


Abstract
We present a theory of pluralistic and stochastic gene regulation. To bridge the gap between empirical studies and mathematical models, we integrate pre-existing observations with our meta-analyses of the ENCODE ChIP-Seq experiments. Earlier evidence includes fluctuations in levels, location, activity, and binding of transcription factors, variable DNA motifs, and bursts in gene expression. Stochastic regulation is also indicated by frequently subdued effects of knockout mutants of regulators, their evolutionary losses/gains and massive rewiring of regulatory sites. We report wide-spread pluralistic regulation in ≈800 000 tightly co-expressed pairs of diverse human genes. Typically, half of ≈50 observed regulators bind to both genes reproducibly, twice more than in independently expressed gene pairs. We also examine the largest set of co-expressed genes, which code for cytoplasmic ribosomal proteins. Numerous regulatory complexes are highly significant enriched in ribosomal genes compared to highly expressed non-ribosomal genes. We could not find any DNA-associated, strict sense master regulator. Despite major fluctuations in transcription factor binding, our machine learning model accurately predicted transcript levels using binding sites of 20+ regulators. Our pluralistic and stochastic theory is consistent with partially random binding patterns, redundancy, stochastic regulator binding, burst-like expression, degeneracy of binding motifs and massive regulatory rewiring during evolution.
INTRODUCTION
Most disease-associated mutations are located outside of protein coding regions, likely affecting transcriptional regulation or chromosomal organization (1,2). To draw objective and consistent biological and clinical conclusions from the over two million human genomes to be sequenced by 2020 (3), we need new models and theories of gene regulation that are highly consistent with observations and minimally biased (4). Almost inherent biases include the number and selection of transcriptional regulators (TRs), knockout mutants, amplification and sequencing bias. However, we can avoid biased interpretation. Struggling with vast complexity, human perception is naturally biased toward simplifications. Many simplifications had been practical before the Encyclopedia of DNA Elements (ENCODE) Project (5) probed the complexity of transcriptional regulation. In the lac operon and similar prokaryotic models, only a few agents regulate each target gene (6). These models were extrapolated to higher eukaryotes, which regulate gene expression by over a thousand sequence- or shape-specific transcription factors, histone modifying enzymes and chaperones (for brevity, TRs; 7). To handle this complexity, diverse concepts of master regulators were introduced. This term occurs in over 28 700 publications, two-thirds of which are related to cancer or cellular differentiation according to our full-text Scopus search. We present multiple lines of evidence that typically, rather than singular master regulators or oligarchies, large numbers of TRs regulate genes. We report and test our pluralistic and stochastic, minimally biased computational models. Stochastic is defined as ‘partially randomly determined; a process that follows some random probability distribution or pattern, so that its behavior may be analyzed statistically but not predicted precisely’ (8) (quoted verbatim in the Oxford English Dictionary as well). At first glance, stochastic processes may appear vague. Inherently, they are more difficult to understand, reproduce and verify than comparable deterministic processes. Hence demanding high reproducibility leads to ignoring mid-to-low probability events. However, stochastic models allow for more accurate predictions than deterministic simplifications. For example, differentiated fibroblasts can be reprogrammed into pluripotent stem cells in multiple ways (9). OCT4 and SOX2, two essential but insufficient agents, along with either KLF4 and MYC (10) or NANOG and LIN28 (11) can induce such reprogramming. Stochasticity means that either KLF4 and MYC or NANOG and LIN28 can bind in partially random processes (but with similar effects). These four TRs bind to pluripotency targets with probabilities much below certainty but higher than those TRs that cannot induce pluripotency. In this well-established example, deterministic master regulators were replaced by stochastic regulation (12). Similar probabilistic patterns form the very essence of this publication.
A theory of transcriptional regulation is presented which is consistent with our new results reported here:
20–25 TRs bind reproducibly in ≈800 000 co-expressed gene pairs, indicating pluralistic regulation.
20 or more TRs are needed to predict transcript levels of cytoplasmic ribosomal protein genes (cRPGs).
TR binding shows stochastic enrichment patterns in cRPGs compared to high-expression non-ribosomal genes (HE-NRGs).
Pluralistic and stochastic gene regulation is also supported by a novel synthesis of earlier observations:
Cellular levels, location, activity and binding of TRs and polymerases undergo major fluctuations
Transcription bursts and pauses even in the genes of TRs themselves (11,13–16)
A wide variety of ≈1700 human DNA-associated proteins have evolved and been preserved (7)
Transcription factors bind with different strength and regulatory effect to highly variable DNA motifs/shapes (17)
Several double knockout mutants of TRs are viable (18)
Several TRs have been replaced during evolution (Table 1) and their binding sites have been rewired even between human and mouse (19).
Table 1. Few regulators of human cRPGs have orthologs in Saccharomyces cerevisiae. A few double knockout mutants of the orthologous mouse genes are still viable.
| Regulator | Mouse mutant phenotype | ||
|---|---|---|---|
| human | Scer | -/- | +/- |
| IRF1 | phenotype | phenotype | |
| SIX5 | phenotype | phenotype | |
| BRCA1 | phenotype | phenotype | |
| MYC | lethal | phenotype | |
| KAT2A | GCN5 | lethal | reduced transcription elongation |
| ETS1 | partially lethal | phenotype | |
| ELK1 | mostly normal | normal | |
| GTF2B | SUA7 | lethal | ? |
| ZZZ3 | lethal | lethal | |
| TAF1 | TAF1 | phenotype | |
| TAF7 | TAF7 | lethal | phenotype |
| ATF2 | ATF2 | phenotype | phenotype |
| HDAC6 | HDA1 | normal | normal |
| RCOR1 | SNT1 | lethal | phenotype |
| NFKB1 | phenotype | phenotype | |
| CEPB | lethal | normal | |
| CHD1 | CHD1 | phenotype | phenotype |
| CJUN | phenotype | phenotype | |
| EJUN | phenotype | phenotype | |
| JUNB | phenotype | phenotype | |
| CTCF | CTCF | lethal | phenotype |
| MAFF | lethal | phenotype | |
| NELFE | ? | phenotype | |
| NFYB | HAP3 | ? | phenotype |
| NRF1 | lethal | phenotype | |
| RFX5 | phenotype | phenotype | |
| SETDB1 | SET2 | lethal | phenotype/lethal |
Surprisingly, as we will show in the Discussion, concepts of master regulators have already evolved from strict hierarchies to more participative regulation. We continue this trend by integrating the above observations with highly sophisticated stochastic models and computational simulations of transcriptional regulation (9,13,20–26), which were partly validated by experiments (16). To help the experimental community to embrace stochastic gene regulation, we propose a theory of widespread pluralistic and stochastic regulation based on the above wide spectrum of evidence.
MATERIALS AND METHODS
We analyzed six human and two murine cell lines for which twenty or more regulators have been mapped by the (mouse) ENCODE Project to the hg19, hg38 and mm9 genome assemblies. Two pairs of cell lines are comparable across human and mouse: myelogeneous leukemia (K562 and MEL) and lymphoblastoid (GM12878 and CH12.LX). Additional human cell types include embryonic stem cells (h1-hESC1), hepatocarcinoma (HepG2), adenocarcinoma (A549) and cervical cancer (HeLa-S3) cells. Pseudogenes were eliminated, leaving 98 human and 87 mouse cRPGs and 84 human and 76 mouse mRPGs (Supplementary Table S1). cRPGs and mRPGs were compared to either all non-ribosomal genes (NRGs), or their subset, the HE-NRGs (Supplementary Table S2). We compare cRPGs to 169 human and 107 mouse HE-NRGs, the latter defined as genes expressed at higher levels than the least intensively expressed 25% of cRPGs in the Expression Atlas (27). All data have been stored in our MySQL relational database and queried by a PERL Database Interface library and scripts. The human part of the MySQL Database, its documentation, and all the gene pairs with the number of jointly bound and separately bound TRs, are available at our web site: https://git.unl.edu/sladunga2/genereg/tree/master. Other data can be obtained upon request.
TR binding site observations derived from chromatin immunoprecipitation combined with deep sequencing (ChIP-Seq) were downloaded from the ENCODE web sites (https://www.encodeproject.org). Gene coordinates and annotations were taken from the ENSEMBL annotations (Homo_sapiens.GRCh37.59.gtf and Mus_musculus.NCBIM37.67.gtf). From among overlapping gene annotations, the longest splice variant was chosen. Binding sites were mapped to genes as follows: when a binding site was localized within a gene's coding sequence or its up- or downstream 5000 base pair environment (excluding potentially overlapping genes), the binding site was associated with the gene. Five thousand base pairs represent a compromise between the inclusion of not overly distant enhancer regions and the minimization of the number of TRs that do not affect the transcription of the particular gene. To examine the impact of selecting the longest coding regions with 5000 base pair upstream and downstream segments (Gene5kb), we compared the results to the most frequent transcripts and to predicted 600 base pair promoter regions in K562 cells (Supplementary Information, Figure S4 and Tables S4 and S5). The predicted promoter regions largely reproduced the Gene5kb patterns of enrichments although with higher fold changes.
Statistical analyses
High genewise counts of single TRs allowed evaluating the statistical significance of enrichment using the Wilcoxon–Mann–Whitney test. Due to the lower genewise counts of TR dimers and trimers, enrichment was assessed using Fisher's exact test. Both tests are robust against large differences in sample size. Unless otherwise noted, all results reported here are statistically significant at the 0.01 level after multiple test correction by tailwise False Discovery Rate (28).
Statistical/machine learning models
How many TRs are necessary to relatively accurately predict transcript levels from TR binding sites? To answer this question, we use Least Angle Regression (LARS) (29). LARS applies ordinary least squares to minimize the sum of the absolute values of weights assigned to generalized linear models. This parsimonious feature works as Occam's razor by regressing transcript levels using the fewest possible TRs. LARS performs cross-validation, i.e. training the models on one subset of input data and testing performance on the complementary subset. Transcript levels were taken from the Genevestigator database (30) and from the human and mouse ENCODE experiments (31).
RESULTS
Genome-wide functional pluralistic TR binding in the human genome
We found that TR binding sites were about twice as highly reproducible in 799 695 co-expressed human gene pairs than in 100 000 independently expressed genes in six human cell types. We compared five co-expressed and one independently expressed gene sets: all 4851 gene pairs within cRPGs (Supplementary Table S1), all 3486 pairs of mitochondrial ribosomal protein genes (mRPGs, Supplementary Table S1) and all 14 196 pairs high-expression NRGs (HE-NRG's, Supplementary Table S2). Pseudogenes were eliminated. We considered a gene as a HE-NRG if its median RNA-seq transcript level exceeds the 25th percentile of the transcript levels in cRPGs in the Expression Atlas Database (32) (Supplementary Table S2, Materials and Methods). We also analyzed 17 846 pairs of very strongly co-expressed genes (R ≥ 0.9, NRG_A's); 759 316 pairs of strongly co-expressed (0.9 > R ≥ 0.8) genes (NRG_B's), and a sample of 100 000 independently expressed (abs(R) < 0.1) gene pairs (NRG_C's, Figures 1 and 2). Co-expression was measured by Pearson correlation coefficients of transcript levels for each pair of human protein-coding gene over 120 diverse samples in the Expression Atlas (27) (see Materials and Methods).
Figure 1.

High reproducibility of ChIP-Seq peaks in pairs of co-expressed genes. Jaccard coefficients show reproducibility for the following sets of gene pairs: cRPGs (n = 4851 pairs); mRPGs (n = 3486); HE_NRGs (high-expression NRGs, n = 14 196, see Materials and Methods); NRG_A's (diverse gene pairs co-expressed with R ≥ 0.9, n = 17 846); NRG_B's (diverse gene pairs co-expressed with 0.9 > R ≥ 0.8, n = 759 316); and NRG_C's (a sample of independently expressed, diverse gene pairs, abs(R) < 0.1, n = 100 000). TR binding in NRG_C's is about 50% less reproducible than in co-expressed gene sets, indicating that a large portion of the binding events in gene regions is functional.
Figure 2.

TRs bind with similar reproducibility in diverse human cells. Box plots show the distribution of Jaccard coefficients for individual TR. Sets of gene pairs are defined in Figure 1. In all co-expressed sets of gene pairs, over 25 TRs bind with a median reproducibility exceeding 0.5. In independently expressed gene pairs, reproducibility is only about 0.22, corresponding to the magnitude of nonspecific TR binding. Highly significant differences between co-expressed and independently expressed gene sets (P < 10−256, Wilcoxon–Mann–Whitney test) indicate that even those TRs, which bind in highly stochastic processes, may have biological roles.
We quantified the reproducibility of binding for each TR using a simple adaptation of the Jaccard coefficient: 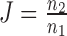 Here n2 is the number of gene pairs where the TR in question is observed in both genes of the pair and n1 is the number of gene pairs where the TR is observed in at least one of the two genes.
Here n2 is the number of gene pairs where the TR in question is observed in both genes of the pair and n1 is the number of gene pairs where the TR is observed in at least one of the two genes.
First, we examined individual TRs and their binding sites. Binding events of PolII, YY1, (C)MYC, KDM5B, TAF1, MAX, PHF8, ELF1 and MAZ are highly reproducible (J ≥ 0.9) in NRG_A's, cRPGs and HE-NRGs (Figure 1). This high between-gene reproducibility is the lower bound of ChIP-Seq reproducibility in the ENCODE experiments (33), as discussed below. In NRG_C's however, the reproducibility of most TRs remains below 0.25. The only three exceptions are RUNX3, CTCF and RAD21.
Which TRs bind most reproducibly across cell types? To answer this question, we compared the cell-wise distributions of the 50 most reproducibly bound TRs in the six gene pair sets (Figure 2). In the five co-expressed sets of gene pairs, about 25 TRs bind with a median reproducibility exceeding 0.5 compared to 0.23 in independently expressed gene pairs. The interquartile ranges show that reproducibility is very similar in all cell types studied for RUNX3, PolII, PHF8, IRF1, KDM5B, POU2F2, CTCF, RAD21 and CREB1, indicating largely cell-independent functions.
We found that twice as many regulators mapped reproducibly in co-expressed pairs than in independently expressed gene pairs (Figure 3). This difference persists independently of gene length (Supplementary Information and Figure S1). While co-regulated gene pairs tend to be expressed at higher levels than independently transcribed ones, the effect of co-regulation on reproducibility is much stronger than the level of expression (Supplementary Information and Figure S2). In K562 cells, at least one gene of an independently expressed (NRG_C) pair binds to a median of 62 TRs (Figure 3). Only 10 of these TRs bind to both genes (J = 0.16). In highly co-expressed pairs (NRG_A), at least one gene of a pair binds to a median of 75 TRs. Of these, 37 TRs bind to both genes (J = 0.49). The significantly more reproducible binding (P < 10−16, Fisher's exact test) in co-expressed versus independently expressed gene pairs indicates markedly pluralistic regulation.
Figure 3.

(A). More TRs bind to both genes in co-expressed gene pairs than in independently expressed pairs (NRG_C's, max(P) < 10−32, Wilcoxon test). (B) Conversely, fewer TRs bind to only one gene in co-expressed gene pairs than in NRG_C's (max(P) < 10−32). The number of TRs that may be associated with co-regulation depends on the TRs mapped in a cell type. The number of TRs implicated in co-regulation ranges from 25 (in A549 and GM12878 cells) to over 50 (in HeLaS3, HepG2 and K562 cells).
Specific model: co-regulation of 98 cRPGS
We present a model based primarily on binding sites of less than three hundred TRs in six human and two murine cell types (Materials and Methods), gain/loss-of-function mutants and evolutionary studies. The ENCODE Consortium mapped these TR binding sites to the human and mouse genomes using Chromatin ImmunoPrecipitation followed by deep sequencing (ChIP-Seq; 5). Despite the strong co-expression of ribosomal protein genes (RPGs, see below), the observed binding patterns of TRs show differences between genes and cell types (Figure 5 and Supplementary Figure S1). As we discuss in Supplementary Information, these differences are largely due to stochastic TR binding, not to experimental error.
Figure 5.

Stochastic TR binding to DNA does not show evident master regulators. The unfiltered numbers of observed binding sites for individual TRs (c) in cytoplasmic and mitochondrial RPGs in human K562 cells. Statistical preferences for several TRs emerge despite considerable randomness, which is partly due to experimental noise. For scalability, log2(c +1) values are shown. Stochastic TR binding is also confirmed for all other analyzed human and mouse cell types (Supplementary Figure S3). The network of cRPG regulation also shows rich and highly variable binding of TRs to diverse cRPGs (Supplementary Figure S5).
To increase confidence and to estimate experimental error in TR binding site observations and to narrow the gray zone, it would be ideal to map all regulators in tens of ChIP-Seq replicates for several cell types. However, such a megaproject would cost multiples of the ENCODE Project Consortium's budget. To increase confidence without astronomic costs, we analyze a relatively homogeneous subpopulation of genes, which are tightly co-regulated to minimize waste in synthesizing stoichiometric amounts of ribosomal proteins (34). Each gene serves as an experimental unit, analogously to clinical trials, where individual patients are not replicates but experimental units, which also facilitate drawing robust conclusions (35). Dispersed across 22 chromosomes, the 98 cytoplasmic RPGs (cRPGs) form the largest co-expressed gene network in the human genome (Figure 4 and Supplementary Table S1) (36,37). Their vital importance is another major advantage: viable null mutants of a TR indicate that the TR is not necessary for cRPG expression.
Figure 4.

Confirmation of tight RPG co-expression across a wide range of conditions and cell types. (A) Base 2 logarithms of transcript levels (horizontal axis) are shown in arbitrary but normalized units from 28,032 Affymetrix microarrays from the Genevestigator Database (30). Transcripts are over hundredfold more abundant in cRPGs than in mRPGs and also vary between families of RPGs. (B) Pearson correlation coefficients (R) for cRPG transcript levels for each RPG pair indicate that variations in transcript levels are reproducible and tightly correlated. The high median correlation of 0.7875 for all cRPGs is very likely due to co-regulation. High co-expression is in accordance with the earlier observation that only a small proportion of RP molecules are located outside the ribosome and the nucleolus (39).
RPG co-regulation has been reported a quarter century ago (36) and in 2006 (37) based on very limited data sets. As the tight co-expression of RPGs is critical to our results, it is necessary to confirm and quantify RPG co-expression by Pearson correlation coefficients on a large data set. For every possible pairs of RPGs across 28 032 microarray samples in the Genevestigator Database (30), the median of the correlation coefficients is as high as 0.788 for cRPGs and 0.514 for mRPGs (Figure 4). The probability of such co-expression across 28K samples is less than 10−256. Its most plausible cause is co-regulation. Imperfect correlations are likely due to possible translational efficiencies and the about one hundred extraribosomal functions that RPs perform (38). However, as extraribosomal RP accumulation evokes nucleolar stress and potentially, cell cycle arrest, most cytoplasmic RP molecules are constrained to the ribosomes and the nucleoli (39). Co-expression is not due to constitutive expression as cells repress or induce RPG transcription in response to changes in energy levels and nutrient availability (37,40,41). Were co-regulation a deterministic process, translational efficacy identical, extraribosomal function, experimental error and nonfunctional binding absent, identical TRs would bind in identical amounts to all of the 98 cRPGs. In sharp contrast, the observed distribution of TRs in six cell types shows a mixture of experimental error and highly stochastic binding of TRs in diverse human cells (Figure 5 and Supplementary Figure S1).
We systematically compared TR binding sites in cRPG to those of all non-ribosomal genes (NRGs) as well as HE-NRGs. We searched for DNA-bound master regulators for cRPGs but could find none. We looked for strong correlations between TR binding and cRPG transcript levels but none exceeded 0.45. Binding sites of ≈20 regulators were needed to accurately predict cRPG transcript levels by machine learning. Most TR knockout mutants in mice are viable (18) indicating that these cannot be necessary controllers of protein synthesis. On the evolutionary scale, the most important RPG regulators in S. cerevisiae does not have mammalian orthologs and the two third of the mammalian cRPG regulators do not have orthologs in fungi (Table 1). Instead of masters, we found that only RNA Polymerase II (POL2), CTCF, MYC, YY1 and IRF1 bind to most cytoplasmic and mitochondrial RPGs in K562 cells (Figure 5) and other human and mouse cells (Supplementary Figure S3). None of these TRs are specific to cRPGs and the rest of TRs bind to RPGs in stochastic patterns (Figure 5).
We examined peaks of binding sites for each TR separately, regardless of overlapping peaks predicted from ChIP-Seq experiments (5), and for brevity, called them single TRs. We also analyzed pairs and triplets of overlapping peaks of distinct TRs and named them putative dimers and trimers, regardless of overlaps with yet other TRs. We called them putative as individual peaks are ‘snapshots’ taken at different times and from different samples, hence some of these binding events may occur in different times.
Enriched TR complexes indicate pluralistic and stochastic regulation and signal integration
We observed statistically highly significant enrichment of several TRs, hundreds of heterodimers and tens of thousands of trimers in cRPGs as compared to HE-NRGs and/or all NRGs (Figure 6 and Supplementary Tables S6–S11). Unless otherwise mentioned, we compared cRPGs to HE-NRGs and all comparisons were significant at the P ≤ 0.01 level (Wilcoxon or Fisher's Exact test, see Materials and Methods) followed by multiple test correction using tail-wise False Discovery Rate (28). We present evidence that these enrichment patterns indicate pluralistic and stochastic integration of external and cellular signals and regulatory mechanisms that are far more complex than earlier reported cis-regulatory modules (42).
Figure 6.


cRPG regulatory binding events show highly specific and statistically significant patterns of enrichment or depletion of single transcriptional regulators, putative TR heterodimers and heterotrimers. Human cRPGs are compared to HE-NRGs and NRGs in separate panels. For single TRs, the significance of enrichment was assessed by the Wilcoxon–Mann–Whitney test, for dimers and trimers, by Fisher's Exact Test. Multiple test corrections were performed using Benjamini and Hochberg's False Discovery Rate (28). Numerical data are available in Supplementary Tables S4–S8. (A) Single TRs, cRPGs versus HE-NRGs. (B) Single TRs, cRPGs versus all NRGs. (C) Heterodimers, cRPGs versus HE-NRGs. (D) The 50 most highly enriched heterodimers, cRPGs versus HE-NRGs. (E) Heterodimers, cRPGs versus all NRGs. (F) The 50 most highly enriched heterodimers, cRPGs versus all NRGs. (G) Heterotrimers, cRPGs versus HE-NRGs. (H) The 50 most highly enriched heterotrimers, cRPGs versus HE-NRGs. (I) Heterotrimers, cRPGs versus all NRGs. (J) The 50 most highly enriched heterotrimers, cRPGs versus all NRGs.
For the biological roles of these complexes, we extrapolated from the roles of individual TRs based on previous experiments to the functions of the multimolecular complexes. These extrapolations provide a reasonably informed hypothetical framework to guide future experiments.
Importantly, the enriched di- and trimers include several well-studied TRs that have not yet been implicated in RPG regulation. Of these, SIX5 (a.k.a. DMAHP or BOR2) preferentially binds together with MYC, CHD1, TAF7, GTF2B, and with cohesin constituents including RAD21, CTCF, SMC3 and ZNF143. Consistent with SIX5 roles in a wide array of disorders (43,44), in one or more of the six human cell types studied, SIX5 binds to 6,779 protein-coding genes. One could expect that homozygous knockout mutants of such a wide-spectrum TR to be lethal. However, both the murine (44) and Drosophila (45) null mutants are impaired in organ development but still viable. Because ribogenesis is critical to protein synthesis, viable null mutants indicate that the ribosome-specific functions of SIX5 can be substituted by other TRs. This and the highly significant enrichment of SIX5 and its complexes show that SIX5 has a stochastic contribution to cRPG regulation, which is robust against SIX5 mutations.
Similarly, the highly enriched BRCA1 (Figure 6) has not been implicated in direct RPG regulation. Indirectly, BRCA1 is known to interact with the nucleoli and the ribosomal protein RPSA (46) to suppress the cell cycle upon DNA damage (47). Preferential co-binding of BRCA1 with CTCF and RAD21 indicates a role in modulating chromosome conformation (Figure 6). Preferred association with GABP1, a known integrator of cellular signaling pathways (48) suggests that GABP1 may interact with BRCA1 to downregulate cRPGs under adverse conditions.
Specific pre-initiation complexes (PICs)
We extend RPG-specific PICs reported earlier (49) with several novel stochastic patterns. The strong enrichment of the transcriptional activator KAT2A (GCN5) indicates that it contributes more to the regulation of cRPGs than to most other genes including HE-NRGs. By acetylating histones, KAT2A prepares the chromatin for intensive transcription. Like HDAC6, one of its antagonists, KAT2A has been conserved between yeast and mammals (50). GCN5, its ortholog in yeast, is known to regulate RPGs directly (51). KAT2A is scaffolded to histones by the similarly enriched ZZZ3. This latter protein is specific to ATAC and only either ATAC or S(T)AGA, but not both, can bind to a highly expressed gene (52). Thus, ZZZ3 enrichment (Figure 6, and Supplementary Tables S6–S11) indicates strong preference for the ATAC complex in cRPGs. As intrinsically disordered regions within KAT2A are known to initiate the formation of PICs (53), we speculate that KAT2A's preferential associations may orchestrate the formation of RPG-specific PIC's. Preferential association with MYC (Figure 6 and Supplementary Tables S8–S11) is consistent with the need for KAT2A-mediated histone acetylation to recruit MYC (54). MYC, a widespread nonspecific regulator of RPGs in vertebrates, has similarly extensive disordered transactivation domains (55). In vitro, these domains can recruit hundreds of regulators but in vivo, the interactors are constrained by the co-bound partners and adjacent DNA motifs (15,56). MYC, its activator, MAX, and repressor, MXI1 appear to interact with TAF7, HDAC6, REST, NELF (RDBP) and BRCA1 (Figure 6, Supplementary Tables S6–S11). Such complex binding events indicate a network far exceeding the MAX/MYC/MXI1 axis for the regulation and deregulation of oncogenic activity. In a positive feedback loop, KAT2A acetylates histones in the genes of MYC, Yin Yang 1 (YY1) and other direct regulators of RPGs. Subsequently, MYC induces the KAT2A gene (57). Enriched complexes of the histone deacetylase HDAC6 or similar agents can break this positive feedback loop.
The robust enrichment of HDAC6 (Figure 6, Supplementary Tables S6–S11) raises the possibility that its influence on the cell cycle (58) may be partly mediated via the regulation of ribogenesis. HDAC6 preferentially co-binds with PolII, P300, estrogen receptor, RUNX2, NFκB and HSP90; an activity likely to be organized by the ubiquitin-binding domain of HDAC6 (59). Despite the fundamental roles of HDAC6, its null mutants display normal phenotype both in Drosophila (60) and mouse (61), strongly indicating that other enzymes, possibly paralogous HDAC family members, can effectively perform HDAC6 functions.
The strongly enriched overlapping peaks of KAT2A and chromodomain helicase DNA binding protein 1 (CHD1) may indicate coupled histone acetylation and methylation (62). This dimer and its superset with ZZZ3 are known to evict nucleosomes to facilitate the passing of the transcriptional machinery (63). KAT2A also forms enriched di- and trimers with TBP/TRF2-associated factors TAF1 and TAF7. These factors form enriched complexes with KAT2A, MYC, MAX, MXI1, Pol II, SIX5, YY1, G2F2B, ZZZ3, IRF1, CHD1 and ZNF143 (Figure 6).
GTF2 subunits are among the most enriched regulators (Figure 6). GTF2 is known to link the TFIID complex to Pol II (64). We found that several joint members of the quantitative regulator complexes TFIID and S(T)AGA complexes are enriched in RPG promoters (Figure 6). Taken together, these observations show that PICs of RPGs display significantly different distributions of regulators than PICs of other genes including HE-NRGs.
Pol II is enriched in cRPGs compared to NRGs, but not compared to HE-NRGs (Figure 6 and Supplementary Tables S6–S11). An inducer of polymerase pausing, RD RNA binding protein (RDBP a.k.a. NELF) is enriched in cRPGs relative to NRGs (Supplementary Tables S6–S11) but not compared to HE-NRGs. Pausing is known to counteract nucleosome reconstitution hence to prepare the chromatin for active transcription (65). On this basis, we speculate that RPG-specific PICs and transcriptional machinery modulate polymerase performance and pausing. Relief from pausing allows rapid RPG induction in timely response to improved growth conditions. In cancer cells, the MAX-MYC dimer relieves Pol II from pausing and amplifies transcription (66). In both malignant transformation and experimental overexpression, MYC and MAX may overinduce thousands of active genes by interacting with members of the basal transcriptional machinery during PIC formation (67). Under such conditions, MYC and MAX indeed act as master regulators. We found that MYC and MAX bind to 148 of the 183 human RPGs in at least one of the six major cell lines (Figure 5 and Supplementary Figure S1). Unlike MAX, MYC by itself is enriched in RPGs relative to both HE-NRGs and NRGs. In differentiated cells, MAX forms the four most enriched pairs with REST, G2F2F1, KAT2A and ZNF143, followed by KAT2A_ MAX (Figure 6). PIC constituents MYC, MAX, KAT2A, TAF1, TAF7 and SIX5 form the most enriched triplets with the sole exception of A549 cells (Figure 6). These observations and the presence of MYC and MAX in mRPGs, which are expressed at hundredfold lower levels than cRPGs (Figure 4) indicate MYC and MAX functions that are not related to intensive transcription.
Stochasticity is the most plausible resolution for the ostensible controversy regarding the ternary complex factor ELK1. Despite its high enrichment, ELK1 is redundant for the regulation of ribogenesis and other processes. Its deletion mutants in mice are not impaired in immune reaction, brain and spleen function (68). Were ELK1 roles deterministic, it would be either enriched and essential or unenriched and unnecessary. Instead, we observed enrichment because it frequently but not necessarily regulates cRPGs. TRs like ELK1 can be substituted by other TRs in stochastic processes.
The tumor suppressor REST is enriched in cRPGs as compared to NRGs both as a monomer and when co-bound with MYC, STAT5A, Pol II, TAF7, TCF3 and TAF1 (Figure 6 and Supplementary Tables S6–S11). The enriched complex of IRF1, yet another tumor suppressor, with MYC and Pol II may counteract the hyperactivation of the cell cycle by inhibiting MYC (69). These negative feedback mechanisms are critical to cRPG regulation.
Machine learning models of transcriptional regulation
Next we asked: how many TRs bound to cRPGs can predict the observed transcript levels? To avoid inaccurate predictions on untrained observations (known as overtraining), machine learning methods need to be trained on about three-to-five times fewer carefully selected TRs than genes. For example, K562 cells transcribe 98 cRPGs. Therefore we had to select subsets of 98/4–25 or fewer TRs to maximize prediction accuracy for untrained observations. For this purpose, Least Angle Regression (LARS) (29) provided for the highest accuracy (see Materials and Methods). Note that TR selection (in computer science terms, feature selection) methods maximize regression accuracy, not the biological importance of the TRs. For example, if two or more TRs, such as the mandatory components of the PIC or transcriptional machinery, bind to similar genes in similar quantity under similar conditions, only one is necessary for regression despite similar biological necessity of the other proteins. For this reason, regression typically demands fewer TRs than transcriptional regulation, making our estimates for the numbers of necessary TRs conservative.
LARS achieved 74% cross-validation prediction accuracy for K562 cells using binding sites of 20 TRs (Figure 7, Supplementary Table S5). Higher accuracy would demand more TRs but the number of cRPGs limits the number of TRs that can account for robust predictions. These findings implicate a minimum of twenty TRs in the regulation of cRPGs in human and mouse. As several TRs correlate moderately (0.3 < R < 0.45) with transcript levels (Figure 8), were these TRs acting independently, four TRs would account for almost all of the regulation and would allow for accurate predictions. This is not the case, indicating strongly interdependent effects of these agents. Therefore none of the above TRs is sufficient for the regulation of cRPGs under the conditions of the ChIP-Seq experiments. Despite moderate correlations between the binding of individual TRs and transcript levels, the complexes that regulate cRPG transcription contain no less than 29 different TRs which are both enriched and predictive for cRPG expression in one or more cell type studied.
Figure 7.

Accurate prediction of transcript levels by Least Angle Regression (29) models (see Materials and Methods) requires binding sites of no less than 20 TRs in human and mouse cell types. Cross-validation prediction accuracy is shown in the function of the number of TRs selected by Least Angle Regression.
Figure 8.

No master regulator emerges from the moderate correlations between TR binding sites and transcript levels in cRPGs. Transcript levels for human K562 and GM12878 cells were taken as the average transcript levels from the Genevestigator Database (30); for mouse MEL and CH12.LX cells RNA sequencing transcript levels were calculated from raw data of the mouse ENCODE Project (31).
DISCUSSION
We report a general theory of pluralistic and stochastic regulation of PolII-mediated transcription in human. This theory is a synthesis of our above results with a broader spectrum of published evidence. In our studies, most of the ≈800 000 tightly co-expressed gene pairs are bound reproducibly by over twenty TRs, indicating widespread pluralistic regulation. In RPGs, the largest co-expressed network of genes in human, observed TR binding sites vary greatly among individual RPGs (Figure 5 and Supplementary Figure S1) despite their tight co-expression (36,37). A particular RPG in a particular cell type can be regulated by very diverse TRs. This variation significantly exceeds the level of ChIP-Seq error. We searched for DNA-associated master regulators of cRPGs but could find none. We looked for strong correlations between TR binding and cRPG transcript levels but none exceeded 0.45. Binding sites of ≈20 regulators were needed to accurately predict cRPG transcript levels by machine learning. Most TR knockout mutants in mice are viable (18) indicating that these cannot be necessary controllers of protein synthesis. On the evolutionary scale, the most important RPG regulators in S. cerevisiae does not have mammalian orthologs and the two third of the mammalian cRPG regulators do not have orthologs in fungi (Table 1). Instead of master regulators, we found significant enrichment of 41 individual TRs, 700 putative dimers, and 9827 trimers in cRPGs compared to HE-NRGs (Supplementary Tables S6–S11). The probability of the random occurrence for such strong patterns is close to zero. This enrichment shows that a large number of TRs, dimers, trimers, and likely higher order complexes collectively regulate cRPGs. MYC, NFκB and other widely bound TRs act as regulatory hubs recruiting other TRs. Under most normal, stress and disease conditions, repressors like HDAC6, MXI1, NELF1, REST, IRF1 and BRCA1 prevent regulatory hubs from becoming uncontrolled master regulators. When negative feedback fails, MYC and MAX may become master regulators and amplify the transcription of thousands of genes in cancer (66). The observed stochastic TR binding and interactions are more robust against regulatory malfunctions (such as mutations and evolutionary substitution of regulators and their binding sites) than rigid hierarchies controlled by masters (70).
We also found that accurate predictions of cRPG transcript levels demand a minimum of 20 regulators. This is in concordance with the viability of null mutants for several orthologous murine (18) and Drosophila TRs (45). These observations show that TRs regulating cRPGs can substitute each other to a large extent. A high number of TRs, even some of those that bind to thousands of genes, are not critical to survival (71). In a study of the control of growth arrest and differentiation in a leukemia cell line, none of the 52 TRs knocked down by short interfering RNAs proved to be as necessary (master) regulators (72).
Many regulators disappear and others emerge during evolution. Despite the vital role of ribogenesis and the strong conservation of most RPs in eukaryotes (73), their regulators and regulator binding sites have evolved rapidly (74). We have implicated 27 TRs in the regulation of cRPGs. This is the union of highly predictive TRs in machine learning experiments and the enriched single TRs in all six human cell types. We hypothesize that several other TRs contribute to the governance of cRPG expression. The confirmation of this hypothesis requires additional experiments. Of the 27 TRs implicated here, only 11 have apparent orthologs in Saccharomyces cerevisiae (CTCF, KAT2A, GTF2, TBP, TAF1, TAF7, ATF2, HDAC6, RCOR1, NFYB and SETDB1; Supplementary Table S6). MYC binds to most RPGs in mammals (56) but neither MYC nor its prime interactors, MAX and MXI1, nor the also widely bound BRCA1 have detectable homologs in yeast (Supplementary Table S6, Wu, Y.-C., Bansal, M.S., Rasmussen, M.D., Herrero, J. and Kellis, M. (2014) Phylogenetic Identification and Functional Characterization of Orthologs and Paralogs across Human, Mouse, Fly and Worm. bioRxiv, doi:10.1101/005736). In S. cerevisiae, RAP1 is one of the most important regulators of RPGs; whereas in another yeast, Candida albicans, TBF1 plays a similar role (75). Neither have detectable homologs in humans. Such extensive gains and losses of TRs have not caused lethal impairments to protein synthesis in the ancestors of contemporary species, indicating that these TRs were not necessary at some times and in some lineages. Compared to prokaryotes and yeasts, over 1200 additional TRs evolved in the lineage of mammals (7). The resulting vast combinatorics also facilitates sophisticated responses to internal and external signals. For example, the mTORC1 kinase complex governs RPGs by directly or indirectly phosphorylating MYC, YY1, STAT's, JUN, histone deacetylases, BRCA1, RAD21, ZZZ3, KDM5A and TAF1 (76,77). To a limited extent, even the mTORC1 kinase complex can be substituted by the phosphatidylinositol 3-kinase and ERK-MAP pathways (78,79).
DNA binding sites of transcription factors evolve very fast. The resulting variability modulates the strength and the regulatory effects of individual sites (17). As old binding sites are transferred to new loci or deteriorate and new sites emerge, a large part of the regulatory network changes during evolution. Only 36% of the mouse regulatory regions (DNase hypersensitivity sites) maps to human regions and only 14% of them are conserved in both content and position (19,31). Such mutations in regulatory regions are responsible for massive evolutionary rewiring of the regulatory networks.
In RPGs and 800 000 co-expressed gene pairs, regulatory specificity is generated by as many as 20–25 TRs. The distribution of TR binding sites and associations follow statistical patterns ranging from strictly preferential to highly random. A wide spectrum of stochastic protein–protein and protein–DNA interactions are promoted by an unusual abundance of intrinsically disordered domains in transcription factors (15). TRs like MYC and KAT2A with exceptionally large intrinsically disordered domains (15) may bind to a large variety of other regulators. Widely bound TRs such as MYC and NFκB recruit other TRs. These interactions are built and broken in a matter of seconds (80), further increasing probabilistic binding, which is the plausible cause for burst-like expression patterns (14). Stochastic TR binding may cause stochastic regulatory effects, including pauses and bursts of transcription. The binding of different TRs is considerably but not fully preferential and is affected by random effects such as the availability and Brownian motion of TRs in the nucleus (81).
Surprisingly, pluralistic and stochastic gene regulation can be reconciled with what most recent authors call master regulators. Masters can be defined as inducers of a cascade of regulatory events that guide the cell cycle, cellular differentiation and other biological processes (82). Note that this definition requires neither necessity nor rigorous sufficiency of the master for inducing a process or a phenotype. As more and more TRs are implicated in the governance of animal development, Chan and Kyba (83) pointed out that ‘the genome might have more masters than servants’. Hence the metaphor of masters taken from human societies may lose its relevance.
With the emergence of regulatory information, authors relaxed the concepts of master regulators. Originally, a master regulator was defined as ‘a gene which… should not be under the regulatory influence of any other gene’ (84). However, the ENCODE Project (85) demonstrated that even TR genes are bound by numerous other TRs. According to a somewhat later definition, master regulators are necessary and sufficient agents for producing a phenotype or differential gene expression (72). Necessity means that no other TR is sufficient and sufficiency means that no other TR is necessary. As the number of genes is strictly controlled in Metazoa (86), hundreds of nonfunctional TR genes would have been eliminated. Second, being known targets of signaling pathways, many of the implied ‘servants’ integrate and convey a wide variety of cellular information to improve regulatory decisions (87).
Such a ‘dictatorial’ concept may be overly strict and several proposed masters were not verified rigorously. Necessity can be validated using homozygous knockout mutants (18). For example, the transcription factor BCL11A is necessary for the developmental stage-specific downregulation of the γ-globin gene as shown using BCL11A−/− transgenic mice (88). However, its sufficiency remains unproven as DEAD and/or SIX6 may also be necessary for downregulation (88). Proving the sufficiency of individual candidates by overinducing the expression of their genes can be problematic (89). In an ideal overinduction experiment, no other specific regulator would bind to synthetic promoters and enhancers, and no cofactors would be associated with the candidate master. As such in vivo experiments are hardly feasible in higher eukaryotes given that co-binding specific regulators confound practical sufficiency tests. At far beyond physiological levels, MYC and MAX flood low-affinity DNA sites and outcompete repressors. This low-specificity upregulation of several thousand genes (90,91) is named as transcriptional amplification (66). Under such conditions, MYC and MAX act as strict sense master regulators.
Hierarchy may exist in the regulation of the transcriptional regulators themselves. This complex network problem requires additional studies. Necessary and sufficient masters including MyoD (92) and SCL (93) do exist even under physiological conditions. However, their number could be far lower than previously thought (83). Considerably random effects were found even in the action of classic masters including Bicoid, Hunchback, Caudal and Nanos, which orchestrate the segmentation of Drosophila embryos (94). Therefore, compared to deterministic approaches, thermodynamic models of multiple TRs predict the transcription of segmentation-related genes more accurately (95).
The concept of master regulators can be extended to sets of a few TRs (96), which we call ‘oligarchies’. To prevent the uncontrolled growth of oligarchies, we require that none of the individual oligarchs is sufficient to induce a phenotype but each of them is necessary. These criteria disqualify several previous claims for master regulators and oligarchies. In the fibroblast reprogramming example (9) mentioned in the Introduction, OCT4 and SOX2 are essential but insufficient for reprogramming and none of the four TRs, KLF4 and MYC (10) or NANOG and LIN28 (11) is necessary. Hence none of these six TRs is a strict sense master regulator.
According to a Scopus search, over 28 700 publications mention master regulators. Most authors use this metaphor solely to indicate the well-established differential importance of TRs (85). Calling the most important TRs as ‘masters’ may be somewhat inaccurate, but this does not conflict with stochastic and pluralistic regulation.
We recognize the limitations of our insight into the vast complexity of transcriptional regulation. As of August 2015, the ENCODE Project (5) mapped about three hundred of the ≈1700 DNA-associated proteins and a fraction of histone modifications in human cells (7). False negative observations and unknown distal enhancer regions (97) may lead to overlooking numerous regulator binding sites. To a lesser extent, false positives also present a concern. We have limited information about the differences in the stability, lifespan and regulatory effects of TR binding sites. Our stochastic and pluralistic model of gene regulation is biased toward highly expressed genes with specific transcriptional machinery. Another potential bias is that cRPGs are governed by a higher number of TRs including a larger percentage of general regulators than most medium-to-low-expression genes. ENCODE's selection of TRs, amplification bias in ChIP-Seq, and phantom peaks may further bias analyses including ours. However, using objective statistics, machine learning methods, evolutionary observations and gain-/loss-of-function mutants, we reduced interpretational and simplification bias.
In summary, the stochastic distribution of TR binding sites across the human genome, the viability of null mutants for most TRs and the evolutionary rewiring of the regulatory networks indicate the wide extent of stochastic regulation. TRs bind to DNA and associate with each other in partially random manner but with probabilistic preferences. Deterministic regulation cannot produce stochastic, burst-like transcription. Stochastic mechanisms have a major evolutionary advantage over rigid, deterministic systems. Positive Darwinian selection for increasingly adaptive regulators (98) and binding site patterns improves adaptation to new environments and elevate organismal complexity during evolution. Neither positive selection for mutants with improved fitness nor negative selection against less adaptive mutants was able to eliminate stochastic regulation. Also, degrading most of the ≈1700 human TRs to mere ‘servants’ would eliminate robustness against mutations, the large majority of regulatory repertoire, and hence the pool for evolutionary adaptation. The ≈800 000 co-regulated gene pairs and the cRPGs indicate that pluralistic and stochastic mechanisms are widespread in the human and likely other genomes. This does not contradict most of the ≈28 700 publications that discuss master regulators in some relaxed sense. We recommend evaluating both stochastic and more or less hierarchical regulation as well. Sophisticated and minimally biased interpretations of transcriptional regulation will guide us to understand the effects of regulatory mutations in the millions of human genomes soon to be sequenced (3) and to design therapeutic interventions.
AVAILABILITY
Key data are available in Supplementary Tables. All data have been stored in our MySQL relational database. The human part of the MySQL Database and its documentation are available at our web site: https://git.unl.edu/sladunga2/genereg/tree/master. Other data can be obtained upon request.
ACCESSION NUMBERS
Are available in Supplementary Tables S1 and S2.
Supplementary Material
Acknowledgments
We acknowledge support by Drs J. J. M. Riethoven, Bioinformatics Core Research Facility; K.M. Eskridge; D. Swanson, T. Harvill, A. Caprez, A. Guru and J. Thieltges from UNL's Holland Computing Center.
Authors’ contributions: Designed the study: I.L. Analyzed the data: I.L., M.F.C. and J.S. Wrote the manuscript: I.L., D.P.W. and E.N.S. All authors read and approved the final manuscript.
Footnotes
Present addresses:
Elisa N. Salas, Perrigo Animal Health, Omaha, NE 68138, USA.
Jiang Shu, Department of Computer Science and Engineering, University of Nebraska-Lincoln, NE 68588, USA.
Matyas F. Cserhati, Department of Genetics, Cell Biology & Anatomy, University of Nebraska Medical Center, Omaha, NE 68198, USA.
FUNDING
University of Nebraska-Lincoln's Institute of Agricultural and Natural Resources [I.L. and D.P.W.]; National Science Foundation [MCB-0952533 and EPSCoR-1004094 to D.P.W.]. Funding for open access charge: University of Nebraska-Lincoln's Institute of Agricultural and Natural Resources; NSF [MCB-0952533 and EPSCoR-1004094].
Conflict of interest statement. None declared.
REFERENCES
- 1.Montgomery S.B., Dermitzakis E.T. From expression QTLs to personalized transcriptomics. Nat. Rev. Genet. 2011;12:277–282. doi: 10.1038/nrg2969. [DOI] [PubMed] [Google Scholar]
- 2.Pickrell J.K. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am. J. Hum. Genet. 2014;94:559–573. doi: 10.1016/j.ajhg.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stephens Z.D., Lee S.Y., Faghri F., Campbell R.H., Zhai C., Efron M.J., Iyer R., Schatz M.C., Sinha S., Robinson G.E. Big Data: Astronomical or Genomical? PLoS Biol. 2015;13:e1002195. doi: 10.1371/journal.pbio.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cookson W., Liang L., Abecasis G., Moffatt M., Lathrop M. Mapping complex disease traits with global gene expression. Nat. Rev. Genet. 2009;10:184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.ENCODE Project Consortium. Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J., et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monod J., Jacob F. Teleonomic mechanisms in cellular metabolism, growth, and differentiation. Cold Spring Harb. Symp. Quant. Biol. 1961;26:389–401. doi: 10.1101/sqb.1961.026.01.048. [DOI] [PubMed] [Google Scholar]
- 7.Vaquerizas J.M., Kummerfeld S.K., Teichmann S.A., Luscombe N.M. A census of human transcription factors: function, expression and evolution. Nat. Rev. Genet. 2009;10:252–263. doi: 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- 8.Karlin S., Taylor H.M. A first course in stochastic processes. 2nd edn. NY: Academic Press; 1975. [Google Scholar]
- 9.Zhang B., Wolynes P.G. Stem cell differentiation as a many-body problem. Proc. Natl. Acad. Sci. U.S.A. 2014;111:10185–10190. doi: 10.1073/pnas.1408561111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 11.Yu J., Vodyanik M.A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J.L., Tian S., Nie J., Jonsdottir G.A., Ruotti V., Stewart R., et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 12.Jullien J., Pasque V., Halley-Stott R.P., Miyamoto K., Gurdon J.B. Mechanisms of nuclear reprogramming by eggs and oocytes: a deterministic process? Nat. Rev. Mol. Cell. Biol. 2011;12:453–459. doi: 10.1038/nrm3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elowitz M.B., Levine A.J., Siggia E.D., Swain P.S. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 14.Pennington K.L., Marr S.K., Chirn G.W., Marr M.T. 2nd. Holo-TFIID controls the magnitude of a transcription burst and fine-tuning of transcription. Proc. Natl. Acad. Sci. U.S.A. 2013;110:7678–7683. doi: 10.1073/pnas.1221712110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andresen C., Helander S., Lemak A., Fares C., Csizmok V., Carlsson J., Penn L.Z., Forman-Kay J.D., Arrowsmith C.H., Lundstrom P., et al. Transient structure and dynamics in the disordered c-Myc transactivation domain affect Bin1 binding. Nucleic Acids Res. 2012;40:6353–6366. doi: 10.1093/nar/gks263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neuert G., Munsky B., Tan R.Z., Teytelman L., Khammash M., van Oudenaarden A. Systematic identification of signal-activated stochastic gene regulation. Science. 2013;339:584–587. doi: 10.1126/science.1231456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bais A.S., Kaminski N., Benos P.V. Finding subtypes of transcription factor motif pairs with distinct regulatory roles. Nucleic Acids Res. 2011;39:e76. doi: 10.1093/nar/gkr205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White J.K., Gerdin A.K., Karp N.A., Ryder E., Buljan M., Bussell J.N., Salisbury J., Clare S., Ingham N.J., Podrini C., et al. Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell. 2013;154:452–464. doi: 10.1016/j.cell.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vierstra J., Rynes E., Sandstrom R., Zhang M., Canfield T., Hansen R.S., Stehling-Sun S., Sabo P.J., Byron R., Humbert R., et al. Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science. 2014;346:1007–1012. doi: 10.1126/science.1246426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sasai M., Wolynes P.G. Stochastic gene expression as a many-body problem. Proc. Natl. Acad. Sci. U.S.A. 2003;100:2374–2379. doi: 10.1073/pnas.2627987100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lillacci G., Khammash M. The signal within the noise: efficient inference of stochastic gene regulation models using fluorescence histograms and stochastic simulations. Bioinformatics. 2013;29:2311–2319. doi: 10.1093/bioinformatics/btt380. [DOI] [PubMed] [Google Scholar]
- 22.Lyons D.B., Lomvardas S. Repressive histone methylation: a case study in deterministic versus stochastic gene regulation. Biochim. Biophys. Acta. 2014;1839:1373–1384. doi: 10.1016/j.bbagrm.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 23.Nevozhay D., Adams R.M., Van Itallie E., Bennett M.R., Balazsi G. Mapping the environmental fitness landscape of a synthetic gene circuit. PLoS Comput. Biol. 2012;8:e1002480. doi: 10.1371/journal.pcbi.1002480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stavreva D.A., Varticovski L., Hager G.L. Complex dynamics of transcription regulation. Biochim. Biophys. Acta. 2012;1819:657–666. doi: 10.1016/j.bbagrm.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monteiro P.T., Dumas E., Besson B., Mateescu R., Page M., Freitas A.T., de Jong H. A service-oriented architecture for integrating the modeling and formal verification of genetic regulatory networks. BMC Bioinformatics. 2009;10:450. doi: 10.1186/1471-2105-10-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Jong H., Gouze J.L., Hernandez C., Page M., Sari T., Geiselmann J. Qualitative simulation of genetic regulatory networks using piecewise-linear models. Bull. Math. Biol. 2004;66:301–340. doi: 10.1016/j.bulm.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 27.Petryszak R., Burdett T., Fiorelli B., Fonseca N.A., Gonzalez-Porta M., Hastings E., Huber W., Jupp S., Keays M., Kryvych N., et al. Expression Atlas update–a database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic Acids Res. 2014;42:D926–D932. doi: 10.1093/nar/gkt1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple hypothesis testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 29.Efron B., Hastie T., Johnstone I., Tibshirani R. Least angle regression. Annu. Stat. 2004;32:407–451. [Google Scholar]
- 30.Hruz T., Laule O., Szabo G., Wessendorp F., Bleuler S., Oertle L., Widmayer P., Gruissem W., Zimmermann P. Genevestigator V3: a reference expression database for the meta-analysis of transcriptomes. Adv. Bioinform. 2008;2008:420747. doi: 10.1155/2008/420747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B.D., et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolesnikov N., Hastings E., Keays M., Melnichuk O., Tang Y.A., Williams E., Dylag M., Kurbatova N., Brandizi M., Burdett T., et al. ArrayExpress update-simplifying data submissions. Nucleic Acids Res. 2015;43:D1113–D1116. doi: 10.1093/nar/gku1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P., et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li B., Nierras C.R., Warner J.R. Transcriptional elements involved in the repression of ribosomal protein synthesis. Mol. Cell. Biol. 1999;19:5393–5404. doi: 10.1128/mcb.19.8.5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raftery J., Young A., Stanton L., Milne R., Cook A., Turner D., Davidson P. Clinical trial metadata: defining and extracting metadata on the design, conduct, results and costs of 125 randomised clinical trials funded by the National Institute for Health Research Health Technology Assessment programme. Health Technol. Assess. 2015;19:1–138. doi: 10.3310/hta19110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perry R.P., Meyuhas O. Translational control of ribosomal protein production in mammalian cells. Enzyme. 1990;44:83–92. doi: 10.1159/000468749. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Y., McIntosh K.B., Rudra D., Schawalder S., Shore D., Warner J.R. Fine-structure analysis of ribosomal protein gene transcription. Mol. Cell. Biol. 2006;26:4853–4862. doi: 10.1128/MCB.02367-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Warner J.R., McIntosh K.B. How common are extraribosomal functions of ribosomal proteins. Mol. Cell. 2009;34:3–11. doi: 10.1016/j.molcel.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y., Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell. 2009;16:369–377. doi: 10.1016/j.ccr.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brueggeman A.J., Gangadharaiah D.S., Cserhati M.F., Casero D., Weeks D.P., Ladunga I. Activation of the carbon concentrating mechanism by CO2 deprivation coincides with massive transcriptional restructuring in Chlamydomonas reinhardtii. Plant Cell. 2012;24:1860–1875. doi: 10.1105/tpc.111.093435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Worley J., Luo X., Capaldi A.P. Inositol pyrophosphates regulate cell growth and the environmental stress response by activating the HDAC Rpd3L. Cell Rep. 2013;3:1476–1482. doi: 10.1016/j.celrep.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suryamohan K., Halfon M.S. Identifying transcriptional cis-regulatory modules in animal genomes. Wiley Interdiscip. Rev. Dev. Biol. 2015;4:59–84. doi: 10.1002/wdev.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoskins B.E., Cramer C.H., Silvius D., Zou D., Raymond R.M., Orten D.J., Kimberling W.J., Smith R.J., Weil D., Petit C., et al. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am. J. Hum. Genet. 2007;80:800–804. doi: 10.1086/513322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarkar P.S., Appukuttan B., Han J., Ito Y., Ai C., Tsai W., Chai Y., Stout J.T., Reddy S. Heterozygous loss of Six5 in mice is sufficient to cause ocular cataracts. Nat. Genet. 2000;25:110–114. doi: 10.1038/75500. [DOI] [PubMed] [Google Scholar]
- 45.Kirby R.J., Hamilton G.M., Finnegan D.J., Johnson K.J., Jarman A.P. Drosophila homolog of the myotonic dystrophy-associated gene, SIX5, is required for muscle and gonad development. Curr. Biol. 2001;11:1044–1049. doi: 10.1016/s0960-9822(01)00319-0. [DOI] [PubMed] [Google Scholar]
- 46.Guerra-Rebollo M., Mateo F., Franke K., Huen M.S., Lopitz-Otsoa F., Rodriguez M.S., Plans V., Thomson T.M. Nucleolar exit of RNF8 and BRCA1 in response to DNA damage. Exp. Cell Res. 2012;318:2365–2376. doi: 10.1016/j.yexcr.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 47.Ciccia A., Elledge S.J. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosmarin A.G., Resendes K.K., Yang Z., McMillan J.N., Fleming S.L. GA-binding protein transcription factor: a review of GABP as an integrator of intracellular signaling and protein-protein interactions. Blood Cells Mol. Dis. 2004;32:143–154. doi: 10.1016/j.bcmd.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 49.Parry T.J., Theisen J.W., Hsu J.Y., Wang Y.L., Corcoran D.L., Eustice M., Ohler U., Kadonaga J.T. The TCT motif, a key component of an RNA polymerase II transcription system for the translational machinery. Genes Dev. 2010;24:2013–2018. doi: 10.1101/gad.1951110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weichhart T., Costantino G., Poglitsch M., Rosner M., Zeyda M., Stuhlmeier K.M., Kolbe T., Stulnig T.M., Horl W.H., Hengstschlager M., et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008;29:565–577. doi: 10.1016/j.immuni.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 51.Ohtsuki K., Kasahara K., Shirahige K., Kokubo T. Genome-wide localization analysis of a complete set of Tafs reveals a specific effect of the taf1 mutation on Taf2 occupancy and provides indirect evidence for different TFIID conformations at different promoters. Nucleic Acids Res. 2010;38:1805–1820. doi: 10.1093/nar/gkp1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krebs A.R., Karmodiya K., Lindahl-Allen M., Struhl K., Tora L. SAGA and ATAC histone acetyl transferase complexes regulate distinct sets of genes and ATAC defines a class of p300-independent enhancers. Mol. Cell. 2011;44:410–423. doi: 10.1016/j.molcel.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schuetz A., Bernstein G., Dong A., Antoshenko T., Wu H., Loppnau P., Bochkarev A., Plotnikov A.N. Crystal structure of a binary complex between human GCN5 histone acetyltransferase domain and acetyl coenzyme A. Proteins. 2007;68:403–407. doi: 10.1002/prot.21407. [DOI] [PubMed] [Google Scholar]
- 54.Patel J.H., Du Y., Ard P.G., Phillips C., Carella B., Chen C.J., Rakowski C., Chatterjee C., Lieberman P.M., Lane W.S., et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 2004;24:10826–10834. doi: 10.1128/MCB.24.24.10826-10834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hay N., Takimoto M., Bishop J.M. A FOS protein is present in a complex that binds a negative regulator of MYC. Genes Dev. 1989;3:293–303. doi: 10.1101/gad.3.3.293. [DOI] [PubMed] [Google Scholar]
- 56.Tu W.B., Helander S., Pilstal R., Hickman K.A., Lourenco C., Jurisica I., Raught B., Wallner B., Sunnerhagen M., Penn L.Z. Myc and its interactors take shape. Biochim. Biophys. Acta. 2014;1849:469–483. doi: 10.1016/j.bbagrm.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 57.Knoepfler P.S., Zhang X.Y., Cheng P.F., Gafken P.R., McMahon S.B., Eisenman R.N. Myc influences global chromatin structure. EMBO J. 2006;25:2723–2734. doi: 10.1038/sj.emboj.7601152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J., Sprung R., Pei J., Tan X., Kim S., Zhu H., Liu C.F., Grishin N.V., Zhao Y. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol. Cell. Proteomics. 2009;8:215–225. doi: 10.1074/mcp.M800187-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boyault C., Gilquin B., Zhang Y., Rybin V., Garman E., Meyer-Klaucke W., Matthias P., Muller C.W., Khochbin S. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 2006;25:3357–3366. doi: 10.1038/sj.emboj.7601210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miskiewicz K., Jose L.E., Yeshaw W.M., Valadas J.S., Swerts J., Munck S., Feiguin F., Dermaut B., Verstreken P. HDAC6 is a Bruchpilot deacetylase that facilitates neurotransmitter release. Cell Rep. 2014;8:94–102. doi: 10.1016/j.celrep.2014.05.051. [DOI] [PubMed] [Google Scholar]
- 61.Williams K.A., Zhang M., Xiang S., Hu C., Wu J.Y., Zhang S., Ryan M., Cox A.D., Der C.J., Fang B., et al. Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J. Biol. Chem. 2013;288:33156–33170. doi: 10.1074/jbc.M113.472506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pray-Grant M.G., Daniel J.A., Schieltz D., Yates J.R. 3rd, Grant P.A. Chd1 chromodomain links histone H3 methylation with SAGA- and SLIK-dependent acetylation. Nature. 2005;433:434–438. doi: 10.1038/nature03242. [DOI] [PubMed] [Google Scholar]
- 63.Suganuma T., Gutierrez J.L., Li B., Florens L., Swanson S.K., Washburn M.P., Abmayr S.M., Workman J.L. ATAC is a double histone acetyltransferase complex that stimulates nucleosome sliding. Nat. Struct. Mol. Biol. 2008;15:364–372. doi: 10.1038/nsmb.1397. [DOI] [PubMed] [Google Scholar]
- 64.Lee T.I., Young R.A. Transcription of eukaryotic protein-coding genes. Annu. Rev. Genet. 2000;34:77–137. doi: 10.1146/annurev.genet.34.1.77. [DOI] [PubMed] [Google Scholar]
- 65.Gilchrist D.A., Nechaev S., Lee C., Ghosh S.K., Collins J.B., Li L., Gilmour D.S., Adelman K. NELF-mediated stalling of Pol II can enhance gene expression by blocking promoter-proximal nucleosome assembly. Genes Dev. 2008;22:1921–1933. doi: 10.1101/gad.1643208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin C.Y., Loven J., Rahl P.B., Paranal R.M., Burge C.B., Bradner J.E., Lee T.I., Young R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nie Z., Hu G., Wei G., Cui K., Yamane A., Resch W., Wang R., Green D.R., Tessarollo L., Casellas R., et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151:68–79. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cesari F., Brecht S., Vintersten K., Vuong L.G., Hofmann M., Klingel K., Schnorr J.J., Arsenian S., Schild H., Herdegen T., et al. Mice deficient for the ETS transcription factor ELK-1 show normal immune responses and mildly impaired neuronal gene activation. Mol. Cell. Biol. 2004;24:294–305. doi: 10.1128/MCB.24.1.294-305.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murtas D., Maric D., De Giorgi V., Reinboth J., Worschech A., Fetsch P., Filie A., Ascierto M.L., Bedognetti D., Liu Q., et al. IRF-1 responsiveness to IFN-gamma predicts different cancer immune phenotypes. Br. J. Cancer. 2013;109:76–82. doi: 10.1038/bjc.2013.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barabasi A.-L., Oltvai Z.N. Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 71.Schmid S., Mordstein M., Kochs G., Garcia-Sastre A., tenOever B.R. Transcription factor redundancy ensures induction of the antiviral state. J. Biol. Chem. 2010;285:42013–42022. doi: 10.1074/jbc.M110.165936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Suzuki H., Forrest A.R., van Nimwegen E., Daub C.O., Balwierz P.J., Irvine K.M., Lassmann T., Ravasi T., Hasegawa Y., de Hoon M.J., et al. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat. Genet. 2009;41:553–562. doi: 10.1038/ng.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ramakrishnan V., White S.W. Ribosomal protein structures: insights into the architecture, machinery and evolution of the ribosome. Trends Biochem. Sci. 1998;23:208–212. doi: 10.1016/s0968-0004(98)01214-6. [DOI] [PubMed] [Google Scholar]
- 74.Zheng W., Zhao H., Mancera E., Steinmetz L.M., Snyder M. Genetic analysis of variation in transcription factor binding in yeast. Nature. 2010;464:1187–1191. doi: 10.1038/nature08934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mallick J., Whiteway M. The evolutionary rewiring of the ribosomal protein transcription pathway modifies the interaction of transcription factor heteromer Ifh1-Fhl1 (interacts with forkhead 1-forkhead-like 1) with the DNA-binding specificity element. J. Biol. Chem. 2013;288:17508–17519. doi: 10.1074/jbc.M112.436683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hsu P.P., Kang S.A., Rameseder J., Zhang Y., Ottina K.A., Lim D., Peterson T.R., Choi Y., Gray N.S., Yaffe M.B., et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332:1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu Y., Yoon S.O., Poulogiannis G., Yang Q., Ma X.M., Villen J., Kubica N., Hoffman G.R., Cantley L.C., Gygi S.P., et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332:1322–1326. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shah O.J., Wang Z., Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 2004;14:1650–1656. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 79.Carracedo A., Ma L., Teruya-Feldstein J., Rojo F., Salmena L., Alimonti A., Egia A., Sasaki A.T., Thomas G., Kozma S.C., et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hager G.L., McNally J.G., Misteli T. Transcription dynamics. Mol. Cell. 2009;35:741–753. doi: 10.1016/j.molcel.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lomholt M.A., van den Broek B., Kalisch S.M., Wuite G.J., Metzler R. Facilitated diffusion with DNA coiling. Proc. Natl. Acad. Sci. U.S.A. 2009;106:8204–8208. doi: 10.1073/pnas.0903293106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Herwig S., Strauss M. The retinoblastoma protein: a master regulator of cell cycle, differentiation and apoptosis. Eur. J. Biochem. 1997;246:581–601. doi: 10.1111/j.1432-1033.1997.t01-2-00581.x. [DOI] [PubMed] [Google Scholar]
- 83.Chan S.S., Kyba M. What is a Master Regulator? J. Stem Cell Res. Ther. 2013;3:e114. doi: 10.4172/2157-7633.1000e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ohno S. Major sex-determining genes. Monogr. Endocrinol. 1978;11:1–140. [PubMed] [Google Scholar]
- 85.Gerstein M.B., Kundaje A., Hariharan M., Landt S.G., Yan K.K., Cheng C., Mu X.J., Khurana E., Rozowsky J., Alexander R., et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Levine M., Tjian R. Transcription regulation and animal diversity. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- 87.Waltermann C., Klipp E. Signal integration in budding yeast. Biochem. Soc. Trans. 2010;38:1257–1264. doi: 10.1042/BST0381257. [DOI] [PubMed] [Google Scholar]
- 88.Yu Y., Wang J., Khaled W., Burke S., Li P., Chen X., Yang W., Jenkins N.A., Copeland N.G., Zhang S., et al. Bcl11a is essential for lymphoid development and negatively regulates p53. J. Exp. Med. 2012;209:2467–2483. doi: 10.1084/jem.20121846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zeigler R.D., Cohen B.A. Discrimination between thermodynamic models of cis-regulation using transcription factor occupancy data. Nucleic Acids Res. 2014;42:2224–2234. doi: 10.1093/nar/gkt1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rosenbauer F., Koschmieder S., Steidl U., Tenen D.G. Effect of transcription-factor concentrations on leukemic stem cells. Blood. 2005;106:1519–1524. doi: 10.1182/blood-2005-02-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sabo A., Amati B. Genome recognition by MYC. Cold Spring Harb. Perspect. Med. 2014;4 doi: 10.1101/cshperspect.a014191. pii: a014191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tapscott S.J., Davis R.L., Thayer M.J., Cheng P.F., Weintraub H., Lassar A.B. MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science. 1988;242:405–411. doi: 10.1126/science.3175662. [DOI] [PubMed] [Google Scholar]
- 93.Porcher C., Swat W., Rockwell K., Fujiwara Y., Alt F.W., Orkin S.H. The T cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic lineages. Cell. 1996;86:47–57. doi: 10.1016/s0092-8674(00)80076-8. [DOI] [PubMed] [Google Scholar]
- 94.Driever W., Nusslein-Volhard C. A gradient of bicoid protein in Drosophila embryos. Cell. 1988;54:83–93. doi: 10.1016/0092-8674(88)90182-1. [DOI] [PubMed] [Google Scholar]
- 95.Segal E., Raveh-Sadka T., Schroeder M., Unnerstall U., Gaul U. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature. 2008;451:535–540. doi: 10.1038/nature06496. [DOI] [PubMed] [Google Scholar]
- 96.Ulirsch J.C., Lacy J.N., An X., Mohandas N., Mikkelsen T.S., Sankaran V.G. Altered chromatin occupancy of master regulators underlies evolutionary divergence in the transcriptional landscape of erythroid differentiation. PLoS Genet. 2014;10:e1004890. doi: 10.1371/journal.pgen.1004890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gibcus J.H., Dekker J. The hierarchy of the 3D genome. Mol. Cell. 2013;49:773–782. doi: 10.1016/j.molcel.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mattick J.S., Taft R.J., Faulkner G.J. A global view of genomic information–moving beyond the gene and the master regulator. Trends Genet. 2010;26:21–28. doi: 10.1016/j.tig.2009.11.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.