

Summary
Organisms respond to mitochondrial stress through the upregulation of an array of protective genes, often perpetuating an early response to metabolic dysfunction across a lifetime. We find that mitochondrial stress causes widespread changes in chromatin structure through histone H3K9 di-methylation marks traditionally associated with gene silencing. Mitochondrial stress response activation requires the di-methylation of histone H3K9 through the activity of the histone methyltransferase met-2 and the nuclear co-factor lin-65. While globally the chromatin becomes silenced by these marks, remaining portions of the chromatin open up, at which point the binding of canonical stress responsive factors such as DVE-1 occurs. Thus, a metabolic stress response is established and propagated into adulthood of animals through specific epigenetic modifications that allow for selective gene expression and lifespan extension.
Introduction
The fitness of an organism depends upon its capacity to adapt its metabolism in accordance with anticipated energetic demands and nutrient availability. In support of this requirement are observations that acute metabolic stresses during early development can cause persistent, widespread changes in epigenetic patterning and gene expression late in life, potentially increasing the preparedness of the organism for additional metabolic stressors. Changes in methylation patterns of chromatin caused by metabolic perturbations are often expansive and enduring, affecting long-term organismal function and occasionally permeating into subsequent generations (Jiménez-Chillarón et al., 2012). In humans, for example, in utero deficiency of a single nutrient, such as folate or choline, results in a massive restructuring of chromatin methylation patterns that remain visible throughout childhood (Choi and Friso, 2010). Susceptibility to obesity and diabetes in adults can be linked to dietary intake during early development and is associated with prominent modifications in chromatin (Vrachnis et al., 2012). Evidence in invertebrates further suggests that dietary-driven epigenetic modifications may permeate for generations (Rechavi et al., 2014). In each of these instances, the forced persistence of metabolic restructuring and gene expression must have a hidden adaptive value that offsets a potential loss in metabolic elasticity.
A logical regulatory center for these epigenetic phenomena lies in the mitochondria, the metabolic centers of the cell. Mitochondria are responsible for both coordinating the synthesis of NADH-donating substrates through the TCA cycle and for the production of the energy that is derived from oxidative phosphorylation. Several pieces of evidence suggest that effects of early mitochondrial dysfunction persist throughout the lifespan of the organism. For example, researchers have reported a beneficial effect of mild mitochondrial dysfunction during specific developmental stages on the lifespan of multiple organisms including the nematode C. elegans (Copeland et al., 2009; Dillin et al., 2002; Feng et al., 2001; Lee et al., 2003).
An important biomarker of the cellular sensing and response to this stress is a massive and persistent restructuring in gene expression patterns, as evidenced in analyses of long-lived mitochondrial mutant animals (Cristina et al., 2009; Yee et al., 2014). The upregulation of a significant portion of mitochondrial stress-responsive transcripts in C. elegans depends upon the activity of the transcription factor atfs-1, which appears to be required for the regulation of approximately half of these genes (Nargund et al., 2012). Genes regulated by atfs-1 are classified as part of the mitochondrial unfolded protein response, or UPRmt, which includes the upregulation of mitochondrial chaperones, quality control proteases, and xenobiotic response pathway components (Nargund et al., 2012). UPRmt activation also causes substantial metabolic restructuring, activating genes associated with glycolysis and repressing those associated with oxidative phosphorylation (Nargund et al., 2015). Additionally, full induction of the UPRmt also requires the nuclear localization of the transcription factor dve-1 and co-factor ubl-5, as well as the activity of the quality control protease clpp-1 (Benedetti et al., 2006; Haynes et al., 2007). While the role for each of these genes in UPRmt activation is well-described, the interactions among these various components at a molecular level remains ambiguous.
Histone H3K9 methylation plays a prominent role in regulating gene expression across a wide range of species (Nakayama et al., 2001; Jaenisch and Bird, 2003; González-Aguilera et al., 2014). In C. elegans, histone H3K9 is methylated sequentially by the methyltransferases MET-2/SETDB1 and SET-25 (Bessler et al., 2010; Towbin et al., 2012). MET-2 mono- and di-methylates H3K9 while it is still in the cytoplasm, prior to its incorporation into nucleosomes (Towbin et al., 2012). In contrast, SET-25, which is localized in the nucleus, acts as a trimethylation methyltransferase to H3K9 (Towbin et al., 2012). Collectively, these marks initiate chromatin anchoring and silencing, and are strongly associated with gene repression. H3K9me1/2 has previously been found to be critical in the appropriate regulation of energy expenditure and fat storage (Meister et al., 2011). Additionally, subtelomeric gene silencing, mediated by the H3K36 demethylase Rph1p, is induced by mitochondrial ROS and is implicated in yeast chronological lifespan (Schroeder et al., 2013).
We find that mitochondrial stress in C. elegans causes widespread changes in chromatin structure, dependent on marks traditionally associated with heterochromatin. The remodeling of chromatin during stress is required for the full activation of UPRmt. Chromatin reorganization requires activity of the histone methyltransferase met-2 and a nuclear co-factor lin-65. While globally the chromatin becomes silenced by these marks, remaining portions of the chromatin open up, at which point binding of canonical stress responsive factors such as DVE-1 occurs. Furthermore, we find that chromatin modifications work synergistically with atfs-1. Collectively, our results point towards a mechanism by which early metabolic stress remodels chromatin and establishes persistent gene expression patterns.
Results
Identification of lin-65 as a requirement for the appropriate activation of mitochondrial stress responses
We have observed that the expression of a Q40 polyglutamine tract in the neurons of C. elegans (rgef-1p∷Q40∷yfp) (Brignull et al., 2006) activates UPRmt in non-neuronal tissues (Figure 1A, S1A). To identify molecular components required for this stress response, we undertook an unbiased, large-scale screen to identify genes that are required for the PolyQ-dependent activation of the UPRmt. To this end, we subjected rgef-1p∷Q40∷yfp; hsp-6p∷gfp animals to EMS mutagenesis and analyzed 2400 mutagenized genomes for suppression of hsp-6p∷gfp reporter expression (an indicator of UPRmt activation). We isolated 16 mutant strains that exhibited a partial or complete suppression of PolyQ dependent hsp-6p∷gfp expression in the F2 generation. The strong and stable suppression of the reporter GFP expression in a strain carrying the uth2 mutation caused us to prioritize it for additional characterization (Figure S1A).
Figure 1. Neuronal polyglutamine expression induces a lin-65 dependent activation of the UPRmt.
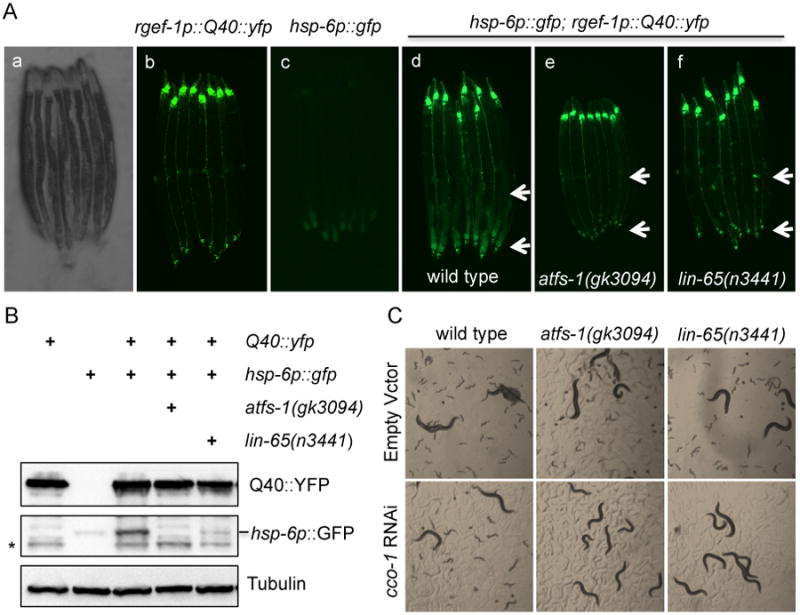
(A) Representative photomicrographs demonstrating: a) bright field image of aligned, wild type worms; b) YFP expression in the neurons; c) hsp-6p∷gfp expression in wild type (WT) animals; d) hsp-6p∷gfp is induced in the intestine in rgef-1p∷Q40∷yfp animals; e) hsp-6p∷GFP suppression in atfs-1 animals; f) hsp-6p∷GFP suppression in lin-65 animals. Arrows indicate the posterior region of the intestine where hsp-6p∷gfp is induced or suppressed. See also Figure S1.
(B) Immunoblots of GFP expression in hsp-6p∷gfp; rgef-1p∷Q40∷yfp animals in the presence or absence of atfs-1 or lin-65 mutation. Anti-tubulin serves as a loading control.
* indicates a cleaved band from Q40∷YFP that is recognized by GFP antibody.
(C) Representative photomicrographs of WT, atfs-1 and lin-65 animals grown on EV or cco-1 RNAi from hatch.
Day 2 of adulthood animals were used for imaging and western blots analyses.
Whole genome deep sequencing (Bigelow et al., 2009; Sarin et al., 2008) identified a mutation in the uth2 allele at amino acid 367 (GAG→AAG[Glu→Lys]) in the gene lin-65. Using the characterized lin-65 mutant allele n3441, we confirmed that lin-65 is required for the UPRmt in response to rgef-1p∷Q40∷yfp expression (Figure 1A). We found that the loss in PolyQ-dependent hsp-6p∷gfp expression found in lin-65 mutants was similar to that observed in atfs-1(gk3094) deletion strains (Figure 1A). Western blot analyses confirmed the loss of hsp-6p∷gfp expression in lin-65 mutants, while PolyQ expression remained unaffected (Figure 1B). The lin-65 mutation strongly attenuated the upregulation of the UPRmt in animals fed RNAi targeting the cytochrome c oxidase-1 subunit Vb/COX4 of the electron transport chain, cco-1, or animals fed RNAi targeting phb-2, the ortholog of prohibitin PHB2 (Figure S1B). Furthermore, lin-65 mutants exhibited a strong synthetic sterile phenotype when combined with cco-1 RNAi (Figure 1C). The synthetic sterility was similar to that which is caused by cco-1 RNAi in combination with loss of function of atfs-1 (Figure 1C). Collectively, these results indicate that lin-65 is required for UPRmt activation across a range of mitochondrial perturbations, and that lin-65 plays an important role in the regulation of mitochondrial homeostasis.
lin-65 encodes a proline-rich protein with a low complexity domain that was originally identified as a synmuv B class gene required for vulval development in C. elegans (Ceol et al., 2006). LIN-65 generally lacks obvious homologous counterparts in non-nematode species, aligning most closely to a human ACRC (acidic repeat-containing) protein. To test whether lin-65 was acting to specifically affect mitochondrial homeostasis, we examined whether lin-65 also played a role in the regulation of compartment-specific UPRs in either the endoplasmic reticulum (ER) or cytoplasm. We crossed the lin-65(n3441) mutation into animals harboring a reporter (hsp-4p∷gfp) for an ER-specific stress response, the UPRer (Ron and Walter, 2007). Animals were then treated with the ER-specific stressor tunicamycin, which blocks N-linked glycosylation and induces the UPRer (Heifetz et al., 1979). lin-65 did not affect the upregulation of the hsp-4p∷gfp reporter by tunicamycin (Figure S1C). Similarly, we observed that lin-65 animals were fully capable of a cytoplasmic response to a heat shock through upregulation of an hsp-16.2p∷gfp reporter (Figure S1D) (Link et al., 1999). These results indicate that lin-65 plays a specific role in the activation of mitochondrial stress responses.
Mitochondrial stress induces LIN-65 nuclear distribution
The observation that lin-65 is specifically required for UPRmt activation suggests that the expression and/or localization of lin-65 may be directly regulated by mitochondrial stress. To test this hypothesis, we constructed a transgenic strain expressing the 1.3kb promoter region of lin-65 fused to the mCherry protein. We found that lin-65 was expressed predominantly in the intestine, with weaker expression in several head neurons (Figure S2A). While lin-65 expression was slightly upregulated in Day 1 adult animals upon cco-1 RNAi treatment, by Day 2 this effect had dissipated, suggesting that a transcriptional upregulation of lin-65 is not the primary mechanism by which the UPRmt affects lin-65 mediated functions (Figure S2B).
Next, to determine whether lin-65 is post-transcriptionally regulated, we created animals in which we fused the lin-65 cDNA between its native promoter and the mCherry fluorescent protein. lin-65p∷lin-65∷mCherry plasmid was able to rescue the suppressed UPRmt induction in lin-65(n3441) mutants, suggesting that the fusion protein is functional (Figure S2C). Under normal conditions, intestinal lin-65p∷lin-65∷mCherry expression was diffuse in the cytosol (Figure 2A and Figure S2D). Upon cco-1 RNAi treatment, however, lin-65p∷lin-65∷mCherry fluorescence signals strengthened and became strongly localized to the nucleus (Figure 2A). Immunoblots against transgenics in which LIN-65 was translationally fused to an HA tag further confirmed that cco-1 RNAi was sufficient to induce LIN-65 protein expression by Day 2 of adulthood (Figure 2B) and that the signal was abolished with lin-65 RNAi (Figure S2E). Altogether, these data suggest that the localization and protein levels of LIN-65 are regulated by mitochondrial stress.
Figure 2. The C. elegans histone H3K9me1/2 methyltransferase met-2 is required for LIN-65 nuclear accumulation upon cco-1 RNAi.
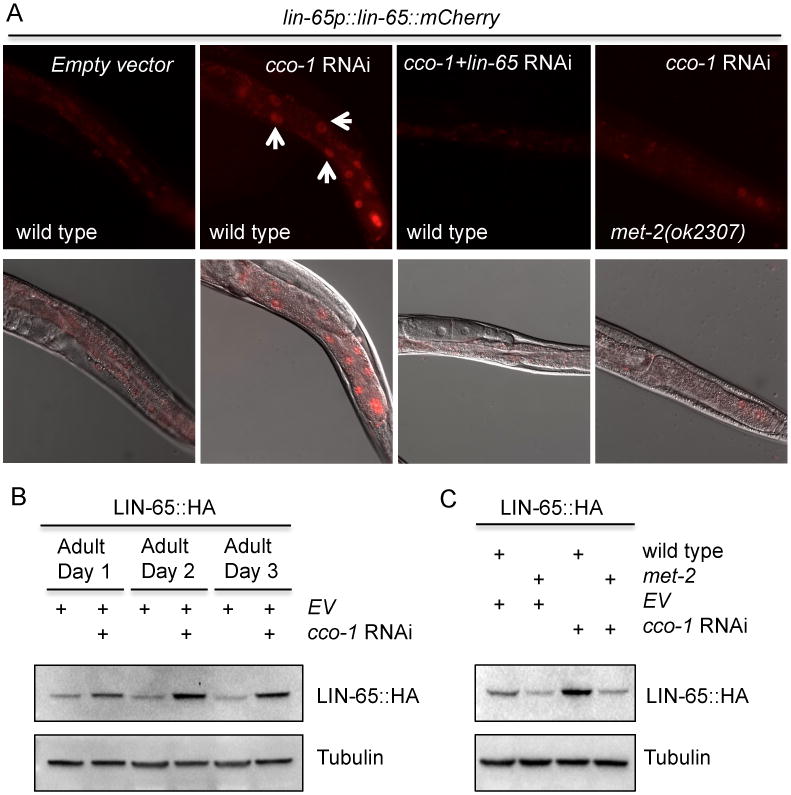
(A) Representative photomicrographs of lin-65p∷lin-65∷mCherry animals in a WT or met-2 mutant background grown on EV, cco-1 RNAi, or cco-1+lin-65 double RNAi bacteria from hatch (as indicated). Images were taken at day 1 of adulthood. Arrows indicate intestinal nuclei.
(B) Immunoblot of LIN-65∷HA expression in animals grown on EV and EV+cco-1 RNAi from hatch as indicated. Animals were collected at Day 1, 2 and 3 of adulthood. Anti-tubulin serves as a loading control.
(C) Immunoblot of LIN-65∷HA expression in WT or met-2 mutants grown on EV or cco-1 RNAi (as indicated) from hatch. Animals were collected at Day 2 of adulthood. Anti-tubulin serves as a loading control.
The H3K9 methyltransferase met-2 is required for LIN-65 nuclear accumulation induced by mitochondrial stress
Previously, lin-65 was identified as a synmuv B class gene along with a group of chromatin modifiers (Couteau et al., 2002; Poulin et al., 2005; Ceol et al., 2006; Andersen and Horvitz, 2007). We hypothesized that one or more of these genetic interactors might influence the localization of LIN-65 in response to mitochondrial stress. Using a candidate approach against known lin-65 genetic interactors, we examined synmuv genes for their effect on LIN-65 localization after cco-1 RNAi treatment. We found that met-2, a homolog of the mammalian H3K9 methyltransferase SETDB1 (Schultz et al., 2002; Bessler et al, 2010), was required for LIN-65 nuclear accumulation during induction of the UPRmt. met-2(ok2307) mutation strongly blocked the nuclear accumulation of lin-65p∷lin-65∷mCherry in the intestine upon cco-1 RNAi treatment (Figure 2A). The LIN-65∷HA fusion protein no longer accumulated in met-2 mutants after treatment with cco-1 RNAi (Figure 2C). The requirement of met-2 for LIN-65 nuclear localization and expression suggested that epigenetic modification might also be involved in UPRmt regulation. Indeed, the met-2 mutation heavily attenuated the UPRmt, in both cco-1 and phb-2 RNAi treated animals, as measured by hsp-6p∷gfp induction (Figure S1B).
Global chromatin reorganization induced by mitochondrial stress is dependent on met-2 and lin-65
MET-2 specifically deposits mono- and di- methyl groups on H3K9 in C. elegans (Towbin et al., 2012). We therefore examined whether animals with reduced ETC function exhibit changes in methylation patterns. To this end, we performed immunohistochemistry against H3K9me2 marks in germlines carrying mutations in met-2 or lin-65. In agreement with previous reports, H3K9me2 level was almost undetectable in met-2 mutants (Figure S3A). Intriguingly, however, we found that lin-65 mutation severely reduced H3K9me2 levels as well (Figure S3A). In contrast, consistent with a role for set-25 in H3K9me3, we observed an increase in H3K9me2 marks in set-25 mutants (Figure S3A). met-2 or lin-65 mutations also abolished or severely reduced H3K9me2 methylation in intestinal cells under basal or mitochondrial stress conditions compared to wild type animals (Figure 3A and 3B). Surprisingly, we also observed striking changes in chromatin structure in intestinal nuclei upon mitochondrial stress. The intestinal nuclei appeared smaller and much more condensed in cco-1 RNAi treated animals, while in contrast, met-2 and lin-65 animals had enlarged nuclei and a loose chromatin structure that remained relatively loose in structure even after mitochondrial stress was induced by cco-1 RNAi (Figure 3A).
Figure 3. Mitochondrial stress induced chromatin reorganization.
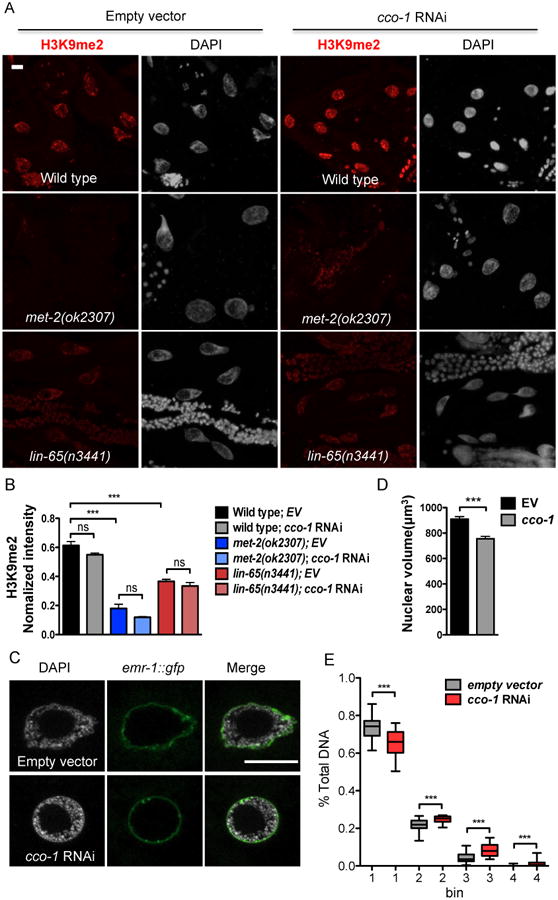
(A) Representative maximal intensity projection images of H3K9me2 immunostaining of intestinal nuclei in Day 1 adult WT, met-2 or lin-65 mutant animals grown on EV or cco-1 RNAi from hatch (as indicated). H3K9me2 (Red); DAPI (Grey). Scale bar represents 10 μm.
(B) Quantification of H3K9me2 level. The genotypes and treatments are as in (A). H3K9me2 intensity is normalized to DAPI intensity. (*** denotes p < 0.0001; ns denotes p > 0.05 via t-test, error bars indicate SEM, n ≥ 15 nuclei)
(C) Representative of 3 center images of DAPI staining with lmn-1p∷emr-1∷gfp of intestinal nuclei in Day 1 adult animals grown on EV or cco-1 RNAi from hatch as indicated. lmn-1p∷emr-1∷gfp (Green); DAPI (Grey). Scale bar represents 10 μm.
(D) Quantification of the intestinal nuclear size at Day 1 adulthood using lmn-1p∷emr-1∷gfp as a marker. Animals grown on EV or cco-1 RNAi from hatch (*** denotes p < 0.0001 via t-test, error bars indicate SEM, n ≥ 20 nuclei)
(E) Quantification of the distribution of DAPI staining signal in intestinal nuclei at Day 1 of adulthood in animals grown on EV or cco-1 RNAi from hatch. The distribution of fractions from the top four bins are shown as boxplots. (*** denotes p < 0.0001 via mann-whitney test, error barsindicate SEM, n ≥ 25 nuclei)
To directly measure nuclear size in the intestine, we used a nuclear membrane marker, lmn-1p∷emr-1∷gfp, to label intestinal nuclei (Figure 3C) (Haithcock et al., 2005). In keeping with our initial observations, we observed that the intestinal nuclear volume was significantly decreased in cco-1 RNAi treated animals (Figure 3D) while the number and DNA content of intestinal nuclei remained unaffected (Figure S3B and S3C). Quantification of binned DAPI fractions confirmed an overall change of chromatin in cco-1 RNAi treated animal intestinal nuclei (Figure 3E). In sum, these data suggest that mitochondrial stress induces global chromatin reorganization in addition to decreased nuclear size.
H3K9me2 is required for mitochondrial stress-induced DVE-1 nuclear puncta formation
To understand how global changes in chromatin structure may regulate UPRmt induction, we next characterized the genetic interaction between the UPRmt regulator DVE-1 and MET-2/LIN-65. dve-1 encodes a homeobox domain transcription factor homologous to SATB1/SATB that translocates from the cytosol to the nucleus during mitochondrial stress (Haynes et al., 2007) (Figure 4A). To understand the interdependency of DVE-1 and LIN-65 in response to mitochondrial stress, we performed epistasis analysis in lin-65(n3441) animals carrying a dve-1p∷dve-1∷gfp reporter and fed cco-1 RNAi. Strikingly, lin-65 mutants abolished DVE-1∷GFP nuclear accumulation in cco-1 RNAi treated animals (Figure 4B). This phenotype was rescued by expression of lin-65 with either its own promoter or an intestinal specific gly-19 promoter (Figure S4A and S4B). These data indicate that lin-65 is not only required but also functions cell autonomously for DVE-1 nuclear distribution under mitochondrial stress in the intestine.
Figure 4. DVE-1 nuclear distribution upon mitochondrial stress is dependent on lin-65 and met-2.
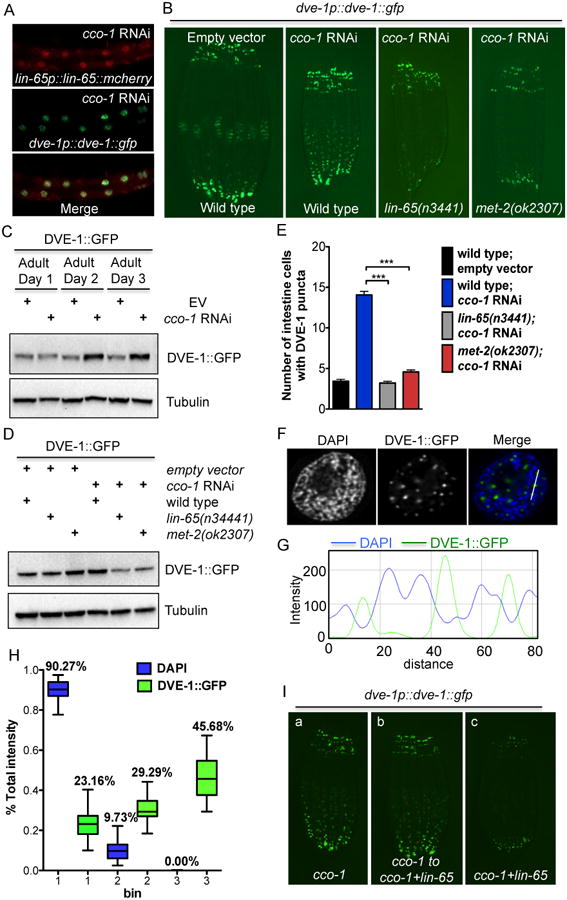
(A) Representative photomicrographs of Day 1 adult animals expressing both lin-65p∷lin-65∷mCherry and dve-1p∷dve-1∷gfp grown on cco-1 RNAi from hatch.
(B) Representative photomicrographs of Day 1 dve-1p∷dve-1∷gfp animals in wild type, lin-65 and met-2 background grown on EV or cco-1 RNAi from hatch.
(C) Immunoblot of GFP expression in WT animals grown on EV or cco-1 RNAi from hatch. Animals were collected at Day 1, 2 and 3 of adulthood. Anti-tubulin serves as a loading control.
(D) Immunoblot of GFP expression in Day 1 adult animals in WT, lin-65 or met-2 mutant background grown on EV or cco-1 RNAi from hatch. Anti-tubulin serves as a loading control.
(E) Quantification of the number of intestinal nuclei with DVE-1 puncta structure per worm. The genotypes and treatments are as in (B). (*** denotes p < 0.0001 via t-test, error bars indicate SEM, n ≥ 30 worms). See also Figure S4D.
(F) Representative photomicrographs of DVE-1∷GFP punctate structure in one intestinal nucleus. DVE-1∷GFP (Green); DAPI (Blue).
(G) The intensity profile of both DAPI and DVE-1∷GFP across the white line as shown in Figure 4F.
(H) Quantification of the distribution of DVE-1∷GFP signal in nuclei with DAPI staining. DVE-1∷GFP animals grown on cco-1 RNAi from hatch. The distribution of fractions of all three bins are shown as boxplots (n≥30 nuclei).
(I) Representative photomicrographs of DVE-1∷GFP expression in animals grown on : (a) cco-1 RNAi for 5 days from hatch; (b) cco-1 RNAi for 3 days from hatch and then transferred to cco-1+lin-65 double RNAi for 2 more days; (c) cco-1+lin-65 double RNAi from L4, F1 generations were examined.
During mitochondrial stress, DVE-1 not only translocates to the nucleus but also accumulates, while its mRNA level remains unaffected (Figure 4C and S4C). To further explore the role of LIN-65 in mediating the DVE-1 nuclear signal, we asked if LIN-65 also plays a role in regulating DVE-1 protein levels. Western blot analyses confirmed that DVE-1∷GFP protein levels decreased in lin-65 animals when cco-1 RNAi was applied (Figure 4D). Similarly, met-2 was required for DVE-1∷GFP nuclear accumulation upon stress (Figure 4B and 4D). Taken together, these data indicated that LIN-65/MET-2 are specifically required for mitochondrial stress-induced DVE-1 nuclear distribution and expression.
During these experiments, we also noticed that the DVE-1∷GFP fusion protein formed punctate structures in the nucleus during mitochondrial stress (Figure 4B and S4D) (Haynes et al., 2007). In comparison, the intensity of nuclear DVE-1∷GFP signal decreased in both lin-65 and met-2 animals fed cco-1 RNAi (Figure S4D) and fewer cells contained detectable DVE-1∷GFP puncta (Figure 4E and Figure S4D). To resolve the specific areas in which DVE-1 puncta were localized in the nucleus, we performed immunohistochemistry with H3K9me2 antibody in DVE-1∷GFP animals fed cco-1 RNAi. We did not observe co-localization between DVE-1 puncta and the H3K9me2 signal (Figure S4E). Instead, however, we found that the DVE-1∷GFP puncta reside in the weak DAPI staining region, surrounded by condensed chromatin that exhibit strong DAPI staining (Figure 4F, 4G). To globally quantify this phenotype, we analyzed the relative ratios of GFP and DAPI intensity in different regions of the nucleus. Consistently, DVE-1∷GFP signals were significantly enriched in regions of the nucleus exhibiting low levels of DAPI signal, while depleted from regions exhibiting strong DAPI staining signals (Figure 4H). Collectively, these results indicate that DVE-1∷GFP puncta are preferentially localized in nuclear compartments devoid of condensed chromatin where gene expression is predicted to be most active.
Because both H3K9me2 and H3K9me3 are important for heterochromatin formation and gene silencing, we tested whether set-25 has a similar effect on DVE-1 localization upon mitochondrial stress. To this end, we monitored nuclear DVE-1∷GFP localization in set-25(n5021) mutants in which H3K9me2 levels are elevated (Figure S3A). Remarkably, we observed an opposing phenotype: set-25 mutants increased DVE-1∷GFP nuclear localization even in the absence of mitochondrial stress (Figure S5A). The DVE-1∷GFP accumulation in set-25 mutants, however, was neither required for cco-1 RNAi induced longevity nor sufficient for lifespan extension (Figure S5B and Table S1). These results suggest that H3K9me2 is specifically required for DVE-1 nuclear accumulation in response to mitochondrial stress.
lin-65 functions in development to establish UPRmt induction
To understand whether LIN-65 is required for establishing or perpetuating UPRmt into adulthood, we tested the timing requirements of lin-65 in mediating DVE-1 puncta formation. Animals in which mitochondrial stress was induced during development (cco-1 RNAi during development), but which had lin-65 reduced only during adulthood retained nuclear DVE-1∷GFP puncta formation (Figure 4I). In contrast, animals with both mitochondrial stress and reduced lin-65 activity only during development completely suppressed DVE-1∷GFP puncta formation in adulthood (Figure 4I). These data indicate that lin-65 likely establishes an epigenetic mark on H3K9 during development, allowing for DVE-1 puncta formation in response to mitochondrial stress.
lin-65 and met-2 are required for regulating the transcriptional response to mitochondrial stress
To understand the role of lin-65 and met-2 in regulating the transcriptional response to mitochondrial stress, we performed RNA-seq on wild type, lin-65 and met-2 animals, each in the presence or absence of cco-1 RNAi. In wild type animals, our analysis identified 1312 genes that were differentially expressed in the cco-1 RNAi treated condition relative to animals grown on empty vector bacteria (Benjamini adjusted p-value < 0.05) (Figure 5A, S6A, Table S2). Using qPCR, we confirmed that hsp-6 and timm-23 induction by cco-1 RNAi were perpetuated throughout adulthood (Figure 5B), consistent with epigenetic modifications induced by cco-1 being sufficient to ensure target gene expression long after establishing chromatin marks during development. In the absence of lin-65 and met-2, many cco-1 regulated genes exhibited wide variance in expression patterns and failed to become significantly changed from their expression on empty vector. In fact, a majority of the gene expression changes (884 up-regulated genes and 380 down-regulated genes) induced by cco-1 RNAi were abrogated in lin-65 or met-2 mutant animals (Figure 5A, S6A, S6B and Table S2) (see Discussion). These data demonstrate that proper regulation of gene expression in response to cco-1 RNAi is dependent upon the chromatin reorganization activities of lin-65 and met-2 and that the global changes in gene expression due to mitochondrial stress are dependent upon the function of LIN-65 and/or MET-2.
Figure 5. Gene expression changes upon cco-1 RNAi treatment is strongly dependent upon lin-65 and met-2.

(A) Venn diagram of numbers of differentially expressed genes in wild type, lin-65 and met-2 mutant animals grown on cco-1 RNAi for 2 days. Genes with an adjusted P-value < 0.05 were selected as differentially expressed genes. See also Table S2.
(B) Quantitative PCR of hsp-6 and timm-23 mRNA level. Synchronized Day1, 2 and 3 adult animals grown on EV or EV+cco-1 RNAi from hatch were collected for qPCR. (* denotes p < 0.05, ** denotes p < 0.01 via t-test, error bar indicates the SEM from three biological replicates)
The nuclear localization of LIN-65 is partially dependent on CLPP-1/DVE-1 and independent of ATFS-1
We next asked whether the function of additional UPRmt components were required for LIN-65's expression and/or nuclear localization. The most proximal responders to mitochondrial stress required to induce the UPRmt are the mitochondrial protease, clpp-1, and the transcription factor dve-1 (Haynes et al., 2007). We found that RNAi against clpp-1 or dve-1 strongly suppressed LIN-65 nuclear translocation as well as protein expression under both normal and mitochondrial stress conditions (Figure 6A, 6B and S7A). This suggested that a reciprocal, interdependent relationship exists between DVE-1 and LIN-65 for their expression and localization.
Figure 6. Mitochondrial stress-induced LIN-65 nuclear translocation is partially dependent on clpp-1 and dve-1, but independent of atfs-1.
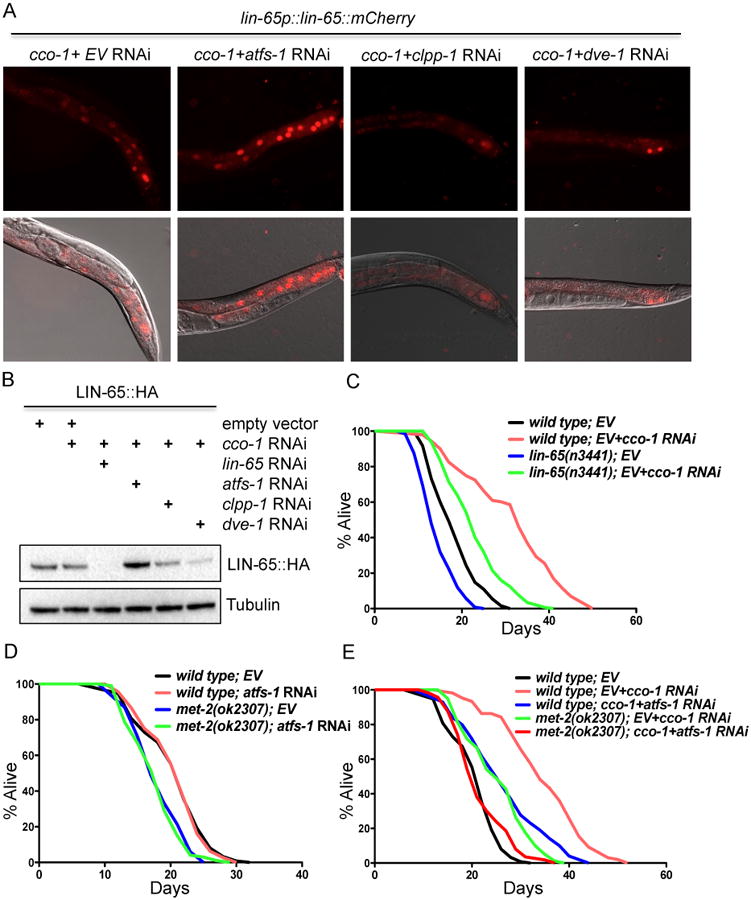
(A) Representative photomicrographs of Day 1 adult animals expressing lin-65p∷lin-65∷mCherry grown on RNAi from hatch as indicated.
(B) Immunoblot of against HA-tag in LIN-65∷HA animals grown on RNAi bacteria from hatch (as described in (A)). Animals were collected at Day 1 of adulthood. Anti-tubulin serves as a loading control.
(C) Survival analyses of lin-65(n3441) animals on EV or EV+cco-1 RNAi. See Table S1 for lifespan statistics.
(D) Survival analyses of met-2(ok2307) animals on EV or atfs-1 RNAi. See Table S1 for lifespan statistics.
(E) Survival analyses of met-2(ok2307) and atfs-1 RNAi treated animals under mitochondrial stress condition with cco-1 RNAi. See Table S1 for lifespan statistics.
Much like LIN-65, ATFS-1 exhibits dual localization in the cell, increasing its propensity toward nuclear localization under mitochondrial stress, during which it is responsible for the transcriptional upregulation of an array of genes that protect from protein unfolding, bacterial pathogenesis, and cholesterol deprivation (Nargund et al., 2012). Intriguingly, we found that loss of atfs-1, which is sufficient to abolish UPRmt, does not affect the nuclear localization of LIN-65 or chromatin compaction induced by cco-1 RNAi (Figure 6A and S7B). In fact, atfs-1 RNAi actually enhanced LIN-65 protein levels under both normal and mitochondrial stress conditions (Figure 6B and S7A). These data suggest that multiple parallel pathways, regulated by both CLPP-1/LIN-65/DVE-1 and ATFS-1, are required for overall regulation of UPRmt.
lin-65/met-2 and atfs-1 are required for mitochondrial stress-induced longevity
Mitochondrial stress induced by cco-1 RNAi during development increases lifespan and induces the UPRmt (Dillin et al., 2002, Durieux et al., 2011). To test whether lin-65 is required for lifespan extension upon mitochondrial stress, we assayed the lifespan of lin-65 animals with or without cco-1 RNAi. Both lin-65 and met-2 animals are short lived under normal conditions. As predicted, both lin-65(n3441) and met-2(ok2307) only partially suppressed the lifespan extension of cco-1 RNAi treated animals (Figure 6C-D and Table S1). We hypothesized that perhaps a redundancy with atfs-1 function on lifespan was driving the incomplete suppression of lifespan in these experiments. To test this, we assayed the lifespan of met-2 animals with or without cco-1 RNAi in combination with atfs-1 RNAi. Like met-2 and lin-65, atfs-1 RNAi alone was incapable of fully suppressing the extension of lifespan in cco-1 RNAi animals (Figure 6D and Table S1). afts-1 RNAi also did not further decrease the lifespan of wild type or met-2 mutant animals under normal conditions (Figure 6D and Table S1). Importantly, however, atfs-1 RNAi further decreased the lifespan extension of met-2; cco-1 RNAi treated animals to the level of wild type animals (Figure 6E and Table S1), suggesting that met-2 and atfs-1 function synergistically to mediate mitochondrial stress-induced longevity.
Together, these data suggest that mitochondrial stress during development is sufficient to initiate a cascade of events, including met-2 dependent H3K9me2 methylation, LIN-65 nuclear accumulation, chromatin reorganization and DVE-1 nuclear punctate structure formation, in addition to nuclear localization of the mitochondrial stress specific transcription factor ATFS-1 (Figure 7A-G). Collectively, this cascade of responses works to initiate beneficial changes in gene expression that are perpetuated into adulthood. Thus, chromatin reorganization and induction of the UPRmt function synergistically to regulate mitochondrial stress-induced longevity.
Figure 7. Model for mitochondrial stress signaling pathway.
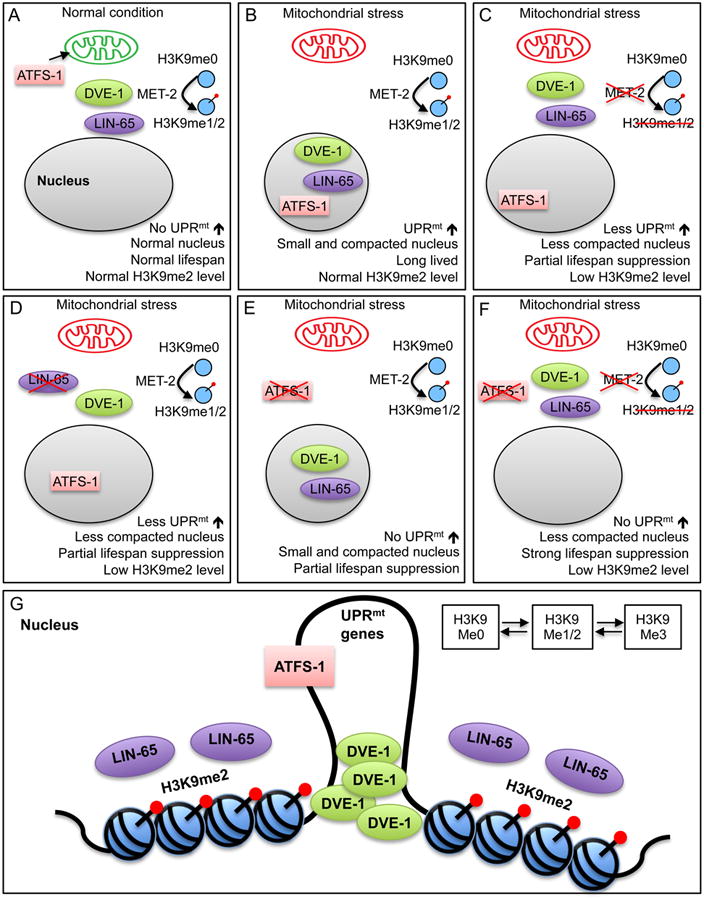
(A) Under non-stressed conditions, MET-2 produces H3K9me1/2 histone subunits in the cytoplasm. ATFS-1 translocates to the mitochondria and is degraded. DVE-1 and LIN-65 do not accumulate in the nucleus and the UPRmt is not induced, animals are normal lived and the nucleus is not compacted (Figure 3A).
(B) During mitochondrial stress, MET-2 continues to produce H3K9me2 histone subunits, ATFS-1 now translocates to the nucleus to induce UPRmt. DVE-1 and LIN-65 accumulate in the nucleus (Figure 2A-C and Figure 4B-D). Animals are long-lived, the UPRmt is induced, nuclei become compacted (Figure 3A), and H3K9me2 levels remain unchanged (Figure 3B).
(C) Loss of met-2 during mitochondrial stress results in reduced nuclear H3K9me2 levels, nuclei that are less compacted (Figure 3A), reduced DVE-1 and LIN-65 nuclear accumulation (Figure 2A and Figure 4B), reduced UPRmt induction (Figure S1B) and partial suppression of increased lifespan (Figure 6E).
(D) Loss of lin-65 during mitochondrial stress results in reduced nuclear H3K9me2 levels (Figure 3B), nuclei that are less compacted (Figure 3A), reduced DVE-1 nuclear accumulation (Figure 4B), reduced UPRmt induction (Figure 3A and Figure S1B) and partial suppression of increased lifespan (Figure 6C).
(E) Loss of atfs-1 during mitochondrial stress results in suppressed UPRmt induction (Figure 1A) and partial suppression of lifespan extension (Figure 6E). This is independent of compacted nuclei (Figure S7B) and nuclear accumulation of DVE-1 and LIN-65 (Figure 6A and 6B).
(F) Loss of both met-2 and atfs-1 during mitochondrial stress results in nuclei that are less compacted, reduced nuclear accumulation of DVE-1 and LIN-65, no UPRmt induction and complete suppression of lifespan extension. (Figure 6E).
(G) During mitochondrial stress, LIN-65 and DVE-1 accumulate in the nucleus, chromatin becomes remodeled, and DVE-1 is able to form puncta at the loose regions of the chromatin, activating transcription of UPRmt targets. This remodeling works in parallel to the relocation of mitochondrial specific transcription factor ATFS-1 to initiate UPRmt and regulate longevity.
Discussion
We have taken advantage of a cell non-autonomous mitochondrial stress model that elicits mild mitochondrial dysfunction to identify the genetic requirements of UPRmt induction, identifying lin-65/met-2 as integral parts of the UPRmt signaling pathway. We believe that the model by which this occurs will be complex: due to the fact that LIN-65 translocates from the cytosol to the nucleus upon mitochondrial stress as well as H3K9me2 being produced in the cytosol where MET-2 is present (Towbin et al., 2012) and the possibility that lin-65 plays a pivotal role in nucleosome/histone exchange in the nucleus in order for the H3K9me2 marks to appear on the chromatin at the proper loci upon mitochondrial stress. Whether LIN-65 is able to directly bind histone H3 in the cytoplasm, and how it is involved in H3K9 di-methylation are yet to be determined.
Nuclear H3K9me2 levels do not increase in the presence of mitochondrial stress, although nuclear LIN-65 accumulation does. One possible explanation for this observance is that LIN-65's function is not limited to the potential shuttling of H3K9me2 subunits to the nucleus, but plays a more definitive role in the deposition or localization of the H3K9me2 subunit onto the chromatin under mitochondrial stress. Therefore, it is not the total accumulation of H3K9me2 in the nucleus that determines UPRmt induction and longevity, but rather the appropriate localization and utilization of this subunit to form precise chromatin structures for gene regulation. Likewise, in the absence of mitochondrial stress, loss of lin-65 results in a significant reduction of nuclear H3K9me2, although LIN-65 has not accumulated in the nucleus. If indeed LIN-65 is required to shuttle and distribute H3K9me2 subunits in the nucleus, under non-stressed conditions LIN-65 continues to shuttle to the nucleus, but is not retained and does not accumulate. The lack of LIN-65 in the nucleus during non-stressed conditions could result in release of the H3K9me2 subunits and their conversion to H3K9me1/3 or degradation. Alternatively, loss of lin-65 may result in a subset of H3K9me2 subunits in the cytoplasm accumulating that negatively impact MET-2 activity.
DVE-1 was previously identified as an UPRmt regulator found at the promoters of UPRmt target genes in mitochondrial-stressed C. elegans (Haynes et al., 2007). In this study, we find there is interdependency between LIN-65 and DVE-1 for their nuclear localization and to maintain their protein levels during mitochondrial stress. It is possible that cells may benefit from this dependency to elicit intricate chromatin remodeling and gene regulation in response to the specific stress signals. Future studies will help determine the relative contributions of protein stability and translation on LIN-65 and DVE-1 expression patterns.
Under energy depleted conditions, silencing of gene expression by active global chromatin condensation will conserve energy required for the timely activation of mitochondrial stress related genes, thereby ensuring an adequate stress response optimal for survival. Interestingly, we noticed that DVE-1 forms puncta upon mitochondrial stress, which only reside in the loose chromatin regions where DAPI staining is weak. Similarly, a special AT-rich binding protein SATB1 (a mammalian homolog of DVE-1) also locates in regions devoid of condensed chromatin (Cai et al., 2003). SATB1 plays a key role in organization of the chromatin “loopscape” that allows for the regulation of gene expression at a distance (Galande et al., 2007, Yasui et al., 2002). It is likely that mitochondrial stress induces global changes in high order chromatin structure through H3K9me2, which passively forces DVE-1 to the more assessable regions on the chromatin open for gene expression. Alternatively, other histone modifications or chromatin modifying factors might be directly recruiting DVE-1 to the determined positions.
Studies in yeast have shown that subtelomeric silencing can mediate mitochondrial ROS-induced chronological longevity, propagating heterochromatin formation near telomere regions and regulating post-mitotic cellular health (Schroeder et al., 2013). Because the chromatin marks found in yeast undergoing ROS-induced damage are distinct from the chromatin marks found in this study using reduced ETC, we hypothesize that mitochondrial stressors may act via highly specific mechanisms to coordinate the activation of the protective pathways best suited to the mitochondrial damage at hand. In future studies, it will be interesting to compare the chromatin status across different forms of mitochondrial stress to understand the extent to which they correlate with lifespan extension.
Our RNA-Seq analysis identified a large set of genes that were differentially expressed in response to cco-1 RNAi treatment, establishing this set of genes as being pivotal for animals to thrive in the face of mitochondrial stress. Although the overwhelming majority of cco-1 induced gene expression changes were abrogated upon loss of lin-65 or met-2, many exhibited mean expression changes that were qualitatively similar to that seen in wild type animals (Figure S6A). This is particularly true for the down-regulated genes in the lin-65 mutant (Figure S6A). Importantly, many of the genes we analyzed exhibited increased variability within biological replicates in the lin-65 and met-2 mutant animals (Figure S6B, Table S2), limiting the statistical significance for many gene expression changes. In general, the variance among biological replicates was increased in the lin-65 and met-2 mutants relative to wild type, despite careful handling and harvesting of each of the replicate sets together. Among the possible explanations for increased variance in these mutants, we favor a model where loss of lin-65 or met-2 results in increased transcriptional noise, which is consistent with previous reports that the chromatin environment can epigenetically regulate transcriptional noise in diverse systems (Hansen and O'Shea, 2013, Anderson et al., 2014, Lavigne et al., 2015, Dey et al., 2015). While it will be important to formally address this hypothesis in the future, the data presented here clearly emphasize the importance of lin-65 and met-2 in properly orchestrating the chromatin in order to mount an appropriate response to mitochondrial stress.
The impacts of early life stress on the long-term well-being of an organism, especially in the progression of the aging process, have been long debated. The fact that the mitochondrial stress experienced early during development extends lifespan indicates that these animals might possess an epigenetic mechanism to ensure the beneficial effects from such stress are maintained throughout the lifespan. Such a mechanism seems to be carried out by both H3K9 methylation and DVE-1 mediated chromatin structure changes in peripheral tissues.
Experimental Procedures
Strains
SJ4100 (zcIs13[hsp-6p∷gfp]), CL2070 (dvIs70[hsp-16.2p∷gfp]), SJ4005 (zcIs4[hsp-4p∷gfp]), SJ4197 (zcIs39[dve-1p∷dve-1∷gfp]), MR142 (cdc-25.1(rr31); rrIs1 [elt-2∷gfp + unc-119(+)]), MT13232 (lin-65(n3441)), RB1789 (met-2(ok2307)), MT17463 (set-25(n5021)), LW699 (jjIs699[lmn-1p∷emr-1∷gfp] and N2 wild type strains were obtained from the Caenorhabditis Genetics Center (Minneapolis, MN). AM101 (rmIs101[rgef-1p∷Q40∷yfp]) was a generous gift from Rick Morimoto. VC3201 (atfs-1(gk3094)) was obtained from the National BioResource Project (Tokyo, Japan).
AGD1491 (uthIs400 [lin-65p∷mCherry∷unc-54 3′UTR; pRF4(rol-6)]) (6×),
AGD1492 (uthIs401 [lin-65p∷lin-65cDNA∷mCherry∷unc-54 3′UTR; pRF(rol-6)]) (6×),
AGD1540 (uthIs432 [lin-65p∷lin-65cDNA∷HA∷unc-54 3′UTR; pRF4(rol-6)]) (3×),
AGD1540 (uthIs433 [lin-65p∷lin-65cDNA∷HA∷unc-54 3′UTR; pRF4(rol-6)]) (3×)
For generation of lin-65p∷mCherry strain, the lin-65 1.3kb promoter was PCR amplified from genomic DNA and cloned into pNB23(sur-5p∷mCherry) using SphI and XmaI. For the lin-65p∷lin-65cDNA∷mCherry strain, the lin-65 cDNA(2184bp) without stop codon was inserted into lin-65p∷mCherry construct with XmaI and XbaI. lin-65 cDNA was a gift from Robert Horvitz. For the generation of lin-65p∷lin-65cDNA∷HA transgenic worms, the mCherry∷unc-54 3′UTR was replaced by HA-tag∷unc-54 3′UTR using XbaI and ApaI. To replace the lin-65 promoter, the gly-19 promoter was cloned in place of the lin-65 promoter in the lin-65p∷lin-65 cDNA∷mCherry plasmid. Transgenic strains were generated by microinjecting target constructs (70ng/ul) mixed with a pRF4(rol-6) (50ng/ul) co-injection marker. Extrachromosomal arrays were integrated using irradiation and backcrossed 3-6 times.
RNAi feeding
Worms were grown from hatch on HT115 E. coli containing an empty vector control or expressing double-stranded RNA. RNAi strains were from the Vidal library if present, or the Ahringer library if absent from the Vidal library.
Western blot Analysis
Age synchronized worms were grown on plates containing HT115 RNAi bacteria at 20°C. For large-scale experiments, worms were washed off the plate with M9 buffer and worm pellets were frozen in liquid nitrogen for further processing. Worm extracts were generated by glass bead disruption in non-denaturing lysis buffer [150 mM NaCl, 50 mM Hepes at pH 7.4, 1mM EDTA, 1% Triton X100, protease inhibitor cocktail III (Calbiochem)]. For small-scale experiments, 120 worms were picked into 14ul M9 buffer and frozen in liquid nitrogen. 6×SDS loading buffer and 10× reducing reagent were added to each sample. Samples were boiled for 15 mins and resolved by NuPAGE Bis-Tris mini gels.
Antibodies
Antibodies used for Western blot and IF were as follows: anti-GFP antibody (Roche 11814460001), anti-Tubulin antibody (Sigma T6074); anti-HA-Tag (C29F4) Rabbit mAb #3724; anti-Histone H3 (di methyl K9) antibody (mAbcam 1220)
Lifespan analysis
Lifespan experiments were performed at 20°C as previously described (Dillin et al., 2002). Worms were synchronized by egg bleach and grown on E. coli HT115 for RNAi from hatch. Worms were scored every second day. Prism5 software was used for statistical analysis. Log-rank (Mantel-Cox) method was used to determine the significance difference.
Supplementary Material
Acknowledgments
We thank the Caenorhabditis Genetic Center and Shohei Mitani of the National BioResource Project for providing strains. We also thank Dr. Robert Horvitz for providing the lin-65 cDNA plasmid. We are grateful to Tao Cai at NIBS for the help with whole genome deep sequencing data analyses. This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 Instrumentation Grants S10RR029668 and S10RR027303. This work used the CRL Molecular Imaging Center, supported by Gordon and Betty Moore Foundation. Y.T. was supported by the Glenn Foundation for Medical Research Postdoctoral Fellowship. K.K.S. was supported by a grant from the Jane Coffin Childs Memorial Fund for Medical Research. S.W. received support from the Glenn Foundation for Medical Research. This work was also supported by the NIH grant (R37 AG024365) and HHMI. A.D. is a founder of Proteostasis Therapeutics and Mitobridge, inc. and declares no financial interest.
Footnotes
Author Contributions: Y.T. and A.D. conceived the study. Y.T. designed and performed the experiments. Y.T., G.G. and Q.B. performed immunohistochemistry experiments and data analyses on chromatin samples. Y.T., K.K.S., L.J. prepared the samples for RNA-seq and K.K.S. performed data analyses. B.M. provided expertise and ideas on chromatin analysis. Y.T., S.W. and A.D. wrote the manuscript with contributions from other authors. A.D. supervised the research.
Accession Numbers: The RNA-Seq data described in this publication have been deposited in NCBI's Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO series accession number GSE78252.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersen EC, Horvitz HR. Two C. elegans histone methyltransferases repress lin-3 EGF transcription to inhibit vulval development. Dev Camb Engl. 2007;134:2991–2999. doi: 10.1242/dev.009373. [DOI] [PubMed] [Google Scholar]
- Anderson MZ, Gerstein AC, Wigen L, Baller JA, Berman J. Silencing is noisy: population and cell level noise in telomere-adjacent genes is dependent on telomere position and sir2. PLoS Genet. 2014;10:e1004436. doi: 10.1371/journal.pgen.1004436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006;174:229–239. doi: 10.1534/genetics.106.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessler JB, Andersen EC, Villeneuve AM. Differential localization and independent acquisition of the H3K9me2 and H3K9me3 chromatin modifications in the Caenorhabditis elegans adult germ line. PLoS Genet. 2010;6:e1000830. doi: 10.1371/journal.pgen.1000830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigelow H, Doitsidou M, Sarin S, Hobert O. MAQGene: software to facilitate C. elegans mutant genome sequence analysis. Nat Methods. 2009;6:549. doi: 10.1038/nmeth.f.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignull HR, Moore FE, Tang SJ, Morimoto RI. Polyglutamine proteins at the pathogenic threshold display neuron-specific aggregation in a pan-neuronal Caenorhabditis elegans model. J Neurosci Off J Soc Neurosci. 2006;26:7597–7606. doi: 10.1523/JNEUROSCI.0990-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S, Han HJ, Kohwi-Shigematsu T. Tissue-specific nuclear architecture and gene expression regulated by SATB1. Nat Genet. 2003;34:42–51. doi: 10.1038/ng1146. [DOI] [PubMed] [Google Scholar]
- Ceol CJ, Stegmeier F, Harrison MM, Horvitz HR. Identification and Classification of Genes That Act Antagonistically to let-60 Ras Signaling in Caenorhabditis elegans Vulval Development. Genetics. 2006;173:709–726. doi: 10.1534/genetics.106.056465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SW, Friso S. Epigenetics: A New Bridge between Nutrition and Health. Adv Nutr Int Rev J. 2010;1:8–16. doi: 10.3945/an.110.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland JM, Cho J, Lo T, Jr, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW. Extension of Drosophila Life Span by RNAi of the Mitochondrial Respiratory Chain. Curr Biol. 2009;19:1591–1598. doi: 10.1016/j.cub.2009.08.016. [DOI] [PubMed] [Google Scholar]
- Couteau F, Guerry F, Müller F, Palladino F. A heterochromatin protein 1 homologue in Caenorhabditis elegans acts in germline and vulval development. EMBO Rep. 2002;3:235–241. doi: 10.1093/embo-reports/kvf051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristina D, Cary M, Lunceford A, Clarke C, Kenyon C. A Regulated Response to Impaired Respiration Slows Behavioral Rates and Increases Lifespan in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000450. doi: 10.1371/journal.pgen.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey SS, Foley JE, Limsirichai P, Schaffer DV, Arkin AP. Orthogonal control of expression mean and variance by epigenetic features at different genomic loci. Mol Syst Biol. 2015;11:806. doi: 10.15252/msb.20145704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Rates of Behavior and Aging Specified by Mitochondrial Function During Development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- Durieux J, Wolff S, Dillin A. The Cell Non-Autonomous Nature of Electron Transport Chain-Mediated Longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Bussière F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell. 2001;1:633–644. doi: 10.1016/s1534-5807(01)00071-5. [DOI] [PubMed] [Google Scholar]
- Galande S, Purbey PK, Notani D, Kumar PP. The third dimension of gene regulation: organization of dynamic chromatin loopscape by SATB1. Curr Opin Genet Dev. 2007;17:408–414. doi: 10.1016/j.gde.2007.08.003. [DOI] [PubMed] [Google Scholar]
- González-Aguilera C, Palladino F, Askjaer P. C. elegans epigenetic regulation in development and aging. Brief Funct Genomics. 2014;13:223–234. doi: 10.1093/bfgp/elt048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haithcock E, Dayani Y, Neufeld E, Zahand AJ, Feinstein N, Mattout A, Gruenbaum Y, Liu J. Age-related changes of nuclear architecture in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2005;102:16690–16695. doi: 10.1073/pnas.0506955102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AS, O'Shea EK. Promoter decoding of transcription factor dynamics involves a trade-off between noise and control of gene expression. Mol Syst Biol. 2013;9:704. doi: 10.1038/msb.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13:467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Heifetz A, Keenan RW, Elbein AD. Mechanism of action of tunicamycin on the UDP-GlcNAc:dolichyl-phosphate GlcNAc-1-phosphate transferase. Biochemistry (Mosc) 1979;18:2186–2192. doi: 10.1021/bi00578a008. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Jiménez-Chillarón JC, Díaz R, Martínez D, Pentinat T, Ramón-Krauel M, Ribó S, Plösch T. The role of nutrition on epigenetic modifications and their implications on health. Biochimie. 2012;94:2242–2263. doi: 10.1016/j.biochi.2012.06.012. [DOI] [PubMed] [Google Scholar]
- Lavigne MD, Vatsellas G, Polyzos A, Mantouvalou E, Sianidis G, Maraziotis I, Agelopoulos M, Thanos D. Composite macroH2A/NRF-1 Nucleosomes Suppress Noise and Generate Robustness in Gene Expression. Cell Rep. 2015;11:1090–1101. doi: 10.1016/j.celrep.2015.04.022. [DOI] [PubMed] [Google Scholar]
- Lee SS, Lee RYN, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003;33:40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- Link CD, Cypser JR, Johnson CJ, Johnson TE. Direct observation of stress response in Caenorhabditis elegans using a reporter transgene. Cell Stress Chaperones. 1999;4:235–242. doi: 10.1379/1466-1268(1999)004<0235:doosri>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister P, Schott S, Bedet C, Xiao Y, Rohner S, Bodennec S, Hudry B, Molin L, Solari F, Gasser SM, et al. Caenorhabditis elegans Heterochromatin protein 1 (HPL-2) links developmental plasticity, longevity and lipid metabolism. Genome Biol. 2011;12:R123. doi: 10.1186/gb-2011-12-12-r123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SIS. Role of Histone H3 Lysine 9 Methylation in Epigenetic Control of Heterochromatin Assembly. Science. 2001;292:110–113. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and Nuclear Accumulation of the Transcription Factor ATFS-1 Promotes OXPHOS Recovery during the UPR(mt) Mol Cell. 2015;58:123–133. doi: 10.1016/j.molcel.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin G, Dong Y, Fraser AG, Hopper NA, Ahringer J. Chromatin regulation and sumoylation in the inhibition of Ras-induced vulval development in Caenorhabditis elegans. EMBO J. 2005;24:2613–2623. doi: 10.1038/sj.emboj.7600726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechavi O, Houri-Ze'evi L, Anava S, Goh WSS, Kerk SY, Hannon GJ, Hobert O. Starvation-Induced Transgenerational Inheritance of Small RNAs in C. elegans. Cell. 2014;158:277–287. doi: 10.1016/j.cell.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Sarin S, Prabhu S, O'Meara MM, Pe'er I, Hobert O. Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat Methods. 2008;5:865–867. doi: 10.1038/nmeth.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder EA, Raimundo N, Shadel GS. Epigenetic Silencing Mediates Mitochondria Stress-induced Longevity. Cell Metab. 2013;17:954–964. doi: 10.1016/j.cmet.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919–932. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin BD, González-Aguilera C, Sack R, Gaidatzis D, Kalck V, Meister P, Askjaer P, Gasser SM. Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell. 2012;150:934–947. doi: 10.1016/j.cell.2012.06.051. [DOI] [PubMed] [Google Scholar]
- Vrachnis N, Antonakopoulos N, Iliodromiti Z, Dafopoulos K, Siristatidis C, Pappa KI, Deligeoroglou E, Vitoratos N. Impact of Maternal Diabetes on Epigenetic Modifications Leading to Diseases in the Offspring. J Diabetes Res. 2012;2012:e538474. doi: 10.1155/2012/538474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui D, Miyano M, Cai S, Varga-Weisz P, Kohwi-Shigematsu T. SATB1 targets chromatin remodelling to regulate genes over long distances. Nature. 2002;419:641–645. doi: 10.1038/nature01084. [DOI] [PubMed] [Google Scholar]
- Yee C, Yang W, Hekimi S. The Intrinsic Apoptosis Pathway Mediates the Pro-Longevity Response to Mitochondrial ROS in C. elegans. Cell. 2014;157:897–909. doi: 10.1016/j.cell.2014.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.