H treatment are given on the left. (A) Mean values for wells of respective cell lines ± SD; [(B) to (E)] means ± SD for each of the three groups. *P < 0.05; ** P < 0.01; ***P <0.001 by analysis of variance (ANOVA) [(B) and (C)]; plasma clearance, unpaired t test (E).
To evaluate the physiological impact of the P376L variant on HDL-C levels and catabolism in vivo, we used adeno-associated virus (AAV) vectors to direct hepatic overexpression of WT SR-BI or the P376L variant in mice with depleted Scarb1 [Scarb1 knockout (KO) mice]. The two groups of mice demonstrated similar hepatic expression levels of Scarb1 mRNA (fig. S5A) and SR-BI protein (fig. S5B). Mice expressing WT Scarb1 demonstrated a robust 73% decrease in HDL-C. In contrast, mice expressing the P376L variant had no reduction in HDL-C; their HDL-C levels were comparable to those in the control AAV-null injected mice (Fig. 2B). Although the clearance of 125I-labeled HDL protein was not different among the three groups, the clearance of [3H]HDL-CE was much slower in mice expressing the P376L variant compared with those expressing WT SR-BI and was comparable to that in the control mice (Fig. 2, C and D). Selective HDL-CE clearance from plasma was increased by WT SR-BI but was undetectable in the P376L-expressing mice (Fig. 2E and fig. S5C), as was hepatic uptake of [3H]CE at 24 hours (fig. S5D). This indicates that the P376L sequence variant results in complete loss of the canonical function of SR-BI—namely, selective uptake of HDL-CE.
We hypothesized that the markedly reduced HDL-CE uptake could be because of aberrant processing of the P376L SR-BI protein, which leads to impaired cell surface localization. To test this, we isolated cell surface proteins from COS7 cells transfected with WT and P376L SR-BI using biotinylation and found markedly reduced cell surface SR-BI in the P376L transfected cell lysates after streptavidin cell surface protein pull-down assays (fig. S4E). Given that SR-BI undergoes N-glycosylation in the endoplasmic reticulum concomitant with proper folding, we hypothesized that altered posttranslational modification may underlie its reduced cell surface localization (27–29). We measured the molecular weights of SR-BI forms after endoglycosidase-H (Endo-H) treatment of transfected COS7 (fig. S4E) and iPSC-derived HLC lysates, as well as mouse liver lysates expressing WT or mutant SR-BI (Fig. 2, F and G). Higher-molecular-weight forms represent N-glycosylation modified Endo-H–resistant and partially sensitive forms at the cell surface after modification by alpha-mannosidase II in the Golgi apparatus (28).In the iPSC-derived differentiated HCLs from the P376L homozygote (Fig. 2F), we found much less total cellular SR-BI in the mutant cell lines relative to that of WT cells, despite comparable SCARB1 gene expression (fig. S3C). After Endo-H treatment, the SR-BI from SCARB1 WT cell and liver lysates across models was predominantly the partially sensitive form, along with small amounts of the fully resistant form. In contrast, the SR-BI from cell and tissue lysates across P376L-expressing groups was all the immature, fully Endo-H–sensitive form (Fig. 2, F and G, and fig. S4F). Together, these data are consistent with a model in which the P376L sequence variant alters the endogenous posttranslational N-glycosylation of SR-BI to prevent either transit from the ER to the Golgi or further posttranslational modifications in the Golgi, which ultimately result in reduced cell surface expression.
SCARB1 P376L is associated with increased risk of CHD in humans
Despite a profound increase in HDL-C, SR-BI deficiency in mice causes accelerated atherosclerosis (17–20). The relationship of reduced SR-BI function to atherosclerotic cardiovascular disease in humans has not been established. The P376L homozygous subject did not have clinical CHD, but her carotid intimal-medial thickness (cIMT) was 0.789 mm (left-right average), which is in the >75th percentile for females of her age; in addition, she had detectable plaque throughout the left internal carotid artery and at the bifurcation of her right internal carotid artery. cIMT measurements were not significantly different in the P376L heterozygotes compared with both groups of controls (fig. S8), but because of small sample size, the statistical power is limited.
To achieve greater statistical power to address this question, we performed a meta-analysis of large exome array genotyping studies of CHD cases and healthy controls to determine the relationship of the P376L variant with risk of CHD (Table 3). Among 16 sample sets from two consortia [the CARDIoGRAM Exome Consortium and the CHD Exome+ Consortium], we tested the association between P376L carrier status and CHD in 137,995 individuals. Across 49,846 CHD cases and 88,149 CHD controls, we found that P376L carriers had a significantly higher risk of CHD compared with noncarriers [odds ratio for disease among carriers = 1.79; P = 0.018] (Table 3). Thus, carriers of this SCARB1 P376L variant have significantly increased HDL-C levels and a significantly increased risk of CHD.
Table 3. Meta-analysis of association of SCARB1 P376L variant with CHD.
CHD cases and healthy controls across the CARDIoGRAM Exome Consortium and CHD Exome+ Consortium were genotyped for the SCARB1 P376Lvariant by using the exome array. BioVU, Vanderbilt University Medical Center Biorepository; BHF, British Heart Foundation; GoDARTS-CAD, Genetics of Diabetes and Audit Research Tayside Study; MHI, Montreal Heart Institute; North German, German North Coronary Artery Disease Study; Ottawa, Ottawa Heart Study; PAS, Premature Atherosclerosis Study—Academic Medical Center—Amsterdam; Penn, University of Pennsylvania CHD Cohort; South German, German South Coronary Artery Disease Study; WHI-EA, Women’s Health Initiative—American Cohort; CCHS, Copenhagen City Heart Study; CIHDS/CGPS, gen Ischemic Heart Disease Study/Copenhagen General Population Study; EPIC-CVD, European Prospective Investigation into Cancer and Nutrition— Cardiovascular Disease Study; MORGAM, MOnica Risk, Genetics, Archiving and Monograph Project; PROSPER, Prospective Study of Pravastatin in the Elderly at Risk Study; WOSCOPS, West of Scotland Coronary Prevention Study. The association of the P376L variant with CHD cases was determined using a Mantel-Haenszel fixed-effects Meta-analysis; results were odds ratio = 1.79; P = 0.018.
| Consortium or study cohort | P376L carriers | Total | Frequency | |||
|---|---|---|---|---|---|---|
| Cases | Controls | CHD cases | Controls | Cases | Controls | |
| CARDIoGRAM Exome Consortium | ||||||
| BioVU | 6 | 10 | 4587 | 16546 | 0.0013 | 0.0006 |
| BHF | 1 | 0 | 2833 | 5912 | 0.0004 | 0 |
| GoDARTS-CAD | 1 | 0 | 1568 | 2772 | 0.0006 | 0 |
| MHI | 0 | 4 | 2483 | 8085 | 0 | 0.0005 |
| North German | 0 | 1 | 4464 | 2886 | 0 | 0.0004 |
| Ottawa | 0 | 1 | 1024 | 2267 | 0 | 0.0004 |
| PAS | 1 | 1 | 728 | 808 | 0.0014 | 0.0012 |
| Penn | 3 | 0 | 683 | 156 | 0.0044 | 0 |
| South German | 4 | 0 | 5255 | 2921 | 0.0008 | 0 |
| WHI-EA | 8 | 29 | 2860 | 14929 | 0.0028 | 0.0019 |
| CHD Exome+ Consortium | ||||||
| CCHS | 1 | 1 | 2020 | 6087 | 0.0003 | 0.0001 |
| CIHDS/CGPS | 4 | 3 | 8079 | 10367 | 0.0003 | 0.0001 |
| EPIC-CVD | 4 | 2 | 9810 | 10970 | 0.0002 | 0.0001 |
| MORGAM | 0 | 0 | 2153 | 2118 | 0 | 0 |
| PROSPER | 1 | 0 | 640 | 638 | 0.0008 | 0 |
| WOSCOPS | 0 | 0 | 659 | 687 | 0 | 0 |
| Total | 34 | 52 | 49846 | 88149 | 0.00068 | 0.00059 |
Discussion
Studies of mice have provided important insights into the effects of SR-BI on HDL metabolism, RCT, and atherosclerosis. These studies revealed that overexpression of SR-BI reduces HDL-C (7–10) and reduces atherosclerosis (14–16), whereas gene deletion of SR-BI increases HDL-C (11–13) and accelerates atherosclerosis (17–20). The clinical relevance of these findings has remained uncertain, however. Studies of injected labeled HDL-CE in humans suggested that the majority of the HDL-CE was transported to the liver via CETP-mediated exchange to apoB-containing lipoproteins rather than by direct uptake from HDL by the liver (30), which brings into question the importance of hepatic SR-BI in human physiology. Common genetic variants near the SCARB1 locus were found to be significantly associated with plasma HDL-C levels, which suggests that SR-BI may play a role in HDL metabolism in humans (22, 31). A family with a rare SCARB1 variant in which serine replaces proline 297 (P297S) was reported in which the heterozygous carriers of the variant had modestly elevated HDL-C levels (31). However, the variant retains substantial SR-BI activity, no homozygotes were identified, the apparent effect on HDL-C was modest, and there was insufficient power to address its effects on atherosclerosis.
Through sequencing of subjects with extremely high plasma levels of HDL-C, we identified a homozygote for a P376L variant in SR-BI. Our complementary approaches consistently demonstrated that this variant confers virtually complete loss of function of SR-BI. Our results demonstrate many similarities in the consequences of SR-BI deficiency on HDL composition between mice and humans, including a shift toward large, buoyant HDL particles and a significant increase in apoA-I, but not apoA-II, in plasma and HDL (12, 32, 33). The homozygote is a woman who had two healthy children without fertility issues or delivery complications, which suggests that, in humans, SR-BI deficiency may not impair reproductive function in the same manner as it does in mice (18, 34). In mice, SR-BI–mediated CE uptake from HDL is a critical process underlying steroid hormone synthesis in adrenal and gonadal tissues, and SR-BI deficiency alters adrenal cholesterol content, impairs adrenal glucocorticoid response under stress, and can lead to fasting-induced hypoglycemia (6, 35, 36). We did not observe any differences in fasting glucose, serum cortisol, adrenocorticotropic hormone, or female gonadal hormones in P376L heterozygous subjects versus controls, and we saw only a modest increase in testosterone in male P376L heterozygotes relative to noncarriers. We postulate that differences in expression or capacity for up-regulation of apoB-containing lipoprotein receptors relative to SR-BI between mouse models and humans in steroidogenic tissues may account, at least partially, for the lack of recapitulation of some of the phenotypes of SR-BI deficiency in mice. We also observed no differences in platelet levels, cholesterol content, and activation from the P376L carriers, despite reports of thrombocytopenia and altered platelet activity in Scarb1 KO mice (31). These results suggest a relatively different contribution of SR-BI to platelet function between mice and humans. Note that the phenotypes of human SCARB1 P376L homozygote (elevated HDL-C and large HDL particles but relatively normal steroidogenesis, reproductive viability, and platelet function) are comparable to those observed in mice lacking PDZ domain containing 1 (PDZK1), an adaptor protein for SR-BI (37).
Perhaps the most important finding of our study is that, despite the elevation in HDL-C, P376L carriers exhibit increased risk of CHD, as do Scarb1 KO mice. Our results are consistent with a growing theme in HDL biology that steady-state more important than absolute levels. Using an in vivo assay of macrophage RCT, we previously showed that Scarb1 KO mice have impaired macrophage RCT even though they have elevated HDL-C levels (21). Our results suggest that reduced hepatic SR-BI function in humans causes impaired RCT, which leads to increased risk of CHD despite elevation in HDL-C levels. However, SR-BI is also expressed in vascular cell types, including endothelial cells, vascular smooth muscle cells, and macrophages, where it could have protective effects against atherosclerosis as well (38, 39). Our results are also consistent with the previously suggested concept (39) that up-regulation or enhancement of SR-BI could be a novel therapeutic approach to reducing CHD risk in the general population.
Supplementary Material
Acknowledgments
We appreciate the participation and support of participants of the deep clinical phenotyping studies. We thank E. Mohler for assistance in interpretation of cIMT results and J. Billheimer and E. Pashos for helpful discussions. We also acknowledge J. Tabita-Martinez for expert assistance with clinical phenotyping studies. This work was supported in part by an award from the National Center for Research Resources (grant TL1RR024133) and National Center for Advancing Translational Sciences of the NIH (grant TL1R000138) to support patient recruitment. D.B.L. was supported by a fellowship from the Doris Duke Charitable Foundation. S.K. has financial relationships with Novartis, Aegerion, Bristol-Myers Squibb, Sanofi, AstraZeneca, Alnylam, Eli Lilly, Leerink Partners, Merck, Catabasis, Regeneron Genetic Center, San Therapeutics, and Celera. H.S. has financial relationships with MSD Sharp and Dohme, Sanofi-Aventis, and Amgen. S.B. has financial relationships with Boehringer Ingelheim, Bayer, Novartis, Roche, and Thermo Fisher. N.S has financial relationships with Amgen, Sanofi, Astrazeneca, and MSD Sharp and Dohme. A.K. has a financial relationship with Amgen. J.D. has a financial relationship with Novartis. A.T.-H. has financial relationships with Eli Lilly and LGC Genomics. Sequencing data have been deposited in GenBank (SRX1458096). Genotyping data have been deposited in the Gene Expression Omnibus (GSE76065).
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/351/6278/1166/suppl/DC1
Materials and Methods
References (40–76)
Consortia and Study Author Lists
REFERENCES AND NOTES
- 1.Rader DJ, Tall AR. Nat Med. 2012;18:1344–1346. doi: 10.1038/nm.2937. [DOI] [PubMed] [Google Scholar]
- 2.Haase CL, et al. J Clin Endocrinol Metab. 2012;97:E248–E256. doi: 10.1210/jc.2011-1846. [DOI] [PubMed] [Google Scholar]
- 3.Frikke-Schmidt R, et al. JAMA. 2008;299:2524–2532. doi: 10.1001/jama.299.21.2524. [DOI] [PubMed] [Google Scholar]
- 4.Voight BF, et al. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Acton S, et al. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 6.Hoekstra M, Van Berkel TJ, Van Eck M. World J Gastroenterol. 2010;16:5916–5924. doi: 10.3748/wjg.v16.i47.5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang N, Arai T, Ji Y, Rinninger F, Tall AR. J Biol Chem. 1998;273:32920–32926. doi: 10.1074/jbc.273.49.32920. [DOI] [PubMed] [Google Scholar]
- 8.Ueda Y, et al. J Biol Chem. 1999;274:7165–7171. doi: 10.1074/jbc.274.11.7165. [DOI] [PubMed] [Google Scholar]
- 9.Kozarsky KF, et al. Nature. 1997;387:414–417. doi: 10.1038/387414a0. [DOI] [PubMed] [Google Scholar]
- 10.Ji Y, et al. J Biol Chem. 1999;274:33398–33402. doi: 10.1074/jbc.274.47.33398. [DOI] [PubMed] [Google Scholar]
- 11.Varban ML, et al. Proc Natl Acad Sci USA. 1998;95:4619–4624. doi: 10.1073/pnas.95.8.4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rigotti A, et al. Proc Natl Acad Sci USA. 1997;94:12610–12615. doi: 10.1073/pnas.94.23.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brundert M, et al. Arterioscler Thromb Vasc Biol. 2005;25:143–148. doi: 10.1161/01.ATV.0000149381.16166.c6. [DOI] [PubMed] [Google Scholar]
- 14.Ueda Y, et al. J Biol Chem. 2000;275:20368–20373. doi: 10.1074/jbc.M000730200. [DOI] [PubMed] [Google Scholar]
- 15.Kozarsky KF, Donahee MH, Glick JM, Krieger M, Rader DJ. Arterioscler Thromb Vasc Biol. 2000;20:721–727. doi: 10.1161/01.atv.20.3.721. [DOI] [PubMed] [Google Scholar]
- 16.Arai T, Wang N, Bezouevski M, Welch C, Tall AR. J Biol Chem. 1999;274:2366–2371. doi: 10.1074/jbc.274.4.2366. [DOI] [PubMed] [Google Scholar]
- 17.Van Eck M, et al. J Biol Chem. 2003;278:23699–23705. doi: 10.1074/jbc.M211233200. [DOI] [PubMed] [Google Scholar]
- 18.Trigatti B, et al. Proc Natl Acad Sci USA. 1999;96:9322–9327. doi: 10.1073/pnas.96.16.9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huszar D, et al. Arterioscler Thromb Vasc Biol. 2000;20:1068–1073. doi: 10.1161/01.atv.20.4.1068. [DOI] [PubMed] [Google Scholar]
- 20.Braun A, et al. Circ Res. 2002;90:270–276. doi: 10.1161/hh0302.104462. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, et al. J Clin Invest. 2005;115:2870–2874. doi: 10.1172/JCI25327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teslovich TM, et al. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghodsizadeh A, et al. Stem Cell Rev. 2010;6:622–632. doi: 10.1007/s12015-010-9189-3. [DOI] [PubMed] [Google Scholar]
- 24.Cayo MA, et al. Hepatology. 2012;56:2163–2171. doi: 10.1002/hep.25871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mallanna SK, Duncan SA. Curr Protocol Stem Cell Biol. 2013;26(Unit 1G):4. doi: 10.1002/9780470151808.sc01g04s26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Si-Tayeb K, et al. Hepatology. 2010;51:297–305. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Babitt J, et al. J Biol Chem. 1997;272:13242–13249. doi: 10.1074/jbc.272.20.13242. [DOI] [PubMed] [Google Scholar]
- 28.Viñals M, Xu S, Vasile E, Krieger M. J Biol Chem. 2003;278:5325–5332. doi: 10.1074/jbc.M211073200. [DOI] [PubMed] [Google Scholar]
- 29.Calvo D, Gómez-Coronado D, Lasunción MA, Vega MA. Arterioscler Thromb Vasc Biol. 1997;17:2341–2349. doi: 10.1161/01.atv.17.11.2341. [DOI] [PubMed] [Google Scholar]
- 30.Schwartz CC, VandenBroek JM, Cooper PS. J Lipid Res. 2004;45:1594–1607. doi: 10.1194/jlr.M300511-JLR200. [DOI] [PubMed] [Google Scholar]
- 31.Vergeer M, et al. N Engl J Med. 2011;364:136–145. doi: 10.1056/NEJMoa0907687. [DOI] [PubMed] [Google Scholar]
- 32.Hildebrand RB, et al. Arterioscler Thromb Vasc Biol. 2010;30:1439–1445. doi: 10.1161/ATVBAHA.110.205153. [DOI] [PubMed] [Google Scholar]
- 33.Lagrost L, et al. Arterioscler Thromb. 1993;13:815–825. doi: 10.1161/01.atv.13.6.815. [DOI] [PubMed] [Google Scholar]
- 34.Miettinen HE, Rayburn H, Krieger M. J Clin Invest. 2001;108:1717–1722. doi: 10.1172/JCI13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoekstra M, et al. J Lipid Res. 2008;49:738–745. doi: 10.1194/jlr.M700475-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Hoekstra M, et al. J Lipid Res. 2009;50:1039–1046. doi: 10.1194/jlr.M800410-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kocher O, et al. J Biol Chem. 2003;278:52820–52825. doi: 10.1074/jbc.M310482200. [DOI] [PubMed] [Google Scholar]
- 38.Mineo C, Shaul PW. Curr Opin Lipidol. 2012;23:487–493. doi: 10.1097/MOL.0b013e328357ba61. [DOI] [PubMed] [Google Scholar]
- 39.Rigotti A, Miettinen HE, Krieger M. Endocr Rev. 2003;24:357–387. doi: 10.1210/er.2001-0037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.