

Abstract
The PTPN11 gene, encoding the tyrosine phosphatase SHP-2, is overexpressed in rheumatoid arthritis (RA) fibroblast-like synoviocytes (FLS) compared with osteoarthritis (OA) FLS and promotes RA FLS invasiveness. Here, we explored the molecular basis for PTPN11 overexpression in RA FLS and the role of SHP-2 in RA pathogenesis. Using computational methods, we identified a putative enhancer in PTPN11 intron 1, which contained a glucocorticoid receptor–binding (GR-binding) motif. This region displayed enhancer function in RA FLS and contained 2 hypermethylation sites in RA compared with OA FLS. RA FLS stimulation with the glucocorticoid dexamethasone induced GR binding to the enhancer and PTPN11 expression. Glucocorticoid responsiveness of PTPN11 was significantly higher in RA FLS than OA FLS and required the differentially methylated CpGs for full enhancer function. SHP-2 expression was enriched in the RA synovial lining, and heterozygous Ptpn11 deletion in radioresistant or innate immune cells attenuated K/BxN serum transfer arthritis in mice. Treatment with SHP-2 inhibitor 11a-1 reduced RA FLS migration and responsiveness to TNF and IL-1β stimulation and reduced arthritis severity in mice. Our findings demonstrate how abnormal epigenetic regulation of a pathogenic gene determines FLS behavior and demonstrate that targeting SHP-2 or the SHP-2 pathway could be a therapeutic strategy for RA.
PTPN11, encoding the tyrosine phosphatase SHP-2, is overexpressed in rheumatoid arthritis fibroblast-like synoviocytes due to abnormal methylation of an intragenic enhancer and promotes inflammatory arthritis.
Introduction
Rheumatoid arthritis (RA) is a chronic immune-mediated disease characterized by synovial inflammation and joint damage (1). Although a plethora of novel therapies have been introduced in recent years, a significant percentage of patients continue to experience persistent synovitis (2). Targeting fibroblast-like synoviocytes (FLS), the cells that form the synovial intimal lining of diarthrodial joints, represents an approach that could address this unmet medical need (3–6). In RA, FLS display unique aggressive behavior, invading the articular cartilage and promoting inflammation through production of cytokines, chemokines, and proteases. Abnormal RA FLS function is potentially related to several factors, including a characteristic DNA methylation signature (7, 8) and dysregulation of genes encoding proteins, such as sentrin (9), PTEN (10), and p53 (11).
We recently reported that the PTPN11 gene is overexpressed in RA FLS compared with osteoarthritis (OA) FLS, suggesting that this dysregulated gene contributes to the aggressive rheumatoid phenotype (12). PTPN11 is a proto-oncogene whose gain-of-function mutations lead to leukemias, breast cancer, and other neoplastic syndromes (13). PTPN11 encodes the Src homology 2 domain–containing protein tyrosine phosphatase 2 (SHP-2), which is currently considered a drug target for cancer (14, 15). SHP-2 mediates cellular responses to various growth factors, hormones, and cytokines (13) and is critical for migration and invasion of fibroblasts and tumor cells (16–18). We previously showed that SHP-2 promotes RA FLS aggressiveness by enhancing RA FLS survival, invasiveness, and responsiveness to PDGF and TNF through activation of focal adhesion kinase (FAK) (12). These findings, together with reports of RA risk loci associated with the PTPN11 locus (19–21), suggest that SHP-2 contributes to the pathogenesis of RA.
In the current study, we explored why PTPN11 expression is dysregulated in RA FLS by evaluating for the presence of regulatory regions and RA-specific epigenetic changes. We discovered an intronic glucocorticoid receptor–responsive (GR-responsive) enhancer in PTPN11 that is highly activated in RA FLS. This region includes 2 CpG sites that are hypermethylated in RA FLS and are required for full enhancer activity. These data suggest that differential methylation of the 2 CpG sites could account for the overexpression of PTPN11 in RA FLS. To understand the functional consequences of SHP-2 dysregulation, we showed that genetic deficiency of SHP-2 in radioresistant cells and innate immune cells reduces arthritis severity in vivo and that inhibition of SHP-2 activity reduces RA FLS aggressiveness and attenuates arthritis in mice. We propose that SHP-2 is abnormally regulated in RA due to epigenetic marks that control a GR-responsive enhancer and that targeting the SHP-2 pathway could be a new strategy for treating RA.
Results
Identification of a putative intragenic PTPN11 enhancer displaying epigenetic alterations in RA FLS.
We previously reported that RA FLS overexpress the PTPN11 gene compared with OA FLS (12). Because we discovered that this gene is critical to the aggressive phenotype of RA FLS (12), we explored the mechanisms underlying PTPN11 dysregulation in RA. Through genome-wide assessment of DNA methylation, we recently identified a DNA methylation pattern that distinguishes RA FLS from non-RA FLS and contributes to the pathogenic behavior of RA FLS (8, 22). Although we did not identify abnormal methylation signals in the PTPN11 locus in our previous studies, the array platform used to detect methylation was focused on promoter regions and only included a small subset of enhancer sites (23). We thus hypothesized that overexpression of PTPN11 in RA FLS could be due, in part, to abnormal methylation of a PTPN11 enhancer. To identify putative enhancers for the PTPN11 gene, we assessed the epigenetic profile of PTPN11 and its chromatin state segmentation (ChromHMM) using the Encyclopedia of DNA Elements (ENCODE) (http://genome.ucsc.edu/). No ChromHMM data in FLS are available, so data in normal human lung fibroblasts (NHLF) were used as a surrogate. We identified a 1.4-kb DNA region (chr 12:112859818-112861217) described as a potential enhancer by ChromHMM in intron 1 of the PTPN11 gene (Figure 1A). This fragment included a DNase I hypersensitivity cluster, which suggested the presence of open chromatin available for transcription factor binding. This region was enriched with enhancer markers in NHLF, including histone modification H3K4me1, according to ENCODE ChIP sequencing (ChIP-seq) data in NHLF (Figure 1A).
Figure 1. Identification of a putative intragenic PTPN11 enhancer displaying epigenetic alterations in rheumatoid arthritis fibroblast-like synoviocytes versus osteoarthritis fibroblast-like synoviocytes.

(A) Scheme of the putative intragenic enhancer in chromosomal region 12q24.13, depicting an ENCODE-derived DNase I hypersensitivity cluster, chromatin state annotation tracks produced from ChromHMM, H3K4me1 marker, transcription factor ChIP sequencing, and human/mouse/rat conserved transcription factor–binding sites. Data were downloaded from the UCSC Genome Browser (http://genome.ucsc.edu/; assembly GRCh37/hg19). (B) After serum starving rheumatoid arthritis (RA) fibroblast-like synoviocytes (FLS) for 24 hours, ChIP-PCR was performed using anti-H3K4me1 antibody on the PTPN11 enhancer or MYT1 exon 1 as a control or control (Ctrl) IgG on the PTPN11 enhancer. The occupancy of H3K4me1 or Ctrl IgG is shown as the percentage of input. Box-and-whisker plots depict median (line within box), 25th percentile and 75th percentile (bottom and top borders), and range of minimum to maximum values (whiskers) from 4 independent experiments with different RA FLS lines. Data were analyzed using the 2-tailed paired t test. (C) Sequence of DNase I hypersensitivity region in the PTPN11 enhancer (chr12:112860281-112860755) with 8 CpG dinucleotides. (D) Analysis of differentially methylated loci between RA (n = 8) and osteoarthritis (OA) (n = 6) FLS in the PTPN11 DNase I hypersensitivity region through pyrosequencing. Box-and-whisker plots depict the percentage of methylation of each CpG site. Data were analyzed using the 2-tailed unpaired t test with Welch’s correction.
To independently identify potential enhancer function of this region in RA FLS, we performed site-directed ChIP for the H3K4me1 epigenetic marker, which identifies enhancers (24). H3K4me1 binding was determined by amplifying a 158-bp region within the enhancer. As a negative control, qPCR primers were designed to amplify the MYT1 gene, which showed no reported H3K4me1 in NHLF in ChromHMM. We observed significant enrichment in the ChIP assay in the putative PTPN11 enhancer compared with MYT1 using the anti-H3K4me1 antibody (Figure 1B), which supports that the candidate region is an enhancer in FLS. We then determined the methylation state of 8 CpG sites in the DNase I hypersensitivity cluster of the putative enhancer by pyrosequencing in RA and OA FLS (Figure 1, C and D). As shown in Figure 1D, 2 sites were significantly hypermethylated in RA FLS compared with OA FLS: CpG#6 (chr12:112860522) and CpG#7 (chr12:112860601).
The putative PTPN11 enhancer drives gene expression and is regulated by the GR.
Having identified a putative intragenic PTPN11 enhancer displaying aberrant methylation in RA FLS compared with OA FLS, we next set out to determine whether the enhancer modulates PTPN11 expression in RA FLS. We cloned the 1.4-kb intronic fragment into a luciferase reporter construct driven by a minimal promoter. Enhancer activity of the fragment was assessed by a luciferase reporter assay in transfected RA FLS. As shown in Figure 2A, luciferase activity was significantly increased in RA FLS transfected with the putative PTPN11 enhancer compared with cells transfected with empty luciferase vector (mean ± SEM relative luciferase activity 1.59 ± 0.21 for cells transfected with enhancer and 0.67 ± 0.14 for cells transfected with empty vector, P = 0.012). Therefore, the 1.4-kb region acts as an enhancer in RA FLS.
Figure 2. The putative PTPN11 enhancer region drives gene expression and is regulated by the glucocorticoid receptor.

(A) PTPN11 enhancer luciferase reporter vector or empty control vector was cotransfected into rheumatoid arthritis (RA) fibroblast-like synoviocytes (FLS) with renilla plasmid. Firefly luciferase activity was measured and normalized to renilla luciferase activity 24 hours after transfection. Box-and-whisker plots depict median (line within box), 25th percentile and 75th percentile (bottom and top borders), and range of minimum to maximum values (whiskers) from 4 independent experiments with different RA FLS lines. Data were analyzed using the 2-tailed paired t test. (B) Scheme of location and sequences of the PTPN11 enhancer glucocorticoid receptor–binding (GR-binding) motif and primers used in C. (C) After serum starvation for 24 hours, RA FLS were treated with ethanol (EtOH) or 100 nM dexamethasone (DEX) for 90 minutes. ChIP-PCR was performed using anti-GR antibody or control IgG. Values were normalized to corresponding input DNA and are expressed as fold enrichment relative to control IgG. Scatter plot shows the mean ± SEM of 3 independent experiments with different RA FLS lines. Data were analyzed using the 2-tailed paired t test. (D) PTPN11 enhancer luciferase reporter vector or empty control vector was cotransfected into RA FLS with renilla plasmid. After serum starvation for 24 hours, RA FLS were treated with medium or 100 nM DEX for 24 hours. Firefly luciferase activity was measured and normalized to renilla luciferase activity. Box-and-whisker plots depict the ratio of relative luciferase activity of DEX-treated cells versus medium-treated cells from 14 independent experiments with different RA FLS lines. Data were analyzed using the 2-tailed paired t test.
Using transcription factor ChIP-seq data from ENCODE and data from human/mouse/rat conserved transcription factor–binding sites from the Transfac Matrix Database, we then identified several potential binding sites for relevant transcription factors in the region, including the GR, the zinc finger protein 263 (ZNF263), and the Sry-related HMG box transcription factor 9 (SOX9). We performed ChIP-PCR to determine if any of these transcription factors bind to the enhancer in RA FLS using primers designed to amplify 150–200 bp in the enhancer region containing each binding site (Figure 2B). Significant GR binding was observed when cells were cultured with 100 nM of the glucocorticoid dexamethasone (DEX) (Figure 2C; mean ± SEM fold enrichment 5.62 ± 0.38 for DEX-treated cells, P = 0.007 compared with control). No detectable binding was observed for ZNF263 or SOX9. We next examined the effect of GR activation on enhancer activity in RA FLS using the luciferase reporter assay. As shown in Figure 2D, DEX markedly increased luciferase activity. These data show that the PTPN11 intronic enhancer is glucocorticoid responsive.
PTPN11 expression in RA FLS is glucocorticoid responsive.
To confirm that the GR physiologically regulates PTPN11 expression in intact cells, we determined the effect of DEX stimulation on PTPN11 expression in RA FLS. As shown in Figure 3A, PTPN11 mRNA transcript levels in RA FLS were significantly increased in response to DEX. To confirm in vivo relevance, we also found that treatment of C57BL/6 (B6) mice with 1 μg DEX significantly increased Ptpn11 expression in ankle joints (Figure 3B). Next, we examined whether GR is responsible for DEX-induced PTPN11 expression by depleting GR using siRNA. Treatment of RA FLS with 400 nM GR siRNA for 72 hours reduced GR mRNA levels by ~90% (Figure 3C). As shown in Figure 3C, GR depletion in RA FLS using siRNA abolished the induction of PTPN11 mRNA by DEX. Because the PTPN11 enhancer also contains possible binding motifs for the estrogen receptor (ER) and androgen receptor (AR), which potentially overlap with the GR-binding motif, we also examined the effect of ER and AR activation on PTPN11 expression by incubating cells with 17β-estradiol or Cl-4AS-1, respectively. However, unlike stimulation with DEX, neither 17β-estradiol nor Cl-4AS-1 stimulation affected PTPN11 expression (Figure 3D).
Figure 3. PTPN11 expression in rheumatoid arthritis fibroblast-like synoviocytes is glucocorticoid responsive.

(A) After serum starvation for 24 hours, RA FLS were treated with increasing concentrations of dexamethasone (DEX) for 24 hours. PTPN11 mRNA expression was analyzed by qPCR, and results were normalized to GAPDH mRNA expression. Box-and-whisker plots depict median (line within box), 25th percentile and 75th percentile (bottom and top borders), and range of minimum to maximum values (whiskers) from 5 independent experiments with different rheumatoid arthritis (RA) fibroblast-like synoviocytes (FLS) lines. Data were analyzed using Dunnett’s multiple comparisons test. (B) Female C57BL/6 mice (6–8 weeks of age) were administered 1 μg DEX or vehicle (n = 4 per group) by i.p. injection. Ptpn11 mRNA expression in ankle homogenates was measured after 24 hours by qPCR performed in triplicate. Box-and-whisker plots depict Ptpn11 expression normalized to Gapdh expression. Data were analyzed using the 2-tailed unpaired t test. (C) RA FLS were transfected with control siRNA or siRNA against glucocorticoid receptor (GR) mRNA. After 24 hours, RA FLS were serum starved for 24 hours and then cultured in the absence (left) or presence (right) of 100 nM DEX for 24 hours. qPCR was performed to assess transcript levels of GR and PTPN11. Results were normalized to GAPDH expression. Box-and-whisker plots depict 4 independent experiments with different RA FLS lines. Data were analyzed using the 2-tailed paired t test. (D) After serum starvation for 24 hours, RA FLS were treated with 100 nM β-estradiol (E2), 100 nM Cl-4AS-1, or 100 nM DEX for 24 hours. PTPN11 mRNA expression was analyzed by qPCR. Results were normalized to GAPDH mRNA. Scatter plot shows the mean ± SEM of 3 independent experiments with different RA FLS lines. Data were analyzed using the 2-tailed paired t test.
Glucocorticoid responsiveness of PTPN11 is higher in RA FLS than OA FLS and requires the differentially methylated CpG sites in the PTPN11 enhancer.
We next examined whether DEX-mediated PTPN11 expression is differentially regulated in RA versus OA FLS. Interestingly, we found that DEX-mediated PTPN11 induction was significantly greater in RA FLS than OA FLS (Figure 4A; mean ± SEM PTPN11 induction 2.16 ± 0.19 for RA FLS and 1.60 ± 0.09 for OA FLS, P = 0.025), suggesting that dysregulated GR-induced enhancer activity contributes to the higher expression of PTPN11 in RA FLS compared with OA FLS. To assess whether the differentially methylated CpGs #6 and #7 are required for glucocorticoid responsiveness of the PTPN11 enhancer, we deleted the region containing these 2 CpGs in the PTPN11 enhancer luciferase reporter plasmid. Removal of these sequences significantly reduced DEX-induced enhancer activity in the reporter assay (Figure 4B). Additionally, we observed a significant positive correlation in FLS between extent of methylation of CpGs #6 and #7 and DEX-induced PTPN11 expression (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/jci.insight.86580DS1). This trend was not observed for any of the other CpGs within the PTPN11 enhancer region. We concluded that the region containing CpGs #6 and #7 is required for full enhancer response to GR activation, which supports the concept that altered methylation of the PTPN11 intragenic enhancer between RA and OA FLS promotes higher PTPN11 expression in RA FLS via enhanced glucocorticoid responsiveness.
Figure 4. Glucocorticoid responsiveness of PTPN11 is higher in rheumatoid arthritis fibroblast-like synoviocytes versus osteoarthritis fibroblast-like synoviocytes and depends on the differentially methylated CpG sites in the PTPN11 enhancer.
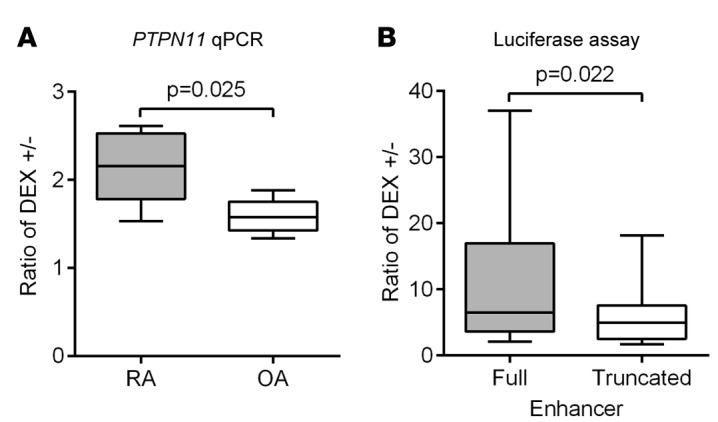
(A) After serum starvation for 24 hours, rheumatoid arthritis (RA) or osteoarthritis (OA) fibroblast-like synoviocytes (FLS) were treated with 100 nM dexamethasone (DEX) for 24 hours. PTPN11 mRNA expression was analyzed by qPCR and normalized to GAPDH expression. Box-and-whisker plots depict median (line within box), 25th percentile and 75th percentile (bottom and top borders), and range of minimum to maximum values (whiskers) of the ratio of relative PTPN11 expression in DEX-treated versus untreated cells from 5 independent experiments with different RA or OA FLS lines. Data were analyzed using the 2-tailed unpaired t test. (B) Truncated PTPN11 enhancer luciferase reporter vector was made by deleting the 81-bp region between the 2 hypermethylated CpG sites (CpGs #6 and #7; shown in Figure 1C) from the PTPN11 enhancer luciferase reporter vector. The full or truncated PTPN11 enhancer luciferase reporter vector was cotransfected into RA FLS with renilla plasmid. After serum starvation for 24 hours, RA FLS were treated with medium or 100 nM DEX for 24 hours, and firefly luciferase activity was measured and normalized to renilla luciferase activity. Box-and-whisker plots depict the ratio of relative luciferase activity in DEX-treated cells versus untreated cells of independent experiments with 12 different RA FLS lines. Data were analyzed using the 2-tailed paired t test.
Expression of SHP-2 in FLS promotes arthritis in vivo.
We then turned our attention to understanding how SHP-2 expression in FLS contributes to inflammatory arthritis in vivo. First we confirmed that SHP-2 is expressed in situ in the RA synovium by performing IHC. This study showed that SHP-2 is expressed throughout the rheumatoid synovium and is enriched in the synovial intimal lining where FLS reside (Figure 5A).
Figure 5. SHP-2 is expressed in the rheumatoid arthritis synovium and promotes fibroblast-like synoviocyte aggressiveness through its phosphatase activity.

(A) IHC staining of rheumatoid arthritis (RA) synovial sections using anti–SHP-2 or control (Ctrl) IgG antibodies. Scale bars: 50 μm. Images from 2 independent experiments are shown. (B) WT (n = 10) and Ptpn11 heterozygous (Het, n = 8) mice were lethally irradiated at 10 to 13 weeks of age and administered bone marrow from WT congenic CD45.1 donor mice (12 weeks of age). After 10 weeks, arthritis was induced in recipients by i.p. injection of K/BxN sera. Mean ± SEM ankle swelling is shown. Data were analyzed using 2-way ANOVA. (C) After a 48-hour serum starvation, RA fibroblast-like synoviocytes (FLS) were allowed to migrate through Transwell chambers in response to 5% FBS for 4 hours in the presence of 25 μM 11a-1 or DMSO. Box-and-whisker plots depict median (line within box), 25th percentile and 75th percentile (bottom and top borders), and range of minimum to maximum values (whiskers) of mean cells per field from 3 independent experiments with RA FLS from different donors. Data were analyzed using the 2-tailed Mann-Whitney test. (D) After a 48-hour serum starvation, RA FLS were stimulated with 50 ng/ml TNF or 2 ng/ml IL-1β for 24 hours in the presence of 25 μM 11a-1 or DMSO. mRNA expression was analyzed for the indicated genes by qPCR performed in triplicate. Box-and-whisker plots depict mRNA expression in RA FLS from 4 different donors normalized to GAPDH expression. Data were analyzed using the 2-tailed Mann-Whitney test.
Next, we used the K/BxN serum transfer arthritis model, which depends on the actions of innate immune cells and FLS (25, 26), to examine the role of SHP-2 in FLS-mediated RA inflammation. Because global knockout of SHP-2 is embryonically lethal, we used heterozygous (Het) Ptpn11+/– mice for these experiments. To isolate the contribution of radioresistant cells, such as FLS, to disease severity, we transplanted bone marrow from WT mice into irradiated WT or Ptpn11 Het mice and subjected the mice to K/BxN serum transfer arthritis. As shown in Figure 5B, when reconstituted with bone marrow from WT mice, arthritis severity was significantly reduced in Ptpn11 Het recipient mice compared with WT recipient mice. While the contribution of additional radioresistant cell populations, including endothelial cells and resident macrophages, cannot be ruled out, the data suggest that a partial reduction of Ptpn11 expression in non-bone marrow–derived cells, such as FLS, is sufficient to inhibit the development of inflammatory arthritis in vivo.
Promotion of FLS invasiveness by SHP-2 requires its phosphatase activity.
We previously reported that induced SHP-2 deficiency impairs RA FLS invasiveness by reducing RA FLS migration in response to growth factors and gene expression in response to TNF stimulation (12). To assess whether the phosphatase activity of SHP-2 is necessary for FLS invasiveness, we evaluated the effect of the SHP-2 small-molecule inhibitor 11a-1 (27) on FLS function in vitro. As shown in Figure 5C, 11a-1 treatment significantly reduced RA FLS migration in a Transwell migration assay (mean ± SEM cells per field 18 ± 3.1 for DMSO-treated cells and 2.8 ± 0.4 for 11a-1–treated cells, P = 0.0003). 11a-1 treatment also significantly reduced induction of several genes implicated in cell migration and invasion (including CXCL10, MMP1, MMP3, MMP9, and MMP13) in response to TNF or IL-1β (Figure 5D). Thus, treatment of FLS with an inhibitor of SHP-2 activity mimicked knockdown of PTPN11 expression (12) in the same cells, suggesting that the phosphatase activity of SHP-2 is required for increased FLS invasiveness.
In vivo inhibition of SHP-2 attenuates inflammatory arthritis.
SHP-2 is also expressed in cells of the immune system, where it promotes antigen receptor signaling in T cells and B cells (28, 29), regulates TLR signaling in myeloid cells (30), and is required for myeloid cell development (31). Partial SHP-2 deletion following inducible haploinsufficiency of Ptpn11 does not result in hematologic deficiency or other side effects in mice (31, 32). Using mice heterozygous for a floxed allele of Ptpn11 and expressing cre recombinase under control of the type 1 IFN–inducible Mx1 promoter, we assessed the severity of passive K/BxN arthritis after inducing haploinsufficiency of Ptpn11. As shown in Figure 6A, inducible partial deletion of SHP-2 resulted in protection from arthritis development. Because inducible Mx1-cre–mediated gene deletion mainly occurs in hematopoietic cells and is ineffective in FLS (N. Bottini and S.M. Stanford, unpublished observations), we concluded that partial inactivation of SHP-2 in non-FLS cells is sufficient to inhibit innate immunity-driven arthritis in mice.
Figure 6. In vivo inhibition of SHP-2 attenuates arthritis in mice.

(A) At 6 to 10 weeks of age, female Ptpn11fl/+-Mx1 cre– (n = 3) and cre+ (n = 4) mice were injected i.p. with 3 doses of 300 μg polyinosinic-polycytidylic acid [poly(I:C)] every other day. Ten days after the last poly(I:C) treatment, mice were injected i.p. with 200 μl K/BxN sera to induce arthritis. Ankle thickness was measured every other day after serum transfer. Mean ± SEM ankle swelling is shown. *P < 0.05, 1-tailed unpaired t test. (B–D) Female C57BL/6 mice were administered 200 μl K/BxN sera at 8 weeks of age and administered 7.5 mg/kg 11a-1 (n = 5) or vehicle (n = 4) i.p. daily for 14 days. (B) Mean ± SEM ankle swelling is shown. Data were analyzed using 2-way ANOVA. (C and D) Histological analyses of ankles stained with H&E or Safranin-O at the end of the disease course. (C) Scatter plots show the mean ± SEM histological score of each ankle. Data were analyzed using the 2-tailed Mann-Whitney test. (D) Representative images of H&E-stained (yellow arrows indicate regions of inflammatory infiltrate) or Safranin-O–stained (red arrows indicate regions of cartilage erosion) joints are shown. Scale bars: 200 μm.
Since partial inactivation of Ptpn11 in non-bone marrow–derived cells also protected mice from arthritis (as seen in Figure 5B), we hypothesized that global partial inhibition of SHP-2 might protect mice from arthritis by inhibiting both immune and FLS-dependent pathogenic actions. To achieve inhibition of SHP-2 activity, we treated mice with the selective SHP-2 inhibitor 11a-1, injecting mice daily with 7.5 mg/kg i.p. This dose, based on pharmacokinetic data, achieves in vivo 11a-1 concentration in the vicinity of the IC50 value and does not cause clinically evident toxicity in mice after 4 weeks of administration (Z.Y. Zhang and L. Zeng, unpublished observations). As shown in Figure 6, B–D, daily i.p. administration of 7.5 mg/kg 11a-1 significantly attenuated arthritis severity and joint damage. These data suggest that SHP-2 promotes inflammatory arthritis pathogenesis through its phosphatase activity and that partial pharmacologic inhibition of SHP-2 could be a safe and effective strategy for treatment of RA.
Discussion
Aberrant epigenetic regulation of gene expression, including DNA methylation, is emerging as a major influence in the pathogenesis of several autoimmune diseases (33). We recently showed that RA FLS display a distinctive DNA methylation pattern that contributes to their pathogenic action in RA (8, 22). We also recently found that expression of PTPN11, encoding the tyrosine phosphatase SHP-2, is significantly greater in RA FLS compared with OA FLS and demonstrated a novel role for SHP-2 as a mediator of the aggressive phenotype of RA FLS (12). Additionally, a recent genome-wide association study revealed an RA risk locus (dbSNP ID = rs10774624) associated with the PTPN11 gene, suggesting a role for PTPN11 in RA (21). These data point towards SHP-2 as an important player in RA FLS pathogenic action and suggest that further insight into the biology and regulation of PTPN11 in RA is warranted. In the present study, we addressed two questions: (a) what is the molecular basis for the overexpression of PTPN11 in RA FLS, and (b) does FLS-expressed SHP-2 promote the pathogenesis of RA?
To answer the first question, we applied an integrative computational and experimental approach. We identified an intronic enhancer in the PTPN11 gene that is specifically responsive to GR and includes 2 CpG sites (CpGs #6 and #7) that are hypermethylated in RA FLS versus OA FLS. We also showed that hypermethylation of CpGs #6 and #7 is associated with increased responsiveness of RA FLS to GR-activating DEX stimulation and that CpGs #6 and #7 are required for full responsiveness of the PTPN11 enhancer to GR.
The data suggest that hypermethylation of CpGs #6 and #7 in the PTPN11 enhancer contributes to the observed overexpression of SHP-2 in RA FLS by enhancing GR-induced PTPN11 expression. Unlike promoter regions, in which methylation typically suppresses transcription (34), methylation of enhancers as an epigenetic mechanism for regulation has not yet been explored in detail. We found a positive correlation between GR-induced PTPN11 expression in FLS from different donors and increasing methylation at CpGs #6 and #7 sites in the PTPN11 enhancer. A similar correlation was not observed for any of the other CpG sites within the PTPN11 enhancer. Based on these data, we concluded that hypermethylation of CpGs #6 and #7 promotes PTPN11 transcription by increasing enhancer activation by endogenous glucocorticoids, although the molecular mechanism remains uncertain. One possibility is that methylation at these sites disrupts enhancer binding by a transcriptional repressor, poising the enhancer for GR activation. The mechanisms that cause hypermethylation of CpGs #6 and #7 in RA FLS are also undefined and likely depend on alterations of factors that direct DNA methyltransferases to these CpGs. These could include nontranscribed RNA, conformational restrictions of genomic DNA, or steric hindrance due to transcription factor binding to DNA, all of which potentially alter access of the modification machinery to DNA (35, 36).
Our findings also suggest that endogenous glucocorticoids in the joint could regulate PTPN11 expression in FLS. In RA, abnormal methylation of the PTPN11 enhancer contributes to joint pathology by increasing PTPN11 expression in response to endogenous glucocorticoids, resulting in enhanced FLS invasiveness and joint destruction. These findings might seem paradoxical given the well-known antiinflammatory effects of pharmacologic doses of corticosteroids; this function might be specific to the low endogenous concentrations in the joint microenvironment or to FLS. An interesting corollary of the observed GR dependence of PTPN11 expression in FLS is that, by enhancing FLS invasiveness, the antiinflammatory action of exogenous corticosteroids is partially counteracted. This phenomenon suggests that corticosteroids can activate some pathogenic mechanisms in addition to their antiinflammatory action and might explain why corticosteroids often have unexpectedly modest effects on joint damage, despite dramatically decreasing synovitis (37). These findings also suggest that inhibition of SHP-2 could augment the antiinflammatory effect of glucocorticoids in RA.
To answer the second question, we assessed whether partial inactivation of SHP-2 in radioresistant cells protects mice from arthritis development. We used the K/BxN serum transfer arthritis model, in which disease pathogenesis is primarily dependent on innate immune cells and FLS (25, 26). Through bone marrow reconstitution experiments, we showed that expression of SHP-2 in radioresistant cells promotes K/BxN arthritis, supporting the idea that FLS-expressed SHP-2 promotes RA. Although further experimentation is needed to rule out an effect of SHP-2 in non-FLS radioresistant cell types that are known to mediate joint inflammation in this model (38), the data suggest that targeting PTPN11 and its pathway in RA FLS might decrease RA severity as well as enhance the efficacy of steroids in this disease.
We recently reported that SHP-2 mediates the aggressive phenotype of RA FLS by promoting activation of FAK (12). SHP-2 is known to promote signaling through both activity-dependent and -independent mechanisms. Here, we showed that a selective inhibitor of SHP-2 activity reduced RA FLS aggressiveness, suggesting that promotion of FAK activation in FLS is dependent on SHP-2 phosphatase activity.
Our data show that partial deletion of SHP-2 is sufficient to reduce the pathogenic action of FLS in RA. Since SHP-2 promotes immune cell signaling, and inhibitors of SHP-2 are being developed as antileukemic agents (15), we propose that partial inhibition of SHP-2 could be a strategy for combined targeting of immune cells and FLS in RA. We demonstrated that partial inactivation of SHP-2 in non-FLS cells and global partial inhibition of SHP-2 using a chemical inhibitor effectively decreases arthritis severity in mice.
In addition to direct targeting of SHP-2, other molecules controlling FLS aggressiveness within the SHP-2 pathway represent alternative candidate targets for RA therapies. For example, recent studies support that anomalous FAK activation significantly contributes to RA FLS aggressiveness. FAK phosphorylation levels are increased in RA synovial lining cells compared with normal synovial tissue (39), and DNA methylation abnormalities were identified within the FAK pathway in RA FLS (8). Additionally, we recently reported that another tyrosine phosphatase that promotes FAK activation — receptor PTP-α — also mediates the aggressiveness of RA FLS and promotes arthritis in mice (40).
In conclusion, our study demonstrates that a combination of computational and biologic methods can be used to dissect the critical abnormalities of RA FLS and define candidate molecular or pathway targets for therapeutic intervention. We provide both genetic and pharmacological evidence that partial inhibition of SHP-2 activity is beneficial for RA treatment. We suggest that further exploration of the SHP-2/FAK pathway may provide additional options for development of new RA therapies targeted to FLS or with combined action on FLS and the immune system.
Methods
Reagents.
TNF and IL-1β were purchased from eBioscience. The development and synthesis of the SHP-2 inhibitor 11a-1 are described in ref. 27. 11a-1 inhibits the enzymatic activity of SHP-2 with an IC50 of 200 nM, displays greater than 5-fold selectivity against classical mammalian tyrosine phosphatases, and inhibits SHP-2 function in cancer cells (27). Unless specified, other reagents were purchased from Sigma-Aldrich.
Computational methods.
Analysis of PTPN11 (accession NM_002834.3; Entrez Gene ID 5781) regulatory regions was performed using the UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway; assembly GRCh37/hg19, February 2009). The ENCODE database was used to identify DNase I hypersensitivity regions assayed in a large collection of cell types, ChromHMM profiles in NHLF, and histone marks, such as H3K4me1, in NHLF. Putative transcription factor–binding sites were identified using the ChIP-seq database from ENCODE and the human/mouse/rat conserved transcription factor–binding sites from the Transfac Matrix Database.
Preparation of human synovial tissue and FLS.
Synovial tissue was obtained from patients with OA and RA at the time of total joint replacement or synovectomy as previously described (41). The diagnosis of RA conformed to American College of Rheumatology 1987 revised criteria (1). For preparation of FLS, the synovium was minced and incubated with 1 mg/ml collagenase type VIII in serum-free RPMI 1640 (Gibco BRL) for 1 hour at 37°C, filtered, extensively washed, and cultured in DMEM (Gibco BRL) supplemented with 10% FBS (Gemini Bio Products), 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μg/ml gentamicin, and 6 mM glutamine in a humidified 5% CO2 atmosphere. Cells were allowed to adhere overnight and then nonadherent cells were removed and adherent FLS were split at 1:3 dilution when greater than 70% confluent. FLS were used from passages 4 to 10, during which time they are a homogeneous population of cells (<1% CD11b positive, <1% phagocytic, and <1% FcγRII and FcγRIII receptor positive). Cells in this study were synchronized in serum starvation media (FLS media with 0.1% FBS) for 24 to 48 hours before addition of the appropriate stimuli.
ChIP PCR.
ChIP assay was performed using the Zymo-Spin ChIP kit according to the manufacturer’s protocol (Zymo Research). Briefly, FLS were serum starved for 24 hours and then fixed in 1% formaldehyde (Thermo Fisher Scientific) for 8 minutes at room temperature. After sonication, chromatin was immunoprecipitated with specific antibodies overnight at 4°C. The antibodies used were anti-H3K4me1 (ab8895, Abcam), anti-GR (catalog 12041, Cell Signaling Technology), and rabbit IgG (catalog 2729, Cell Signaling Technology). Complexes were then immunoprecipitated with protein A magnetic beads for 1 hour at 4°C and reverse-crosslinked at 65°C for 2 hours. The eluted DNA was purified and used as template for qPCR. A 10% input sample was used as control. The enhancer-specific primers, 5′-CTGAGACTGGAGGCTTTTGG-3′ (forward) and 5′-GACATCCCAGTTTCCACTGC-3′ (reverse), were designed using Primer3 software (42). MYT1 exon 1 primers were purchased from Cell Signaling Technology (catalog 4493).
qPCR.
RA FLS were serum starved for 24 to 48 hours prior to stimulation. Where indicated, cells were treated with 50 ng/ml TNF or 2 ng/ml IL-1β for 24 hours. For stimulation with 17β-estradiol or Cl-4AS-1 (Tocris Bioscience), cells were first treated with hormone-depleted, phenol red–free complete medium (Gibco) for 48 hours and synchronized in 0.1% charcoal-stripped FBS for 24 hours before addition of the stimulations. Total RNA and qPCR were performed as previously described (43). Data were normalized to the expression levels of the housekeeping gene GAPDH.
Constructs, transfections, and luciferase reporter assays.
A 1.4-kb enhancer fragment was amplified from genomic DNA of RA FLS with specific primers, 5′-CTGAGCTCGCTAGCCTCGAGCTATTTTCATGGCTGAGAGA-3′ (forward) and 5′-ATACCCTCTAGTGTCTAAGCTTCTCAGGAGACTGAGGCA-3′ (reverse), and subcloned into the pGL4.23 vector with a minimal promoter (Promega). The truncated enhancer plasmid was constructed by removing the sequence between the CpG sites at chr12:112860522 and 112860601 in the enhancer using the Q5 Site Directed Mutagenesis Kit (New England Biolabs). Primers for Q5-mediated mutagenesis were 5′-TGTCCATGGTCAATGTGG-3′ (forward) and 5′-TTAATAAAGACATCCCAGTTTC-3′ (reverse). For luciferase assays, 1 μg of empty vector, enhancer plasmid, or truncated enhancer plasmid was cotransfected with 50 ng renilla plasmid into RA FLS using the Human Dermal Fibroblast Nucleofector Kit (Lonza). Cells were lysed and assayed for luciferase activity using the Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase activity was normalized to renilla activity.
Gene knockdown.
siRNA for GR and other transcription factors and control siRNA (ON-TARGET plus nontargeting pool) were purchased from Dharmacon. siRNAs (400 nM) were introduced into cultured cells using the Human Dermal Fibroblast Nucleofector Kit (Lonza). After 24 hours, cells were serum starved for 24 hours and then treated with DEX or vehicle for 24 hours prior to analysis.
Bisulfite pyrosequencing.
Genomic DNA prepared from RA and OA FLS was bisulfite modified using the EpiTect Bisulfite Kit (Qiagen). Pyrosequencing of the bisulfite-converted DNA was performed by EpigenDx.
IHC of synovial tissue.
The rabbit anti–SHP-2 antibody was purchased from Abcam (ab32083). The control rabbit IgG antibody (unconjugated goat anti-rabbit IgG) was purchased from Vector Labs (AI-1000). Paraffin-embedded slides of human RA synovial tissues were deparaffinated, rehydrated, and pretreated for 10 minutes with boiling citrate antigen retrieval buffer (1.9 mM citric acid, 10 mM Tris-sodium citrate, pH 6.0), and treated with 3% H2O2 for 10 minutes. Slides were blocked with 5% goat serum for 1 hour at room temperature and then incubated with anti–SHP-2 antibody or control rabbit IgG (1:200 in 5% BSA) overnight at 4°C. Slides were washed and incubated with SignalStain Boost IHC Detection reagent (HRP, rabbit; Cell Signaling Technologies) for 30 minutes, incubated for 2 minutes with 3,3′-diaminobenzidine substrate, and counterstained with hematoxylin. Slide images were obtained using an Axio Scan.Z1 slide scanner (Carl Zeiss Microscopy).
Transwell migration assay.
RA FLS migration was assessed in Transwell chambers (Corning) as previously described (12, 40). Each experiment included 3 membranes per sample, and 4 fields were analyzed per membrane.
Mice.
Generation of Ptpn11fl mice was previously described (44). Ptpn11fl mice were backcrossed onto the B6 background for greater than 7 generations. Mice carrying heterozygous deletion of Ptpn11 were generated by crossing with mice carrying cre recombinase under the CMV promoter (Jackson Laboratory). The resulting cre+ offspring carrying excision of the Ptpn11fl allele were crossed with B6 mice (Jackson Laboratory) to generate cre– mice carrying germline heterozygous excision of the Ptpn11fl allele (Het mice). To induce Ptpn11 haploinsufficiency in hematopoietic cells, Ptpn11fl mice were crossed with mice carrying cre recombinase under the Mx1 promoter (Jackson Laboratory) to generate Ptpn11fl/+-Mx1-cre mice. At 6 weeks of age, mice were injected i.p. with 3 doses of 300 μg polyinosinic-polycytidylic acid every other day. B6 KRN mice were provided by Christophe Benoist (Harvard Medical School, Boston, Massachusetts, USA) and were crossed with NOD mice (Jackson Laboratory) to obtain arthritic offspring (K/BxN mice) whose sera was pooled for use in the K/BxN passive serum transfer arthritis model (45). Congenic CD45.1 B6 mice were purchased from Taconic Biosciences.
Reciprocal bone marrow transplantation.
WT and Ptpn11 Het mice were lethally irradiated with 2 doses of 55 Gy and administered bone marrow from gender-matched WT congenic CD45.1 donor mice (40). KBxN pooled sera was administered to induce arthritis (see below) 10 weeks after irradiation. The percentage of engrafted cells in Het recipient mice reconstituted with WT bone marrow was greater than 80%.
K/BxN serum transfer arthritis model.
Arthritis was induced by i.p. injection of 200 μl pooled sera from K/BxN mice. Every 2 days, ankle thickness was measured using a digital caliper (45). For experiments involving administration of the SHP-2 inhibitor, 11a-1 was resuspended in a solution of 5% DMSO, 5% Tween-20, and 90% H2O, and mice were administered 7.5 mg/kg 11a-1 or vehicle daily by i.p. injection starting at the time of arthritis induction.
Histological analysis of arthritic joints.
Hind paws were fixed in 10% neutral-buffered formalin, decalcified, and embedded in paraffin. Sections were prepared from tissue blocks (HistoTox) and stained with H&E and Safranin-O/Fast Green/Hematoxylin (Sigma-Aldrich). Histopathological scoring for inflammation, bone erosion, and cartilage erosion was performed as previously described (40, 46). Histologic analyses were performed in a blinded manner by 2 operators.
Statistics.
The 2-way ANOVA, Mann-Whitney test, Dunnett’s multiple comparisons test, Wilcoxon matched-pairs signed-rank test, Pearson correlation, 2-tailed paired t test, and 1- or 2-tailed unpaired t test were performed using GraphPad Prism software. A comparison was considered significant if P was less than 0.05.
Study approval.
Human studies were approved by the Institutional Review Board of the University of California, San Diego School of Medicine, and informed consent was obtained from all participants. Animal experiments were conducted in accordance with a La Jolla Institute for Allergy and Immunology Institutional Animal Care and Use Committee–approved protocol (AP140-NB4).
Author contributions
KM, SMS, DH, G-SF, JWW, ZYZ, WW, NB, and GSF conceived and designed the study. KM, SMS, DH, CS, LZ, RA, VZ, and GRAM acquired the data. KM, SMS, DH, CS, LZ, RA, VZ, DLB, GRAM, JWW, ZYZ, WW, NB, and GSF analyzed and interpreted the data. All authors were involved in drafting the manuscript or revising it critically for important intellectual content, and all authors approved the final version to be published. GSF and NB had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Supplementary Material
Acknowledgments
This work was supported in part by the Rheumatology Research Foundation (Disease Targeted Innovative Research grant to G.S. Firestein), the Arthritis Foundation (grant to G.S. Firestein), the National Institute of Arthritis and Musculoskeletal and Skin Diseases NIH (grants R01-AR-065466 to G.S. Firestein and R01-AR-066053 to N. Bottini), Janssen R&D of Johnson & Johnson (grant to G.S. Firestein), the Arthritis National Research Foundation (grant to S.M. Stanford), and the National Cancer Institute NIH (grant R01-CA-069202 to Z.Y. Zhang). This is manuscript #1887 from the La Jolla Institute for Allergy and Immunology.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(7):e86580. doi:10.1172/jci.insight.86580.
Contributor Information
Keisuke Maeshima, Email: kmaeshima@ucsd.edu.
Deepa Hammaker, Email: dhammaker@ucsd.edu.
Cristiano Sacchetti, Email: cristianos@liai.org.
Li-fan Zeng, Email: zengl@iupui.edu.
Rizi Ai, Email: riai@mail.ucsd.edu.
Vida Zhang, Email: vida@lji.org.
Gen-Sheng Feng, Email: gfeng@ucsd.edu.
Zhong-Yin Zhang, Email: zyzhang@iupui.edu.
Wei Wang, Email: wei-wang@ucsd.edu.
Nunzio Bottini, Email: nunzio@liai.org.
References
- 1.Arnett FC, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheumatol. 1988;31(3):315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 2.Choy EH, Kavanaugh AF, Jones SA. The problem of choice: current biologic agents and future prospects in RA. Nat Rev Rheumatol. 2013;9(3):154–163. doi: 10.1038/nrrheum.2013.8. [DOI] [PubMed] [Google Scholar]
- 3.Stanczyk J, Ospelt C, Gay RE, Gay S. Synovial cell activation. Curr Opin Rheumatol. 2006;18(3):262–267. doi: 10.1097/01.bor.0000218947.42730.dd. [DOI] [PubMed] [Google Scholar]
- 4.Lefevre S, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med. 2009;15(12):1414–1420. doi: 10.1038/nm.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neumann E, Lefevre S, Zimmermann B, Gay S, Muller-Ladner U. Rheumatoid arthritis progression mediated by activated synovial fibroblasts. Trends Mol Med. 2010;16(10):458–468. doi: 10.1016/j.molmed.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 6.Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9(1):24–33. doi: 10.1038/nrrheum.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakano K, Boyle DL, Firestein GS. Regulation of DNA methylation in rheumatoid arthritis synoviocytes. J Immunol. 2013;190(3):1297–1303. doi: 10.4049/jimmunol.1202572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakano K, Whitaker JW, Boyle DL, Wang W, Firestein GS. DNA methylome signature in rheumatoid arthritis. Ann Rheum Dis. 2013;72(1):110–117. doi: 10.1136/annrheumdis-2012-201526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franz JK, et al. Expression of sentrin, a novel antiapoptotic molecule, at sites of synovial invasion in rheumatoid arthritis. Arthritis Rheumatol. 2000;43(3):599–607. doi: 10.1002/1529-0131(200003)43:3<599::AID-ANR17>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 10.Pap T, Franz JK, Hummel KM, Jeisy E, Gay R, Gay S. Activation of synovial fibroblasts in rheumatoid arthritis: lack of Expression of the tumour suppressor PTEN at sites of invasive growth and destruction. Arthritis Res. 2000;2(1):59–64. doi: 10.1186/ar69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tak PP, Zvaifler NJ, Green DR, Firestein GS. Rheumatoid arthritis and p53: how oxidative stress might alter the course of inflammatory diseases. Immunol Today. 2000;21(2):78–82. doi: 10.1016/S0167-5699(99)01552-2. [DOI] [PubMed] [Google Scholar]
- 12.Stanford SM, et al. Protein tyrosine phosphatase expression profile of rheumatoid arthritis fibroblast-like synoviocytes: a novel role of SH2 domain-containing phosphatase 2 as a modulator of invasion and survival. Arthritis Rheumatol. 2013;65(5):1171–1180. doi: 10.1002/art.37872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008;27(2):179–192. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 14.Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28(6):284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 15.Jiang ZX, Zhang ZY. Targeting PTPs with small molecule inhibitors in cancer treatment. Cancer Metastasis Rev. 2008;27(2):263–272. doi: 10.1007/s10555-008-9113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu DH, Qu CK, Henegariu O, Lu X, Feng GS. Protein-tyrosine phosphatase Shp-2 regulates cell spreading, migration, and focal adhesion. J Biol Chem. 1998;273(33):21125–21131. doi: 10.1074/jbc.273.33.21125. [DOI] [PubMed] [Google Scholar]
- 17.Wang FM, Liu HQ, Liu SR, Tang SP, Yang L, Feng GS. SHP-2 promoting migration and metastasis of MCF-7 with loss of E-cadherin, dephosphorylation of FAK and secretion of MMP-9 induced by IL-1beta in vivo and in vitro. Breast Cancer Res Treat. 2005;89(1):5–14. doi: 10.1007/s10549-004-1002-z. [DOI] [PubMed] [Google Scholar]
- 18.Feng H, et al. Dynamin 2 mediates PDGFRalpha-SHP-2-promoted glioblastoma growth and invasion. Oncogene. 2012;31(21):2691–2702. doi: 10.1038/onc.2011.436. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Zhernakova A, et al. Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS Genet. 2011;7(2):e86580. doi: 10.1371/journal.pgen.1002004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scott IC, et al. Do genetic susceptibility variants associate with disease severity in early active rheumatoid arthritis? J Rheumatol. 2015;42(7):1131–1140. doi: 10.3899/jrheum.141211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okada Y, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506(7488):376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whitaker JW, et al. An imprinted rheumatoid arthritis methylome signature reflects pathogenic phenotype. Genome Med. 2013;5(4):e86580. doi: 10.1186/gm444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whitaker JW, et al. Integrative omics analysis of rheumatoid arthritis identifies non-obvious therapeutic targets. PloS One. 2015;10(4):e86580. doi: 10.1371/journal.pone.0124254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shlyueva D, et al. Hormone-responsive enhancer-activity maps reveal predictive motifs, indirect repression, and targeting of closed chromatin. Mol Cell. 2014;54(1):180–192. doi: 10.1016/j.molcel.2014.02.026. [DOI] [PubMed] [Google Scholar]
- 25.Ji H, et al. Arthritis critically dependent on innate immune system players. Immunity. 2002;16(2):157–168. doi: 10.1016/S1074-7613(02)00275-3. [DOI] [PubMed] [Google Scholar]
- 26.Lee DM, et al. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315(5814):1006–1010. doi: 10.1126/science.1137306. [DOI] [PubMed] [Google Scholar]
- 27.Zeng LF, et al. Therapeutic potential of targeting the oncogenic SHP2 phosphatase. J Med Chem. 2014;57(15):6594–6609. doi: 10.1021/jm5006176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen TV, Ke Y, Zhang EE, Feng GS. Conditional deletion of Shp2 tyrosine phosphatase in thymocytes suppresses both pre-TCR and TCR signals. J Immunol. 2006;177(9):5990–5996. doi: 10.4049/jimmunol.177.9.5990. [DOI] [PubMed] [Google Scholar]
- 29.Tamir I, Dal Porto JM, Cambier JC. Cytoplasmic protein tyrosine phosphatases SHP-1 and SHP-2: regulators of B cell signal transduction. Curr Opin Immunol. 2000;12(3):307–315. doi: 10.1016/S0952-7915(00)00092-3. [DOI] [PubMed] [Google Scholar]
- 30.An H, et al. SHP-2 phosphatase negatively regulates the TRIF adaptor protein-dependent type I interferon and proinflammatory cytokine production. Immunity. 2006;25(6):919–928. doi: 10.1016/j.immuni.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 31.Zhu HH, et al. Kit-Shp2-Kit signaling acts to maintain a functional hematopoietic stem and progenitor cell pool. Blood. 2011;117(20):5350–5361. doi: 10.1182/blood-2011-01-333476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bauler TJ, et al. Development of severe skeletal defects in induced SHP-2-deficient adult mice: a model of skeletal malformation in humans with SHP-2 mutations. Dis Model Mech. 2011;4(2):228–239. doi: 10.1242/dmm.006130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lei W, et al. Abnormal DNA methylation in CD4+ T cells from patients with systemic lupus erythematosus, systemic sclerosis, and dermatomyositis. Scand J Rheumatol. 2009;38(5):369–374. doi: 10.1080/03009740902758875. [DOI] [PubMed] [Google Scholar]
- 34.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31(2):89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 35.Morris KV, Chan SW, Jacobsen SE, Looney DJ. Small interfering RNA-induced transcriptional gene silencing in human cells. Science. 2004;305(5688):1289–1292. doi: 10.1126/science.1101372. [DOI] [PubMed] [Google Scholar]
- 36.Castanotto D, et al. Short hairpin RNA-directed cytosine (CpG) methylation of the RASSF1A gene promoter in HeLa cells. Mol Ther. 2005;12(1):179–183. doi: 10.1016/j.ymthe.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 37.Capell HA, et al. Lack of radiological and clinical benefit over two years of low dose prednisolone for rheumatoid arthritis: results of a randomised controlled trial. Ann Rheum Dis. 2004;63(7):797–803. doi: 10.1136/ard.2003.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Binstadt BA, et al. Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nat Immunol. 2006;7(3):284–292. doi: 10.1038/ni1306. [DOI] [PubMed] [Google Scholar]
- 39.Shahrara S, Castro-Rueda HP, Haines GK, Koch AE. Differential expression of the FAK family kinases in rheumatoid arthritis and osteoarthritis synovial tissues. Arthritis Res Ther. 2007;9(5):e86580. doi: 10.1186/ar2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stanford SM, et al. Receptor protein tyrosine phosphatase α-mediated enhancement of rheumatoid synovial fibroblast signaling and promotion of arthritis in mice. Arthritis Rheumatol. 2016;68(2):359–369. doi: 10.1002/art.39442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alvaro-Gracia JM, Zvaifler NJ, Brown CB, Kaushansky K, Firestein GS. Cytokines in chronic inflammatory arthritis. VI. Analysis of the synovial cells involved in granulocyte-macrophage colony-stimulating factor production and gene expression in rheumatoid arthritis and its regulation by IL-1 and tumor necrosis factor-alpha. J Immunol. 1991;146(10):3365–3371. [PubMed] [Google Scholar]
- 42.Untergasser A, et al. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e86580. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gerlag DM, Boyle DL, Rosengren S, Nash T, Tak PP, Firestein GS. Real-time quantitative PCR to detect changes in synovial gene expression in rheumatoid arthritis after corticosteroid treatment. Ann Rheum Dis. 2007;66(4):545–547. doi: 10.1136/ard.2006.059790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang EE, Chapeau E, Hagihara K, Feng GS. Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. Proc Natl Acad Sci U S A. 2004;101(45):16064–16069. doi: 10.1073/pnas.0405041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Monach PA, Mathis D, Benoist C. The K/BxN arthritis model. Curr Protoc Immunol. 2008;Chapter 15:Unit 15.22. doi: 10.1002/0471142735.im1522s81. [DOI] [PubMed] [Google Scholar]
- 46.Doody KM, et al. Targeting phosphatase-dependent proteoglycan switch for rheumatoid arthritis therapy. Sci Transl Med. 2015;7(288):e86580. doi: 10.1126/scitranslmed.aaa4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.